
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpedient approach for trans-esterification of β-keto esters under solvent free conditions using silica supported boric acid (SiO2–H3BO3) as a recyclable catalyst†
Kamal Das and
Swapan Majumdar*
and
Swapan Majumdar*
Department of Chemistry, Tripura University, Suryamaninagar, 799 022, India. E-mail: smajumdar@tripurauniv.ac.in; Fax: +91-381-2374802; Tel: +91-381-237-9070
First published on 3rd August 2022
Abstract
A highly efficient trans-esterification of β-keto methyl/ethyl esters with primary, secondary, allylic, benzylic and chiral alcohols has been carried out in excellent yields under solvent-free conditions using silica supported boric acid as a heterogeneous catalyst. The surface morphology of the silica-boric acid catalyst (fresh and recycled) has been characterized by SEM and EDX techniques. This sustainable protocol resulted in a remarkable enhancement in the synthetic efficiency (87–95% yield) with high purity and eliminating the use of an environmentally toxic solvent. The work up procedure is very simple and the catalyst has been successfully recovered and recycled. The present methodology is also applicable for trans-esterification with chiral alcohols on a multi-gram scale without compromising the yield. Noteworthy features of this protocol are simple operational procedure, minimizing production of chemical waste, mild reaction conditions, easy preparation of the catalyst and its recyclability up to five cycles without any appreciable loss of catalytic activity.
Introduction
Trans-esterification is an important organic transformation where small esters can be converted to large esters using higher alcohols in the presence of acids, bases or other catalysts.1 The process has wide applications in both academic and industrial research. The poor solubility of most of the carboxylic acids in organic solvents and lower reactivity of alcohols limits the direct synthesis of esters using coupling agents or other catalysts.2 Since methyl or ethyl esters are commercially available they serve conveniently as starting materials for higher esters via the trans-esterification process.3 On the other hand molecules containing multiple reactive sites of electrophilic and nucleophilic nature are valuable and effective species for natural product synthesis, and synthesis of pharmaceutically active products and agro-chemicals. In this respect, β-keto esters are valuable essential synthons in organic synthesis due to the presence of two electrophilic carbonyls, two nucleophilic carbons and additional functional sites (in case of aromatic).4 Their significance arises from the ease with which bonds can be formed at all four carbon atoms in their structural unit because multiple reactive sites of different reactivities can react selectively under the different conditions.5 Therefore, this important class of building blocks have been widely used in the synthesis of a number of complex natural products6 including kermesic and carminic acid esters, paclitaxel, (±)-9-acetoxyfukinanolide, prunustatin A, serricornine, trichodiene and neoantimycin. β-Keto esters are ideal substrates for the synthesis of pyridazine,7 functionalized ketenimines,8 aminopyrazole derivatives and other heterocycles in a variety of traditional chemical processes.9 A representative structure of a β-keto ester with all electrophilic, nucleophilic and additional functionalization sites are shown in Fig. 1.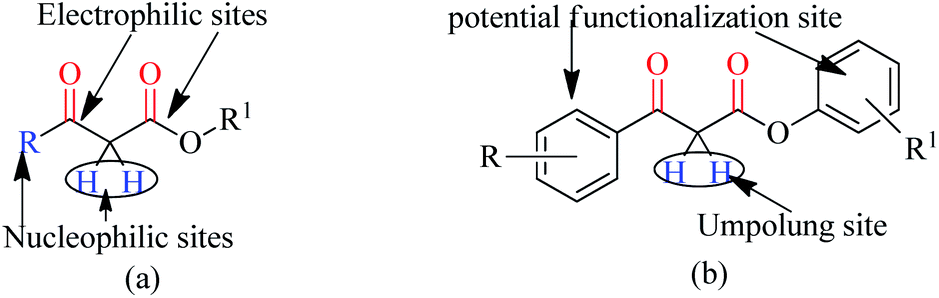 | ||
| Fig. 1 Presence of multiple reactive sites in a β-keto ester system. | ||
Due to the unstable nature of β-keto acid, the direct esterification using traditional reagents is not possible. Alternatively, it could be prepared by decomposition of reactive acylketene with various alcohols.10 In spite of high atom economic nature of this method, the corrosiveness and practical difficulties to generate acylketene limit its utility in organic synthesis. Thus, trans-esterification of simple β-keto esters is a convenient method for modify it to higher ester and widely used in different sphere of synthetic organic chemistry in both academic and industrial research including food and agrochemicals industries. Since trans-esterification is a reversible process,11 various acidic or basic catalysts were employed to make the process irreversible12 and in case of β-keto-esters the reaction proceeds via chelation control enol intermediate.13 Various protic acids, bases, Lewis acids, organic bases and bio-catalysis have been employed successfully in this trans-esterification process.14 Due to the sensitivity of β-keto-esters towards protic acids and bases, various transition, non-transition and lanthanide metal based strategies were extensively studied e.g. molybdenum–zirconium oxide (Mo–ZrO2),15 Niobium oxide (Nb2O5),16 [N,N′-ethylene bis(salicylideneaminato)]manganese(III) chloride,17 CoCl2,18 zinc dust,19 using combination of zinc and iodine(Zn/I2),20 S–SnO2 (ref. 21) and ceria-yttria22 based Lewis acid have been used. Boron based strategy using simple boric acid in xylene,23 B2O3–ZrO2,24 BF3·OEt2 (ref. 25) and aryl boronic acid26 for similar transformations are well documented. Organic bases like DMAP,27 2,6-lutidine28 and triethyl amine in toluene,29 and polyaniline-hydrochloride,30 N,N-diethylaminopropylated silica (NDEAP-SiO2)31 and hexamethylene-tetramine32 were also successfully used for trans-esterification of β-keto esters. Cordova and Janda reported lipase-catalysed trans-esterification of β-keto esters using Candida antarctica lipase B (CALB).33 Natural clays montmorillonite K-10,34 kaolinitic clay,12 H-β-zeolites,35 4 Å molecular sieves,36 N-methyl-2-pyrrolidone hydrogen sulfate,37 N-bromosuccinimide,38 and p-toluene sulfonic acid (p-TSA)39 were also reported. Although, these methods has their own advantages but still many of them suffers from serious shortcomings such as use of expensive reagents, use of high boiling solvents, longer reaction times, tedious workup procedures, toxic waste generation, difficulty in separation and recovery of the catalyst. Hence, it is of great practical importance to develop a more efficient and environmentally benign method that avoids or minimizes all of these shortcomings in cost effective manner. In recent years, the use of reusable heterogeneous catalysts has received considerable attention in organic synthesis as a result of their economic and environmental benefits.40a–c The development of selective and reusable solid support catalysts for organic reactions has been a very active area of research because of their reusability by filtration that not only simplify purification processes but also help in preventing the discharge of toxic reaction residues into the environment. During the last few decades, silica-supported catalysts have achieved considerable attention41 among the synthetic and material sciences community. Silica supported H2SO4, HClO4, PPA, HBF4, NaHSO4, P2O5, FeCl3, H3BO3 etc. were applied in different occasions. Among them silica-FeCl3 and SiO2–H3BO3 are mild in nature while more effective in some specific cases. As a part of our recent activity on the development of C–C and C–N bond forming reaction we are in need of large quantity of β-keto esters. Silica-supported boric acid (SiO2–H3BO3)42 using silica gel of 60–120 mesh has emerged a promising heterogeneous solid acid catalyst in some organic transformations as a cheap, thermally stable and environmentally benign with many other advantages. To this end and in continuation of our ongoing efforts endowed with the finding of new synthetic protocols under the principles of green chemistry, an effective and environment friendly method has been developed for trans-esterification of β-keto esters under solvent free condition using silica (230–400 mesh)-supported boric acid (SiO2–H3BO3) as recyclable heterogeneous catalyst (Scheme 1). The present protocol is applicable for wide ranges of alcohols and β-keto-esters of acyclic and cyclic systems and overcomes the limitations of reported23 protocols of unsupported catalyst. Another important aspect of this new protocol is the synthesis of chiral β-keto-esters in multi-gram scale using various chiral alcohols through trans-esterification of methyl acetoacetate.
