
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA theoretical study based on DFT calculations on the different influence of functional groups on the C–H activation process via Pd-catalysed β-X elimination†
Xin Xiang,
Zeng-Xia Zhao * and
Hong-Xing Zhang*
* and
Hong-Xing Zhang*
Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, College of Chemistry, Jilin University 130023 Changchun, PR China. E-mail: zhaozx@jlu.edu.cn
First published on 15th September 2022
Abstract
We have performed a series of theoretical calculations for palladium-catalyzed β-X elimination reactions. The DFT calculation combined with energy decomposition analysis shows the determining factors of reactivity. Such as, the elemental composition, the structure of different functional groups and the stronger steric repulsions contribution.
Introduction
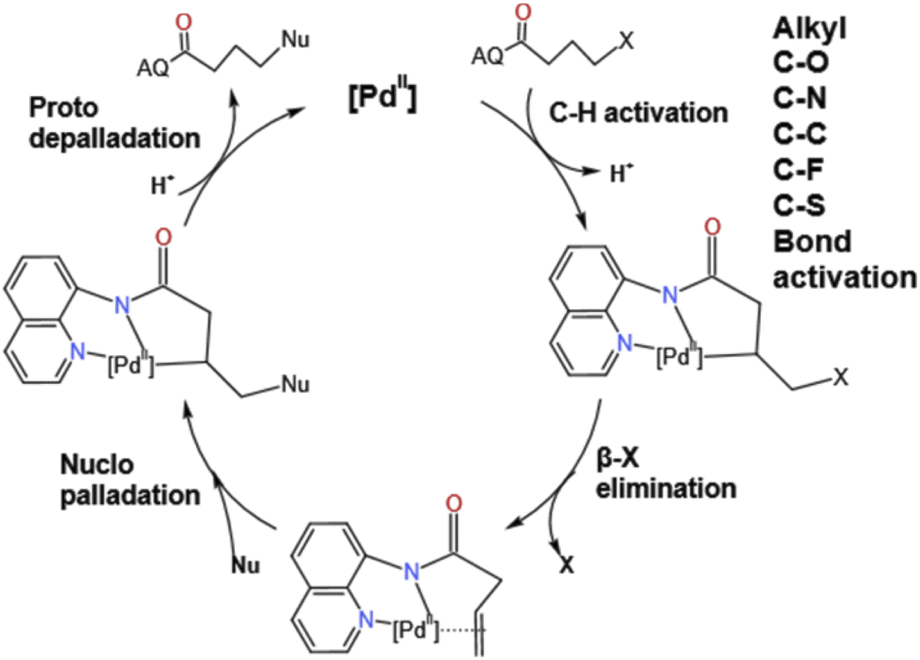
The transition metal catalyzed C–H activation has been very popular in recent years and has attracted a lot of attention from theoretical chemists.1–9 Since the 1970s, the coupling organic reactions with Ni, Ru, Rh,10,11 and Pd4,12–19 as catalysts have been developed rapidly and attracted the attention of many chemists. Pd, as an important part of catalysts, has attracted the attention of many chemists in recent years, such as, the Kumada, Heck, Suzuki and Negishi7 groups. Moreover, the ability to functionalize at C–H bonds in the absence of activation groups has great potential in many synthetic pathways. Among them the strategy of using a transition metal to insert starting materials and substitute the functional group at the target position became the most attractive pathway in synthesizing plenty of substrates used in industrial manufacture, the pharmaceutical field and bioengineering departments. In recent years, Engle's group1,8,14,20–25 has an impressive achievement on the activation of abundant C–O, C–N and C–C bonds via an oxidative addition pathway to form a specific electronically species. To further elucidate the β-X elimination mechanism an experiment was carried out by Engle and co-workers shown in Fig. 1. From these results, we worked out these questions: (i) what is the mechanism of this reaction? (ii) Why is the difference between position of hydroxy made such an influence in 2A-OH and HO-2A of this work? (iii) Why the yield of N-2A the lowest in these substituents? In this work, we theoretically investigated sp3 C–H functionalize of various substrates such as four different nucleophile groups shown in Fig. 2, catalyzed by Pd(II) complex to elucidate characteristic features of this reaction and provide a unique perspective of sp3 C–H functionalization. We selected these substrates to compare reactivity between different functional groups' sp3 C–H bonds, and difference between the presence and the absence of electron with drawing N atom; we made a clear answer to the above-mentioned questions, because this reaction is one of the most important reactions in forming newly redox-neutral approach. C–H provides us a crucial way for making C–C bonds, allowing access to unconventional and structures with high selectivity and efficiency. | ||
