
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePEGylation of Goldbody: PEG-aided conformational engineering of peptides on gold nanoparticles†
Tiange Gao‡
a,
Yuan-Yuan Liu‡a,
Chenxi Loua,
Haifang Wang *a,
Yuanfang Liuab and
Aoneng Cao*a
*a,
Yuanfang Liuab and
Aoneng Cao*a
aInstitute of Nanochemistry and Nanobiology, Shanghai University, Shanghai 200444, China. E-mail: hwang@shu.edu.cn; ancao@shu.edu.cn
bBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 15th September 2022
Abstract
It is still a great challenge to engineer flexible non-functional molecules into special conformations to carry out novel functions. Previously, we successfully restored the native conformations and functions of the flexible complementary-determining regions (CDRs) of antibodies on the surface of gold nanoparticles (AuNPs), and created a class of AuNP-based artificial antibodies, denoted as Goldbodies. Yet, in these Goldbodies, there are dozens of CDRs on one Goldbody. Herein, we show that the number of CDRs per Goldbody could be reduced by more than one order of magnitude, by replacing the majority of the CDRs with polyethylene glycol (PEG) with a molecular weight around 600 Da, while the native conformations and functions of the CDRs could still be restored on AuNPs. Also, we find that the PEG with two terminal –SH groups is much better than the PEG with a single –SH group for aiding the restoration of the native conformation of the CDRs on AuNPs. To demonstrate the potential generic applicability of the PEGylation in aiding conformational engineering of peptides, two PEGylated Goldbodies have been created, which can specifically recognize lysozyme and epidermal growth factor receptor, respectively. The PEGylated Goldbodies further prove the mechanism of conformational engineering and the “Confined Lowest Energy Fragments” (CLEFs) hypothesis, and pave the way for future applications of Goldbodies.
Introduction
For decades, scientists have been conjugating nanoparticles (NPs) with natural proteins to endow NPs with bio-functions.1–5 As an alternative, some protein fragments instead of the whole proteins have also been used to functionalize NPs, given that these fragments are functional by themselves.6–8 Since most fragments of proteins are not structured and are non-functional by themselves, only a very limited number of protein fragments could be used to functionalize NPs by this common conjugation method.After years of efforts, we developed a conformational engineering technique that can restore the native conformations and functions of the flexible fragments of natural proteins on NPs, even though those fragments are unstructured and non-functional by themselves, for example the highly flexible complementary-determining regions (CDRs) of antibodies.9 Thus, we created a new class of AuNP-based artificial antibodies, denoted as Goldbodies.9–11 Goldbodies can specifically recognize the corresponding antigens as the original monoclonal antibodies do, and show much better stability than the monoclonal antibodies. At the same time, Goldbodies do not contain any of the potential immunogenic sequences of the original antibodies except for the short binding CDRs fragments, and thus do not need further humanization as the common therapeutic antibodies do. For this reason, Willson coined a new term of “Goldization” for our technique, comparing it with the widely-used “humanization” technology of therapeutic antibodies.12
The reason that the “Goldization” method is able to control the conformations of the unstructured peptide fragments of proteins is that each CDR peptide is conjugated to AuNPs through two Au–S bonds, instead of the common method of one Au–S bond or one other covalent bond. It is the two Au–S bonds that provide the handles for conformational engineering;9 and the mobility of the Au–S bonds on the surface of AuNPs13,14 makes it possible to adjust the span between the two Au–S bonds of the conjugated peptides. To elucidate the underlying mechanism for the restoration of the native conformations of the peptide fragments, we proposed the “Confined Lowest Energy Structure Fragments” (CLESFs) hypothesis,15 which was then shortened as CLEFs hypothesis.16 This CLEFs hypothesis points out that all natural proteins consist of multiple CLEFs clutched together by a few key strong interactions, and the native conformations of those CLEFs are the most stable ones under the constraints of those key interactions. In other words, the native conformation of a CLEF of a protein is only determined by the CLEF and the related few key interactions, all other amino acids in the protein (in other CLEFs) have almost no effect. Therefore, in Goldbodies, the two Au–S bonds between the CDR and the AuNP resemble the key interactions for the CDR in the original antibody, and the native conformation and the antigen-recognition function of the conjugated CDR are thus resumed.
Since non-functionalized “bare” AuNPs are notoriously known for their strong non-specific interaction with almost any proteins,17,18 in the previous Goldbodies, in order to cover the “sticky” surface of the bare AuNPs, the number of CDRs for one AuNP is much more than necessary for the purpose of binding with the antigens. For example, the anti-lysozyme Goldbody has 60 CDR peptides (Pep1) on one AuNP (∼3.6 nm), while the surface of the Goldbody can only accommodate/bind a few lysozymes.9 The main purpose of the large extra number of CDR peptides is to cover the “bare” surface of AuNPs to prevent the strong non-specific binding with proteins other than the targeted one. As the early proof-of-concept studies,9–11 using solely the same peptide to conjugate AuNPs could eliminate other factors to draw an unambiguous conclusion. However, those extra peptides not only raise the cost of Goldbodies, but also leave a possible doubt that the reported function of Goldbodies might come from the multivalency effect. To eliminate this doubt and to dramatically reduce the cost of Goldbodies, herein, we try to reduce greatly the number of peptides per Goldbody, at the same time use polyethylene glycol (PEG) to passivate the rest of the surface of AuNPs, and still manage to restore the native conformations and functions of the CDRs on AuNPs (Fig. S1†).
PEG has been widely used to conjugate NPs for biomedicine applications.19–25 PEG can be modified and further customized with different groups for different conjugating purposes, and thus PEG functionalization (PEGylation) has almost become a standard procedure for many NPs. In addition, PEG is generally considered safe, and PEGs with molecular weights (MW) between 200 and 9500 Da are FDA-approved for use in human consumer products.26,27 PEG is commonly used in the field of nanotechnology to create stealthy drug carriers with extended circulation time and reduced recognition and clearance by the mononuclear phagocyte system.19,28 Numerous studies have investigated the effect of PEG length and density on the protein adsorption and macrophage uptake of NPs.24,29–37 Generally, as the density of PEG on the surface of NPs and the length of PEG increase, PEGylation is beneficial to inhibiting non-specific protein adsorption and phagocytic uptake of NPs. However, it is not clear whether the PEG conjugated on AuNPs interferes the conformational engineering of peptides on AuNPs. In this study, after comparing the effect of the MW of PEG and the effect of conjugation of PEG to AuNPs with one or two Au–S bonds (Scheme 1 and Fig. S1†), two PEGylated Goldbodies, i.e., the PEGylated anti-lysozyme Goldbody and the PEGylated anti-EGFR (epidermal growth factor receptor) Goldbody, have been successfully synthesized, validating the CLEFs hypothesis and demonstrating the application potential of the PEGylated Goldbodies.