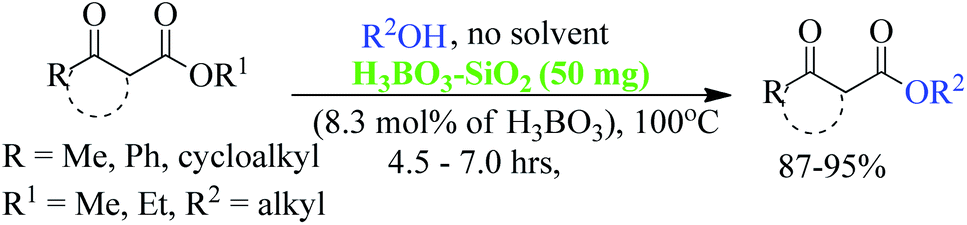 | ||
| Scheme 1 General strategy for solvent free trans esterification of β-keto esters using SiO2–H3BO3 as activator. | ||
Results and discussions
Silica-supported boric acid was prepared with slide modification of the process reported by V. Kumar and co-workers42 and Parveen et al.43 for different occasion. They used silica-gel 60–120 mesh size for the preparation of supported materials. We have prepared silica-supported boric acid using both 230–400 mesh and 60–120 mesh size as we expected that due to the smaller particle size in the case of former it could provide better efficiency than the supported materials prepared using silica-gel grade 60–120. Typically, boric acid (3.0 g) was placed in a 500 mL clean beaker and distilled water (60 mL) was added before the mixture was heated to 60–80 °C for a few minutes. After dissolving the boric acid, 27 g of 230–400 mesh/60–120 mesh silica-gel was added with constant stirring for 5 hours. Water was evaporated under vacuum and the residue was stirred for 6–7 hours at 100 °C to produce free flowing powder of silica–boric acid. Similar procedure was followed for silica grade 60–120 mesh as well. The catalysts were stored at 4 °C for longer shelf life. These supported materials were characterized by SEM and EDX studies. SEM micrographs of the silica–boric acid (60–120 mesh) and silica–boric acid (230–400 mesh) (Fig. 2[A] and [B]) showed that the particles were random size and shape and well dispersed. An EDX spectrum of the catalysts confirmed the presence of Si, B and O elements that suggesting the formation of silica–boric acid catalytic system (ESI).† | ||
| Fig. 2 Characterization of the SiO2–boric acid catalyst: [A] and [B] SEM images of freshly prepared silica–boric acid 60–120 and 230–400 respectively; [C], SEM images of recycled silica (230–400 mesh)–boric acid. | ||
To find out the best condition for trans-esterification of β-keto ester, methyl acetoacetate and benzyl alcohol were selected as a model partner. Initially, a mixture of both substrates was stirred with SiO2–H3BO3 (20 mg, 10% H3BO3) in toluene (2 mL) for 12 h at room temperature but it did not yield any product (Table 1, entry 1). Screening with other solvents like THF, EtOAc, EtOH and CH3CN under similar condition were also unsuccessful (Table 1, entries 2–5). Increasing the temperature of the reaction mixture in toluene to 60 °C leads to 35% yield of benzyl acetoacetate (3a) after 8 h (Table 1, entry 6). Further increase of temperature to 100 °C only 45% yield of 3a was detected after 5 h (Table 1, entry 7). Encouraged by this initial results we decided to increase the amount of catalyst. We were very pleased to observed that in the presence of 50 mg of SiO2–H3BO3 the reaction produced 91% of desired product (3a) after 4.5 h of heating at 100 °C while using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 ratios of methyl acetoacetate and benzyl alcohol results further increased the yield of 3a to 95% (Table 1, entries 8–9).
:1.1 ratios of methyl acetoacetate and benzyl alcohol results further increased the yield of 3a to 95% (Table 1, entries 8–9).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Benzyl alcohol | SiO2–H3BO3 | Temp. (°C) | Solvent | Time (h) | Yielda (%) |
| a Yields refer to isolated yield.b SiO2 (60–120 mesh)–H3BO3 was used. | ||||||
| 1 | 1.0 mmol | 20 mg | rt | Toluene | 12.0 | 0 |
| 2 | 1.0 mmol | 20 mg | rt | THF | 12.0 | 0 |
| 3 | 1.0 mmol | 20 mg | rt | EA | 12.0 | 0 |
| 4 | 1.0 mmol | 20 mg | rt | EtOH | 12.0 | 0 |
| 5 | 1.0 mmol | 20 mg | rt | ACN | 12.0 | 0 |
| 6 | 1.0 mmol | 20 mg | 60 | Toluene | 8.0 | 35 |
| 7 | 1.0 mmol | 20 mg | 100 | Toluene | 5.0 | 45 |
| 8 | 1.0 mmol | 50 mg | 100 | Toluene | 4.5 | 91 |
| 9 | 1.1 mmol | 50 mg | 100 | Toluene | 4.5 | 95 |
| 10 | 1.1 mmol | 50 mg | 100 | No | 4.5 | 95 |
| 11 | 1.1 mmol | 100 mg | 100 | Toluene | 4.5 | 95 |
| 12 | 1.1 mmol | No | 100 | No | 5.0 | 0 |
| 13 | 1.1 mmol | SiO2 | 100 | No | 5.0 | 45 |
| 14 | 1.1 mmol | H3BO3 | 100 | No | 5.0 | 69 |
| 15 | 1.1 mmol | 50 mg | 100 | No | 5.0 | 87b |
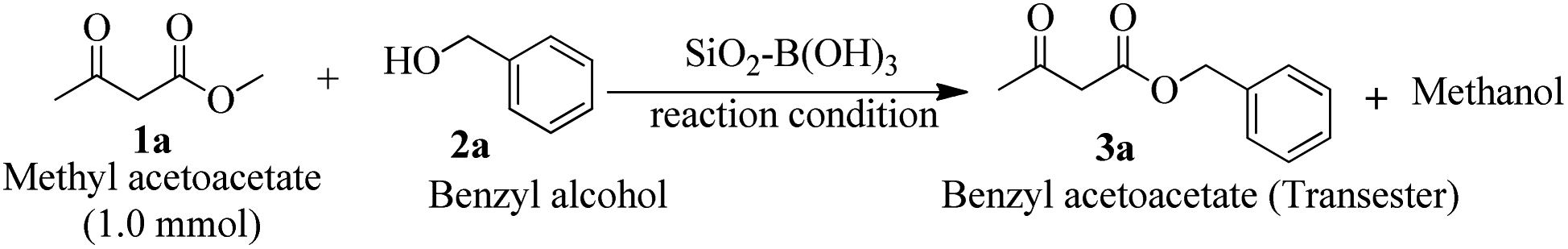
The results of entries 8–9 are very interesting because free boric acid mediated similar transformation23 in xylene produced only 75% yield of 3a after 5 h. This primary result prompted us to investigate the reaction under solvent free condition to save organic volatile solvent and to save our environment. Accordingly, we have conducted the study under solvent free medium by keeping all other condition unchanged. It is worthwhile to mention that the same level of efficiency was found under solvent free condition too (Table 1, entry 10). Increasing the amount of SiO2–H3BO3 from 50 mg to 100 mg yield of 3a was not improved (Table 1, entry 11). Essentiality of the catalyst SiO2–H3BO3 was further confirmed by the cross verifications under solvent free conditions (i) without catalyst (ii) using only silica-gel grade 230–400 mesh and (iii) only H3BO3 (8.3 mol%). Reaction was not proceeds at all under catalyst free condition (Table 1 entry 12), however, other two conditions produced 3a in 45% and 69% yield respectively (Table 1, entries 13–14). Supported materials prepared by using silica of 60–120 mesh produced 87% of 3a under similar condition. Therefore from the results of investigation as assembled in Table 1, it can be concluded that entry 10 is the best condition for of trans-esterification of methyl acetoacetate with benzyl alcohol. Based on the yield of the trans-ester and the catalyst used the turnover number (TON) is roughly estimated to 11585. The high turnover number indicated the high efficiency of the catalytic process.
Work up procedure is very simple and no aqueous work up is required. After completion of reaction (TLC), the mixture was diluted with either diethyl ether or ethyl acetate and filtered. Residue was washed several times and combined filtrate was concentrated under reduced pressure. The product is essentially pure but for analytical sample, it was purified by column chromatography. The identity of 3a was judged by its 1H and 13C NMR; however around 3% of enol content of 3a was seen in 1H NMR recorded in CDCl3.