| Fig. 1 The mechanism of palladium-catalysed reaction of this work. | ||
 | ||
| Fig. 2 The proposed mechanism of the experimental given by the Engle's group. | ||
Computational details
All geometry optimizations without symmetry restriction were performed by the B3LYP/BSI level (BSI designates the basis set combination of LandL2DZ for Pd atom and 6-31G(d) for other nonmetallic atoms) with Grimme's GD3BJ dispersion corrections.26–29 Frequency calculation at the same level were performed to ensure the local minima (no imaginary frequencies) or the transition states (only one imaginary frequency), and to derive the thermochemical corrections for the enthalpies and free energies. The intrinsic reaction coordinate (IRC)30,31 calculations has been employed to track minimum energy paths from transition structure to the corresponding local minima. To evaluate better potential energy, single-point calculations were performed using the M06/BSII level,32–34 using the acetonitrile as the solvent and SMD as the solvation model, where BSII designates the basis set combination SDD for Pd atom and 6-311++G(d,p) for other atoms. Solvation effect (THF) was evaluated with SMD in this work. In all of the figures that contain energy diagrams, calculated relative Gibbs free energies (kcal mol−1) are presented. All these calculations were carried out using Gaussian16 program.35 Multiwfn36 and VMD37 was used to perform electron distribution. The optimized structures of the species under study were illustrated using CYLview.38Results and discussion
We will describe the detailed catalytic reaction process, active species, rate-determination step here. In addition, we will firstly make a comparison of the effect caused by different nucleophilic groups on the whole reaction process. We will make a comparison of reactivity among various reactive substituents and discuss determining factors of reactivity. In the end, the theoretical prediction of an advantaged suggestion will be presented to provide for laboratory reference, while the oxidative addition of C–H bonds is often an essential substrate activation step in important catalytic hydrocarbon functionalization.Overview of catalytic
The details of catalytic reaction mechanism and crucial steps were elucidated here. The overall free energy profiles for the catalytic C–H activation process by palladium catalyst was shown in Fig. 3. This reaction occurs through Pd catalyzed cycle; the first step is Pd-catalyst through C–H activation process added to the nitrogen on the benzene ring of the starting materials 1a's directing group 8-amino-quinoline (AQ); the second step is the rate determining step, it called β-heteroatom/carbon (β-X) elimination in which the leaving groups leave from the M4's geometry to create another stable geometry which contain unsaturated hydrocarbon/alkene; the third step is nucleo-palladation which from species M6 turns into species M9, in which it across a transition state TS4; the final step is the depalladation step also called catalyst regeneration process. Among them we will make a detailed analysis for two important elementary steps: carbon (β-X) elimination and nucleopalladation.