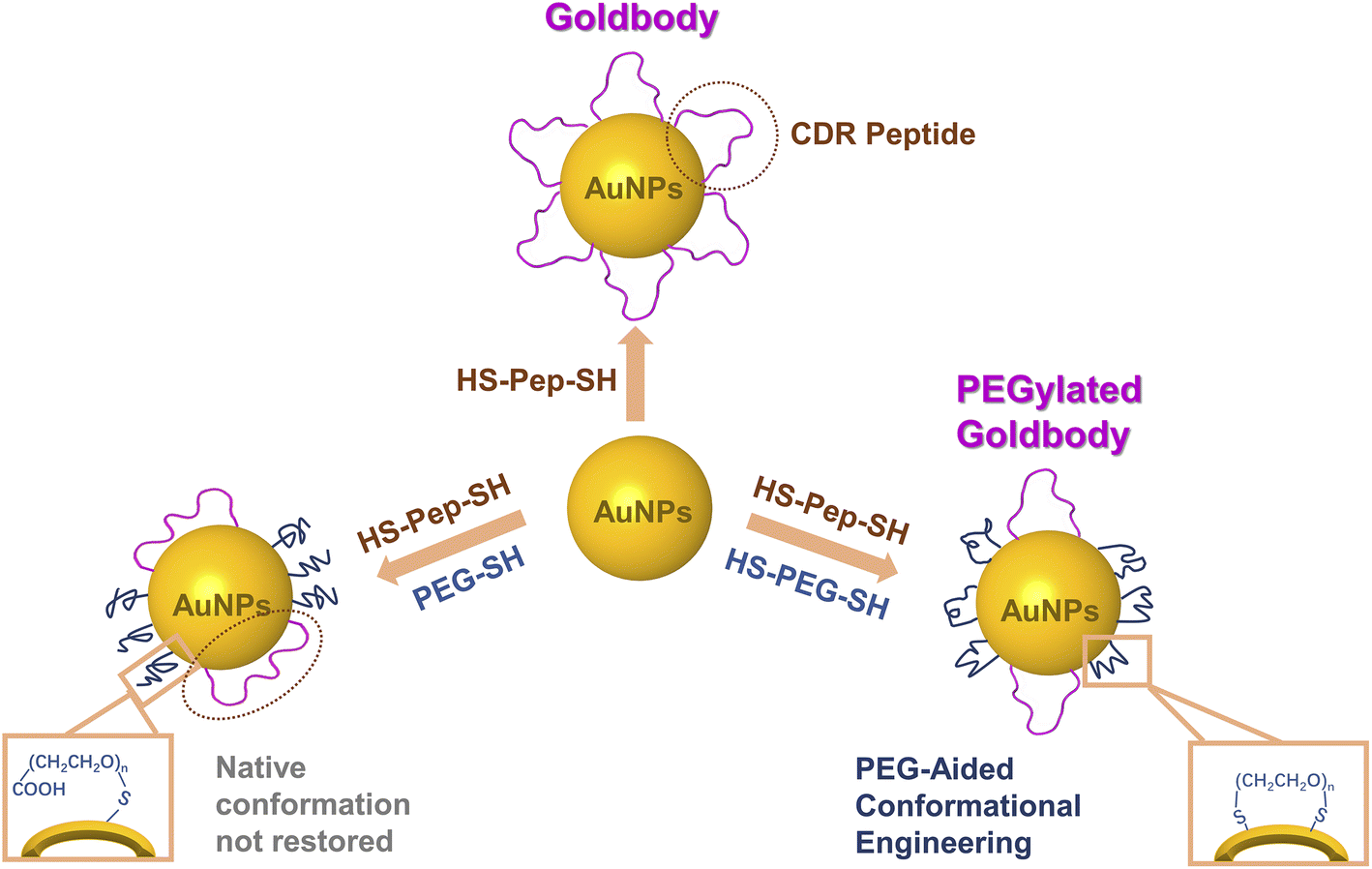 | ||
| Scheme 1 Schematic illustration of the difference between the previous Goldbody (top) and the PEGylated Goldbody (bottom right), and the different effects of PEG–SH (bottom left) or HS–PEG–SH (bottom right) on the conformational engineering of the CDRs on AuNPs. | ||
Experimental
Materials
Chloroauric acid, sodium tetrahydroborate, trisodium citrate dehydrate and sodium hydroxide were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai). Hen egg white lysozyme (HEWL), immunoglobulin G (IgG) and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (USA). Ribonuclease A (RNase A) and Micrococcus lysodeikticus (M. lysodeikticus) cells were purchased from Sangon Biotech (Shanghai) Co., Ltd. Epidermal growth factor (EGF) and the extracellular soluble part of EGFR were purchased from Creative BioMart (USA). Peptides including Pep1, Pep1m, Pep2 and Pep2m (see Table S1†) were of 95% pure and synthesized by Shanghai Science Peptide Biological Technology Co., Ltd (China). All PEGs were obtained from Shanghai Ponsure Biotech, Inc. (China). Amine Coupling Kit (N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), and 1.0 M ethanolamine-HCl, pH 8.5), HBS-EP buffer (10 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 150 mM NaCl, 3 mM EDTA, 0.05% v/v surfactant P20, pH 7.4), acetate buffer (10 mM sodium acetate, pH 4.5), and Series S CM5 sensor chips were purchased from Cytiva (USA). High glucose DMEM medium and trypsin-EDTA were purchased from YOBIBIO (China). Fetal bovine serum (FBS) was purchased from Biological Industries (Israel). Human epithelial cervical cancer cell line HeLa was obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Ultrapure water (Millipore) was used.Synthesis of AuNPs
AuNPs with an average diameter of around 3.6 nm were synthesized following the procedure reported previously.9 The molar concentration of the AuNP solution was calculated from its absorbance at 450 nm measured on an UV-vis spectrophotometer (U-3010, Hitachi, Japan), using an extinction coefficient ε450 nm of 2.768 × 106 M−1 cm−1.9,38 The synthesized AuNPs were stored at room temperature in the dark.Preparation of the PEGylated Goldbodies
The concentration of the peptides was determined using their absorbance at 280 nm (Table S1†).9 To prepare the PEGylated anti-lysozyme Goldbody, the Pep1 solution in phosphate buffer (0.01 M, pH 7.4) was added dropwise to the AuNPs solution in molar ratio of AuNP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pep1 = 1:5. After stirring for 1 h at room temperature, the HS–PEG600–SH aqueous solution was added dropwise to the AuNP–Pep1 solution, and the mixture was stirred for another 1 h at room temperature. The obtained product, AuNP–5Pep1–15(HS–PEG600–SH), abbreviated as AuNP–5Pep1–15PEG is denoted as the PEGylated anti-lysozyme Goldbody. Also, the similar procedure was used to synthesize the controls, including AuNP–5Pep1–x(HS–PEG600–COOH), AuNP–5Pep1–x(HS–PEG300–COOH), AuNP–5Pep1–x(HS–PEG2000–SH), and AuNP–5Pep1m–15(HS–PEG600–SH), where x is the average number of PEG molecules for one AuNP.
:Pep1 = 1:5. After stirring for 1 h at room temperature, the HS–PEG600–SH aqueous solution was added dropwise to the AuNP–Pep1 solution, and the mixture was stirred for another 1 h at room temperature. The obtained product, AuNP–5Pep1–15(HS–PEG600–SH), abbreviated as AuNP–5Pep1–15PEG is denoted as the PEGylated anti-lysozyme Goldbody. Also, the similar procedure was used to synthesize the controls, including AuNP–5Pep1–x(HS–PEG600–COOH), AuNP–5Pep1–x(HS–PEG300–COOH), AuNP–5Pep1–x(HS–PEG2000–SH), and AuNP–5Pep1m–15(HS–PEG600–SH), where x is the average number of PEG molecules for one AuNP.
Similarly, the PEGylated anti-EGFR Goldbody (AuNP–5Pep2–15(HS–PEG600–SH)), abbreviated as AuNP–5Pep2–15PEG was prepared following the same procedure as described above except that the Pep2 solution was used instead of the Pep1 solution. Correspondingly, the controls were synthesized, including AuNP–5Pep2–x(HS–PEG600–COOH), AuNP–5Pep2–x(HS–PEG300–COOH), AuNP–5Pep2–x(HS–PEG2000–SH), and AuNP–5Pep2m–15(HS–PEG600–SH).
Since the conjugation via S–Au bonds is very efficient,9–11 the average numbers of peptides and PEG per AuNP were simply achieved by mixing the reactants in stoichiometric proportions.
Characterization of the PEGylated Goldbodies
The size and morphology of the PEGylated Goldbodies were characterized by using TEM (HT7700, Hitachi) and UV-vis spectrophotometer (U-3010, Hitachi). Before TEM characterization, the solutions were first diluted to around 20 nM. Then an aliquot of 7 μL of the diluted solution was dropped on a TEM grid. After blotting with a filter paper from the side, the grid was allowed to dry in air. The diameters of NPs were analysed from the obtained TEM images using the ImageJ software.The hydrodynamic diameter distributions and zeta-potentials of the AuNPs and the PEGylated Goldbodies were measured by dynamic light scattering (DLS) at 90° with a glass cuvette (PCS1115) on a Nanosizer ZS90 (Malvern) at 25 °C. The dispersant was ultrapure water, and a refractive index of 0.295 was used for Au.
Surface plasmon resonance (SPR) experiments
All SPR experiments were carried out at 25 °C on a Biacore T200 instrument (Cytiva). The HBS-EP buffer was selected as the running buffer. HEWL, EGFR, RNase A, BSA and IgG were separately immobilized on different channels of the Series S CM5 sensor chips by the standard amine coupling procedure.To measure the bindings of the PEGylated Goldbodies and the corresponding controls with HEWL/EGFR/RNase A/BSA/IgG, 8 nM of the PEGylated Goldbodies or the controls diluted in the HBS-EP buffer were injected into the HEWL/EGFR/RNase A/BSA/IgG-immobilized channels at a flow rate of 30 μL min−1.
To evaluate their thermal stability, the PEGylated Goldbodies were first boiled in a water bath for 30 min and then allowed to cool down to the room temperature. Subsequently, the binding of these pre-treated PEGylated Goldbodies with HEWL or EGFR was measured following the same procedure described above.
In the kinetic experiments, the PEGylated anti-lysozyme Goldbody and the PEGylated anti-EGFR Goldbody were diluted to different concentrations with the HBS-EP buffer and then injected into the HEWL and EGFR channels, respectively. The data were fitted with the 1:1 binding model, and the dissociation constants (KD) were calculated.