In order to explore the scope of the present methodology further, we studied the trans-esterification of various β-keto-esters under standardized condition (Table 1, entry 10). It can be seen from the results assembled in Fig. 3, our protocol is general as the structurally diverse β-keto-esters such as open chain (set 1 and 2), aromatic (set 3) and cyclic (set 4) underwent trans-esterification smoothly with wide range of alcohols like primary, secondary, allylic and benzylic alcohols. Methyl acetoacetate (MAA) underwent facile trans-esterification (set 1) with benzylic alcohols containing electron-donating methoxy and methyl substituents, electron-withdrawing chloro and nitro substituents in the aromatic ring; naphthylic alcohol, homobenzylic; long chain alcohols (octyl, cetyl, alkenyl), phthaloyl ethanol and very reactive cinnamyl alcohol with excellent yields (87–95%) of the corresponding trans-esters 3b–l. Secondary alcohols – cyclohexanol, 1-phenyl ethanol; menthol and 1,2:5,6-diisopropylidene glucose also produced trans-esters 3m–p in excellent yield. Our results indicated that the structure of alcohol has no or very little effect on the present trans-esterification process as very marginal differences were noticed between the yield of the reaction and time required in each case. Similarly, ethyl acetoacetate (EAA) underwent trans-esterification (set 2) with benzyl alcohol, β-phenyl ethanol and 9-decenol produced 3a, 3g and 3j in 95% (95% from MAA), 89% (90% from MAA) and 94% (95% from MAA) isolated yield respectively. Under similar condition ethyl benzoyl acetate was also trans-esterified (set 3) with 4-nitro benzyl alcohol, 9-decenol, menthol and 1,2:5,6-diisopropylidene glucose to the higher ester 4a (90%), 4b (91%), 4c (92%) and 4d (89%) respectively. In contrast to the reported method, for instance tin based21 reagent failed to react with aromatic β-keto-esters, the present methodology is effective for the trans-esterification of aromatic β-keto-ester giving excellent yield of the product. Therefore, this can be considered as significant advantages of our protocol over the existing methods. The present methodology is also applicable to cyclic β-keto-esters e.g. methyl 2-oxo-cyclopentane carboxylate underwent trans-esterification with benzyl alcohol, homobenzylic alcohol and cinnamyl alcohol which yielded the corresponding esters 5a–c in excellent yield (set 4). This trans-esterification process is irreversible and applicable to the β-keto esters having small alkyl group such as methyl or ethyl. The irreversibility of the process was evident from the fact that when β-keto-ester 3j was exposed with methanol by keeping other condition unchanged but no trans-esterified product was detected. Another interesting feature of the present methodology is that the process is specific only for β-keto esters. Normal esters (methyl benzoate) and α-keto ester (methyl pyruvate) failed to produce trans-esterified product. The difference of reactivity of β-keto esters with other esters can be attributed by the fact that under the catalysis of boric-acid-SiO2, β-keto ester or its enolized form activated boronate complex which in turn facilitate nucleophilic attack by alcohol as proposed by Kondaiah et al.23 for unsupported catalyst. It seems that nucleophilic attack to the activated six membered boronate is crucial for the success of trans-esterification process. To examine this, we choosed phenol, substituted phenol, thiophenol and tert-butyl alcohol for our study. However, they did