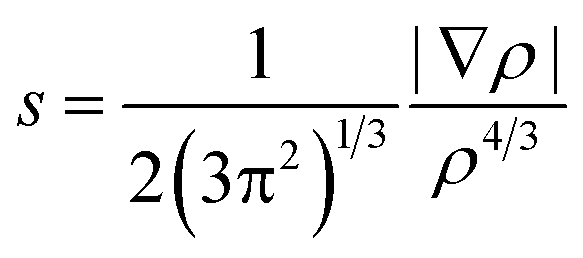DOI:
10.1039/D2RA03945A
(Paper)
RSC Adv., 2022,
12, 23416-23426
Fischer–Helferich glycosidation mechanism of glucose to methyl glycosides over Al-based catalysts in alcoholic media†
Received
27th June 2022
, Accepted 10th August 2022
First published on 17th August 2022
Abstract
The Fischer–Helferich glycosidation reaction is generally the initial step in the conversion of glucose to levulinate in alcohol media. However, the relevant molecular mechanism catalyzed by Al-based catalysts is still not well understood. In this work, the reaction mechanism of the glycosidation from glucose to methyl glycosides catalyzed by Al3+ coordinated with methanol/methoxyl was investigated through density functional theory (DFT) calculations. The whole reaction process includes ring-opening, addition, and ring-closure events. The addition of methanol to the ring-opening structure of glucose makes the electronegativity of C1 site stronger to proceed with the following ring-closure reaction. Among the 28 kinds of ways of ring-closure reaction, the most preferred way is to close the loop through the six-membered ring (O5–C1) to generate methyl glucoside (MDGP). The rate-determining step is the ring-closure and the Al3+ shows a great catalytic effect which is mainly reflected in coordinating with the solvents to transfer protons. The results would be helpful to understanding the Fischer–Helferich glycosidation mechanism catalyzed by Al-based catalysts and comprehend the conversion of glucose to high value-added chemicals.
1. Introduction
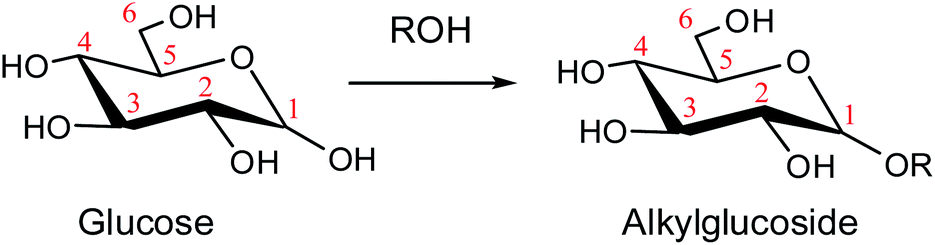
The conversion of biomass resources to valuable platform chemicals is of critical importance according to the diminishing fossil fuels and global warming effects.1,2 Among the various biomass-derived products, glucose is the monomer unit of cellulose which could produce a variety of valuable chemicals, such as 5-hydroxymethyl-furfural and levulinates.3–5 The Fischer–Helferich glycosidation is a simple and cheap method to render alkyl and aryl glycoside from sugars which is also prevalent in the conversion of glucose to levulinate.6 Besides, the carbohydrate derivatives have been widely used in food emulsifiers, cosmetic surfactants, and solvents for membranes.7
As a promising approach, alcoholysis technology has been maturely applied for the conversion of lignocellulose to platform chemicals due to its various advantages. Compared with the conversion of glucose to biofuels in the water medium, alcoholysis could avoid wasting water and decrease humins or chars.8,9 Moreover, the alkyl levulinate generated in alcohol gives a higher yield than levulinic acid obtained in water.10,11 In the previous work, Deng et al. reported that abundant products such as glucose, fructose, and methyl glucoside (MDGP), were detected in methanol, and the yield of MDGP was relatively high because glucose tends to undergo Fischer–Helferich glycosidation to generate MDGP.12 In 2019, Chung et al. described that decyl glucoside was synthesized by direct glucosidation from glucose in 1-decanol using zeolite catalysts.13 The glucose could form alkyl glucoside in the alcohol system through Fischer–Helferich glycosidation which is described in Scheme 1.14,15 Fischer–Helferich glycosidation is one of the most promising choices to prepare alkyl or aryl glycosides from sugars. Most studies have concentrated on the conversion experiments and development of high-efficiency catalysts due to the wide application of high value-added alkyl glucosides. However, the detailed reaction mechanism of Fischer–Helferich glycosidation remains ambiguous.
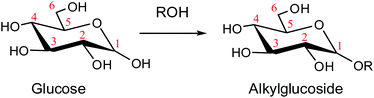 |
| | Scheme 1 The glycosidation of glucose to alkyl glucoside. | |
In the past decades, numerous kinds of catalysts were used for the conversion of glucose in the alcohol media, and the corresponding glycosidation reaction was indicated based on the intermediate alkyl glucosides.11,16 As one kind of sustainable clean catalysts, Al-based catalysts, such as Al2(SO4)3, AlCl3, and AlPW12O40 are efficient for the conversion of biomass carbohydrates in the alcoholic solvents.17,18 Various coordination structures could be formed with the solvents to catalyze many reactions, such as the sugar isomerization, Fischer–Helferich glycosidation, and the dehydration of fructose.19 He et al. investigated the predominant coordination of cyclic β-D-glucose to [Al(OH)(aq)]2+ and [Al(OH)2(aq)]+ ions and gained many stable coordination species, which may aid in understanding the glucose isomerization.20 Norton et al. reported that the hydrolyzed Al3+ complex [Al(H2O)(OH)2]1+ was the active species in the glucose isomerization.21 Furthermore, Saravanamurugan et al. proposed that bifunctional Lewis and Brønsted acid sites in the Al-based catalyst structures were one of the most important reasons to catalyze the glycosidation.22 Recently, as a promising bifunctional catalyst for biomass carbohydrate transformation to methyl levulinate, AlPW12O40 has shown superior activity and the bifunctional Lewis and Brønsted acid site structure [Al(CH3O)2(CH3OH)2]+ can be detected by GC-MS experiments.23 However, the detailed glycosidation mechanism of glucose to methyl glycosides catalyzed by Al-based catalyst is still not clear.
To understand the fundamental reaction mechanism of Fischer–Helferich glycosidation of glucose to methyl glycosides catalyzed by Al-based catalysts in methanol solvent, we investigated in detail the reaction pathways of glucose to methyl glycosides over [Al(CH3O)2(CH3OH)2]+ catalyst by DFT calculations in this work. In the Fischer–Helferich glycosidation process, there are mainly three steps including the ring-opening of glucose, addition, and ring closure reaction. The reaction pathways were explored in detail and activation barriers of each step were compared. The reaction pathways without catalyst and catalyzed by Al-based catalysts in methanol solvent had been also probed. Finally, the Fischer–Helferich glycosidation mechanism of glucose catalyzed by Al-based catalysts had been proposed. The calculated results would provide comprehensive information to understand the glycosidation of glucose to alkyl glycosides in alcoholic solution.
2. Computational method
All the calculations were carried out by using the M06-2X functional,24 which is proposed to be one of the best functionals to accurately calculate energy barrier, with the standard 6-311+G** basis set and Grimme's D3 dispersion corrections,25 as implemented in Gaussian 09 software package.26 The geometries for the reactants, transition states, and products were fully optimized without any constraints in the gas phase and generated by CYL view.27 To account for the solvent effects, single-point calculations were performed in a methanol dielectric by using the SMD solvation model.28 Vibrational frequency calculations were also carried out to verify the optimized structures as minima (zero imaginary frequency) or first-order saddle points (one imaginary frequency) and to provide free energies at 298.15 K, 1 atm. Gibbs free energies were calculated by the sum of energies in the SMD solvent and thermal correction to Gibbs free energies at M06-2X-D3/6-311+G** level. The intrinsic reaction coordinate (IRC) pathways29 of transition states have been traced to confirm that each of them connects the desired reactant and product. Furthermore, the potential energy surface (PES) scans30 were performed to search for the TS structures. The activation energy (Ga) which is the energy barrier and reaction energy (Gr) of the systems are defined as follows:| | |
Ga = GTS − GR, Gr = GP − GR
| (1) |
where GTS, GR, GP represent the Gibbs free energies of transition state, reactant, and product. Noncovalent interactions (NCIs) were characterized by using Reduced Density Gradients (RDGs) approach31 which can be defined as the following:| |
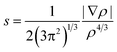 | (2) |
where ρ stands for the electron density and |∇ρ| represents the norm of electron density gradient vector. The RDG is typically plotted against sign(λ2)ρ in a scattering diagram where the sign(λ2) is the sign of the second curvature of the electron density. The topological properties were generated by Multiwfn program32 with the wave functions in M06-2X-D3/6-311+G** level.