Enzymatic activity assay of lysozyme
The enzymatic activity of HEWL was determined by monitoring the first 90 second's digestion of M. lysodeikticus by HEWL with the change of absorbance at 450 nm using the UV-vis spectrophotometer.9 The activity of HEWL is represented by the initial slope of the dynamic absorbance curve. Briefly, the M. lysodeikticus stock solution was freshly prepared by suspending 10 mg of dry bacteria in 30 mL of phosphate buffer (0.1 M, pH 6.2), while HEWL was dissolved in ultrapure water with a concentration of 150 nM. The HEWL solution (0.5 mL) was mixed well with 1 mL of the PEGylated anti-lysozyme Goldbody (60 nM) or the corresponding controls (60 nM) for 1 min. Subsequently, 1 mL of the stock suspension of M. lysodeikticus was added. The mixture was then quickly transferred to a cuvette for the absorbance measurement after a brief vigorous shaking. All assays were conducted at 25 °C.To investigate the specific interaction between the PEGylated anti-lysozyme Goldbody and HEWL, the PEGylated anti-lysozyme Goldbody (1 mL, 60 nM) and the corresponding controls (1 mL, 60 nM) were pre-incubated with RNase A (0.25 mL, 1500 nM) for 1 min, then the HEWL solution (0.25 mL, 300 nM) was added and mixed well for 1 min. Subsequently, 1 mL of the stock suspension of M. lysodeikticus was added to the mixture. The inhibition rates were measured following the above procedure.
To evaluate the thermal stability of the PEGylated anti-lysozyme Goldbody, the PEGylated anti-lysozyme Goldbody solution was first heated in a water bath for 30 min at 50 °C, 75 °C and 100 °C, respectively, and then cooled down to room temperature. Subsequently, the inhibition rates of enzymatic activity of lysozyme by the pre-treated PEGylated anti-lysozyme Goldbody were measured following the same procedure described above.
Inhibition of EGF-induced cell proliferation by the anti-EGFR Goldbody
HeLa cells were seeded in 12-well plates at a density of 3 × 104 per well, and cultured in a CO2 incubator at 37 °C for 24 hours. After the culture medium was discarded, 0.5 mL of fresh serum-free medium or serum-free medium containing 40 nM AuNP–5Pep2 or 40 nM AuNP–5Pep2–15(HS–PEG600–SH) was introduced into the wells. After incubation for 30 min, 0.5 mL of fresh medium (containing 20% fetal bovine serum) with/without EGF (final concentration of 20 nM) was added into the corresponding wells. The cells cultured in 1.0 mL of fresh medium (containing 10% fetal bovine serum) were used as the control. After 72 h of incubation, the cells were collected, re-dispersed in serum-free medium, and counted. Each group had 3 parallel samples, and each sample was counted 3 times. The result is presented as the mean ± standard deviation (SD).Results and discussion
Design and optimization of the PEGylated anti-lysozyme Goldbody
In the original anti-lysozyme Goldbody,9 AuNP was conjugated merely with Pep1, a peptide corresponding to the CDR peptide of an anti-lysozyme antibody, cAB-lys3,39 with a cysteine (Cys) residue added at each of its two terminals (Table S1†). These two Cys residues make it easy to conjugate the peptide onto AuNPs with two Au–S bonds. Since the CDR fragment is very flexible, the free Pep1 is unstructured and non-functional.9 But these two Au–S bonds make it possible to restore the native conformation and the antigen-recognizing function of the CDR fragment. To achieve the highest specific binding with lysozyme and to cover the surface of AuNPs, the optimum peptide density is around 60 Pep1 per AuNP (3.6 nm) in the previous anti-lysozyme Goldbody.To reduce the number of peptides on Goldbody, in this study, we fixed the number of Pep1 on one AuNP to 5, and used different numbers of PEG molecules to tune the conformation of the Pep1 into the active conformation for the specific binding with HEWL.
Considering the MW of the Pep1 peptide on the anti-lysozyme Goldbody (about 1700 Da)9 and the commercial availability of PEG, four different kinds of PEGs have been used to PEGylate the Goldbody. They are: 300 Da PEG with one –SH terminal group (HS–PEG300–COOH), 600 Da PEG with one –SH terminal group (HS–PEG600–COOH), 600 Da PEG with two –SH terminal groups (HS–PEG600–SH), and 2000 Da PEG with two –SH terminal groups (HS–PEG2000–SH).
To prepare the PEGylated anti-lysozyme Goldbody, the AuNPs with an average diameter of 3.6 nm (Fig. 1A) were synthesized as previously reported.9 Then Pep1 and PEGs were conjugated on AuNPs with Au–S bonds between the surface gold atoms and the Cys residues of Pep1 and PEGs (Fig. 1B and S2–S4†). The slight absorbance shift (Fig. 1C), the slight colour change of the solution (inset of Fig. 1C), and the increase of hydrodynamic diameter (Fig. 1D) indicate the successful conjugation of Pep1 and PEG onto AuNPs. No precipitation was observed during the conjugation, indicating the crosslinking due to the dithiol Pep1 and PEG was negligible at the initial concentration of the reactant (AuNP solution) below 80 nM.
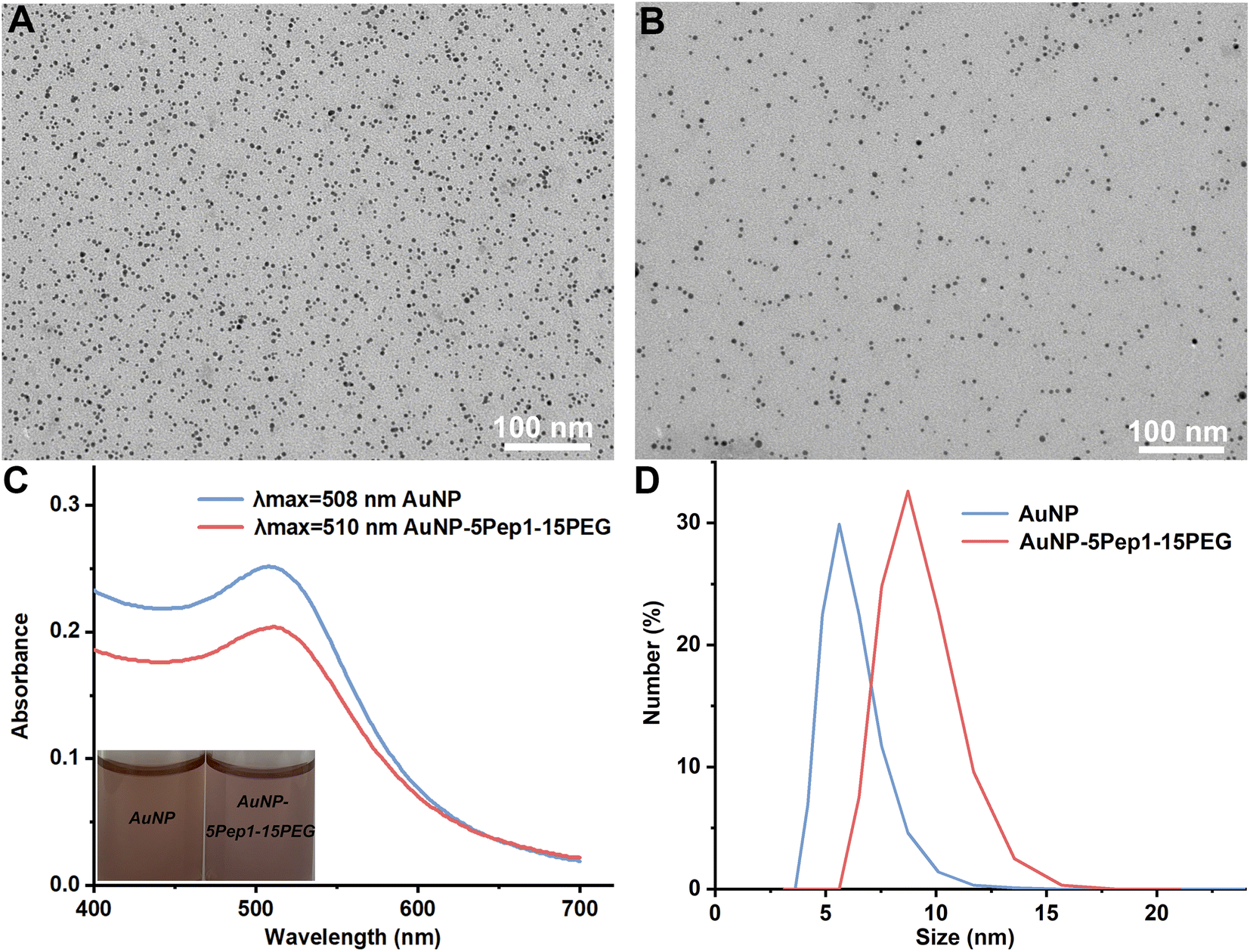 | ||
| Fig. 1 Characterization of the PEGylated anti-lysozyme Goldbody (AuNP–5Pep1–15(HS–PEG600–SH)). (A) TEM image of the as-synthesized bare AuNPs. (B) TEM image of AuNP–5Pep1–15(HS–PEG600–SH). (C) UV-vis spectra of the as-synthesized bare AuNPs and AuNP–5Pep1–15(HS–PEG600–SH), the insets are the optical pictures of the corresponding solutions. (D) DLS size distributions by number of the bare AuNPs (hydrodynamic diameter: 6.0 ± 1.3 nm) and AuNP–5Pep1–15(HS–PEG600–SH) (hydrodynamic diameter: 9.0 ± 1.6 nm). | ||
Since HS–PEG600–SH showed the best effects on assisting the conformational engineering of Pep1 on AuNPs (Fig. 2A), for convenience, unless otherwise stated, PEG in the figures of this paper represents HS–PEG600–SH. The other three different PEGs, i.e., HOOC–PEG300–SH, HOOC–PEG600–SH and HS–PEG2000–SH, were used as controls to demonstrate the critical role of two anchor-points method in conformational engineering and the effect of the length of PEG chain.