not responded trans-esterification when they were allowed to react with methyl acetoacetate due to less nucleophilic character of phenols/thiophenol and steric hindrance of tert-butyl alcohol. These results clearly indicated that the reaction proceed through six membered boronate intermediate not acylketene as proposed by Campbell and Lawrie.10 Based on the above experimental evidences we proposed a six-membered transition state to explain the mechanism of trans-esterification reaction for β-keto esters catalyzed by SiO2–boric acid. Boric acid which is adsorbed on the silica surface become more activated half ester of boronic acid ([A]) that react with enol form of β-keto ester to generate intermediate [B]. Expulsion of small alkoxy group through ring closing of [B] produced six membered cyclic boronate intermediate [C]. Nucleophilic attack by more reactive higher alcohol to [C] produced another intermediate [D] followed by release of product and re-generate the active catalyst for next cycle (Fig. 4).
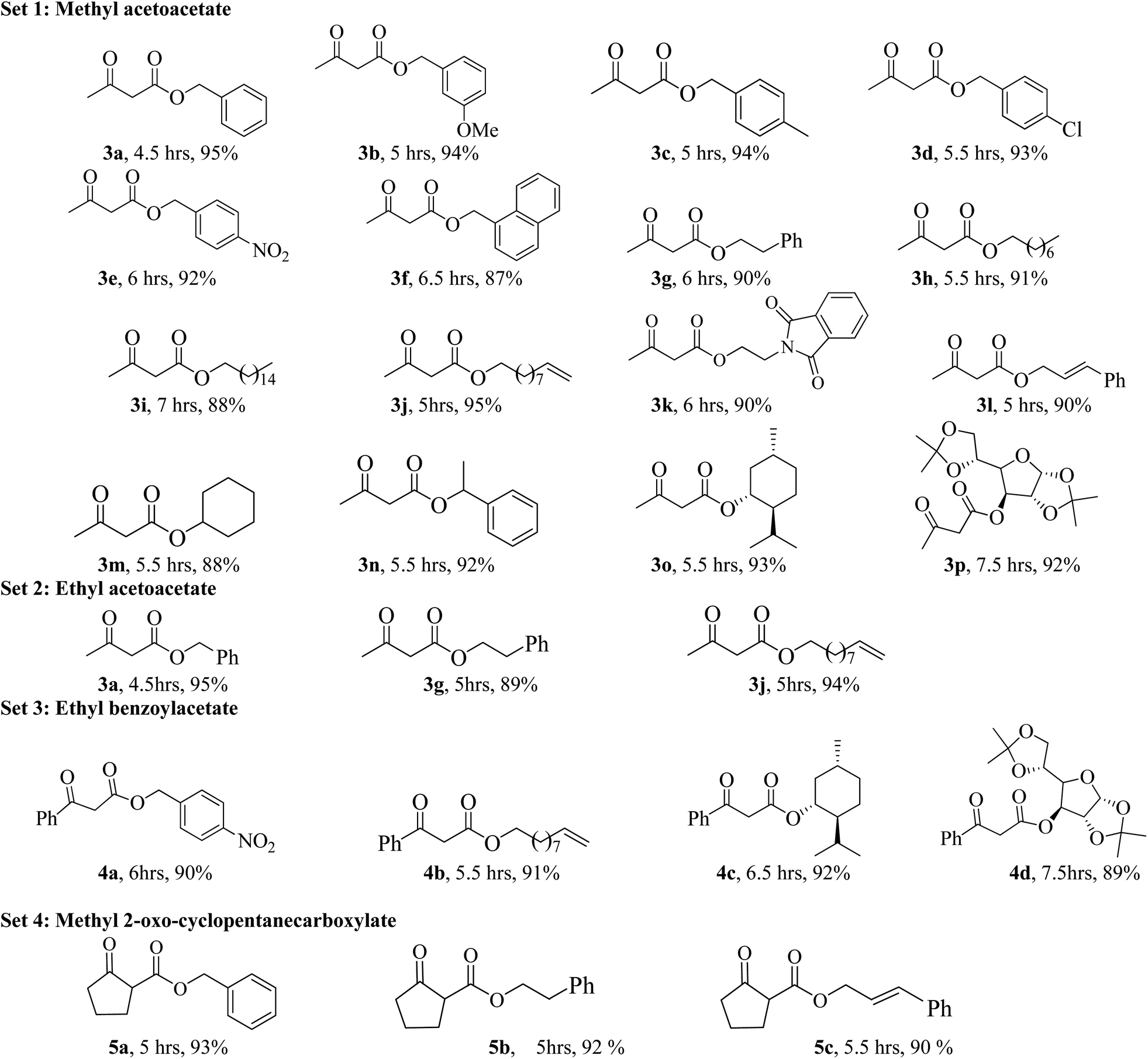 | ||
| Fig. 3 Trans-esterification of various β-keto esters with structurally diverse alcohols. | ||
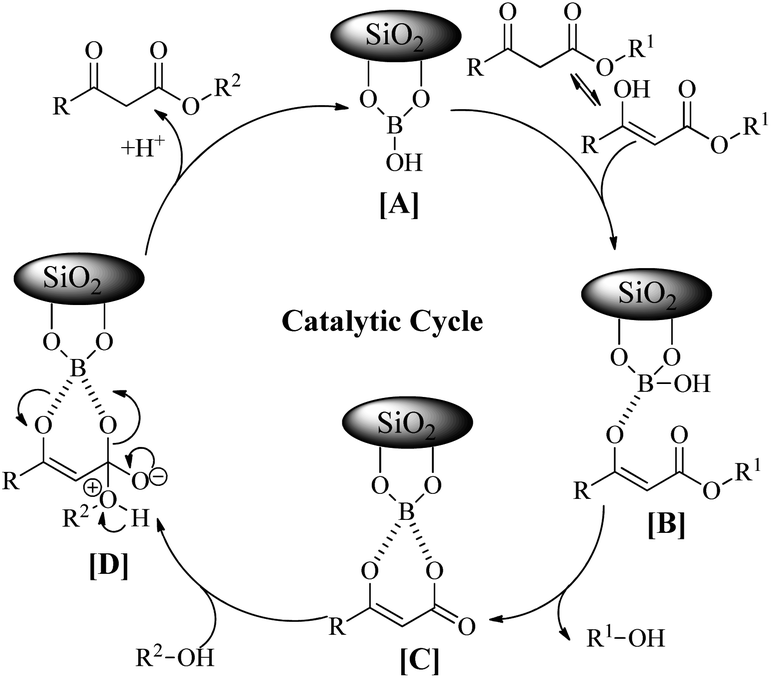 | ||
| Fig. 4 Proposed mechanism of trans-esterification of β-keto esters. | ||
Apart from the study of solvent free reaction, reusability of catalyst is another important credential of green synthesis. So, the reusability of the silica–boric acid catalyst was investigated for present study considering the trans-esterification of methyl acetoacetate with 9-dcenol as model reaction. After completion of reaction diethyl ether was added to the mixture and supported materials was collected by filtration. Yield of the reaction was determined after purification of the product (3j) from ethereal part using silica-gel column. Catalyst was dried and used for subsequent run. Our results indicated that the catalyst (SiO2–H3BO3) can be recycled up to five runs without significant loss its catalytic activity that is reflected in the isolated yield of the product (Fig. 5). We assuming that slight decreasing trends in the yield of the product probably due to the leaching of boric acid from the surface of silica during handling. SEM image of recycled silica–H3BO3 was captured to check any changes occurred on the surface of the catalyst after five cycles. However, more organized morphology has been observed on the surface of the catalyst as shown in Fig. 2[C].
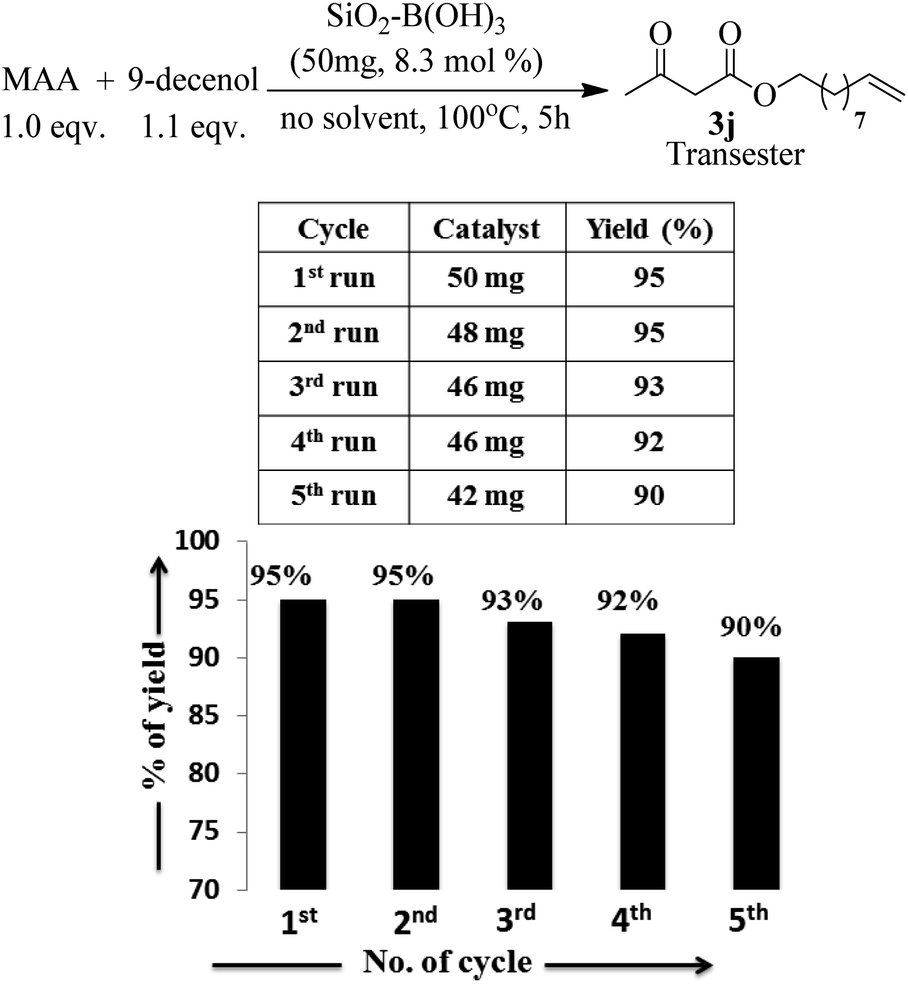 | ||
| Fig. 5 Recyclability test of the catalyst. | ||
Finally, we have demonstrated the scale-up of present methodology towards the trans-esterification of methyl acetoacetate and ethyl benzoyl acetate with chiral alcohol such as available (−) menthol and 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose under the catalysis of silica supported boric acid. Our study indicated that the scale up process (17 mmol scale) required little longer reaction time and produced slightly lesser yield as compared to the 1 mmol scale reaction. For instance, methyl acetoacetate underwent trans-esterification with (−) menthol and di-O-isopropylidene glucose produced 90% and 88% trans-esterified products (3o and 3p) in scale up process whereas in 1 mmol scale the yields are 93% and 92% respectively. Similarly, ethyl benzoyl acetate also produced chiral β-keto esters 4c and 4d in 88% and 85% yield respectively in 17 mmol scale but small scale reaction the yields are 92% and 89% respectively. The results are summarized in Scheme 2.