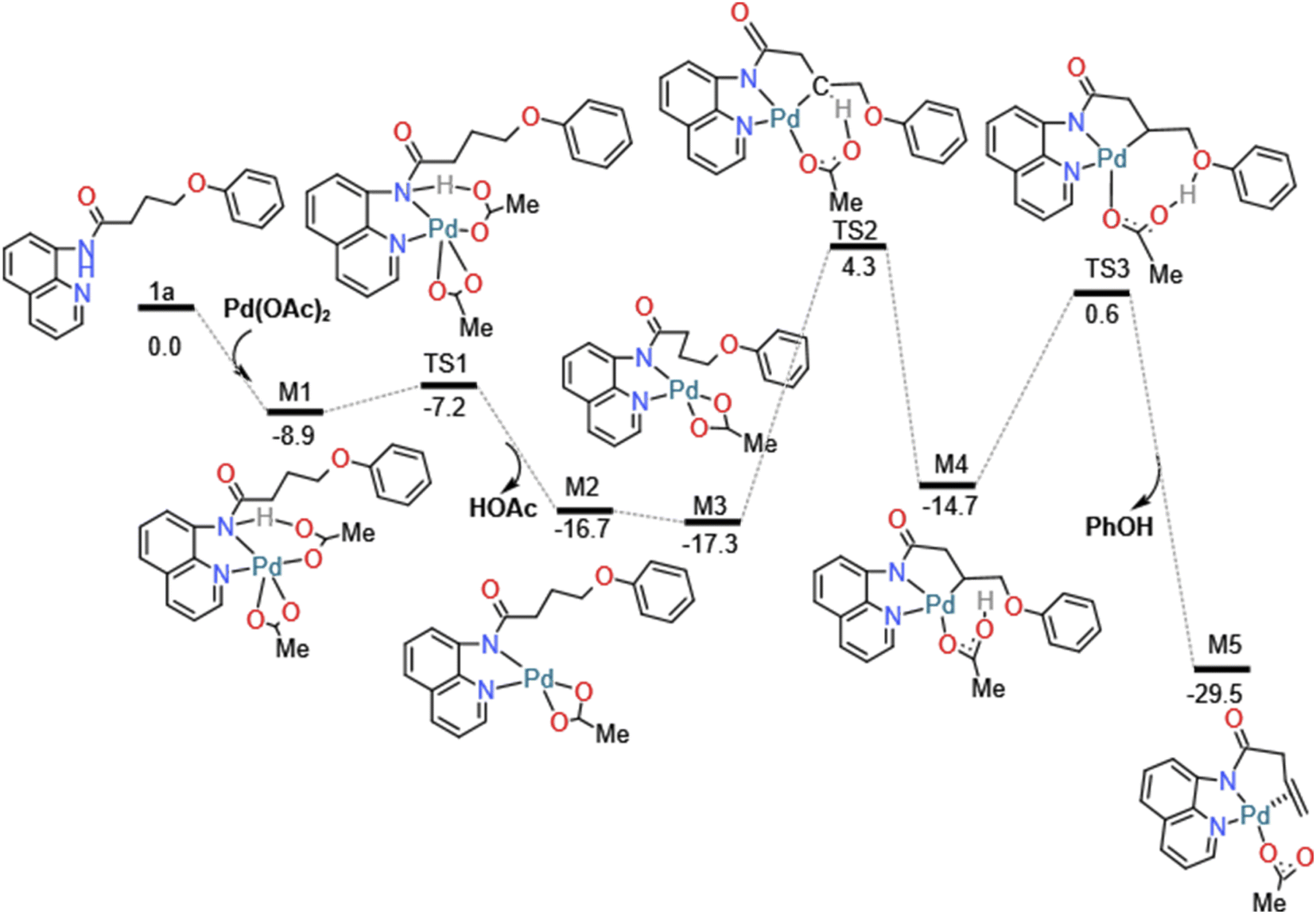 | ||
| Fig. 3 The first part free energy profile for HN-2A catalytic activation. | ||
As shown in Fig. 3, this reaction initiates from the coordination of the directing group 8-amino-quinoline (AQ) of 1a to the active Pd(II) center forming the complex M1 with exoergic by −8.9 kcal mol−1. In order to achieve the relatively remote γ-C's functionalization other than the carbon (α-C) closest to the carbonyl group. By introducing the 8-amino-quinoline (AQ), the directing effect of its bidentate was used to coordinated with pd(II) and activates N–H bond on amide, then followed by the C(sp3)–H activation on carbon chain's γ-C other than α-C to introduce the nucleophilic group to construct C–X bond. In addition, amide bonds are easily hydrolyzed under acidic or alkaline and biological conditions. The N–H activation of the coordinated 1a could take place via the transition state TS1 to form the five-membered metalacyclic intermediate M2 and dissociation of acetic acid (HOAc). This N–H bond activation step is a typical concerted metalation/deprotonation mechanism, with a small barrier of 1.7 kcal mol−1, once the N–H bond is activated and an acetic acid is eliminated with the generation of Pd(II) species. In addition, natural bond orbital (NBO) analysis was performed for TS1 to obtain the atomic charges. The analysis results indicate that the negative charge value of O (−0.711 e) in OAc− is much lower than that of O atom (−0.575 e) in the carbonyl. The N–H activation process can be illustrated by reduced Pd–N bond distances, 2.04 Å in M2 (see Fig. S1, ESI†), which means the chemical bond was formed.
In the following β-heteroatom/carbon (β-X) elimination, M2 isomerize into a more stable intermediate M3 with exoergic by −0.6 kcal mol−1. After generating the metal complex M3, the β-H2 transfer process to the O1 atom of acetic acid can be achieved by a barrier of 21.6 kcal mol−1, accompanied by the formation of Pd–C1 bond. The H transfer process can be illustrated by the gradually elongated C1–H2 bond and reduced Pd–C1 and O1–H2 bond, from the 1.10, 4.29 and 4.38 Å in M3, 1.42, 2.28 and 1.34 Å in TS2, and 2.74, 2.06 and 1.00 Å in M4 in which acetic acid act as a proton shuttle. The following H1 atom can easily attack the O2 atom of the leaving group and reductive elimination from M4 can be facilely accomplished via the regioselectivity-determining transition state TS3 generating the more stable π-alkene complex M5 with exergonic by 14.8 kcal mol−1. In the geometry of M5, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is almost perpendicular to the Pd heterocyclic plane, so the formed five-membered ring and six-membered ring do not have obvious twisted heterocyclic. The barrier for this process is predicted to be 15.2 kcal mol−1, which is accessible for experimental realization under mild conditions.