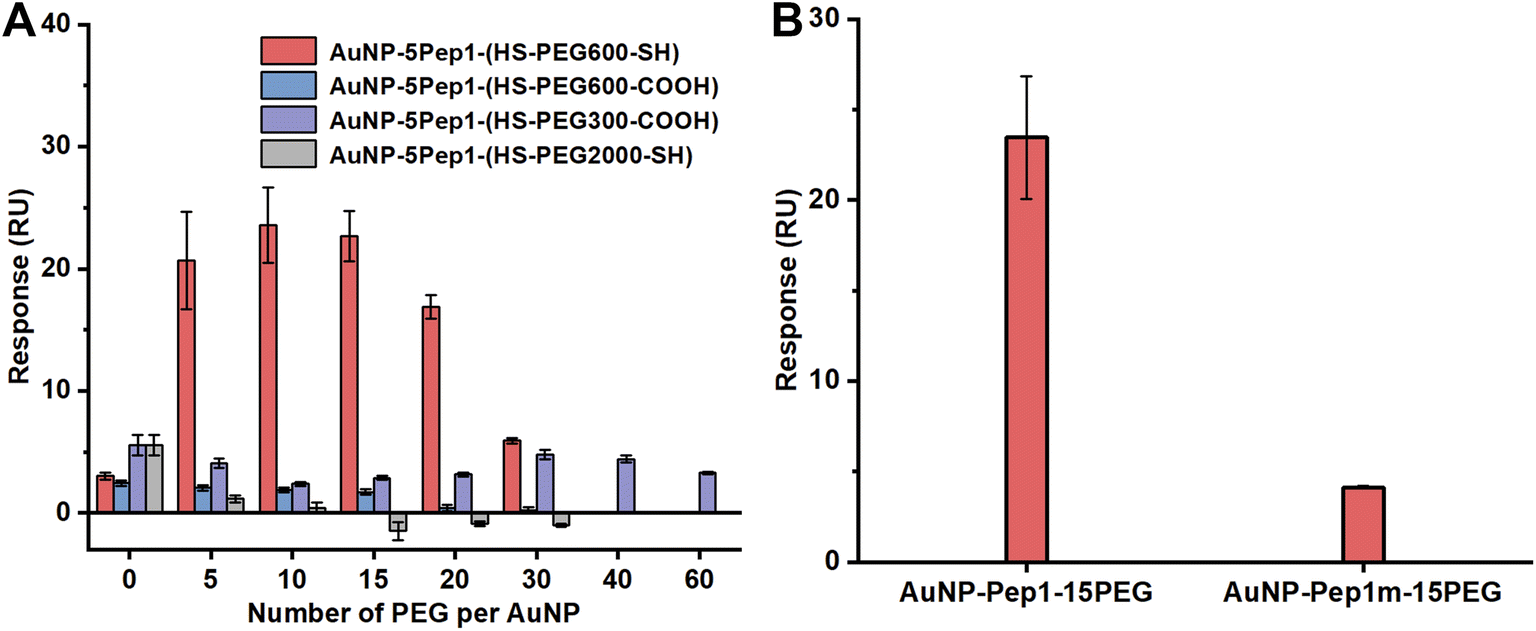 | ||
| Fig. 2 Conformational engineering of the CDR peptides on AuNPs. (A) Effects of the number of PEG per AuNP, the MW of PEG, and the terminal groups of PEG on the binding of the AuNP–5Pep1–PEG conjugates with the immobilized HEWL measured on a Biacore T200. (B) SPR binding signals of AuNP–5Pep1–15(HS–PEG600–SH) and AuNP–5Pep1m–15(HS–PEG600–SH) with the immobilized HEWL. | ||
Similar to the previous report,9 the density of the conjugated molecules, i.e., the average number of PEG per AuNP (as the number of Pep1 is fixed), is apparently a major parameter to tune the conformation of the conjugated Pep1. So, while fixing the number of Pep1, we conjugated different numbers of PEGs on AuNPs, and investigated the effects of the PEG number per AuNP, the MW of PEGs, and the terminal groups of PEGs on the binding of the AuNP–5Pep1–PEG with lysozyme using the standard SPR technique. As can be clearly seen from Fig. 2A, HS–PEG600–SH shows the best effect on restoring the native conformation of the CDR peptide (Pep1), so as to achieve the strongest binding with lysozyme. While the three other PEGs show much less effect on the binding of the AuNP-conjugates with lysozyme. Among AuNP–5Pep1–(HS–PEG600–SH) species with different PEG numbers, 10–15 HS–PEG600–SH molecules per AuNP show the strongest binding affinity with HEWL, suggesting it is the optimal PEG density range for assisting Pep1 to restore the native conformation of the CDR fragment in the original antibody, cAB-lys3 (Fig. 2A). Considering that the bare surface of AuNPs should be covered as much as possible to prevent the non-specific binding with other proteins, and previous studies show that 15 Pep1 per AuNP is enough to suppress the non-specific binding surface of AuNPs,9,10 the AuNP–5Pep1–15(HS–PEG600–SH) or simplified as AuNP–5Pep1–15PEG is the optimal PEGylated anti-lysozyme Goldbody.
To demonstrate that the function of the PEGylated anti-lysozyme Goldbody is really due to the conformational engineering or the restoration of the native/binding conformation of the CDR peptide, Pep1m, a control peptide with almost exactly the same sequence as that of Pep1 but containing only one terminal Cys (Table S1†), was conjugated on AuNPs with the same number of PEG as that for the AuNP–5Pep1–15PEG. As shown in Fig. 2B, the AuNP–5Pep1m–15PEG conjugate shows much weaker binding with lysozyme than the AuNP–5Pep1–15PEG conjugate (the PEGylated anti-lysozyme Goldbody), clearly indicating the necessity of the two Au–S bonds for the conformational engineering of Pep1 and the importance of the correct conformation of Pep1 for the strong binding of the PEGylated anti-lysozyme Goldbody with lysozyme.