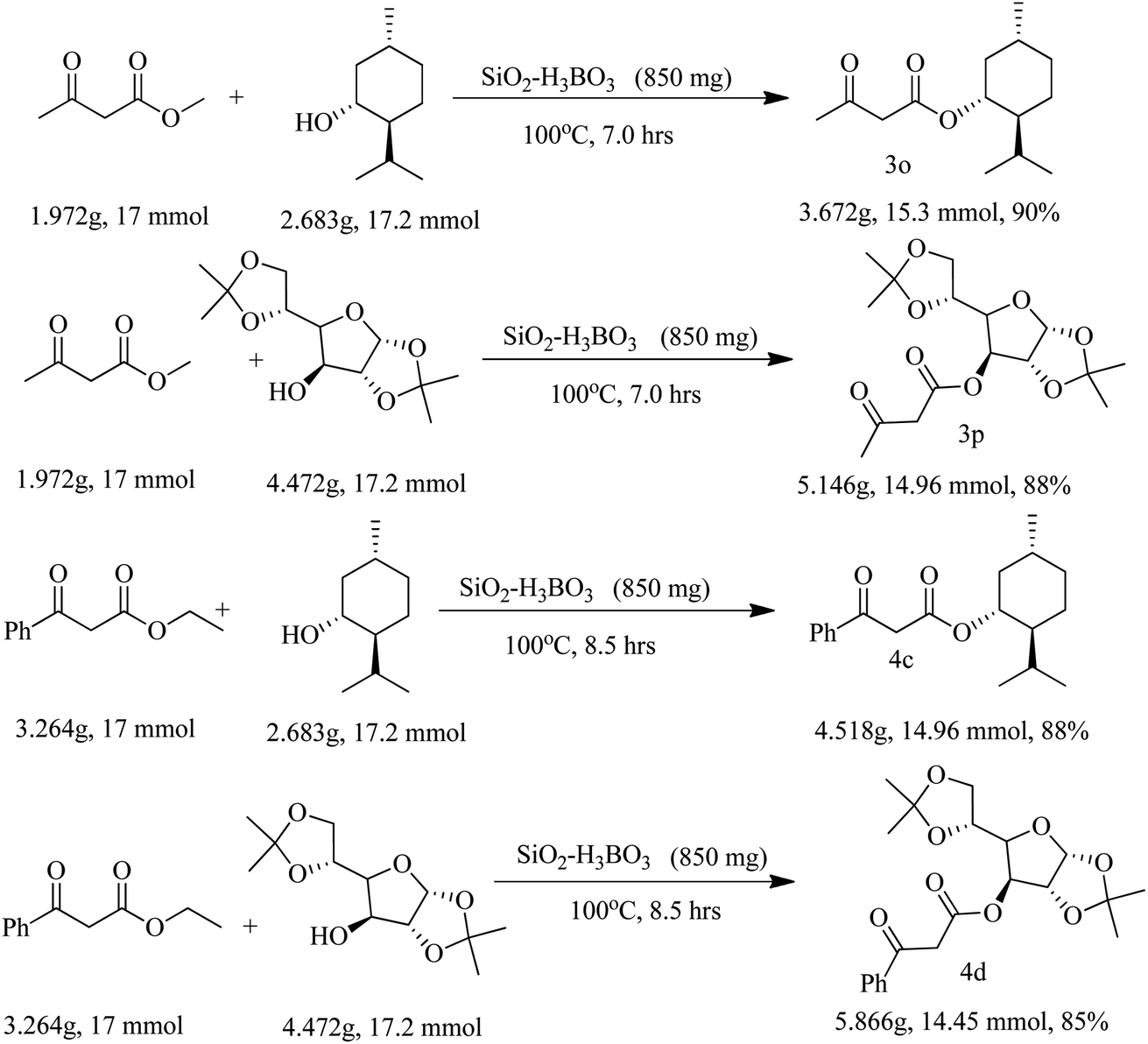 | ||
| Scheme 2 Multi-gram scale trans-esterification to chiral β-keto-esters. | ||
Conclusion
In the present study we have developed a novel route for trans-esterification of methyl/ethyl β-keto esters to wide verities of β-keto-esters using an environmentally friendly and recyclable SiO2–H3BO3 catalyst under solvent free conditions. This protocol offers several advantages over other existing synthetic methodologies in terms of yield, reaction times, operational procedures and environment concern. This cost effective procedure possesses diverse applicability and is compatible to a range of functional groups and equally effective in multi-gram scale. Our results reveals that supported catalyst prepared by using silica-gel 230–400 mesh is more effective than that of known SiO2 (60–120 mesh)–H3BO3 catalyst.Experimental section
All reactions were monitored by thin-layer chromatography carried out on silica coated aluminium plates using UV-light and iodine or Lieberman–Burchard reagent for visualization. Unless otherwise noted, all utilizing reagents were used exactly as received and solvents were freshly distilled before use. Column chromatography was performed on silica gel (60–120 mesh and 100–200) using n-hexane and ethyl acetate as eluent. Evaporation of solvents was conducted under reduced pressure at temperatures less than 50 °C. FTIR spectra (Bruker Alpha FTIR) were recorded in neat. SEM image are recoded on model-Sigma 300 (Carl Zeiss). 1H NMR and 13C NMR spectra (Bruker Ascend™ 400 MHz) were recorded in CDCl3 solvent. Chemical shifts δ and coupling constants J are given in ppm (parts per million) and Hz (hertz) respectively. Chemical shifts are reported relative to residual solvent as an internal standard for 1H and 13C (CDCl3: δ 7.28 ppm for 1H and 77.04 ppm for 13C). Mass spectra (HRMS) were obtained from XEVO G2-XS QTOF (waters) using 70 eV in positive ion mode.General procedure for trans-esterification reaction
A mixture of β-keto-esters (1 mmol), alcohols (1.1 mmol) and silica supported boric acid (SiO2–H3BO3) (50 mg, 8.3 mol%), were charged into a clean reaction vessel equipped with a magnetic stir bar and heated for 5–7 hours at 100 °C temperature. After completion of reaction (TLC), the mixture was cooled, diluted with diethyl ether or ethyl acetate and filtered to remove the catalyst and the crude product was purified using column chromatography with silica gel (100–200 mesh) EtOAc/hexane (1:9, 1:4). Melting point, FTIR data, 1H NMR, 13C NMR and HRMS analyses were used to determine the products identities.
Benzyl 3-oxobutanoate (3a)44
Yield: 95%, colourless oil; IR (neat) νmax 1716, 1261, 1146, 1025, 744 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.40–7.36 (m, 5H), 5.20 (s, 2H), 3.53 (s, 2H), 2.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.4, 167.0, 135.3, 128.6, 128.5, 128.4, 128.1, 67.2, 50.1, 30.2.3-Methoxybenzyl 3-oxobutanoate (3b)44
Yield: 94%, colourless oil; IR (neat) νmax 2951, 1717, 1596, 1453, 1262, 1148, 1036, 782 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.30 (d, J = 8.0 Hz, 1H), 6.97–6.89 (m, 3H), 5.18 (s, 2H), 3.84 (s, 3H), 3.53 (s, 2H), 2.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.4, 166.9, 159.8, 136.8, 129.7, 120.5, 114.1, 113.7, 67.0, 55.3, 50.1, 30.2.4-Methylbenzyl 3-oxobutanoate (3c)45
Yield: 94%, colourless oil; IR (neat) νmax 1718, 1643, 1516, 1312, 1262, 1146, 1026, 966 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 1.6 Hz, 2H), 7.20 (d, J = 8 Hz, 2H), 5.16 (s, 2H), 3.50 (s, 2H), 2.38 (s, 3H), 2.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.5, 167.0, 138.4, 132.3, 129.3, 128.6, 128.3, 89.7, 67.1, 65.7, 50.1, 30.2, 21.2.4-Chlorobenzyl 3-oxobutanoate (3d)26
Yield: 93%, colourless oil; IR (neat) νmax 1717, 1492, 1410, 1363, 1261, 1145, 1091, 1013, 805 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.37–7.28 (m, 4H), 5.15 (s, 2H), 3.52 (s, 2H), 2.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.3, 166.8, 134.4, 133.8, 129.5, 128.8, 89.5, 66.3, 64.8, 50.0, 30.2.4-Nitrobenzyl 3-oxobutanoate (3e)46
Yield: 92%, reddish oil; IR (neat) νmax 1714, 1606, 1519, 1411, 1345, 1257, 1143, 1036, 847 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.25 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.8 Hz, 2H), 5.29 (s, 2H), 3.59 (s, 2H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.0, 176.8, 166.6, 147.8, 142.2, 128.5, 128.2, 123.9, 89.2, 65.5, 64.1, 49.8, 30.3, 21.3.Naphthalen-1-ylmethyl 3-oxobutanoate (3f)
Yield: 87% colourless oil; IR (neat) νmax 1717, 1643, 1511, 1410, 1359, 1309, 1250, 1145, 962, 861, 786 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.0 Hz, 1H), 7.90 (t, J = 8.8 Hz, 2H), 7.62–7.54 (m, 3H), 7.50–7.28 (m, 1H), 5.67 (s, 2H), 3.52 (s, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.4, 176.0, 172.4, 167.1, 133.7, 131.6, 130.8, 129.6, 129.3, 128.8, 127.9, 127.4, 126.7, 126.6, 126.1, 126.0, 125.3, 123.5, 89.7, 65.5, 64.0, 50.1, 30.2, 21.3; HRMS calcd for C15H14O3 + Na (M + Na) 265.0841; found: 265.0840.Phenethyl 3-oxobutanoate (3g)47
Yield: 90%, colourless oil; IR (neat) νmax 1717, 1644, 1413, 1313, 1245, 1148, 1031, 744 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35–7.23 (m, 5H), 4.41–4.35 (m, 2H), 3.45 (s, 2H), 2.99 (t, J = 7.2 Hz, 2H), 2.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.6, 167.1, 137.4, 128.9, 128.6, 128.5, 126.7, 65.8, 64.5, 50.1, 34.9, 30.1.Octyl 3-oxobutanoate (3h)48