C bond is almost perpendicular to the Pd heterocyclic plane, so the formed five-membered ring and six-membered ring do not have obvious twisted heterocyclic. The barrier for this process is predicted to be 15.2 kcal mol−1, which is accessible for experimental realization under mild conditions.
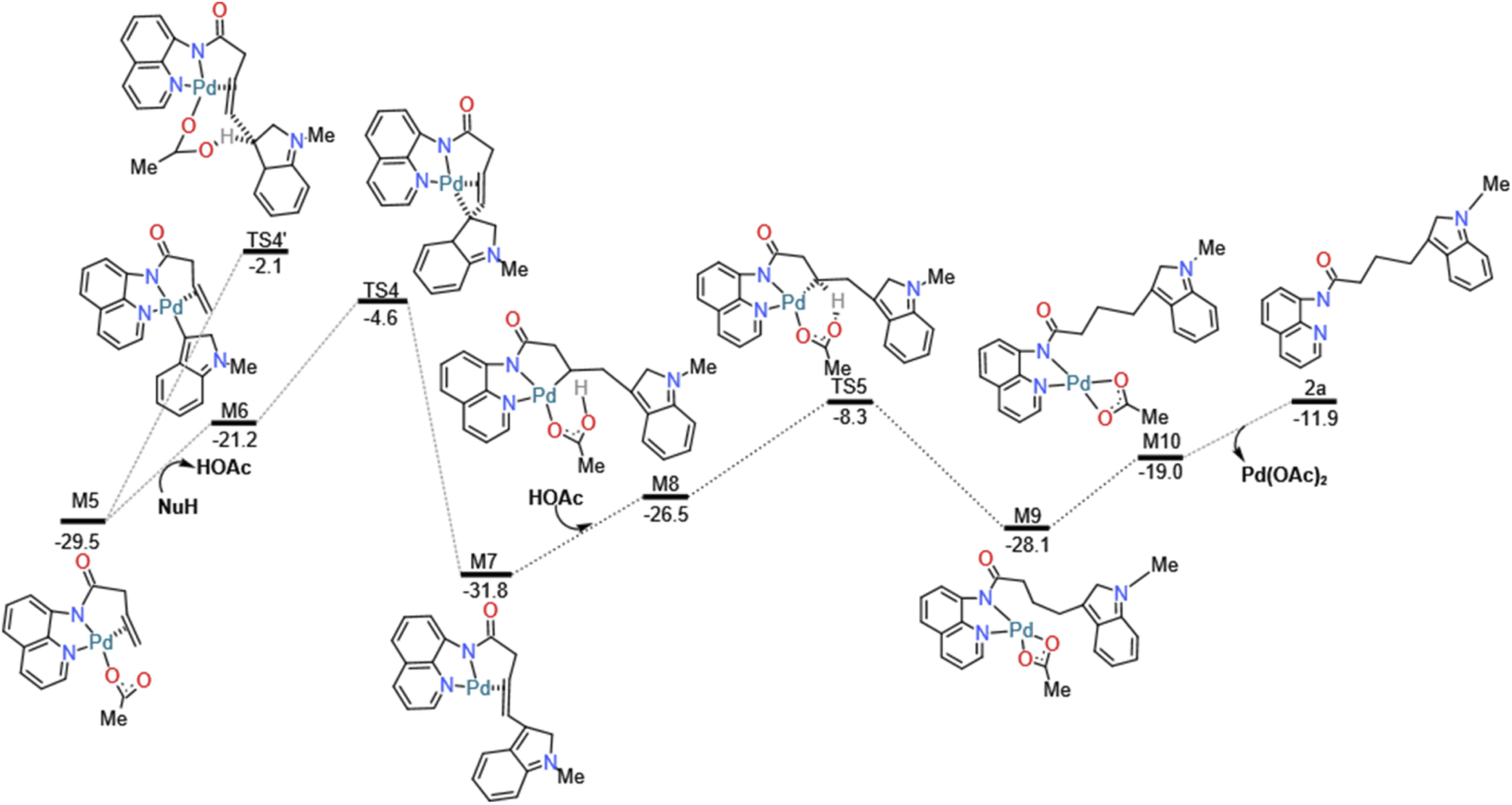
And then we considered the syn-nucleopalladation and anti-nucleopalladation pathways according to the energy profile. In the nucleo-palladation step, the nitrogen nucleophile (NuH) attacks the Pd(II) centre forming the intermediate M6 (in Fig. 4) through ligand exchange. In the meantime, intramolecular syn-nucleopalladation occurs via TS4 (in Table 1) syn-nucleopalladated intermediate M7, which is a [5–5–5] thick ring structure, the CC bond is almost perpendicular to the Pd heterocyclic plane, due to the nucleophilic group takes a position to the unsaturated C atom of olefin, the barrier for this inner-sphere syn-addition pathway is predicted to be 16.6 kcal mol−1, but if measured from the most stable intermediate M5, the barrier should be increased to 24.9 kcal mol−1. On the other hand, we also located the anti-addition pathway (TS4') from the complex M5 is disfavored and requires a barrier of 27.4 kcal mol−1. The formation of the syn-nucleopalladated intermediate M7 is exoergic by 31.8 kcal mol−1, which can provide thermodynamic driving force to move the reaction forward. The syn-nucleopalladation process can be explanation by the reduced Pd–C3 bond (2.11 Å), while Natural Bond Orbital (NBO) clearly showed that Pd (0.175 au) attack C (−0.156 au) to form Pd–C3 bond (2.11 Å), shown in Fig. 5.
 | ||
| Fig. 4 The second part free energy profile for HN-2A catalytic activation. | ||
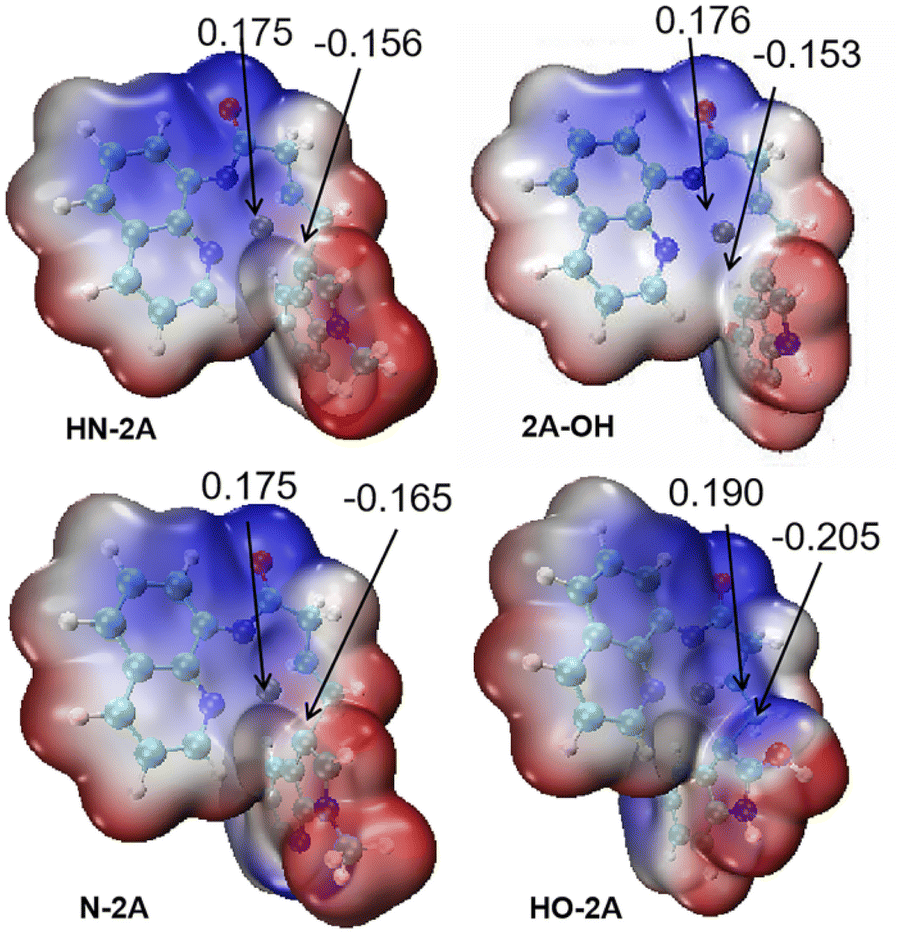 | ||
| Fig. 5 Natural bond orbital (NBO) analysis and electron-static potential map (ESP) for geometry TS4 of different nucleophile groups. | ||
After that, in the deprotonation process, the involvement of the acetic acid solvent to coordinate with the Pd center of the M7 gives the Pd(II) complex M8. Subsequently, the M8 undergo H-transfer process via the transition state TS5 generating the complex M9. The barrier for this H-transfer step is predicted to be 17.2 kcal mol−1. The distorted complex M9 can be readily transformed to the more stable complex M10.
Finally, in the depalladation step, Pd(II) center exchange with acetic acid molecule in the solution, then Pd–N bond eventually dissociates from the N atom of the directing group 8-amino-quinoline (AQ) to release the target nucleophilic substitution 2a, and the palladium-(II) catalyst is regenerated, this catalyst regenerates process is exergonic by 7.1 kcal mol−1.