Specific interaction between the PEGylated anti-lysozyme Goldbody and HEWL
To demonstrate that the interaction between the PEGylated anti-lysozyme Goldbody and HEWL is specific, DLS was used to characterize the formation of possible complexes between the PEGylated anti-lysozyme Goldbody and HEWL (MW: 14.7 kDa, pI: 11), as well as that between the PEGylated anti-lysozyme Goldbody and RNase A (MW: 13.7 kDa, pI: 9.45), a control protein that is very similar to lysozyme in many aspects including size, structural feature and charge. As shown in Fig. 3A, the hydrodynamic diameter of the mixture of AuNP–5Pep1–15PEG and RNase A is almost the same (within experimental errors) as that of the “pure” AuNP–5Pep1–15PEG (Fig. 1D), indicating that no complex was formed between AuNP–5Pep1–15PEG and RNase A; as a dramatic comparison, the hydrodynamic diameter of AuNP–5Pep1–15PEG changed significantly after mixing with HEWL (Fig. 3A), indicating the formation of the PEGylated anti-lysozyme Goldbody/lysozyme complex. As DLS is not accurate in determination of the absolute size values, especially because of lacks of accurate parameters including the refractive index of the complex available for the calculation, the estimated large hydrodynamic diameter does not represent the real size of the lysozyme-PEGylated anti-lysozyme Goldbody complex. Nevertheless, these results demonstrate qualitatively and unambiguously that the PEGylated anti-lysozyme Goldbody can discriminate these two similar proteins.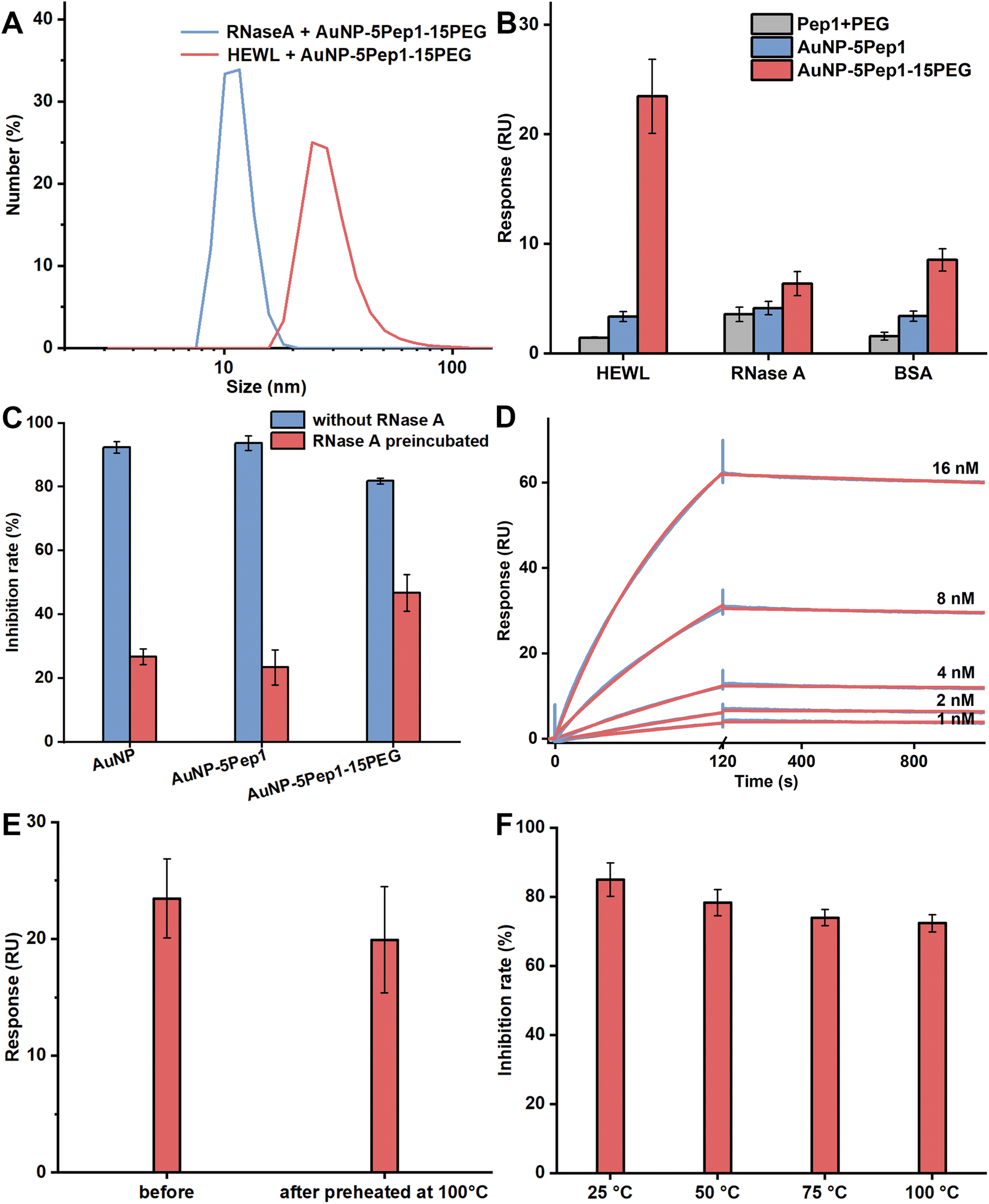 | ||
| Fig. 3 The specific interaction between the PEGylated anti-lysozyme Goldbody and HEWL and the thermal stability of the PEGylated anti-lysozyme Goldbody. (A) DLS size distributions by number of the mixture of AuNP–5Pep1–15(HS–PEG600–SH) and RNase A (hydrodynamic diameter: 11.3 ± 1.8 nm) and the mixture of AuNP–5Pep1–15(HS–PEG600–SH) and HEWL (hydrodynamic diameter: 29.6 ± 9.7 nm). (B) SPR binding signals of AuNP–5Pep1–15(HS–PEG600–SH) (red), AuNP–5Pep1 (blue), and pure Pep1 plus PEG (grey) with the equal concentrations onto the immobilized HEWL, RNase A and BSA. (C) Activities of HEWL inhibited by AuNPs, AuNP–Pep1 and AuNP–5Pep1–15(HS–PEG600–SH) in the absence/presence of RNase A. Blue columns: NPs (final concentration of 24 nM) were incubated with HEWL (final concentration of 30 nM); red columns: NPs (final concentration of 24 nM) were pre-incubated with RNase A (final concentration of 150 nM), and then HEWL (final concentration of 30 nM) was added. Error bars indicate SDs. (D) SPR kinetics of the interaction between AuNP–5Pep1–15(HS–PEG600–SH) and the immobilized HEWL. Red curves are the fitting lines. (E) SPR binding signals of AuNP–5Pep1–15(HS–PEG600–SH) (before and after preheating for 30 min) with the immobilized HEWL. (F) Inhibition of the activity of HEWL by AuNP–5Pep1–15(HS–PEG600–SH) that had been heated for 30 min at different temperatures. | ||
To further demonstrate the binding specificity of the PEGylated anti-lysozyme Goldbody, the SPR technique was used to quantify the binding of the PEGylated anti-lysozyme Goldbody with the immobilized HEWL. Also, RNase A and BSA were immobilized in two different channels as controls. As shown in Fig. 3B, the PEGylated anti-lysozyme Goldbody only strongly binds with HEWL, but binds very weakly with both RNase A and BSA mainly via non-specific binding. Again, these results prove that the PEGylated anti-lysozyme Goldbody can specifically recognize HEWL, the targeted antigen.
Remarkably, the PEGylated anti-lysozyme Goldbody can specifically recognize and inhibit HEWL even in the presence of a high concentration of RNase A. As can be seen from Fig. 3C, the bare AuNPs and the AuNP–5Pep1 can completely inhibit the activity of HEWL, which is due to the strong non-specific binding on the bare surfaces of AuNPs.17,18 This is also an evidence that 5 Pep1 per AuNP is not enough to cover the surfaces of AuNPs. When the bare AuNPs and the AuNP–5Pep1 were pre-incubated with RNase A, the exposed bare surfaces of AuNPs will adsorb and thus be covered by RNase A, losing the capability of binding with other proteins. This is why only little inhibition effect on HEWL was observed for these two species. On the contrary, even after the PEGylated Goldbody being pre-incubated with a high concentration of RNase A, the PEGylated Goldbody can significantly inhibit the activity of HEWL, which is similar to the original anti-lysozyme Goldbody.9
The affinity of the specific binding of the PEGylated anti-lysozyme Goldbody with HEWL was then determined by SPR (Fig. 3D). The binding data can be well fitted with the 1:1 model. The obtained apparent affinity KD value is 5.6 × 10−11 M (kon = 6.1 × 105 M−1 s−1 and koff = 3.4 × 10−5 s−1). Considering the relatively large experimental errors due to the non-homogeneity of NPs, this slightly stronger apparent affinity is basically at the same level as that of the original anti-lysozyme Goldbody (1.5 × 10−10 M).9
Thermal stability of the PEGylated anti-lysozyme Goldbody
Goldbodies consist of a rigid core of AuNP, which endows the Goldbodies with excellent thermal stability.9 The PEGylation does not change the stable core, so, just as expected, the PEGylated anti-lysozyme Goldbody also has excellent thermal stability. Fig. 3E and F show that the PEGylated anti-lysozyme Goldbody maintains its binding capability with lysozyme and its inhibition ability on lysozyme even after incubation at 100 °C for 30 min.Design and characterization of the PEGylated anti-EGFR Goldbody
To verify that the above PEGylation strategy is generally applicable to all Goldbodies, the same procedure was used to PEGylate another Goldbody, the anti-EGFR Goldbody.9 The original anti-EGFR Goldbody was created by conformationally engineering a CDR fragment (Pep2) of an anti-EGFR antibody, 7D12,40 on AuNPs; and the optimal peptide density is 40–60 Pep2 per AuNP (3.6 nm), very similar to that of the anti-lysozyme Goldbody.9 So, for the PEGylated anti-EGFR Goldbody, we also fixed the number of Pep2 per AuNP to 5.Similar to that of the PEGylated anti-lysozyme Goldbody, the Pep2 conjugation and PEGylation of AuNPs resulted in negatively charged and well dispersed conjugates with increased hydrodynamic diameter (Fig. 4A, B and S5–S7†).