3. Results and discussion
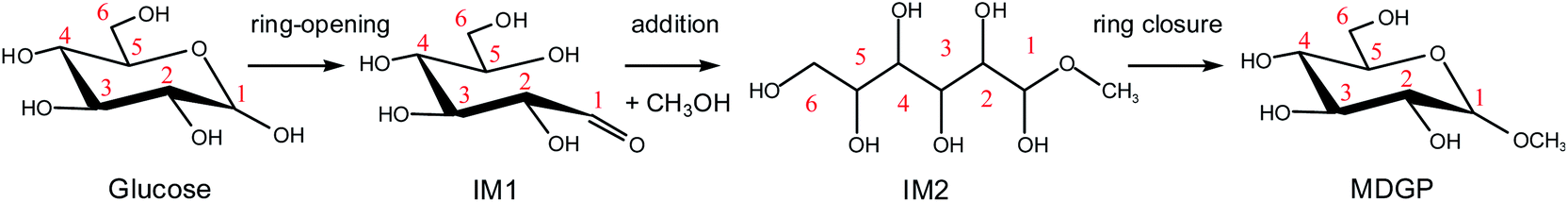
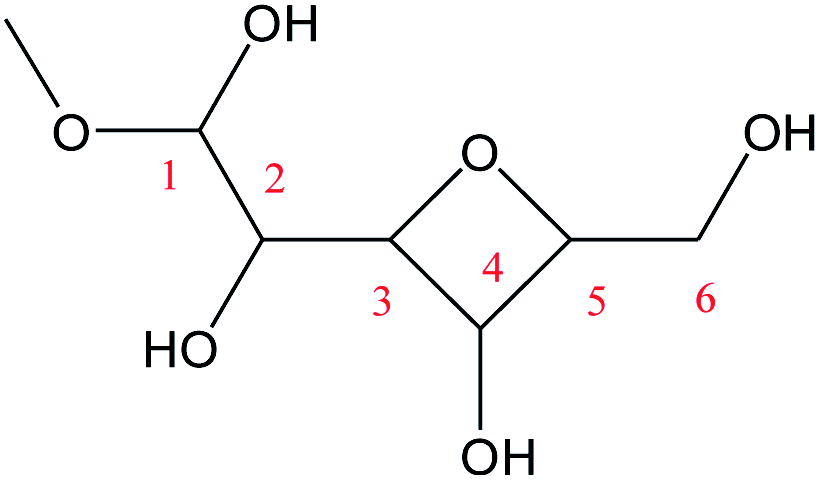
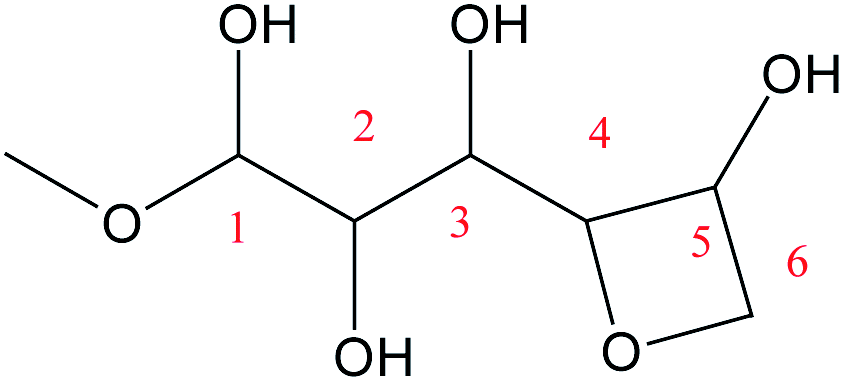
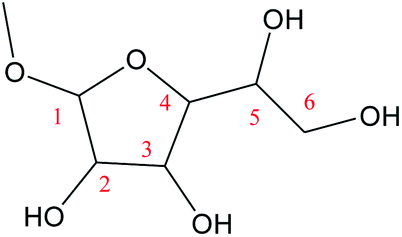
For the Fischer–Helferich glycosidation reaction, it is prevalent in the conversion experiments of glucose under alcohol solvent.33 The detailed reaction pathways were not expounded in most of the studies. The Mayer bond order is a natural extension of the Wiberg bond order, which has proved extremely useful in bonding analysis.34 The Mayer bond order analysis has been made for the C–O of glucose which was shown in Fig. S1.† The Mayer bond order of C1–O5 (the C–O in the ring) and C1–O1 (the C–O in hydroxyl) are 0.86, and 1.36, respectively. It means the C1–O5 bond is easier cleavage than C1–O1. For the direct etherification reaction of methanol (the oxygen of CH3OH attacks C1 of glucose), the potential energy 3D surface scan was made in Fig. S2.† When the methanol attacks the C1 site of glucose, the C1–O1H1 bond length would increase and attract the proton of methanol for dehydration. It can be found that the relative energy will increase for direct etherification and there are also no saddle points in the 3D surface scan. In addition, the ring-opening of glucose with a linear hemiacetal structure (IM1) can be detected in the initial reaction stage by using radioactive sugars.35,36 Based on previous research on the glycosidation of glucose to MDGP,37,38 it is generally recognized that the conversion occurs through the methanol addition to the ring-opening structure of glucose which is shown in Scheme 2. Therefore, the whole glycosidation mechanism generally includes ring-opening, addition, and ring closure events. In the following sections, we will discuss the mechanism of glucose to MDGP without and with [Al(CH3O)2(CH3OH)2]+ catalyst.
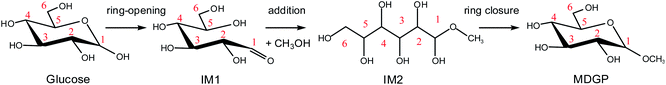 |
| | Scheme 2 The general mechanism for the glycosidation of glucose to methyl glycoside. | |
3.1 The ring-opening reaction of glucose
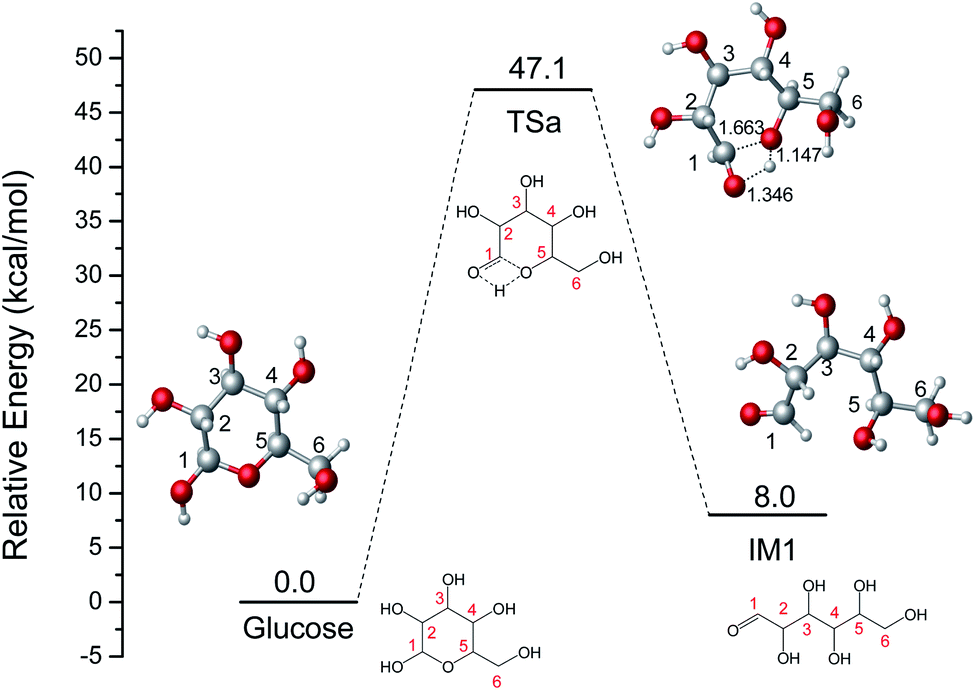
The ring-opening reaction step where the proton from the hydroxyl group at C1 transfers to O6 atom has been reported by some theoretical studies like the isomerization of sugars.39,40 Firstly, the ring-opening reaction of glucose without catalyst was calculated and shown in Fig. 1. The proton at O1 directly transfers to O5 and the length of C1–O5 bond gradually increases cleavage with the structure of TSa. The product (IM1) of the ring-opening reaction will be formed with a 47.1 kcal mol−1 energy barrier. This process is an endothermic reaction of 8.0 kcal mol−1.