Yield: 91% colourless oil; IR (neat) νmax 2926, 2858, 1720, 1643, 1459, 1361, 1313, 1237, 1150, 1034, 728 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.15 (t, J = 6.8 Hz, 2H), 3.46 (s, 2H), 2.29 (s, 3H), 1.69–1.62 (m, 2H), 1.29 (s, 6H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.7, 167.2, 89.8, 65.6, 64.1, 50.2, 31.8, 30.1, 29.2, 28.7, 28.5, 25.9, 22.6, 21.2, 14.1.Hexadecyl 3-oxobutanoate (3i)49
Yield: 88%, colourless oil; IR (neat) νmax 2916, 2848, 1739, 1707, 1465, 1411, 1368, 1315, 1260, 1153, 1153 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.15 (t, J = 6.8 Hz, 2H), 3.47 (s, 2H), 2.30 (s, 3H), 1.66–1.62 (m, 7H), 1.27 (s, 36H), 0.90 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.6, 167.2, 89.8, 65.6, 64.1, 50.2, 31.9, 30.9, 30.1, 29.7, 29.7, 29.6, 29.5, 29.4, 29.2, 28.7, 28.5, 25.9, 25.8, 22.7, 21.2, 14.1.Dec-9-en-1-yl 3-oxobutanoate (3j)47
Yield: 95%, colourless oil; IR (neat) νmax 2926, 2855, 1722, 1641, 1312, 1237, 1151, 1035, 993, 909, 727 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.88–5.78 (m, 1H), 5.03–4.94 (m, 2H), 4.15 (t, J = 6.4 Hz, 2H), 3.47 (s, 2H), 2.29 (s, 3H), 2.08–2.02 (m, 2H), 1.66 (t, J = 7.2 Hz, 3H), 1.32 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 200.6, 167.2, 139.2, 129.1, 114.2, 89.8, 65.6, 64.1, 50.2, 33.8, 30.1, 29.3, 29.1, 29.0, 28.8, 28.7, 28.5, 25.9, 25.8, 21.2.2-(1,3-Dioxoisoindolin-2-yl)ethyl 3-oxobutanoate (3k)50
Yield: 90%, white solid, mp 120 °C [lit = 88–89 °C]; IR (neat) νmax 1701, 1391, 1316, 1145, 997, 795, 714 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 2.4 Hz, 2H), 7.75 (t, J = 2.4 Hz, 2H), 4.39 (t, J = 4.8 Hz, 2H), 3.98 (t, J = 5.2 Hz, 2H), 3.44 (s, 2H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.4, 168.1, 167.0, 134.2, 131.9, 123.4, 89.4, 62.5, 45.7, 37.1, 30.2, 21.3.Cinnamyl 3-oxobutanoate (3l)26
Yield: 90% colourless oil; IR (neat) νmax 1716, 1257, 1146, 962, 741 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.42–7.28 (m, 5H), 6.67 (d, J = 16.0 Hz, 1H), 6.34–6.23 (m, 1H), 4.82 (d, J = 6.8 Hz, 2H), 3.53 (s, 2H), 2.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.5, 166.9, 136.0, 134.9, 128.7, 128.3, 126.7, 123.1, 122.4, 89.6, 66.0, 50.0, 30.2.Cyclohexyl 3-oxobutanoate (3m)46
Yield: 88%, colourless oil; IR (neat) νmax 2935, 2860, 1716, 1643, 1448, 1412, 1241, 1151, 1023, 961, 907, 803 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.84 (m, 1H), 3.43 (s, 2H), 2.27 (s, 3H), 1.88–1.85 (m, 2H), 1.78–1.72 (m, 2H), 1.56–1.47 (m, 1H), 1.45–1.27 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 200.8, 166.6, 90.1, 73.8, 72.2, 31.6, 31.4, 25.2, 23.7, 21.1.1-Phenylethyl 3-oxobutanoate (3n)51
Yield: 92%, colourless oil; IR (neat) νmax 2982, 2933, 1717, 1644, 1495, 1448, 1411, 1358, 1252, 1150, 1061, 1026, 945, 858, 803 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.28 (m, 5H), 5.99 (q, J = 6.4 Hz, 1H), 3.49 (s, 2H), 2.26 (s, 3H), 1.60 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 200.5, 166.4, 140.9, 129.0, 128.6, 128.5, 128.1, 126.2, 124.5, 90.0, 73.6, 50.4, 30.9, 30.1, 22.0.(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 3-oxobutanoate (3o)24
Yield: 93%, colourless oil; IR (neat) νmax 2952, 2867, 1718, 1643, 1454, 1240, 1149, 1029, 975, 913, 843 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.79–4.73 (m, 1H), 3.45 (s, 2H), 2.29 (s, 2H), 2.04 (d, J = 15.2 Hz, 1H), 1.90–1.88 (m, 1H), 1.70 (d, J = 12.0 Hz, 2H), 1.61 (s, 3H), 1.52–1.37 (m, 2H), 1.09–0.97 (m, 3H), 0.92 (t, J = 6.8 Hz, 7H), 0.78 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 200.7, 166.7, 73.7, 50.6, 46.9, 40.7, 34.2, 31.4, 30.1, 26.2, 23.6, 22.0, 20.7, 16.2.(3aR,5R,6S,6aR)-5-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydro furo[2,3-d][1,3]dioxol-6-yl 3-oxobutanoate (3p)52
Yield: 92%, colourless syrup; IR (neat) νmax 2987, 1751, 1718, 1373, 1214, 1152, 1070, 1017, 846, 731 cm−1; 1H NMR (400 MHz, CDCl3) δ 11.90 (s, enol-OH), 5.90 (s, 1H), 5.33 (s, 1H), 4.60 (d, J = 3.6 Hz, 1H), 4.21 (s, 2H), 4.10–4.02 (m, 2H), 3.53 (d, J = 4.4 Hz, 2H), 2.30 (s, 3H), 1.68 (s, 4H), 1.54 (s, 4H), 1.23 (s, 3H), 1.34 (s, 7H); 13C NMR (100 MHz, CDCl3) δ 199.9, 166.2, 165.8, 112.4, 112.3, 109.5, 109.3, 105.1, 105.0, 89.2, 83.4, 83.2, 83.1, 79.7, 79.6, 79.2, 75.7, 72.4, 72.3, 68.4, 67.4, 67.0, 64.4, 50.0, 50.0, 30.9, 30.4, 30.1, 30.0, 26.9, 26.7, 26.7, 26.2, 25.2, 21.3.4-Nitrobenzyl 3-oxo-3-phenylpropanoate (4a)26
Yield: 90%, reddish oil; IR (neat) νmax 1742, 1683, 1605, 1518, 1449, 1339, 1262, 1181, 1141, 986, 846, 744, 686 cm−1; 1H NMR (400 MHz, CDCl3) δ (s, enol-OH), two sets of doublets 8.26 and 8.21 (J = 8.8 Hz, 2H), 7.96–7.94 (m, 2H), two sets of multiplets 7.82–7.80 and 7.66–7.58 (m, 2H), 7.53–7.44 (m, 5H), 5,79 (s, enol-CH), 5.36 and 5.32 (s, 2H), 4.13 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 192.1, 172.5, 167.1, 147.7, 143.2, 142.6, 135.8, 134.0, 133.1, 131.7, 128.9, 128.6, 128.5, 128.4, 128.3, 126.2, 123.9, 123.8, 86.6, 65.5, 64.5, 45.8.Dec-9-en-1-yl 3-oxo-3-phenylpropanoate (4b)53
Yield: 91%, colourless oil; IR (neat) νmax 2926, 2854, 1740, 1687, 1631, 1455, 1412, 1263, 1190, 1076, 993, 910, 764, 688 cm−1; 1H NMR (400 MHz, CDCl3) δ 12.61 (enol-OH), 7.97–7.95 (m, 2H), 7.81–7.79 (m), 7.61 (t, J = 6.4 Hz, 1H), 7.52–7.42 (m, 3H), 5.86–5.77 (m, 1H), 5.69 (s, due enol-CH), 5.03–4.96 (m, 2H), two sets of triplet at 4.21 and 4.16 (J = 6.4 Hz, 2H), 4.01 (s, 2H), 2.06 (q, J = 6.8 Hz, 2H), 1.73 (s, 1H), 1.27 (s, 14H); 13C NMR (100 MHz, CDCl3) δ 192.5, 167.6, 139.2, 136.0, 133.5, 131.2, 128.8, 128.5, 126.1, 114.2, 87.4, 65.7, 64.5, 46.1, 33.8, 29.4, 29.3, 29.2, 29.1, 29.0, 28.9, 28.7, 28.4, 25.7.(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 3-oxo-3-phenyl pro-panoate (4c)54
Yield: 92%, colourless oil; IR (neat) νmax 2952, 2866, 1734, 1687, 1631, 1453, 1408, 1263, 1193, 1147, 1085, 975, 763, 687 cm−1; 1H NMR (400 MHz, CDCl3) δ 12.70 (s, enol 1H, OH), 7.96 (d, J = 7.2 Hz, keto 2H, Ar), 7.79 (d, J = 6.8 Hz, enol 2H, Ar), 7.60 (t, J = 8.0, keto 1H, Ar), 7.51–7.41 (m, keto 2H + enol 3H, Ar), 5.67 (s, enol 1H, C(OH)CHCO), 4.88–4.70 (m, keto 1H + enol 1H, OCH), 4.02 (d, J = 15.6 Hz, 1H, keto 1H, COCH2CO), 3.96 (d, J = 15.6 Hz, 1H, keto 1H, COCH2CO), 2.11–1.30 (m, keto 4H + enol 4H, OCHCHCH, OCHCH2), 1.13–0.07 (m, keto 14H + enol 14H, OCHCH2CHCH2CH2, CH(CH3)2, CH3); 13C NMR (100 MHz, CDCl3) δ 192.6 (keto), 172.9 (enol), 171.3 (enol), 167.1 (keto), 136.1 (keto), 133.6 (keto + enol), 131.2 (enol), 128.7 (keto), 128.5 (keto + enol), 126.0 (enol), 87.7 (enol), 75.6 (keto), 74.2 (enol), 47.1 (enol), 46.8 (keto), 46.6 (keto), 41.1 (enol), 40.6 (keto), 34.2 (enol), 34.1 (keto), 31.4 (keto + enol), 26.3 (enol), 25.9 (keto), 23.5 (enol), 23.2 (keto), 22.0 (enol), 21.9 (keto), 20.7 (keto + enol), 16.4 (enol), 16.0 (keto).(3aR,5R,6S,6aR)-5-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyl tetrahydrofuro [2,3-d][1,3]dioxol-6-yl 3-oxo-3-phenyl propanoate (4d)52
Yield: 89% colourless syrup; IR (neat) νmax 2987, 1750, 1686, 1376, 1256, 1212, 1153, 1069, 1013, 947, 844, 765, 689 cm−1; 1H NMR (400 MHz, CDCl3) δ 12.37 (enol-OH), 7.94 (d, J = 7.6 Hz) and 7.80 (d, J = 7.2 Hz) 7.64 (t, J = 8.0 Hz, 1H), 7.54–7.43 (m, 3H), 5.94 and 5.83 (d, J = 4.0 and 3.2 Hz, 1H), 5.71 (enol-CH) 5.39 and 5.34 (d, J = 2.4 Hz, 1H), 4.61 (m, 1H), 4.22–3.77 (m, 7H), 1.56 and 1.53 (two s, 3H), 1.45 and 1.42 (two s, 3H), 1.34, 1.33 and 1.31 (three s, 6H); 13C NMR (100 MHz, CDCl3) δ 192.1, 172.7, 171.9, 166.3, 135.8, 134.0, 133.0, 131.7, 128.9, 128.6, 128.5, 128.4, 126.2, 112.4, 109.4, 105.1, 86.7, 83.4, 83.1, 79.6, 76.1, 72.4, 72.3, 67.2, 67.1, 45.8, 26.9, 26.7, 26.2, 25.3.Benzyl 2-oxocyclopentane-1-carboxylate (5a)47
Yield: 93%, colourless oil; IR (neat) νmax 2962, 2887, 1720, 1497, 1453, 1380, 1335, 1249, 1175, 1109, 996, 831, 742, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.39–7.34 (m, 5H), 5.20 (s, 2H), 3.23 (t, J = 9.2 Hz, 1H), 2.34–2.26 (m, 4H), 2.19–2.10 (m, 1H), 1.94–1.82 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 212.2, 169.3, 135.6, 128.6, 128.3, 128.1, 67.0, 54.8, 38.1, 27.4, 21.0.Phenethyl 2-oxocyclopentane-1-carboxylate (5b)31
Yield: 92%, colourless oil; IR (neat) νmax 2961, 1719, 1454, 1335, 1248, 1172, 1109, 831, 744, 697 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.35–7.23 (m, 5H), 4.47–4.32 (m, 2H), 3.16 (t, J = 8.8 Hz, 1H), 3.06–2.90 (m, 2H), 2.43–2.24 (m, 4H), 2.14–2.08 (m, 1H), 1.92–1.83 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 212.4, 169.4, 137.6, 129.0, 128.5, 126.6, 65.8, 54.8, 38.1, 35.0, 27.4, 21.0.Cinnamyl 2-oxocyclopentane-1-carboxylate (5c)52
Yield: 90%, colourless oil; IR (neat) νmax 2962, 1719, 1661, 1449, 1389, 1334, 1246, 1176, 1109, 966, 831, 741 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.41–7.26 (m, 5H), 6.70 (d, J = 15.6 Hz, 1H), 6.34–6.27 (m, 1H), 4.84–4.82 (m, 2H), 3.23 (t, J = 9.2 Hz, 1H), 2.38–2.32 (m, 4H), 2.18–2.15 (m, 1H), 1.93–1.89 (m, 1H), 13C NMR (100 MHz, CDCl3) δ 212.3, 169.2, 136.1, 134.5, 128.6, 128.1, 126.7, 122.7, 65.9, 54.8, 38.1, 27.4, 21.0.Conflicts of interest
The authors declare no conflict of interest.Author contributions
S. M. designed the work and K. D. performed all experiments and analyzed the spectral data. S. M. wrote the manuscript with input from K. D.Acknowledgements
We sincerely acknowledged Department of Science and Technology (DST), Govt. of India for providing 400 MHz NMR spectrometer through DST-FIST programme (No SR/FST/CSI-263/2015) and Central Instrumental Center (CIC), Tripura University for instrumental facility. K. D. is thankful to Tripura University for providing Non-NET fellowship.References
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- Z. Khan, F. Javed, Z. Shamair, A. Hafeez, T. Fazal, A. Aslam, W. B. Zimmerman and F. Rehman, Ind. Eng. Chem. Res., 2021, 103, 80–101 CrossRef CAS.
- G. Krishnaiah, B. Sandeep, D. Kondhare, K. C. Rajanna, J. N. Reddy, Y. R. Rao and P. K. Zhubaidha, Tetrahedron Lett., 2013, 54, 703–706 CrossRef CAS.
- R. Das and D. Chakraborty, Appl. Organomet. Chem., 2012, 26, 140–144 CrossRef CAS.
- S. Benetti and R. Romagnoli, Chem. Rev., 1995, 95, 1065–1114 CrossRef CAS.
- B. S. Balaji, M. Sasidharan, R. Kumar and B. Chanda, Chem. Commun., 1996, 707–708 RSC.
- O. A. Attanasi, P. Filippone, C. Fiorucci, E. Foresti and F. Mantellini, J. Org. Chem., 1998, 63, 9880–9887 CrossRef CAS.
- I. Yavari and F. Nourmohammadian, J. Chem. Res., 2000, 5, 218–219 CrossRef.
- S. J. Bingham and J. H. P. Tyman, Org. Prep. Proced. Int., 2001, 33, 357–361 CrossRef CAS.
- D. S. Campbell and C. W. Lawrie, J. Chem. Soc. D, 1971, 7, 355–356 RSC.
- J. Christoffers and N. Önal, Eur. J. Org. Chem., 2000, 8, 1633–1635 CrossRef.
- D. E. Ponde, V. H. Deshpande, V. J. Bulbule, A. Sudalai and A. S. Gajare, J. Org. Chem., 1998, 63, 1058–1063 CrossRef.
- M. Gianotti, G. Martelli, G. Spunta, E. Campana, M. Panunzio and M. Mendozza, Synth. Commun., 2000, 30, 1725–1730 CrossRef CAS.
- M. C. Hennessy and T. P. O'Sullivan, RSC Adv., 2021, 11, 22859–22920 RSC.
- T. J. Broering and G. W. Morrow, Synth. Commun., 1999, 29, 1235–1239 CrossRef.
- M. I. de Sairre, E. S. Bronze-Uhle and P. M. Donate, Tetrahedron Lett., 2005, 46, 2705–2708 CrossRef CAS.
- M. L. Kantam, V. Neeraja, B. Bharathi and C. V. Reddy, Catal. Lett., 1999, 62, 67–69 CrossRef CAS.
- J. Otera, Acc. Chem. Res., 2004, 37, 288–296 CrossRef CAS PubMed.
- B. P. Bandgar, V. S. Sadavarte and L. S. Uppalla, J. Chem. Res., 2001, 16–17 CrossRef CAS.
- S. P. Chavan, K. Shivasankar, R. Sivappa and R. Kale, Tetrahedron Lett., 2002, 43, 8583–8586 CrossRef CAS.
- S. P. Chavan, P. K. Zubaidha, S. W. Dantale, A. Keshavaraja, A. V. Ramamswamy and T. Ravindranathan, Tetrahedron Lett., 1996, 37, 233–236 CrossRef CAS.
- R. K. Pandey and P. Kumar, Catal. Commun., 2007, 8, 1122–1125 CrossRef CAS.
- G. C. M. Kondaiah, L. A. Reddy, K. S. Babu, V. M. Gurav, K. G. Huge, R. Bandichhor, P. P. Reddy, A. Bhattacharyaa and R. V. Anand, Tetrahedron Lett., 2008, 49, 106–109 CrossRef CAS.
- B. R. Madje, P. T. Patil, S. S. Shindalkar, S. B. Benjamin, M. S. Shingare and M. K. Dongare, Catal. Commun., 2004, 5, 353–357 CrossRef CAS.
- J. Yang, C. Ji, Y. Zhao, Y. Li, S. Jiang, Z. Zhang, Y. Ji and W. Liu, Synth. Commun., 2010, 40, 957–963 CrossRef CAS.
- R. H. Tale, A. D. Sagar, H. D. Santan and R. N. Adude, Synlett, 2006, 37, 415–418 CrossRef.
- D. F. Taber, J. C. Amedio and Y. K. Patel, J. Org. Chem., 1985, 50, 3618–3619 CrossRef CAS.
- O. Mhasni and F. Rezgui, Tetrahedron, 2011, 67, 6322–6326 CrossRef CAS.
- O. Mhasni, I. Erray and F. Rezgui, Synth. Commun., 2014, 44, 3320–3327 CrossRef CAS.
- S. Palaniappan and R. C. Shekhar, Polym. Adv. Technol., 2004, 15, 140–143 CrossRef CAS.
- H. Hagiwara, A. Koseki, K. Isobe, K. ichi Shimizu, T. Hoshi and T. Suzuki, Synlett, 2004, 12, 2188–2190 CrossRef.
- R. S. Ribeiro, R. O. M. A. de Souza, M. L. A. A. Vasconcellos, B. L. Oliveira, L. C. Ferreira and L. C. S. Aguiar, Synthesis, 2007, 61–64 CAS.
- A. Córdova and K. D. Janda, J. Org. Chem., 2001, 66, 1906–1909 CrossRef PubMed.
- J. Tongshou, Z. Suling and L. Tongshuang, Green Chem., 2002, 4, 32–34 RSC.
- B. S. Balaji and B. M. Chanda, Tetrahedron, 1998, 54, 13237–13252 CrossRef CAS.
- L. I. Koval, V. I. Dzyuba, O. L. Ilnitska and V. I. Pekhnyo, Tetrahedron Lett., 2008, 49, 1645–1647 CrossRef CAS.
- Z. S. Qureshi, K. M. Deshmukh, M. D. Bhor and B. M. Bhanage, Catal. Commun., 2009, 10, 833–837 CrossRef CAS.
- B. P. Bandgar, L. S. Uppalla and V. S. Sadavarte, Synlett, 2001, 1715–1718 CrossRef CAS.
- Y. M. Ren, Z. C. Wu, R. C. Yang, T. X. Tao, J. J. Shao, Y. G. Gao, S. Zhang and L. Li, Adv. Mater. Res., 2013, 781, 259–262 Search PubMed.
- (a) P. Puthiaraj, A. Ramu and K. Pitchumani, Asian J. Org. Chem., 2014, 3, 784–791 CrossRef CAS; (b) D. Habibi and M. Nasrollahzadeh, Synth. Commun., 2012, 42, 2023–2032 CrossRef CAS; (c) A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS.
- (a) W. Nabgan, A. A. Jalil, B. Nabgan, A. H. Jadhav, M. Ikram, A. Ul-Hamid, M. W. Ali and N. S. Hassana, RSC Adv., 2022, 12, 1604–1627 RSC; (b) D. R. Lathiya, D. V. Bhatt and K. C. Maheria, ChemistrySelect, 2019, 4, 4392–4397 CrossRef CAS; (c) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC; (d) C. Zili, L. Peixia, L. Weitong, Z. Zhe, W. Yuanyuan, Z. Hanqing and Z. Jianjun, Chin. J. Org. Chem., 2020, 40, 1716–1724 CrossRef; (e) M. B. Gawande, R. Hosseinpour and R. Luquec, Curr. Org. Synth., 2014, 11, 526–544 CrossRef CAS; (f) Z. N. Siddiqui, Arabian J. Chem., 2019, 12, 2788–2797 CrossRef CAS; (g) Y. Hemanth Sriram, T. Fatima, M. Satish Kumar, K. C. Rajanna, M. Venkateswarlu, M. Sai Sudhakar and R. M. Raju, Iran. Chem. Commun., 2017, 5, 352–363 Search PubMed; (h) T. Fatima, Y. Hemanth Sriram, M. Satish Kumar, M. Venkateswarlu and K. C. Rajanna, Asian J. Green Chem., 2017, 1, 69–77 Search PubMed.
- V. Kumar, C. Singh, U. Sharma, P. K. Verma, B. Singh and N. Kumar, Indian J. Chem., 2014, 53B, 83–89 CAS.
- M. Parveen, F. Ahmad, A. M. Malla and S. Azaza, New J. Chem., 2015, 39, 2028–2041 RSC.
- M. Gohain, V. Kumar, J. H. V. Tonder, H. C. Swart, O. M. Ntwaeaborwa and B. C. B. Bezuidenhoudt, RSC Adv., 2015, 5, 18972–18976 RSC.
- T. R. Mani, Y. H. Sriram, K. Bhaskar, A. K. Kumar and K. R. Kudle, Chem. Methodol., 2018, 2, 83–92 CAS.
- B. P. Bandgar, V. S. Sadavarte and L. S. Uppalla, Synth. Commun., 2001, 31, 2063–2066 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, A. D. Krishna, C. S. Reddy and A. V. Narsaiah, J. Mol. Catal. A: Chem., 2007, 261, 93–97 CrossRef CAS.
- M. L. Kantam and P. Sreekanth, Catal. Lett., 2001, 77, 241–243 CrossRef CAS.
- A. C. H. Weber, T. C. Batista, B. Gonçalves, C. R. L. Hack, L. M. Porciuncula, T. G. M. Treptow, C. D. R. M. D'Oca, D. Russowsky and M. G. M. D'Oca, J. Am. Oil Chem. Soc., 2016, 93, 1399–1406 CrossRef CAS.
- S. P. Chavan, Y. T. Subbarao, S. W. Dantale and R. Sivappa, Synth. Commun., 2001, 31, 289–294 CrossRef CAS.
- H. Yue, H. Yu, S. Liu and C. Xu, RSC Adv., 2016, 6, 19041–19051 RSC.
- F. C. da Silva, V. F. Ferreira, R. S. Rianelli and W. C. Perreira, Tetrahedron Lett., 2002, 43, 1165–1168 CrossRef CAS.
- B. P. Bandgar, S. S. Pandit and L. S. Uppalla, Org. Prep. Proced. Int., 2003, 35, 219–222 CrossRef CAS.
- K. Agura, Y. Hayashi, M. Wada, D. Nakatake, K. Mashima and T. Ohshima, Chem.–Asian J., 2016, 11, 1548–1554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03855b |
| This journal is © The Royal Society of Chemistry 2022 |