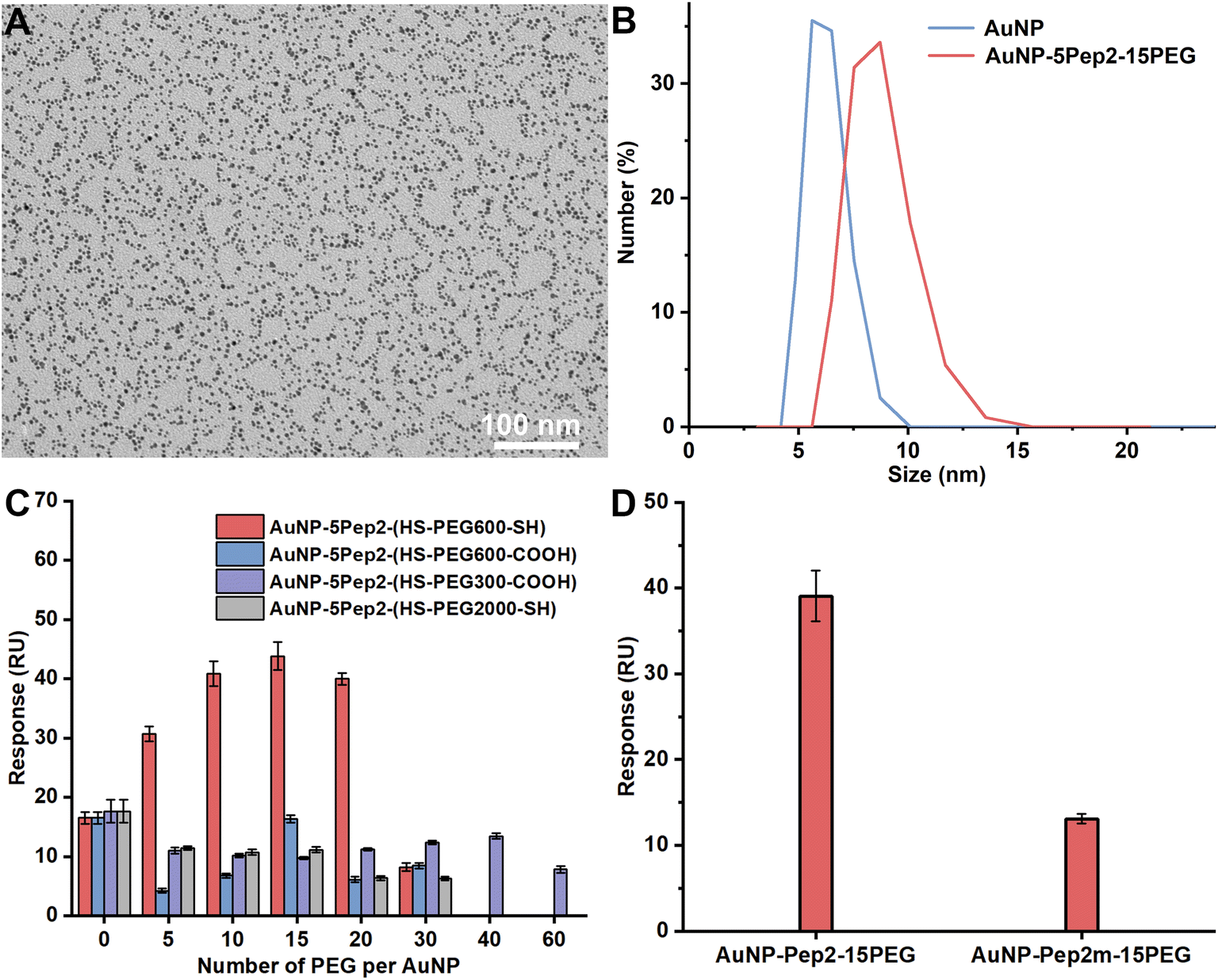 | ||
| Fig. 4 PEGylation of the anti-EGFR Goldbody. (A) TEM image of the PEGylated anti-EGFR Goldbody, i.e., AuNP–5Pep2–15(HS–PEG600–SH). (B) DLS size distributions by number of the as-prepared bare AuNPs (hydrodynamic diameter: 6.2 ± 0.9 nm) and AuNP–5Pep2–15(HS–PEG600–SH) (hydrodynamic diameter: 8.6 ± 1.4 nm). (C) Effects of number, MW, and terminal groups of PEG on the binding of AuNPs–5Pep2–PEG conjugates onto the immobilized EGFR. (D) SPR binding signals of AuNP–5Pep2–15(HS–PEG600–SH) and AuNP–5Pep2m–15(HS–PEG600–SH) with the immobilized EGFR. | ||
Similar to the PEGylation of the anti-lysozyme Goldbody, we also conjugated different numbers of PEG on AuNPs to find the optimal number that assists the restoration of the native conformation and the antigen-recognition function of the CDR peptide (Pep2) on AuNPs. As determined by the SPR binding signals with the immobilized EGFR (Fig. 4C), the best PEG molecule to assist the conformational engineering of Pep2 is still the HS–PEG600–SH, and the optimal PEG number per AuNP (3.6 nm) is around 15. Therefore, the AuNP–5Pep2–15(HS–PEG600–SH) is the PEGylated anti-EGFR Goldbody.
Similarly, to demonstrate that the function of the PEGylated anti-EGFR Goldbody is really due to the restoration of the native/binding conformation of the CDR peptide (Pep2), Pep2m, a control peptide with almost exactly the same sequence as that of Pep2 but containing only one terminal Cys (Table S1†), was conjugated on AuNPs with the same number of PEG as that for the AuNP–5Pep2–15PEG. As shown in Fig. 4D, the AuNP–5Pep1m–15PEG shows much weaker binding with EGFR than the AuNP–5Pep2–15PEG (the PEGylated anti-EGFR Goldbody), indicating again that the function of the PEGylated anti-EGFR Goldbody comes from conformational engineering of the Pep2 peptides, and that the two Au–S bonds are critical for the conformational engineering.
The binding specificity and thermal stability of the PEGylated anti-EGFR Goldbody
To demonstrate that the interaction between the PEGylated anti-EGFR Goldbody and EGFR is specific, the PEGylated anti-EGFR Goldbody was mixed separately with EGFR and BSA, and then DLS was used to characterize the formation of possible complexes. As shown in Fig. 5A, within experimental errors, there is no apparent change of the size of AuNP–5Pep2–15(HS–PEG600–SH) after incubation with BSA (MW: 66.4 kDa, pI: 4.7). While the size of AuNP–5Pep2–15(HS–PEG600–SH) increased significantly after incubation with EGFR (MW: 110 kDa, pI: 6.52), indicating that the PEGylated anti-EGFR Goldbody binds preferably with EGFR.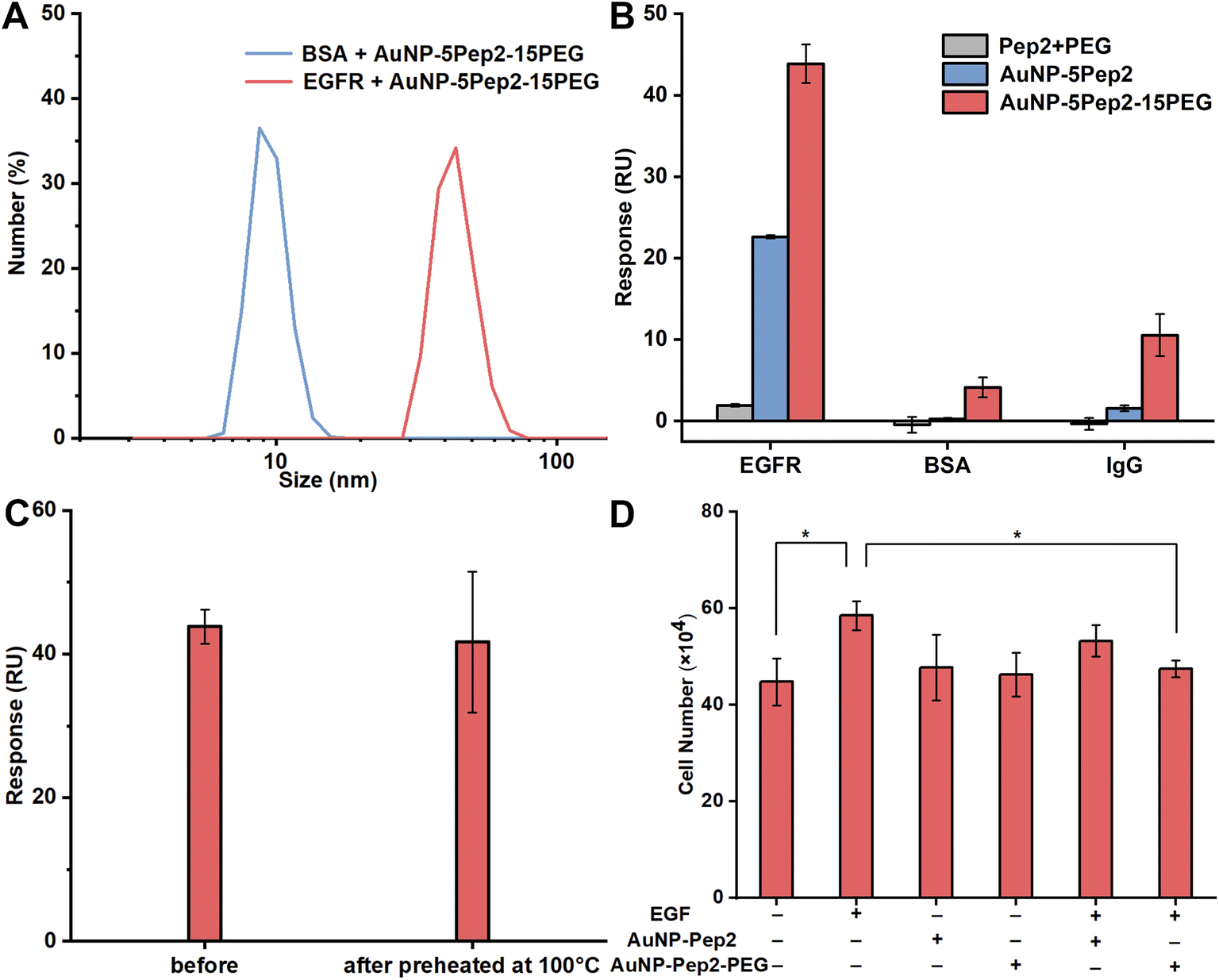 | ||
| Fig. 5 Specific interaction between the PEGylated anti-EGFR Goldbody (AuNP–5Pep2–15(HS–PEG600–SH)) and EGFR. (A) DLS size distributions by number of the mixture of AuNP–5Pep2–15(HS–PEG600–SH) and BSA (hydrodynamic diameter: 9.5 ± 1.4 nm) and the mixture of AuNP–5Pep2–15(HS–PEG600–SH) and EGFR (hydrodynamic diameter: 43.5 ± 7.1 nm). (B) SPR binding signals of AuNP–5Pep2–15(HS–PEG600–SH) (red), AuNP–5Pep2 (blue), and free Pep2 plus PEG (grey) with the immobilized EGFR, BSA, and IgG. All species have the same concentration of peptides. (C) SPR binding signals of AuNP–5Pep2–15(HS–PEG600–SH) (before and after preheated for 30 min) with the immobilized EGFR. (D) Interaction between the PEGylated anti-EGFR Goldbody and EGFR at cellular level, resulting in the inhibition of EGF-induced cell proliferation. Error bars indicate SDs. *P < 0.05. | ||
The specific binding of the PEGylated anti-EGFR Goldbody with EGFR is also confirmed by the SPR binding data. As shown in Fig. 5B, the PEGylated anti-EGFR Goldbody binds strongly with the immobilized EGFR, but only very weakly with the two immobilized control proteins, BSA and IgG.
The binding kinetics between the PEGylated anti-EGFR Goldbody and EGFR was determined by SPR with EGFR immobilized on a CM5 chip and AuNP–Pep2–15(HS–PEG600–SH) of different concentrations as the mobile phase. The binding data (Fig. S8†) can be fitted with the 1:1 model, with an apparent affinity of 8.6 × 10−12 M (kon = 1.4 × 107 M−1 s−1 and koff = 1.2 × 10−4 s−1). Again, this apparent affinity is reasonably at the same level as that of the original anti-EGFR Goldbody binding to EGFR (1.2 × 10−11 M),9 considering the experimental errors due to the non-homogeneity of NPs.
Once again, the PEGylated anti-EGFR Goldbody shows excellent thermal stability. The PEGylated anti-EGFR Goldbody maintained its binding capability after being preheated at 100 °C for 30 min (Fig. 5C).
Inhibition effect of the PEGylated anti-EGFR Goldbody on the EGF-induced cell proliferation