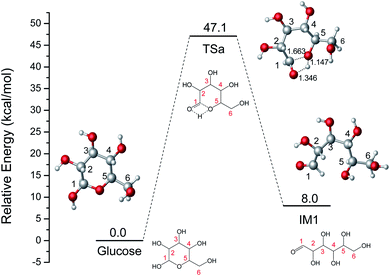 |
| | Fig. 1 The potential energy (kcal mol−1) profile with the structures of reactant, transition state, and product for the ring-opening of glucose without catalyst (bond distances in Å). | |
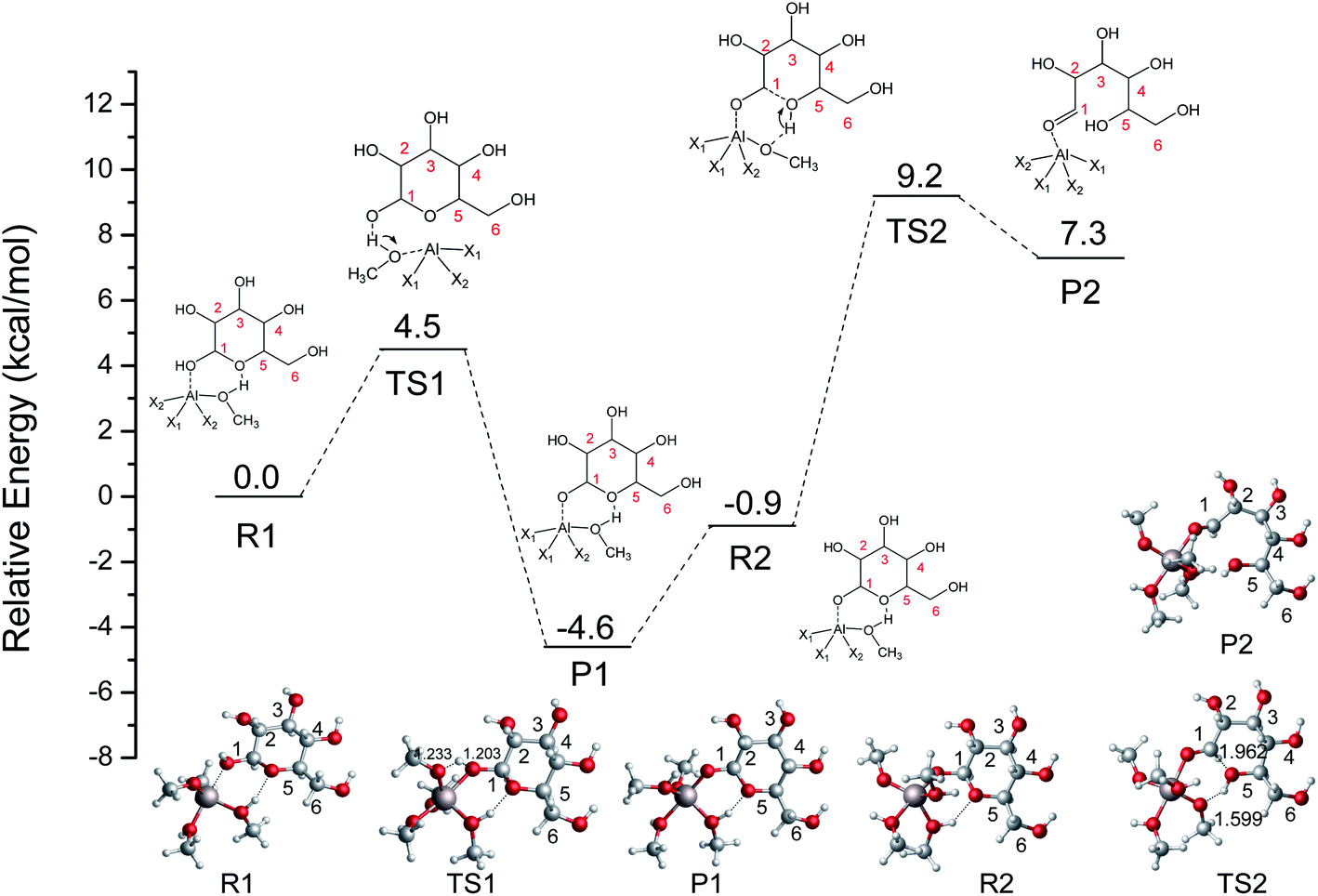
Saravanamurugan et al. reported that Al-based zeolites with combined Brønsted and Lewis acid sites could catalyze the glucose isomerization at relatively low temperatures.22 In aqueous media, the distribution of hydrolyzed AlCl3 species depends on many parameters, e.g., the concentration of AlCl3, pH value, temperature, and so on.41 It is generally believed that Al3+ exists in the form of (1) hexa-coordinated [Al(aq)]3+ in strong acid solution (pH < 3.0), (2) [Al(OH)2(H2O)n−2]+, and [Al(OH)(H2O)n−1]2+ around a pH of 4.0 (where n is the coordination number), (3) [Al(OH)(H2O)n−1]2+ as well as [Al(OH)4]− at pH values of 5.2–6.7, (4) a tetrahedral [Al(OH)4]− structure (pH > 7.0).42,43 Theoretical and experimental research studies indicate that the four-coordination [Al(OCH3)2(CH3OH)2]+ plays a crucial role in the conversion of sugars after the typical stable [Al(CH3O)m(CH3OH)n]3−m (m = 0–6, m + n ≤ 6) species were calculated.19 To gain a further understanding of the Fischer–Helferich glycosidation mechanism, the [Al(CH3O)2(CH3OH)2]+ structure (Fig. 2) containing both bifunctional Lewis and Brønsted acid sites was used and reported in our previous study.19 To investigate the ring-opening reaction of glucose catalyzed by [Al(CH3O)2(CH3OH)2]+, the energy profile and reactants, transition states, and products of this process were probed and presented in Fig. 3. For the ring-opening reaction, the O1H1 of glucose was activated by [Al(CH3O)2(CH3OH)2]+. In R1 ([(η1O1-Glu)Al(CH3O)2(CH3OH)2]+), the Al3+ interacts with the hydroxyl group (O1) at a Al–O bond length of 2.031 Å, and the methanol coordinated with Al3+ forms H-bonds with O5 at a distance of 1.769 Å. The H1 transfers to the methoxy group coordinated with Al3+ via TS1 with a barrier of 4.5 kcal mol−1, which means it is easy to undergo the first step, and it will form P1 (H1 of glucose transfers to OCH3 of [Al(CH3O)2(CH3OH)2]+). This step is exothermic by about 4.6 kcal mol−1. The P1 and R2 are isomers in differently spatial position. Then, the hydrogen of methanol coordinated with Al3+ transfers to O5, and the length of C1–O5 bond gradually becomes longer (TS2). Finally, the ring-opening of the glucose (P2) was formed with the C1–O5 bond cleavage. The second step involves a free energy barrier of 10.1 kcal mol−1 which is 5.5 kcal mol−1 higher than the first step. The ring-opening catalyzed by [Al(CH3O)2(CH3OH)2]+ shows that [Al(CH3O)2(CH3OH)2]+ can significantly decrease the energy barrier through the hydrogen transfer of methanol coordinated with Al3+.
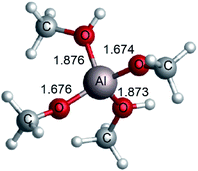 |
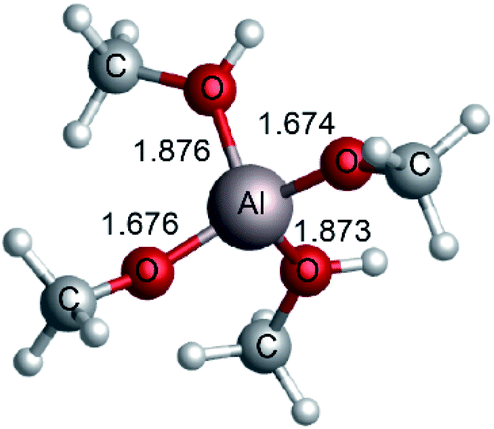
| | Fig. 2 The structure of [Al(CH3O)2(CH3OH)2]+ optimized at M06-2X-D3/6-311+G** level (bond length in Å). | |
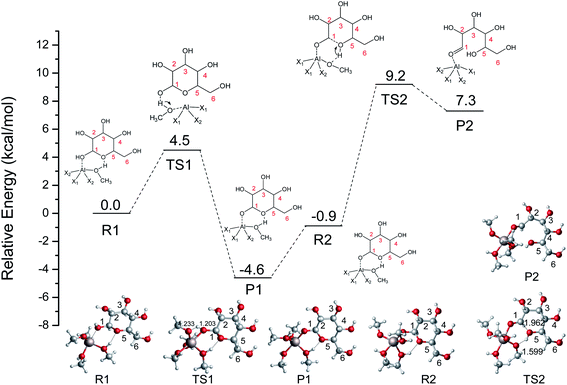 |
| | Fig. 3 The potential energy (kcal mol−1) profile with the structures of reactants, transition states, and products for the ring-opening of glucose catalyzed by [Al(CH3O)2(CH3OH)2]+ (X1 = CH3OH, X2 = CH3O, bond distances in Å). | |
3.2 Addition reaction
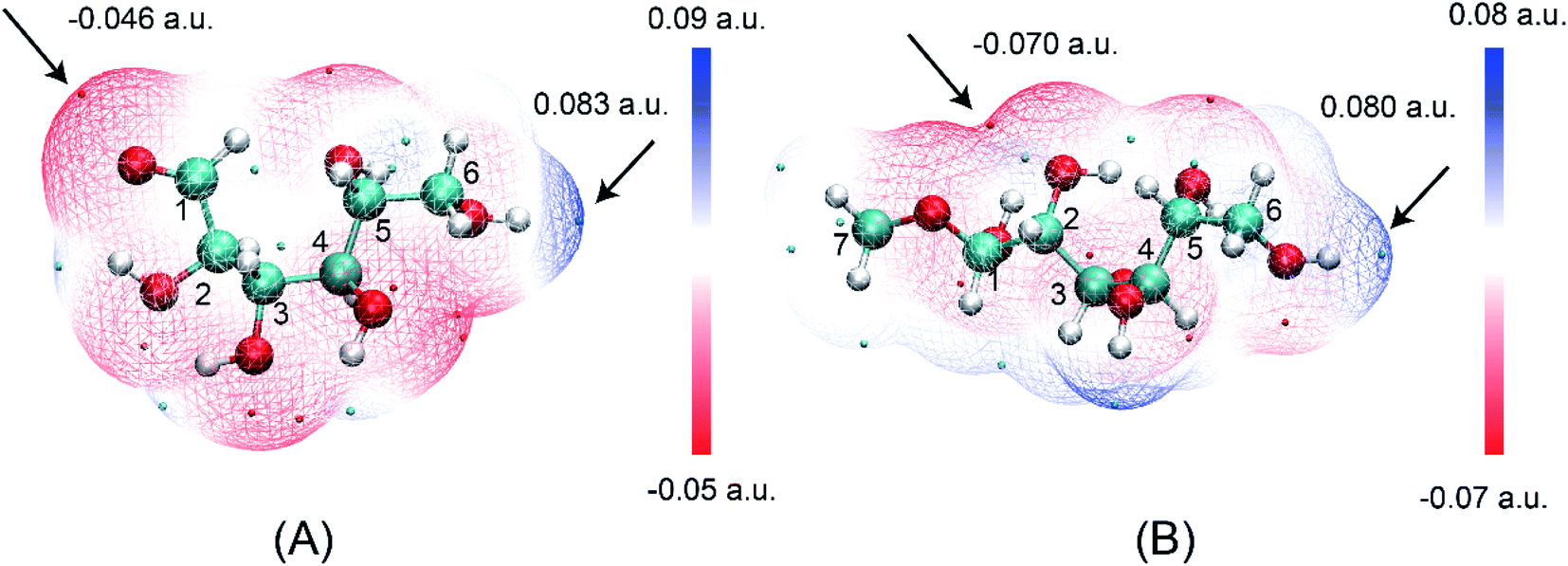
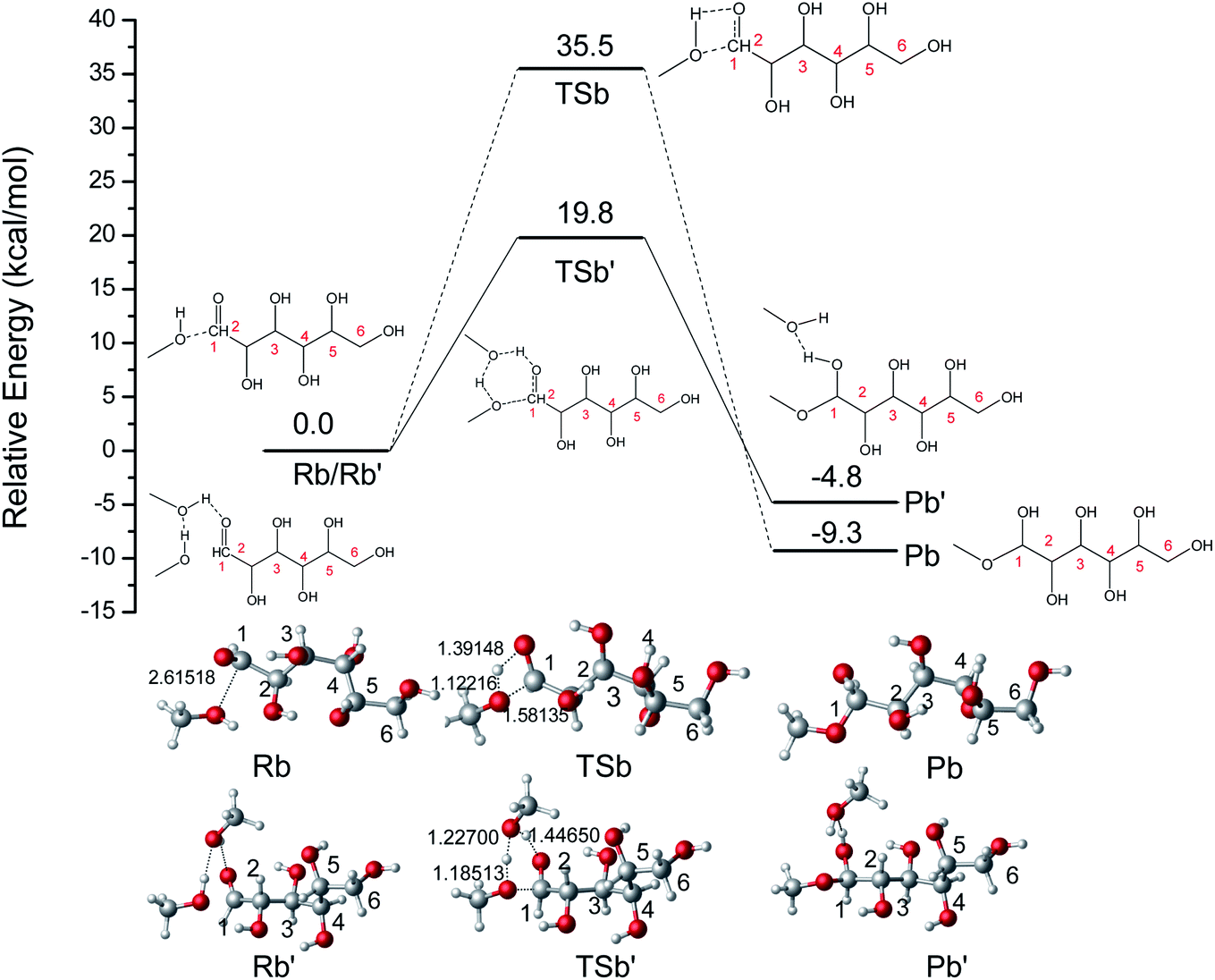