The influence of affinity groups on the reaction process
In this work, several different nucleophile groups were chosen to compare the different influence on nucleo-palladation HN-2A, 2A-OH, HO-2A and N-2A′, respectively. Among these molecules, HN-2A has a pleased yield, it means that this reaction substrate has a higher conversion rate. For this reason, we choose HN-2A as the criterion, of which different nucleophile groups shows different performance, and to figured out the reason why they show such different manner in this mechanism. In contrast to the structure of N-2A and HN-2A, the difference is that the six-membered ring is benzene in HN-2A while the six-membered ring is pyridine in the N-2A. For the HO-2A and 2A-OH, they not only change the methyl group which on nucleophile's pentagon group into H atom which electron-donating ability is lower than before, but also bring in hydroxyl group into the benzene ring in the same time. The difference between those two species is just the positions of the hydroxyl group in their leaving group. As shown in Fig. 6, the three structures all tend to reduce the electron-donating capacity of nucleophile groups, and the difference in the degree of reduction process needs to be further explained.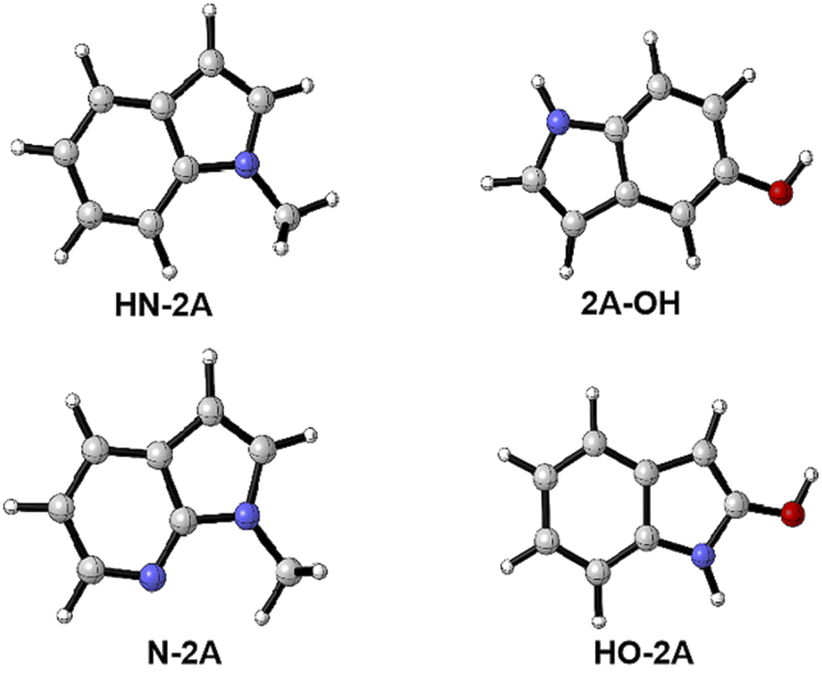 | ||
| Fig. 6 Several different nuclephile groups choosed in this work. | ||
| Species | Molecular | k |
|---|---|---|
| TS1 | Unimolecular | 1.1 × 1014 |
| TS2 | Unimolecular | 1.1 × 1012 |
| TS3 | Unimolecular | 4.9 × 1012 |
| TS4 | Bimolecular | 1.6 × 10−6 |
| TS5 | Unimolecular | 1.8 × 1014 |
First of all, the TS4 energy barriers of HN-2A, 2A-OH, N-2A and HO-2A are −4.6 kcal mol−1, 2.7 kcal mol−1, 2.6 kcal mol−1, and 2.6 kcal mol−1, respectively, when cross through the nucleophilic addition step, the transition state TS4 in which the reaction rate of this are 1.6 × 10−6 cm3 molecula−1 s−1, 8.6 × 10−8 cm3 molecula−1 s−1, 9.0 × 10−8 cm3 molecula−1 s−1, and 9.0 × 10−8 cm3 molecula−1 s−1, respectively, shown in Table 2. All of which go through the same process before they turn into the M6 structure. So, we just comparing the rate constant after the bonding of the nucleophile groups. The rate constant of the reactants through TS4 is roughly scaled down. It can be seen that although 2A-OH is slightly slower than HO-2A when it crosses the transition state TS4 in the decisive step, but the rest of other nucleophile groups' reaction rate is basically the same as the reaction yield. We also found that the transition state TS4 of which N-2A has the highest energy barrier and the lowest rate constant. Because the carbon atoms on the benzene ring of N-2A's nucleophile group is replaced by the N atom who has a stronger electronegativity. In the process of performing transition state TS4, due to the stronger N atom electronic absorption ability, the catalyst does not perform as well as HN-2A to remove nucleophilic group's H atoms, substrates need to cross a much larger barrier of reaction energy, it also makes a lower reaction rate and a lower production rate. Hence, we can conclude that the lone pair electrons strengthen the benzene ring conjugation, which is mainly caused by the weak electron-withdrawing p(N–C)-π conjugation due to the non-planar p orbital on the nitrogen atom at 3-positions of the pyridine-ring have unshared lone pairs, which form a p-π conjugated system with the π electrons of the benzene ring. It is also noted that the electron cloud density of benzene ring is shifted due to a stronger electronegativity of nitrogen atom, resulting in a more unevenly distributed on the conjugated surface and they can increase the electron cloud density of N on the pyridine ring.
| Species | Yield | Energy barrier (kcal mol−1) | kTS4 (s−1) |
|---|---|---|---|
| HN-2A | 90% | −4.6 | 1.6 × 10−6 |
| 2A-OH | 77% | 2.7 | 8.6 × 10−8 |
| N-2A | 19% | 2.6 | 9.0 × 10−8 |
| HO-2A | 6% | 2.6 | 9.0 × 10−8 |
The electron cloud density of pyridine group affects its electron-donating activity, which enhances the difficulty of leaving group to form the substrate M6. In addition, 2A-OH and HO-2A's geometry are similar to each another, they both turn the methyl groups into H atom, meanwhile added a hydroxyl functional group on the nucleophile group, and what worth mentioning is that the difference between them just the position of the hydroxyl group, but there is a big difference in yield, which worth further explanation. First of all, hydroxyl plays different role in 2A-OH and HO-2A, in the state of 2A-OH, the lone pair on O atom plays more as electron-donor than as nucleophile, but in HO-2A situation, the hydroxyl plays as more on nucleophilic groups that have higher electron trapping capacity. Secondly, hydroxyl's electronegativity is larger than methyl, which weaker the nucleophile's H atom leaving capacity, it means the reaction through 2A-OH and HO-2A to forming transition geometry TS4 need more reaction to receive the same result as HN-2A does.
In this work, the deformation energy (EDef-a/EDef-b) of frag-a and frag-b are defined as follows: EDef-a = Et(frag-a)TS − Et(frag-a)opt and EDef-b = Et(frag-b)TS − Et(frag-b)opt, where subscripts “TS” and “opt” denote the geometry in the TS and the optimized stable geometry, respectively. Which is means the energy difference between the frag-a/b moiety in TS4 and that in the equilibrium structure of catalyst. The sum of deformation energies indicates the degree of molecule easy or not to across energy barrier. The result shows that HN-2A, N-2A, HO-2A's deformation energy increase gradually, while Edef-sum is 15.0 kcal mol−1, 15.2 kcal mol−1, 18.5 kcal mol−1, respectively, shown in Table 3. Which indicate that the difficulty of crossing energy barrier is getting harder and harder with the changes in structure, and the results were consistent very well with the yield data.
| Species | Edef-a (kcal mol−1) | Edef-b (kcal mol−1) | Edef-sum (kcal mol−1) |
|---|---|---|---|
| HN-2A | 13.0 | 1.9 | 14.9 |
| 2A-OH | 13.0 | 1.9 | 14.9 |
| N-2A | 13.3 | 1.9 | 15.2 |
| HO-2A | 12.8 | 5.7 | 18.5 |
The energy decomposition analysis (EDA) results are summarized as pie charts shown in Fig. 7 to declare the quantitative contributions of each part of nucleophile–substrate interactions to the overall mechanism in palladation reaction. Each pie chart contains two different effects, steric repulsions and orbital interactions. The weak interactions have been visualized by NICPlots to elucidate the steric effects (see Fig. S8, ESI†). This EDA analysis provides a unique way to identify the dominant factor on how the nucleophile interact on reaction yield. Therefore, a more embedded theoretical investigation on the dominant effect can be performed here to illustrate different effects caused by each nucleophile-substrate. This simple energy decomposition was also performed here. The total energy variation of forming a complex can be decomposed as:
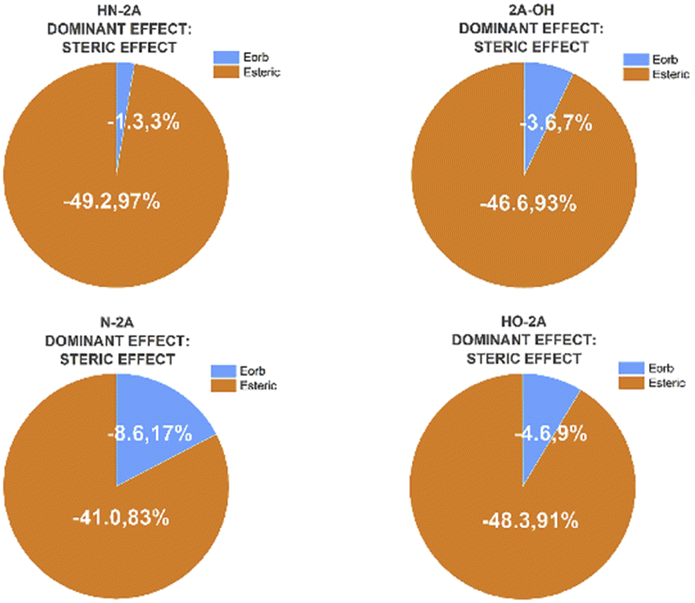 | ||
| Fig. 7 Energy contributions of different types interactions of nucleophile substrate in Pd(OAc)2 catalyzed system. | ||
ΔEorb in above formula is orbital interaction term, it arises from the mix of occupied MOs and virtual MOs, and it exhibits polarization and charge-transfer effects. If the combined wavefunction is used as initial guess for complex, then ΔEorb can be evaluated by subtracting the first SCF iteration energy from the last SCF iteration energy:
| ΔEorb = ESCF,last − ESCF,1st |
Note that ESCF,last = Ecomplex. Obviously, we have below relationship.
While absolute and also relative orbital contributions are lager in the HN-2A complex, with a weaker steric interaction result in the slightly weaker bond strength. The bonding picture of these four species are very different from each other. Without the attractive contributions of −41.0 kcal mol−1 arising from steric interactions, N-2A would not be stable. And the bond dissociation energy of N-2A is slightly larger than the rest of other substrates. The decomposition of the HN-2A, 2A-OH, N-2A, HO-2A's total interaction shows that −49.2 kcal mol−1, −46.5 kcal mol−1, −41.0 kcal mol−1 and −48.3 kcal mol−1 comes from steric effect and −1.3 kcal mol−1, −3.6 kcal mol−1, −8.6 kcal mol−1 and −4.6 kcal mol−1 comes from orbital contribution, respectively, shown in Fig. 8. The EDA shows that the dominant increase come from stronger steric contribution.
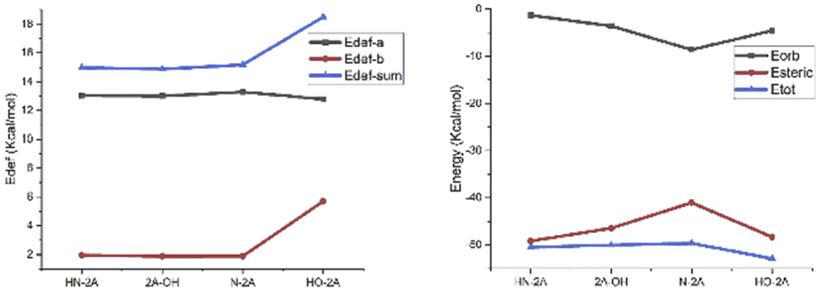 | ||
| Fig. 8 The results of deformation energy analysis and EDA studies performed in this mechanism. | ||
Conclusions
DFT calculation was carried out to simulate the origin of difference manner carried out by nucleophile group, and we get a deeper perspective conversion about these different groups. Firstly, we get the mechanism about the outset of the works, which contain four vital step, N–H activation, β-X elimination, nucleophilic addition and catalyst regeneration. Secondly, we find out the relationship between the structure and reactivity, and how did these differences perform by chemical bond, energy barrier, NBO, BDE. Through the analysis of the chemical bond between the target γ-C and Nu group, we can see that the better the electron donating ability of the Nu group's five-membered ring, has the lower the reaction energy barrier needs to be crossed when combining γ-C atom, and the smaller steric interactions between the target γ-C and Nu groups, and the greater the orbital interactions of the formed chemical bond showed by the EDA analysis. Meanwhile, BDE analysis showed that compared with HO-2A's group with the lowest yield, the bond dissociation energy of other Nu groups was relatively high, indicating that the more stable target chemical bond. Natural Charge Population obtained by NBO analysis showed the same electron flow trend from γ-C to Nu group. Finally, the rate-determining step is the nucleophilic addition step, while to cross TS4 energy barrier, and the EDA shows the steric repulsions contribute to the majority of the intra-molecular interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (grant number 21873038). The authors are very grateful to the editor and reviewers for insightful comments and suggestions.Notes and references
- K. S. Yang, J. A. Gurak, Jr., Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 14705–14712 CrossRef CAS PubMed.
- A. Simaioforidou, M. Papastergiou, A. Margellou, D. Petrakis and M. Louloudi, J. Mol. Catal. A: Chem., 2017, 426, 516–525 CrossRef CAS.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 6, 498–525 CrossRef.
- M. Maffei, G. Giacoia, R. Mancuso, B. Gabriele, E. Motti, M. Costa and N. Della Ca’, J. Mol. Catal. A: Chem., 2017, 426, 435–443 CrossRef CAS.
- H. S. Haniff, L. Knerr, X. Liu, G. Crynen, J. Bostrom, D. Abegg, A. Adibekian, E. Lekah, K. W. Wang, M. D. Cameron, I. Yildirim, M. Lemurell and M. D. Disney, Nat. Chem., 2020, 12, 952–961 CrossRef CAS PubMed.
- Y. Han and H. V. Huynh, Dalton Trans., 2011, 40, 2141–2147 RSC.
- X. Fu, Q. Qi, S. Xu and E.-i. Negishi, Org. Lett., 2021, 23, 8984–8988 CrossRef CAS.
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., 2009, 48, 5094–5115 CrossRef CAS.
- A. N. Bilyachenko, M. S. Dronova, A. I. Yalymov, F. Lamaty, X. Bantreil, J. Martinez, C. Bizet, L. S. Shul'pina, A. A. Korlyukov, D. E. Arkhipov, M. M. Levitsky, E. S. Shubina, A. M. Kirillov and G. B. Shul'pin, Chemistry, 2015, 21, 8758–8770 CrossRef CAS PubMed.
- Y. Wu, H. Yu, Y. Guo, X. Jiang, Y. Qi, B. Sun, H. Li, J. Zheng and X. Li, Chem. Sci., 2019, 10, 10459–10465 RSC.
- S. Ruiz, F. J. Sayago, C. Cativiela and E. P. Urriolabeitia, J. Mol. Catal. A: Chem., 2017, 426, 407–418 CrossRef CAS.
- A. L. Wierschen, N. Romano, S. J. Lee and M. R. Gagne, J. Am. Chem. Soc., 2019, 141, 16024–16032 CrossRef CAS PubMed.
- E. Silarska, M. Majchrzak, B. Marciniec and A. M. Trzeciak, J. Mol. Catal. A: Chem., 2017, 426, 458–464 CrossRef CAS.
- M. L. O'Duill and K. M. Engle, Synthesis, 2018, 50, 4699–4714 CrossRef.
- S. K. Nimmagadda, M. Liu, M. K. Karunananda, D. W. Gao, O. Apolinar, J. S. Chen, P. Liu and K. M. Engle, Angew. Chem., 2019, 58, 3923–3927 CrossRef CAS PubMed.
- Y. Makida, H. Ohmiya and M. Sawamura, Chem. - Asian J., 2011, 6, 410–414 CrossRef CAS.
- C. Lin, F. Gao and L. Shen, Adv. Synth. Catal., 2019, 361, 3915–3924 CrossRef CAS.
- T. He, J. C. Buttner, E. F. Reynolds, J. Pham, J. C. Malek, J. M. Keith and A. R. Chianese, J. Am. Chem. Soc., 2019, 141, 17404–17413 CrossRef CAS PubMed.
- H. Azizollahi, V. P. Mehta and J. A. Garcia-Lopez, Chem. Commun., 2019, 55, 10281–10284 RSC.
- J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS PubMed.
- D. W. Gao, Y. Gao, H. Shao, T. Z. Qiao, X. Wang, B. B. Sanchez, J. S. Chen, P. Liu and K. M. Engle, Nat. Catal., 2020, 3, 23–29 CrossRef CAS.
- J. A. Gurak, V. T. Tran, M. M. Sroda and K. M. Engle, Tetrahedron, 2017, 73, 3636–3642 CrossRef CAS.
- J. A. Gurak, Jr., K. S. Yang, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 5805–5808 CrossRef PubMed.
- L. J. Oxtoby, Z. Q. Li, V. T. Tran, T. G. Erbay, R. Deng, P. Liu and K. M. Engle, Angew. Chem., 2020, 59, 8885–8890 CrossRef CAS PubMed.
- V. T. Tran, J. A. Gurak, Jr., K. S. Yang and K. M. Engle, Nat. Chem., 2018, 10, 1126–1133 CrossRef CAS.
- P. Hay and W. Wadt, Potentials for K to Au Including the Outermost Core Orbitals, J. Chem. Phys., 2021, 82 Search PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- A. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- K. Fukui, J. Phys. Chem., 1997, 74, 4161–4163 CrossRef.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- E. A. P. t. Denis Jacquemin, I. Ciofini, C. Adamo, R. Valero, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071–2085 CrossRef PubMed.
- Y. Zhao and F. Chen, J. Multivariate. Anal., 2008, 99, 215–231 CrossRef.
- Y. Z. a. D. G. Truhla, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- CYLview, 1.0b; Legault, C. Y., Universite de Sherbrooke, 2009, http://www.cylvwe.org.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03506e |
| This journal is © The Royal Society of Chemistry 2022 |