All the above characterizations of the specific interaction between the PEGylated anti-EGFR Goldbody and EGFR were carried out at molecular level using the extracellular soluble part of EGFR. To verify the specific interaction at cellular level, and to pave the way for its future biomedical applications, we also applied the PEGylated anti-EGFR Goldbody to HeLa cells, a cell line with a moderate expression of EGFR on its membrane. As the PEGylated anti-EGFR Goldbody is designed to bind to the domain III of the extracellular part of EGFR, where is also the binding site for EGF, in principle, the PEGylated anti-EGFR Goldbody will compete with EGF for the EGFR of HeLa cells, and thus will inhibit the EGF-induced cell proliferation.9,10 As shown in Fig. 5D, the PEGylated anti-EGFR Goldbody indeed inhibited the EGF-induced cell proliferation, just like the original anti-EGFR Goldbody did.9,10 As a comparison, the AuNP–5Pep2 did not inhibit the EGF-induced cell proliferation, highlighting the important role of PEG for restoration of the native conformation of Pep2.The role of PEG on the conformational engineering of peptides on AuNPs
When the number of CDR peptides on the two Goldbodies was reduced from 60 to 5, a large portion of the surface of AuNPs would be exposed. Since the “bare” surface of the unconjugated AuNPs can bind strongly with almost any proteins,17,18 to prevent unwanted non-specific binding with non-targeted proteins, the exposed bare surface area of AuNP has to be covered with “non-sticky” molecules, for example PEG. The above results show that the optimum compositions for both PEGylated Goldbodies are 5 peptides (Pep1 or Pep2) plus 15 HS–PEG600–SH for one AuNP (3.6 nm), which is basically consistent with the previous result that 15 Pep1 could sufficiently suppress the non-specific binding of AuNPs (3.6 nm).9Covering the bare surface of AuNPs is not the only role of PEG. As the SPR data clearly show, increasing the number of HS–PEG600–SH conjugated on AuNPs could turn the AuNP–5Pep1/Pep2, which did not bind specifically with HEWL/EGFR, into Goldbodies (AuNP–5Pep1/Pep2–15(HS–PEG600–SH)), which can strongly and specifically bind to HEWL/EGFR. There are two possible reasons that the AuNP–5Pep1/Pep2 was inactive. First, some residues of Pep1/Pep2 might form unwanted interactions with the bare surface of AuNPs. Second, the bare surface of AuNPs is too large, leaving too much freedom for the conjugated peptides to move around and adopt numerous other conformations. Therefore, covering the bare surface of AuNPs with PEG could not only significantly prevent the unwanted interactions between peptide residues and the bare surface of AuNPs, but also impose constraints on the movement of the Au–S bonds of the conjugated peptides, and thus reduce the conformational space for the peptides to achieve a beneficial entropy effect for the native conformation of the CDR fragments.
After dramatically reducing the number of CDRs on AuNPs, there is obvious no doubt that the success of Goldbodies is due to conformational engineering, not due to multivalency effects. The fundamental theory for the conformational engineering of the CDR fragments on AuNPs is the CLEFs hypothesis.15,16 According to this hypothesis, when the right constraints are imposed, the CDR fragments (i.e., CLEFs) will automatically fold to their native conformations.
To impose constraints on the conjugated peptides, PEG with two terminal –SH groups is apparently much better than PEG with a single –SH group, because the PEG with two terminal –SH groups can form a loop with both ends immobilized on AuNPs. Therefore, the space between the two immobilization positions of the –S–PEG–S– is also difficult for the peptide to access or pass through. However, when the number of –S–PEG–S– on AuNPs further increases, the surface of AuNPs might become too crowded for the conjugated peptides to adopt their native conformation, as demonstrated by the decreased SPR binding signals after the number of PEG on AuNPs exceeds the optimum number (Fig. 2A and 4C).
Another important factor to influence the conformation engineering is the MW of the PEG. As shown in both Fig. 2A and 4C, HS–PEG2000–SH shows little effect to increase the binding signal for both systems. The possible reason is that the chain of HS–PEG2000–SH is longer than both Pep1 and Pep2, and then the conjugated flexible PEG would partially cover or bury the conjugated peptides, preventing their bindings with the antigens. Therefore, the HS–PEG–SH with a chain length shorter than the conjugated peptide is better to assist the conformational engineering.