The ring-opening structures of glucose (IM1) and MDGP (IM2) are two important intermediates in the Fischer–Helferich glycosidation.44,45 As a useful method to predict the reactive site of the molecular surface, quantitative molecular surface analysis of electronic potential (ESP) had been widely used in many DFT studies.46,47 Based on the structure of reactants optimized at M06-2X-D3/6-311+G** level, the ESP of IM1 and IM2 had been analyzed and listed in Fig. 4. The ESP surface minimum of IM1 and IM2 is located in the O1 area which is −0.046 and −0.070 a.u., respectively. In the IM2, the addition of methanol makes the ESP around the O1 area more negative to undergo the following ring-closure reaction. Besides, the electropositive region of IM1 and IM2 are located around the O6 site. However, the change (0.003 a.u.) is not obvious when the methanol was added to IM1. As one of the most widely used methods to accurately describe the charge distribution in the chemical system, the natural population analysis (NPA) charge was also calculated.48 The NPA charge of IM1 and IM2 was calculated and summarized in Table S1.† In IM2, the NPA charge of C1–C6 are 0.430, 0.073, 0.078, 0.098, 0.074, and −0.054 a.u. The most positive and negative sites are located in C1 and C6, respectively. These changes in the chemical properties when the methanol was added in IM1 would be beneficial for the following ring-closure reaction. The subsequent step is the addition reaction where the ring-opening form glucose (IM1) is transformed into IM2 (Fig. 5). A transition state (TSb) is located to connect Rb (IM1) and Pb (IM2) with a reasonable activation barrier of 35.5 kcal mol−1. In the geometry of TSb, the oxygen of methanol is found to be close to the C1 with a distance of 1.58 Å in the four-membered ring transition structure. In the addition reaction step, the H-transfer assisted by two MeOH molecules had been probed and combined in Fig. 5. A six-membered transition state can be formed with 19.8 kcal mol−1 energy barrier which is lower than that of Rb–Pb.
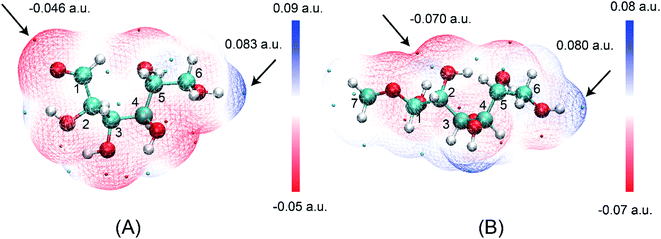 |
| | Fig. 4 The electrostatic potential on the van der Waals surfaces of (A) IM1 and (B) IM2. | |
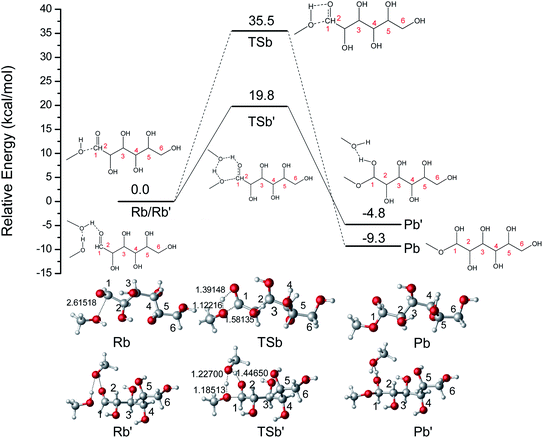 |
| | Fig. 5 The potential energy (kcal mol−1) profile with the structures of reactant, transition state, and product for the addition reaction without catalyst. (bond distances in Å). | |
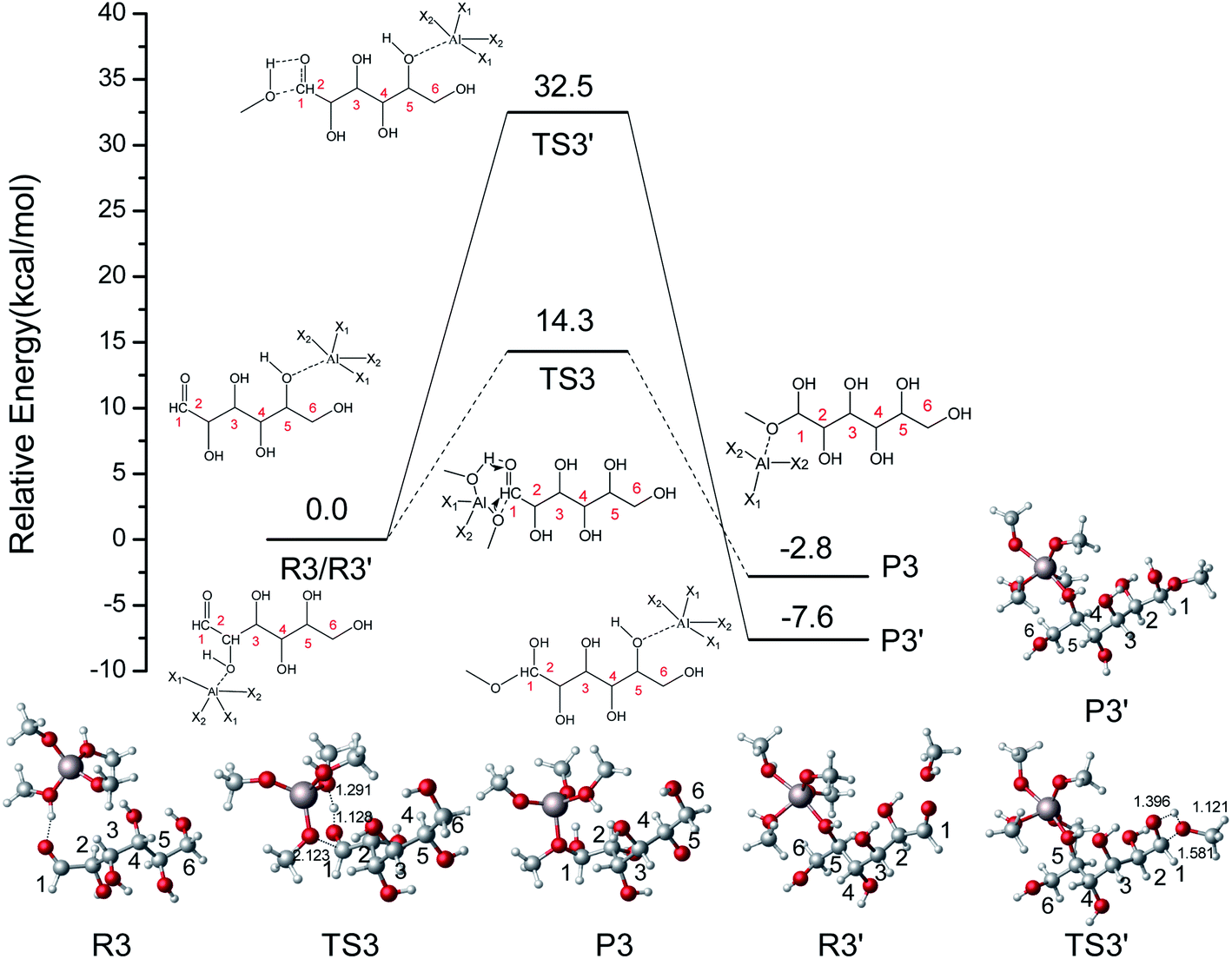
To explore the addition reaction catalyzed by [Al(CH3O)2(CH3OH)2]+, the energy profile with the structures for two different pathways in the addition reaction is shown in Fig. 6. In the R3–P3 reaction pathway, the proton (hydroxyl) of CH3OH coordinated with [Al(CH3O)2(CH3OH)2]+ transfers to O1 of IM1, and the OCH3 coordinated with Al3+ was added to C1 of IM1. The IM1 was active by the coordination of [Al(CH3O)2(CH3OH)2]+ (R3). In TS3, the hydrogen of methanol coordinated with Al3+ transfers to the O1, and the methoxy attacks C1 with only a 14.3 kcal mol−1 energy barrier. This process is exothermic by 2.8 kcal mol−1. In another reaction pathway (R3′–P3′), H-bonds initially can be formed between Al3+ and O5, then H (hydroxyl) of CH3OH transfers to O1, and the methoxy was added to C1 of IM1. The energy barrier of R3′–P3′ is 32.5 kcal mol−1 and higher than that of R3–P3. Therefore, the role of Al3+ is mainly reflected through the hydrogen transfer of coordinated methanol. In the Fischer–Helferich glycosidation, the energy barrier of the addition reaction (R3–P3) is higher than that of the ring-opening step.
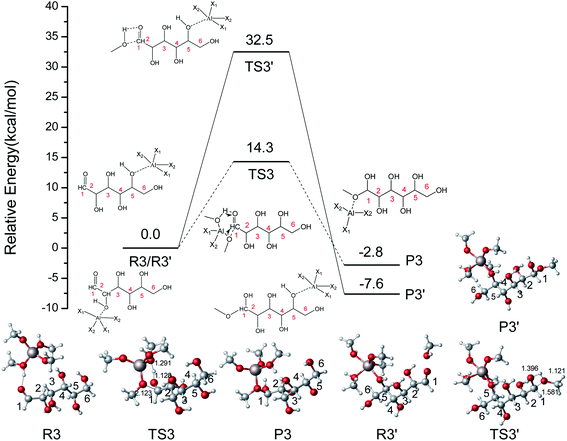 |
| | Fig. 6 The potential energy (kcal mol−1) profile with the structures of reactants, transition states, and products for two different pathways in the addition reaction catalyzed by [Al(CH3O)2(CH3OH)2]+. (X1 = CH3OH, X2 = CH3O, bond distances in Å). | |
3.3 Ring-closure reaction
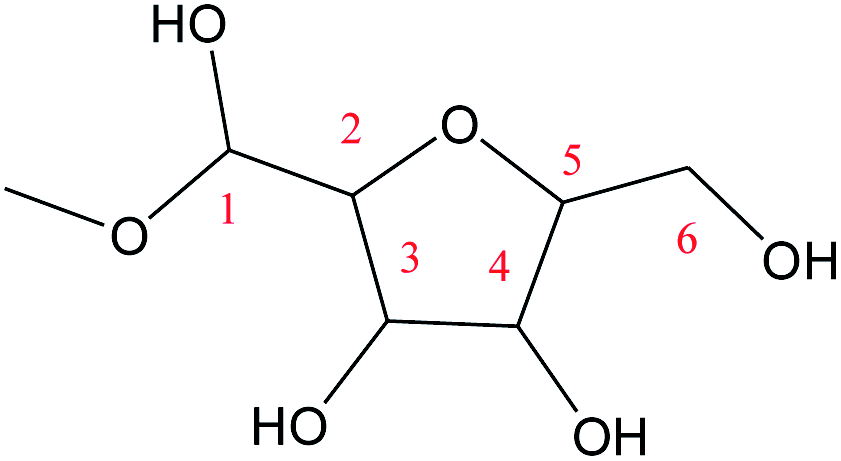
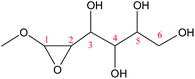
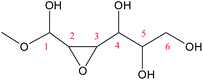
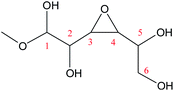
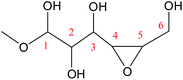
Cyclic anhydrosugars are important intermediates in synthetic carbohydrate chemistry.49 Four different main rings, i.e., three-, four-, five-, and six-membered ring, can be formed due to the six hydroxyls in IM2. For the three-membered rings, there are ten different kinds of ways which are the connection of O1–C2, O2–C3, O3–C4, O4–C5, O5–C6, O2–C1, O3–C2, O4–C3, O5–C4, and O6–C5, respectively. For the four-membered ring, there are eight different types of ways which are O1–C3, O2–C4, O3–C5, O4–C6, O6–C4, O5–C3, O4–C2, and O3–C1. In the five-membered ring reactions, important intermediates would be produced such as methyl fructoside which can provide a referable pathway in the alcohol system. There are six different kinds of ways which are O1–C4, O2–C5, O3–C6, O6–C3, O5–C2, and O4–C1. Also, for the six-membered ring to form meaningful products like MDGP, there are four different ways which are O1–C5, O2–C6, O6–C2, and O5–C1. As a result, there are 28 kinds of ways in the ring-closure reaction step. We had calculated all of the ring-closure reactions discussed above. The corresponding energies and the structures of products are summarized in Table 1. The H5 of O5 transfers to O1H1 and the O5–C1 bond will be formed to produce MDGP based on the pathway with the lowest energy barrier. The detailed discussions of the various ring-closure reactions were put in ESI.†
Table 1 The computed activation energy (Ga), reaction energy (Gr), and the structures of products in the three-(1–10) four-(11–18) five-(19–24) and six-(25–28) membered ring-closure reaction from IM2. (unit kcal mol−1)
| Entry |
Ring-closure |
Ways |
Ga |
Gr |
Structure of products |
| 1 |
Three- |
O1–C2 |
72.3 |
22.7 |
 |
| 2 |
O2–C1 |
53.6 |
16.3 |
| 3 |
O2–C3 |
64.8 |
15.4 |
 |
| 4 |
O3–C2 |
78.5 |
21.8 |
| 5 |
O3–C4 |
80.3 |
17.0 |
 |
| 6 |
O4–C3 |
75.0 |
19.6 |
| 7 |
O4–C5 |
73.7 |
18.3 |
 |
| 8 |
O5–C4 |
72.5 |
16.5 |
| 9 |
O5–C6 |
74.9 |
16.2 |
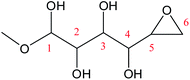 |
| 10 |
O6–C5 |
73.0 |
12.3 |
| 11 |
Four- |
O1–C3 |
77.1 |
19.1 |
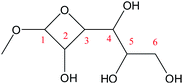 |
| 12 |
O3–C1 |
57.7 |
13.0 |
| 13 |
O2–C4 |
79.9 |
17.4 |
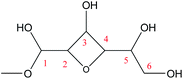 |
| 14 |
O4–C2 |
82.2 |
15.8 |
| 15 |
O3–C5 |
92.3 |
17.5 |
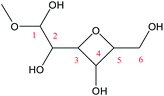 |
| 16 |
O5–C3 |
88.2 |
18.3 |
| 17 |
O4–C6 |
81.3 |
14.8 |
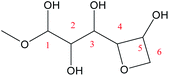 |
| 18 |
O6–C4 |
82.7 |
8.2 |
| 19 |
Five- |
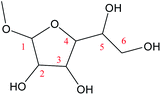
O1–C4 |
65.6 |
−1.3 |
 |
| 20 |
O4–C1 |
65.1 |
−3.5 |
| 21 |
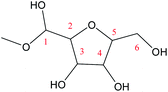
O2–C5 |
70.9 |
−4.6 |
 |
| 22 |
O5–C2 |
74.7 |
−5.5 |
| 23 |
O3–C6 |
76.3 |
−0.8 |
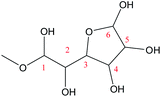 |
| 24 |
O6–C3 |
73.4 |
1.8 |
| 25 |
Six- |
O1–C5 |
70.3 |
−5.4 |
 |
| 26 |
O5–C1 |
52.8 |
−6.5 |
| 27 |
O2–C6 |
70.8 |
−7.8 |
 |
| 28 |
O6–C2 |
71.1 |
−12.0 |
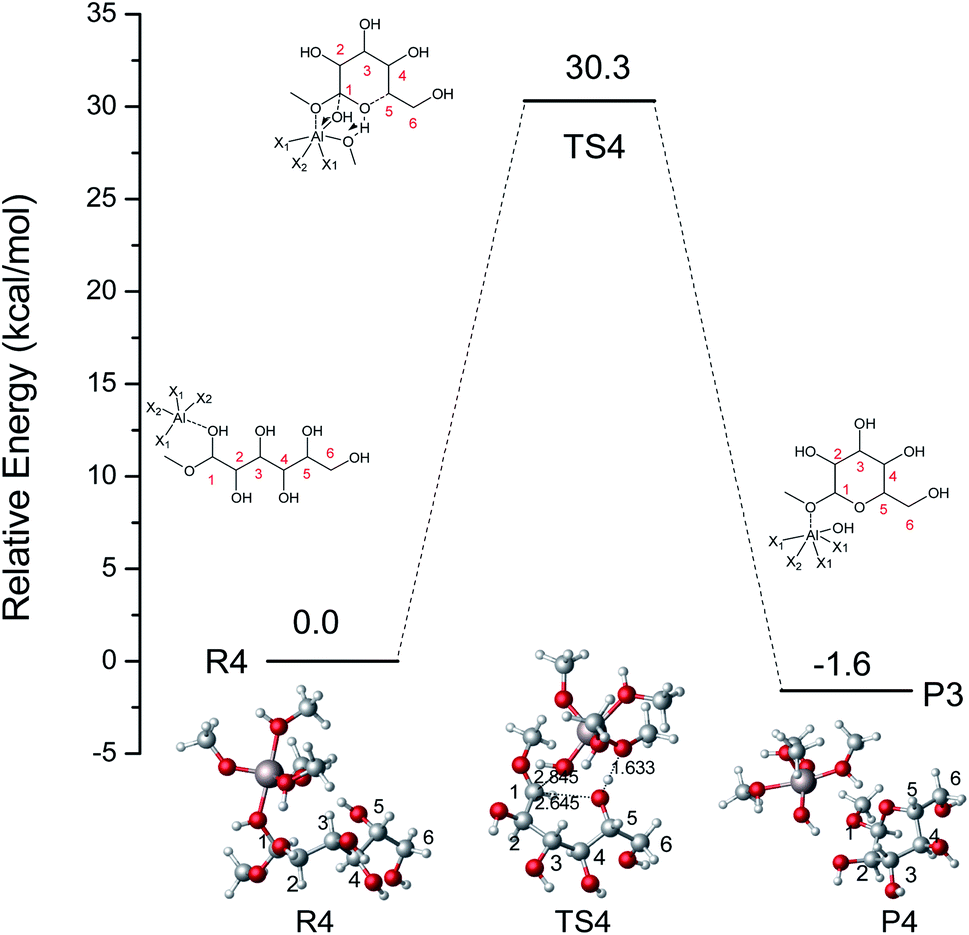
Based on the results mentioned above, the lowest energy barrier (52.8 kcal mol−1) is for the O5–C1 six-membered ring closure reaction which can produce MDGP. After probing the reaction pathways of the ring-closure reaction without a catalyst, we choose the way of O5–C1 six-membered ring closure with lowest energy barrier and the reaction mechanism catalyzed by [Al(CH3O)2(CH3OH)2]+ had been investigated based on the DFT calculations. The lowest energy barrier of IM2 through O5–C1 ring-closure reaction to MDGP is demonstrated in Fig. 7. Initially, H-bonds were formed between Al3+ and O1 with a 1.960 Å bond length. The C1 site was activated by Al3+ as the C1⋯O1H bond elongated by 0.538 Å (original 1.422 Å). In the TS4 structure, the H5 of O5 transfers to the methoxy coordinated with Al3+, and the O1–H1 broke away with the coordination of Al3+. The length of C1–O5 gradually decreased and MDGP was finally formed with 30.3 kcal mol−1. This process is exothermic by 1.6 kcal mol−1. Comparing the energy barrier of each step, the ring-closure reaction is the overall limited step in the Fischer–Helferich glycosidation.
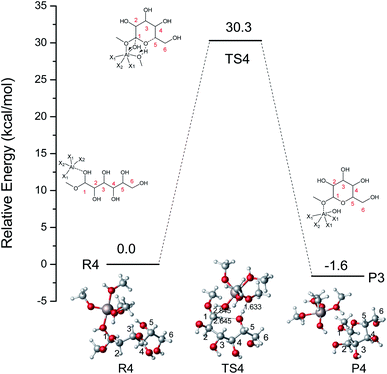 |
| | Fig. 7 The potential energy (kcal mol−1) profile with the structures of reactants, transition states, and products for the O5–C1 ring-closure reaction to MDGP catalyzed by [Al(CH3O)2(CH3OH)2]+. (X1 = CH3OH, X2 = CH3O, bond distances in Å). | |
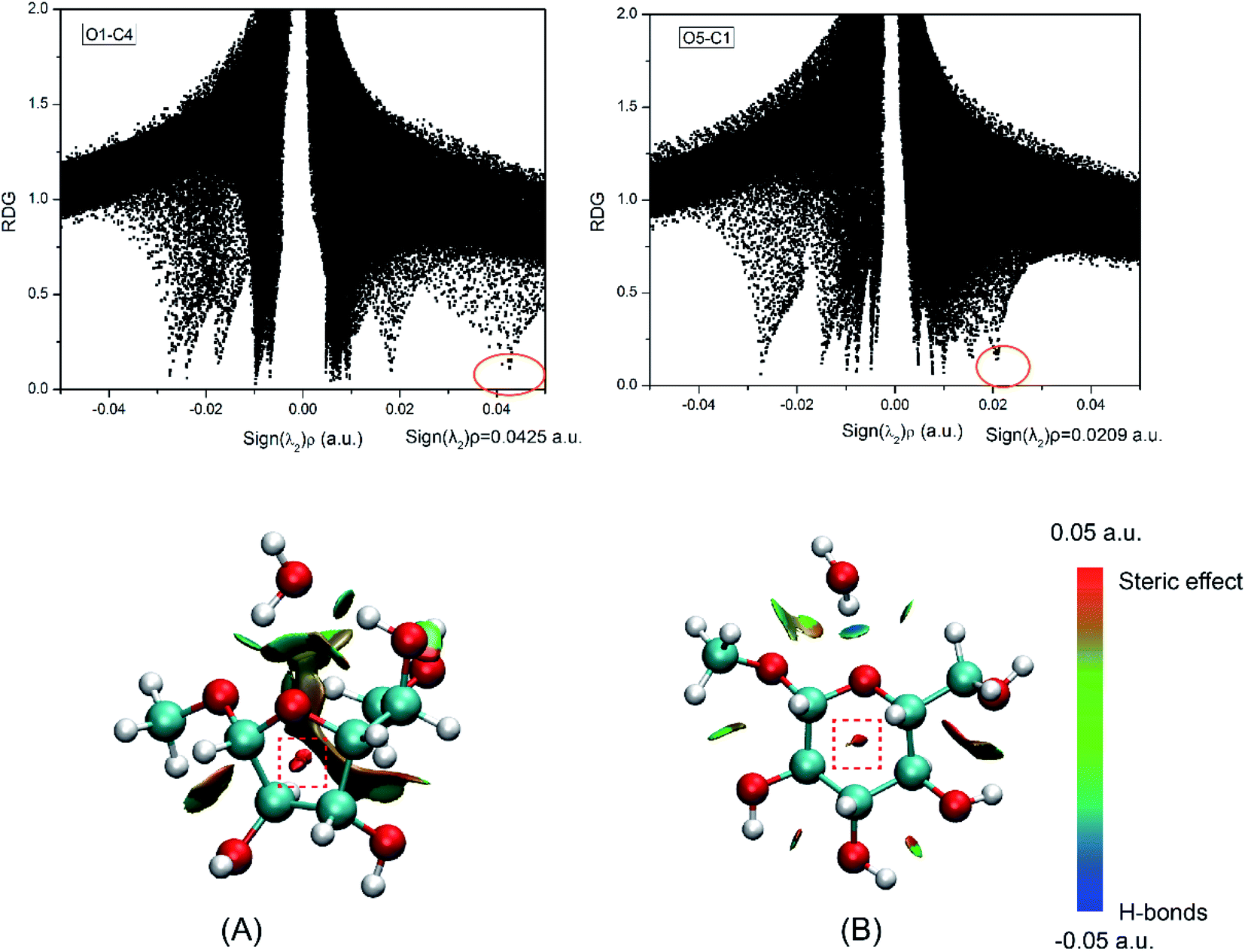
As a practical tool for visualizing interaction types, reduced density gradient (RDG) analysis was proposed by Yang et al. based on the reduced density gradient versus the electron density multiplied by the sign of second Hessian eigenvalue.50 This method has been prevalently used to study non-covalent interaction in many DFT types of research.51,52 To assess the ability of different ring-closure reactions, RDG analysis was performed for the products of five-(O4–C1) and six-(O5–C1) membered ring closure reactions as listed in Fig. 8. In the ring formation, the effects of steric hindrance will become apparent.53,54 The red region (Fig. 8) which was plotted with a red rectangle can be classified as the steric hindrance effect in the ring closure process. The sign(λ2)ρ of the five- and six-membered ring closure products is 0.0425 and 0.0209 a.u., respectively. It indicates that the steric effect in the middle of the five-membered ring is stronger than that of six-.
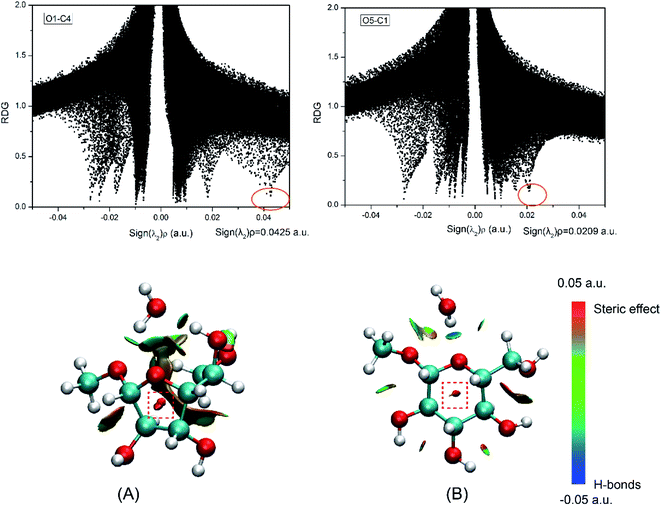 |
| | Fig. 8 RDG analysis for the (A) five-number ring and (B) six-number ring. The isosurfaces are colored on a blue–green–red scale according to values of sign (λ2)ρ, ranging from −0.05 to 0.05 a.u. Blue indicates strong attractive interaction and red indicates steric effect. | |
3.4 The whole glycosidation of glucose to methyl glycosides
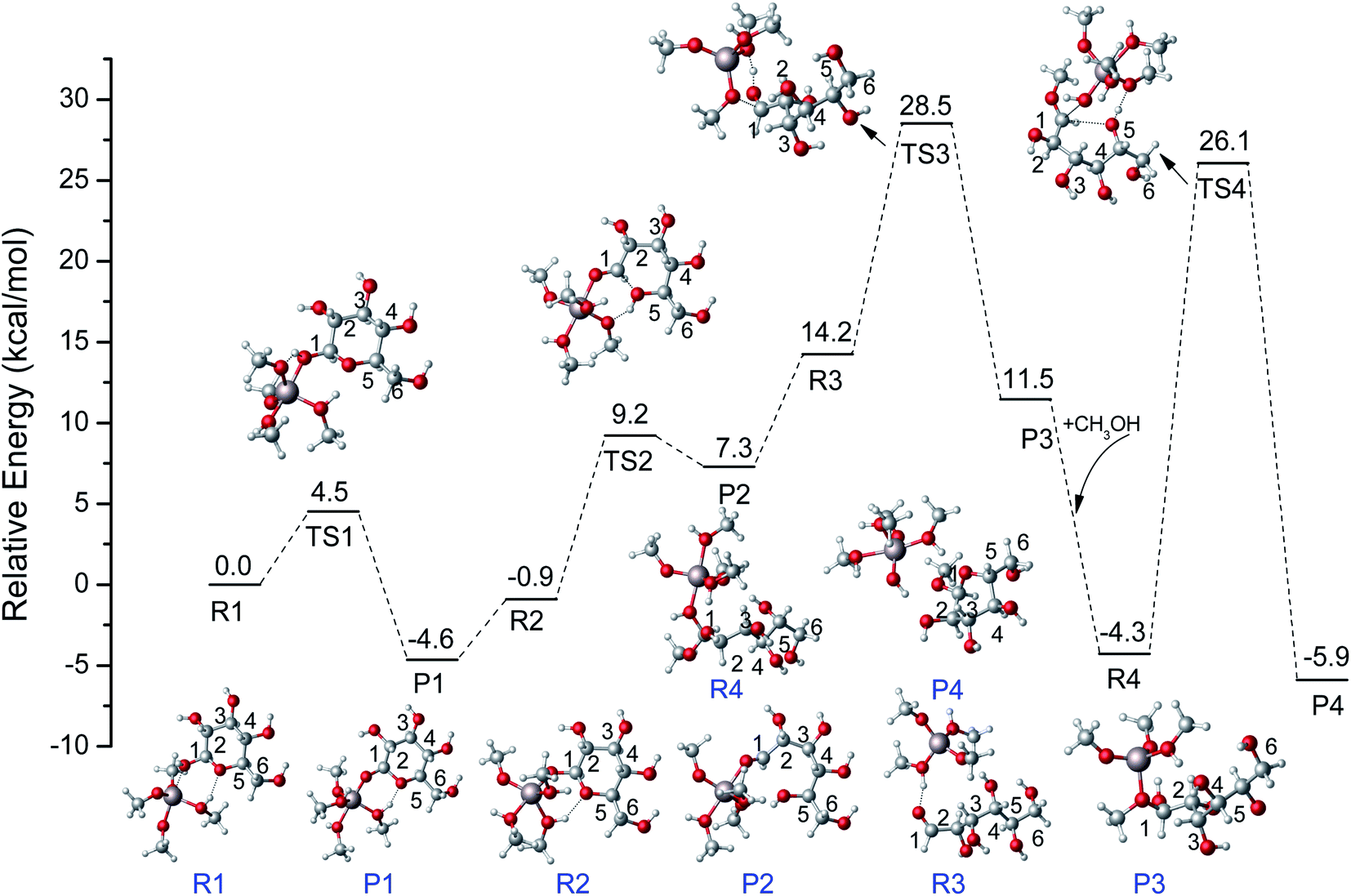
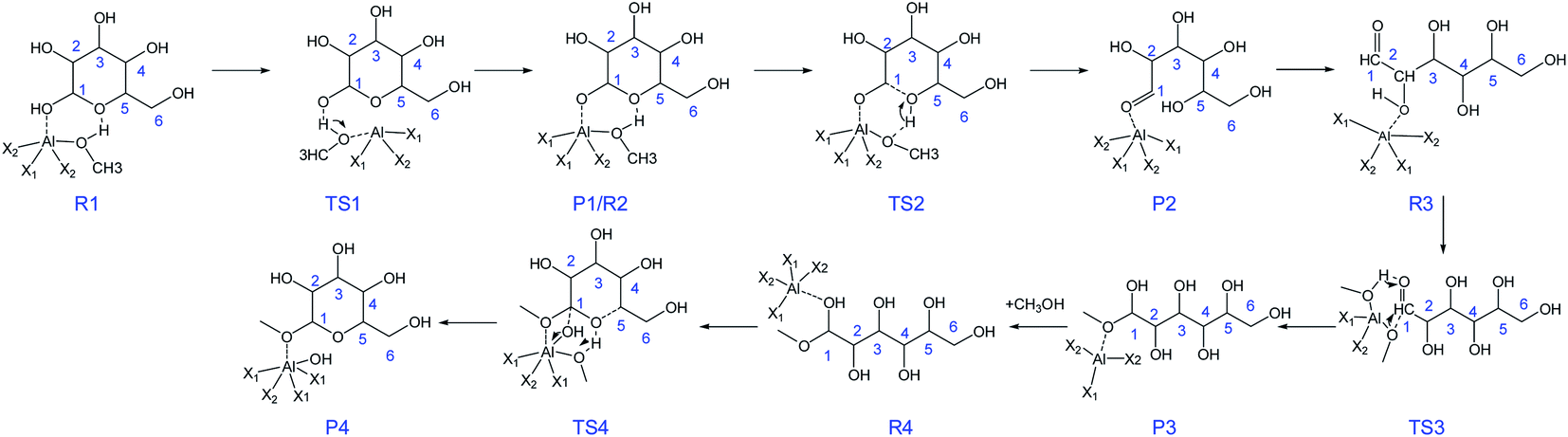
Combining the above DFT calculations, the complete mechanism of glycosidation of glucose to methyl glycosides catalyzed by [Al(CH3O)2(CH3OH)2]+ is depicted in Fig. 9 and the detailed reaction mechanism is presented in Scheme 3 based on the lowest reaction pathways by DFT calculations. The optimized XYZ coordinates of all TSs without catalyst and catalyzed by [Al(CH3O)2(CH3OH)2]+ are listed in Table S2 and S3,† respectively. The first stage of the ring-opening reaction corresponds to the approach of glucose toward the active site resulting in complex R1, in which the O1 strongly bonded to the Al3+ center (Al–O1 = 2.031 Å) and the O5 bonded to the methanol coordinated with Al3+ (OH⋯O5 = 1.769 Å). About 4.5 kcal mol−1 energy barrier needs to be overcome via the transition state structure of TS1. The formation of intermediate P1 where Al3+ coordinated with O1 of glucose will be beneficial for the following ring-opening step. Subsequently, the hydrogen of hydroxyl of methanol coordinated with Al3+ migrates to O5, and the reaction needs to overcome the 10.1 kcal mol−1 energy barrier with the C1–O5 bond cleavage. The formation of the ring-opening structure is endothermic with an energy change of 8.1 kcal mol−1. The structure of P2 was the ring-opening of glucose with the Al3+ interacting O1 site. For the R3, it has the same 2D molecular formula as P2 through the rotation of C–C bond. They are spatial isomers without transient states. For the ring-opening step of glucose, it had been referred by the isomerism of glucose to fructose reported before.55,56 Then, the methoxy (CH3O) coordinated with Al3+ forms CH3O–C1 bond, and the hydrogen of hydroxyl of methanol migrates to the O1 via TS3. The barrier of the addition reaction is 14.3 kcal mol−1 which is relatively higher than the ring-opening step. This step is exothermic with 2.7 kcal mol−1. In the last step, the center Al3+ strongly coordinated with O1, the C1–O1 bond has been activated where the bond length of C1–O1 becomes longer (original 1.409 to 1.453 Å). For the TS4, the O1H will break away from C1, and the H5 transfers to the methoxy coordinated with Al3+, at the same time O5–C1 bond was formed and finally generate MDGP. The energy barrier is 30.5 kcal mol−1 and this step is exothermic with 1.6 kcal mol−1. It can be concluded that the rate-determining step is the ring closure reaction. Compared with the energy profile of no catalyst, the Al3+ can show a great catalytic effect in the glycosidation of glucose to MDGP, and Al3+ plays a critical role in the hydrogen transfer.
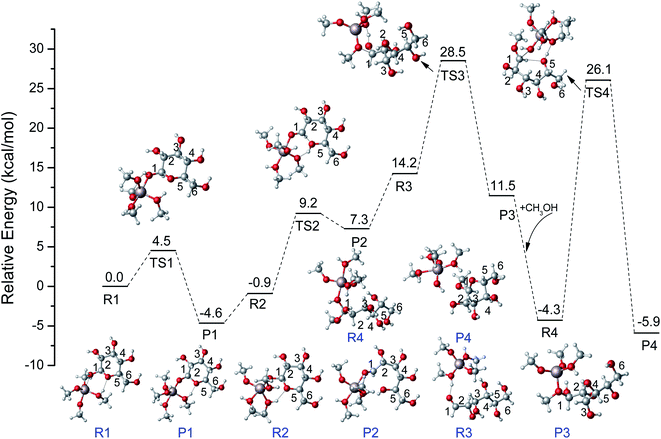 |
| | Fig. 9 The potential energy (kcal mol−1) profiles for glycosidation of glucose to methyl glycosides catalyzed by [Al(CH3O)2(CH3OH)2]+ at M06-2X-D3/6-311+G** level. | |
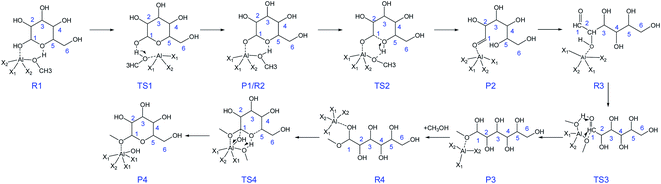 |
| | Scheme 3 The proposed mechanism for glucose to methyl glycosides in methanol solvent catalyzed by [Al(CH3O)2(CH3OH)2]+. (X1 = CH3OH, X2 = CH3O). | |
4. Conclusion
For the methanolysis of carbohydrates into methyl levulinate, Al-based catalysts, such as Al2(SO4)3 and AlPW12O40 have shown great advantages in the conversion process. As a prevalent reaction, Fischer–Helferich glycosidation is easy to proceed from glucose to alkyl glucosides in an alcoholic solvent. DFT calculations have been carried out to reveal the detailed mechanism for the glycosidation of glucose to methyl glycosides by Al-based catalysts. There are mainly three steps for the Fischer–Helferich glycosidation, which are ring-opening, addition, and ring-closure steps. The Al3+ coordinated with methanol/methoxyl could reduce the energy barrier to some extent in the ring-opening step by transferring the proton. In the addition reaction, the electronegativity of C1 increased when the methanol was added to the C1 site. It would be beneficial for the following ring-closure step. For the ring-closure step, there are probably 28 different kinds of ways, and the O5–C1 which rendering the MDGP is more preferred with the lowest energy barrier. In the whole process of Fischer–Helferich glycosidation catalyzed by [Al(CH3O)2(CH3OH)2]+, the ring-closure is the rate-determining step and the Al3+ coordinated with methanol/methoxyl mainly reflects on a proton shuttle to promote the reaction. A novel reaction mechanism of Fischer–Helferich glycosidation was proposed based on our DFT calculations. This work would provide basic aid to understand the mechanism of Fischer–Helferich glycosidation catalyzed by Al-based catalysts and the conversion of glucose in the alcoholic solvent.
Author contributions
Mengting Yu: methodology, writing and editing. Yao Li: funding acquisition, Cheng Zhang and Huaying Luo: project administration and resources, Chengsheng Ge: formal analysis, Xiaobin Chen and Lanlan Fu: data curation, Zhaoyang Ju and Xiaoqian Yao: review and editing. All authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (21908222), the Key R&D Program of Zhejiang Province (2021C02031), and the Research Fund for the Quzhou University (BSYJ202015 and BSYJ202113). The work was carried out at Shanxi Supercomputing Center of Chia, and the calculations were performed on TianHe-2.
Notes and references
- K. Alper, K. Tekin, S. Karagöz and A. J. Ragauskas, Sustainable Energy Fuels, 2020, 4, 4390–4414 RSC.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- D. Di Menno Di Bucchianico, Y. Wang, J.-C. Buvat, Y. Pan, V. Casson Moreno and S. Leveneur, Green Chem., 2022, 24, 614–646 RSC.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- Z. Ju, X. Yao, Z. Luo, M. Cao and W. Xiao, Carbohydr. Res., 2020, 487, 107882 CrossRef CAS PubMed.
- J. Liu, X. Wang, B. Yang, C. Liu, C. Xu and W. Dong, Renewable Energy, 2018, 120, 231–240 CrossRef CAS.
- A. Farrán, C. Cai, M. Sandoval, Y. Xu, J. Liu, M. J. Hernáiz and R. J. Linhardt, Chem. Rev., 2015, 115, 6811–6853 CrossRef PubMed.
- S. Zhu, J. Guo, X. Wang, J. Wang and W. Fan, ChemSusChem, 2017, 10, 2547–2559 CrossRef CAS PubMed.
- H. Chen, J. Liu, X. Chang, D. Chen, Y. Xue, P. Liu, H. Lin and S. Han, Fuel Process. Technol., 2017, 160, 196–206 CrossRef CAS.
- F. Yu, R. Zhong, H. Chong, M. Smet, W. Dehaen and B. F. Sels, Green Chem., 2017, 19, 153–163 RSC.
- D. Ding, J. Xi, J. Wang, X. Liu, G. Lu and Y. Wang, Green Chem., 2015, 17, 4037–4044 RSC.
- W. Deng, L. Mi, Q. Zhang and W. Ye, Catal. Today, 2011, 164, 461–466 CrossRef CAS.
- K. Chung, H. Kim, Y. Park, B. Kim, S. Kim and S. Jung, J. Nanosci. Nanotechnol., 2019, 19, 1172–1175 CrossRef CAS PubMed.
- S. Masui, Y. Manabe, K. Hirao, A. Shimoyama, T. Fukuyama, I. Ryu and K. Fukase, Synlett, 2019, 30, 397–400 CrossRef CAS.
- H. Zhou, B. Wang, X. Guo, X. Zhang, X. Wei, C. Peng, D. R. MacFarlane and Y. Yuan, Chem. Commun., 2018, 54, 11969–11972 RSC.
- W. G. Ramdani, A. Karam, K. D. O. Vigier, S. Rio, A. Ponchel and F. Jérôme, Mol. Catal., 2019, 468, 125–129 CrossRef CAS.
- L. Zhou, H. Zou, J. Nan, L. Wu, X. Yang, Y. Su, T. Lu and J. Xu, Catal. Commun., 2014, 50, 13–16 CrossRef CAS.
- K. Tominaga, K. Nemoto, Y. Kamimura, A. Yamada, Y. Yamamoto and K. Sato, RSC Adv., 2016, 6, 65119–65124 RSC.
- Z. Ju, Y. Zhang, T. Zhao, W. Xiao and X. Yao, ACS Sustainable Chem. Eng., 2019, 7, 14962–14972 CrossRef CAS.
- M. F. He, H. Q. Fu, B. F. Su, H. Q. Yang and C. W. Hu, J. Phys. Chem. B, 2014, 118, 13890–13902 CrossRef CAS PubMed.
- A. M. Norton, H. Nguyen, N. L. Xiao and D. G. Vlachos, RSC Adv., 2018, 8, 17101–17109 RSC.
- S. Saravanamurugan, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 135, 5246–5249 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, X. Lyu, G. Zhao, T. Zhao, L. Han and W. Xiao, J. Cleaner Prod., 2019, 215, 712–720 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- H. Guernon and C. Y. Legault, Organometallics, 2013, 32, 1988–1994 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, Theory Appl. Comput. Chem., 2005, 195–249 CAS.
- D. N. Lande, S. A. Bhadane and S. P. Gejji, J. Phys. Chem. A, 2017, 121, 1814–1824 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. Liu, X. Chen, Y. Zhang, M. Lu, H. Lyu, L. Han and W. Xiao, Energy Fuels, 2020, 34, 7085–7093 CrossRef CAS.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, J. Chem. Soc., Dalton Trans., 2001, 2095–2108, 10.1039/B102094N.
- D. D. Heard and R. Barker, J. Org. Chem., 1968, 33, 740–746 CrossRef.
- R. J. Ferrier and L. R. Hatton, Carbohydr. Res., 1968, 6, 75–86 CrossRef CAS.
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1996, 161, 713–719 CrossRef CAS.
- F. Dwight and J. Mowery, J. Org. Chem., 1961, 26, 3484–3486 CrossRef.
- J. Guan, Q. Cao, X. Guo and X. Mu, Comput. Theor. Chem., 2011, 963, 453–462 CrossRef CAS.
- J. Li, J. Li, D. Zhang and C. Liu, J. Phys. Chem. B, 2015, 119, 13398–13406 CrossRef CAS PubMed.
- J. Tang, X. Guo, L. Zhu and C. Hu, ACS Catal., 2015, 5, 5097–5103 CrossRef CAS.
- N. D. Tzoupanos, A. I. Zouboulis and C. A. Tsoleridis, Colloids Surf., A, 2009, 342, 30–39 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- Y. Yu, C. Hu and M. M. Abu-Omar, Bioresour. Technol., 2012, 116, 190–194 CrossRef PubMed.
- A. V. Demchenko, J. Am. Chem. Soc., 2010, 132, 16297 CrossRef CAS.
- Z. Ju, W. Xiao, X. Yao, X. Tan, B. A. Simmons, K. L. Sale and N. Sun, Phys. Chem. Chem. Phys., 2020, 22, 2878–2886 RSC.
- Z. Ju, S. Feng, L. Ren, T. Lei, H. Cheng, M. Yu and C. Ge, RSC Adv., 2022, 12, 2788–2797 RSC.
- F. Chen and T. Lu, Acta Phys.-Chim. Sin., 2012, 28, 1–18 Search PubMed.
- M. Miljkovic, Carbohydrates: synthesis, mechanisms, and stereoelectronic effects, Springer Science & Business Media, 2009 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- H. Li, Y. Chang, W. Zhu, W. Jiang, M. Zhang, J. Xia, S. Yin and H. Li, J. Phys. Chem. B, 2015, 119, 5995–6009 CrossRef CAS PubMed.
- H. He, S. Zhang, X. Liu, J. Wang, X. Yao and X. Zhang, Fluid Phase Equilib., 2013, 360, 169–179 CrossRef CAS.
- B. D. Marshall, A. Haghmoradi and W. G. Chapman, J. Chem. Phys., 2014, 140, 164101 CrossRef PubMed.
- B. D. Marshall and W. G. Chapman, Phys. Rev. E, 2013, 87, 052307 CrossRef PubMed.
- J. Li, J. Li, D. Zhang and C. Liu, J. Phys. Chem. B, 2015, 119, 13398–13406 CrossRef CAS PubMed.
- Y. Jing, J. Gao, C. Liu and D. Zhang, J. Phys. Chem. B, 2017, 121, 2171–2178 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yao Lib,
Cheng Zhangc,
Huaying Luoc,
Chengsheng Gea,
Xiaobin Chena,
Lanlan Fua,
Zhaoyang Ju
a,
Yao Lib,
Cheng Zhangc,
Huaying Luoc,
Chengsheng Gea,
Xiaobin Chena,
Lanlan Fua,
Zhaoyang Ju