At present, antibodies are widely used in many fields including treatment of many diseases such as cancers.41–44 Compared with widely-applied monoclonal antibodies, Goldbodies have the advantages of excellent stability and no need for humanization. Accumulating evidence shows that “Goldization” will become a general technique to produce more and more Goldbodies targeting various antigens.9–11,45,46 And now we show here that the PEGylation of Goldbodies can dramatically reduce the cost of Goldbodies. It is foreseeable that PEGylation would likely become a standard procedure for Goldbodies for their future potential applications in various fields.
Conclusions
In summary, we have shown here that PEG with two terminal –SH groups could be used to effectively aid the conformational engineering of the CDR fragments of monoclonal antibodies on AuNPs. Two PEGylated Goldbodies, i.e., the PEGylated anti-lysozyme Goldbody and the PEGylated anti-EGFR Goldbody, have been thus created. With the aid of the conjugated PEG, the number of CDRs per Goldbody has been reduced by more than one order of magnitude, and the PEGylated Goldbodies thus eliminate the doubt that the success of Goldbodies might be due to the multivalency effect instead of the conformational engineering. The conjugated PEG not only prevents the strong non-specific binding of the bare surface of AuNPs, but also imposes proper constraints on the freedom of the conjugated peptides to achieve a beneficial entropy effect for the native conformation of the peptides, which further proves the CLEFs hypothesis. PEGylation also dramatically reduces the cost of Goldbodies, thus paves the way for future applications of Goldbodies in various fields.Author contributions
A. C. and H. W. supervised the project. T. G., Y.-Y. L. and C. L. conducted the experiments. All authors analysed the data. A. C., T. G. and H. W. co-wrote manuscript with contribution from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31871007, 32071404, 31771105, and 22071145) and the National Key Research and Development Plan of China (No. 2016YFA0201602).Notes and references
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- I. H. El-Sayed, X. H. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS PubMed.
- B. Huang, Z. Yang, S. Fang, Y. Li, Z. Zhong, R. Zheng, J. Zhang, H. Wang, S. Wang, Q. Zou and L. Wu, Nanoscale, 2020, 12, 5834–5847 RSC.
- Z. Luo, Y. Hu, R. Xin, B. Zhang, J. Li, X. Ding, Y. Hou, L. Yang and K. Cai, J. Biomed. Mater. Res., Part A, 2014, 102, 3781–3794 CrossRef PubMed.
- S. W. Vedakumari, S. J. V. Jancy, Y. R. Pravin, J. Bhoopathy, K. Iyswariya, S. Thomas, R. Rubiya, L. Prabakaran, C. Kumar, P. Prabu and R. Murugesan, Environ. Res., 2022, 209, 112925 CrossRef CAS PubMed.
- N. Mitri, K. Rahme, G. Fracasso and E. Ghanem, Nanotechnology, 2022, 33, 315101 CrossRef.
- A. Quarta, D. Bernareggi, F. Benigni, E. Luison, G. Nano, S. Nitti, M. C. Cesta, L. Di Ciccio, S. Canevari, T. Pellegrino and M. Figini, Nanoscale, 2015, 7, 2336–2351 RSC.
- S. A. Townsend, G. D. Evrony, F. X. Gu, M. P. Schulz, R. H. Brown and R. Langer, Biomaterials, 2007, 28, 5176–5184 CrossRef CAS.
- G. H. Yan, K. Wang, Z. Shao, L. Luo, Z. M. Song, J. Chen, R. Jin, X. Deng, H. Wang, Z. Cao, Y. Liu and A. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E34–E43 CAS.
- L. Luo, Y. Y. Liu, T. Gao, X. Wang, J. Chen, H. Wang, Y. Liu and A. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 34514–34523 CrossRef CAS PubMed.
- Q. Liu, L. Sheng, Y. Y. Liu, T. Gao, H. Wang, Y. Liu and A. Cao, ChemMedChem, 2022, 17, e202100623 CAS.
- R. Willson, Faculty Opinions Recommendation of [G.H. Yan et al.], Proc. Natl. Acad. Sci. U. S. A., 2018, 115(1), E34–E43, DOI:10.3410/f.732327157.793541057 , Faculty Opinions, Online, January 9, 2018..
- M. Yu, N. Bovet, C. J. Satterley, S. Bengió, K. R. J. Lovelock, P. K. Milligan, R. G. Jones, D. P. Woodruff and V. Dhanak, Phys. Rev. Lett., 2006, 97, 166102 CrossRef.
- M. M. Biener, J. Biener and C. M. Friend, Surf. Sci., 2007, 601, 1659–1667 CrossRef CAS.
- A. Cao, Acta Phys.-Chim. Sin., 2020, 36, 1907002 Search PubMed.
- A. Cao, Protein J., 2020, 39, 422–433 CrossRef CAS PubMed.
- S. T. Yang, Y. Liu, Y. W. Wang and A. Cao, Small, 2013, 9, 1635–1653 CrossRef CAS PubMed.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- D. Shi, D. Beasock, A. Fessler, J. Szebeni, J. Ljubimova, K. Afonin and M. Dobrovolskaia, Adv. Drug Delivery Rev., 2022, 180, 114079 CrossRef CAS PubMed.
- S. T. Yang, K. A. S. Fernando, J. H. Liu, J. Wang, H. F. Sun, Y. Liu, M. Chen, Y. Huang, X. Wang, H. Wang and Y. P. Sun, Small, 2008, 4, 940–944 CrossRef CAS PubMed.
- L. W. Shi, J. Q. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- J. Zhao, Z. Qin, J. Wu, L. Li, Q. Jin and J. Ji, Biomater. Sci., 2018, 6, 200–206 RSC.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. de la Fuente, G. Nienhaus and W. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS.
- Y. Du, J. Jin, H. Liang and W. Jiang, Langmuir, 2019, 35, 8316–8324 CAS.
- E. Harrison, J. Nicol, M. Macias-Montero, G. Burke, J. Coulter, B. Meenan and D. Dixon, Mater. Sci. Eng., C, 2016, 62, 710–718 CrossRef CAS.
- USFDA, Center for food safety and applied nutrition, Food additive status list, Polyethylene glycol, 2019.
- S. Alconcel, A. Baas and H. Maynard, Polym. Chem., 2011, 2, 1442 RSC.
- J. Suk, Q. Xu, N. Kim, J. Hanes and L. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- M. Li, S. Jiang, J. Simon, D. Paßlick, M. Frey, M. Wagner, V. Mailänder, D. Crespy and K. Landfester, Nano Lett., 2021, 21, 1591–1598 CrossRef CAS.
- J. L. Perry, K. G. Reuter, M. P. Kai, K. P. Herlihy, S. W. Jones, J. C. Luft, M. Napier, J. E. Bear and J. M. DeSimone, Nano Lett., 2012, 12, 5304–5310 CrossRef CAS PubMed.
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974–1989 CrossRef CAS PubMed.
- Q. Yang, S. W. Jones, C. L. Parker, W. C. Zamboni, J. E. Bear and S. K. Lai, Mol. Pharmaceutics, 2014, 11, 1250–1258 CrossRef CAS PubMed.
- Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085–1092 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- J. Jokerst, T. Lobovkina, R. Zare and S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- J. Stefanick, J. Ashley, T. Kiziltepe and B. Bilgicer, ACS Nano, 2013, 7, 2935–2947 CrossRef CAS.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- W. Haiss, N. Thanh, J. Aveyard and D. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- A. Desmyter, T. R. Transue, M. A. Ghahroudi, M. H. D. Thi, F. Poortmans, R. Hamers, S. Muyldermans and L. Wyns, Nat. Struct. Biol., 1996, 3, 803–811 CrossRef CAS PubMed.
- K. R. Schmitz, A. Bagchi, R. C. Roovers, P. M. P. V. E. Henegouwen and K. M. Ferguson, Structure, 2013, 21, 1214–1224 CrossRef CAS PubMed.
- C. U. Blank, J. B. Haanen, A. Ribas and T. N. Schumacher, Science, 2016, 352, 658–660 CrossRef CAS PubMed.
- M. Dougan, G. Dranoff and S. K. Dougan, Annu. Rev. Cancer Biol., 2019, 3, 55–75 CrossRef.
- P. T. Jones, P. H. Dear, J. Foote, M. S. Neuberger and G. Winter, Nature, 1986, 321, 522–525 CrossRef CAS.
- C. Queen, W. P. Schneider, H. E. Selick, P. W. Payne, N. F. Landolfi, J. F. Duncan, N. M. Avdalovic, M. Levitt, R. P. Junghans and T. A. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 10029–10033 CrossRef CAS.
- L. Zhao, S. Wang, Q. Hu, H. Jia, Y. Xin, L. Luo and F. Meng, Nanoscale, 2022, 14, 2802–2815 RSC.
- Y. Wang, X. Wang, T. Gao, C. Lou, H. Wang, Y. Liu and A. Cao, J. Phys. Chem. B, 2022, 126, 5045–5054 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03903f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |