
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of lipid bilayer encapsulation and lipid composition on the catalytic activity and colloidal stability of hydrophobic palladium nanoparticles in water†
Dominick D. Ortega,
Nicholas Pavlakovich and
Young-Seok Shon *
*
Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Blvd., Long Beach, CA 90840, Unites States. E-mail: ys.shon@csulb.edu
First published on 9th August 2022
Abstract
This article shows the preparation of a lipid-nanoparticle assembly (LNA) which contains hydrophobic palladium nanoparticles (PdNPs) within the hydrophobic regions of the liposomal micelles. To understand the colloidal stability and catalytic activity of LNAs, the structure–property relationships of LNAs are investigated by manipulating the lipid composition and reaction temperature. The studies of LNAs using dynamic light scattering (DLS), differential scanning calorimetry (DSC), and transmission electron microscopy (TEM) show decreased colloidal stability with the incorporation of PdNPs compared to their counterpart 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) liposomes without PdNPs. LNAs with PdNPs catalyze the hydrogenation of 1-octene and its isomers to octane under one atm hydrogen gas and at room temperature within 24 h. The kinetic studies show that the isomerization of 1-octene to 2-octene occurs more favorably in the early stage of the reactions, which is followed by the subsequent hydrogenation of all octene isomers. The studies on temperature effects indicate that there is a significant increase in conversion yield of substrates when the reaction temperature increases from 22 to 37 °C, which correspond to room temperature and biological temperature, respectively. Phase transition of DSPC-PdNP LNAs from gel to liquid crystalline phase changing the fluidity of the bilayer is proposed to be the main reason for dramatic increases in the catalytic activity of the LNAs. It is also found that the rate of hydrogenation is dependent on the lipid composition of LNAs with the presence of cholesterol having a negative influence on the catalytic activity of LNAs while increasing their colloidal stability.
Introduction
Nanoparticles (NPs) in the biomedical field have been of great interest over many years as applications of nanoparticles emerge in a wide variety of fields including as drug carriers, probes, and therapeutics.1–9 In addition to their unique size-dependent properties, the versatility of constructing the nanoparticles with various ligand functionalities has led to additional applications such as in controlled release, immune stimulation, deep tissue imaging, and sensing of cellular behavior.1,10,11 In order to further advance the biomedical applications of nanoparticles, however, the aqueous colloidal science of nanoparticles specific to the influence of biomolecules with different sizes, composition, and chemical properties should be better understood.12There have been recent advancements in implementing metal nanoparticles on, within barriers, and inside biological macrostructures, notably lipid structures.13 Nanoparticle-liposomal hybrid assemblies have gained attention after negatively charged Au nanoparticles were found to stabilize cationic liposome assemblies by coating on the surface and suppressing the fusion of liposomes. The stabilized liposomes could then fuse with bacterial cells and deliver the topical antimicrobial drug encapsulated within the liposomes.14 Examples of nanoparticle hybrid systems have now been expanded to include liposomes with surface-bound nanoparticles, liposomes with bilayer-embedded nanoparticles, liposomes with core-encapsulated nanoparticles, lipid assemblies with hydrophobic-core encapsulated nanoparticles, and lipid bilayer-coated nanoparticles.13–18
The nanoparticles embedded within the bilayer of liposomes are generally smaller than the ones incorporated in previously mentioned examples.19–21 It has been observed that the size and loading concentrations of embedded nanoparticles affect the membrane fluidity, bilayer thickness, and ultimately the phase transition temperature.14,19–23 Investigation by Park et al. has led to the analysis of fluidity of liposome bilayers as a function of nanoparticle loading concentrations. Results showed that the presence of dodecanethiolate-capped Ag nanoparticles could increase the fluidity of bilayer both above and below the phase transition temperature.22,23 Direct application of liposome bilayer-embedded nanoparticles includes effective stimuli-triggered thermal release based on the fusion with target cell membranes. Bilayer-embedded super paramagnetic iron oxide nanoparticles (SPIONS) exposed to alternating magnetic fields (AMFs) generated heat which increases permeability or rupturing of the bilayer, triggering the release of therapeutic agent such as hydrophobic raloxifene, a treatment for osteoporosis, and doxycycline HCl, an antimicrobial for bacterial infections.24 In addition to drug delivery/release, the enhancement of therapeutic effectiveness has also been investigated by Bao et al. in the utilization of Au nanoparticles capped by a derivative of anti-cancer drug paclitaxel (PTX-PEG-SH).25 The examples showed the robust and versatile nature of lipid–metal nanoparticle assemblies in the biomedical applications.
Our research group has shown that alkanethiolate-capped PdNPs exhibit excellent catalytic activity and selectivity for isomerization of unsaturated compounds in organic solvents.26–31 This PdNP was synthesized using the thiosulfate protocol with sodium S-alkyl thiosulfate as ligand precursor. Previous studies have shown that the catalytic reactions of hydrophobic PdNPs in aqueous solvent have not been as successful due to the incompatibility of the catalyst and substrate with polar protic solvents.26 To further examine the utility of hydrophobic ligand-capped PdNPs as efficient catalysts in aqueous environments and for the application of the aforementioned nanoparticles in conjunction with biological macrostructures, our work aimed to utilize hydrophobic PdNPs by embedding them within the bilayer of phosphatidylcholine (PC), a component lipid in the COVID-19 vaccine and other therapeutic agents.32–35 To obtain valuable fundamental information on colloidal stability and catalytic activity of liposomes with bilayer-embedded hydrophobic PdNPs in water, the effects of liposome encapsulation and lipid modulator (cholesterol) presence within lipid-nanoparticle assemblies (LNAs) of distearoylphosphatidylcholine (DSPC) are investigated. The effect of reaction temperature on catalytic activity of LNAs is also studied by performing the catalytic hydrogenation reactions at, below, and above the phase transition temperatures of DSPC liposome. The hydrogenation of organic compounds is a reaction of great industrial importance spanning from petroleum to pharmaceutical industries. The research into structure–function relationships of these biologically compatible catalytic materials for simple model reaction may allow for a more targeted approach when it comes to engineering them for specific purposes. In addition to such fundamental understanding on LNA catalysis, which does not exist in the current literature, the potential of LNAs as an efficient platform for performing organic reactions in aqueous green environments is demonstrated.
Results and discussion
Synthesis of hydrophobic Pd nanoparticles
Hydrophobic PdNPs were synthesized using a previously published method, the thiosulfate protocol as shown in Scheme 1.36–39 Briefly, tetrachloropalladate and S-dodecyl thiosulfate ligands (2 molar equivalents) were transferred to the organic toluene layer by a phase transfer agent, tetraoctylammonium bromide (TOAB). The subsequent reduction of Pd(II) to Pd(0) using sodium borohydride resulted in the formation of dodecanethiolate-capped PdNPs. The synthesized PdNPs were completely soluble in nonpolar solvents such as toluene and chloroform. PdNPs were characterized using UV-vis spectroscopy (Fig. S2†) and 1H NMR (Fig. S3†). UV-vis spectroscopy results confirmed the complete reduction of PdCl42− (Pd(II)) to Pd(0), the absence of any oxidized Pd species, and the successful formation of PdNPs.36 The absence of any other chemical shifts in 1H NMR spectrum besides the ones corresponding to surface-bound dodecanethiolate ligands indicated the high purity of the synthesized PdNPs.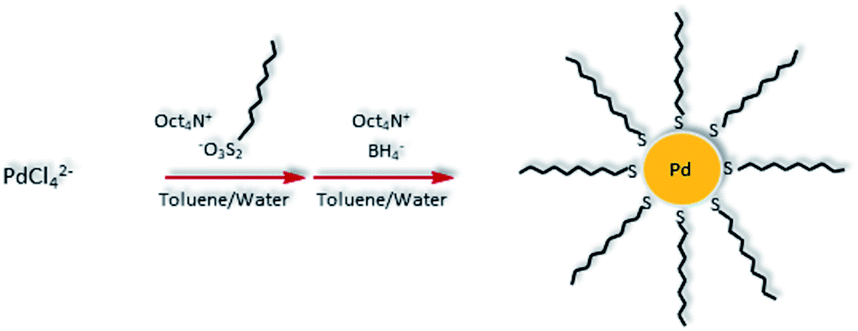 | ||
| Scheme 1 Synthesis of alkanethiolate-capped Pd nanoparticles using the thiosulfate synthetic protocol. | ||
Transmission electron microscopy (TEM) was used to determine the average core size (2.7 ± 1.1 nm) and morphology (spherical) of the nanoparticles (Fig. 1). The organic and Pd core compositions of PdNPs were determined using weight loss analysis (∼33% organic content) of thermogravimetric analysis (TGA) results shown in Fig. S4.† In conjunction with TEM results, the organic content was used for obtaining an estimated molecular formula [∼Pd586(C12H25)150] based on a truncated octahedron model.36 Using % composition of PdNPs, the ligand surface coverage to surface Pd ratio was estimated at ∼0.55. This calculation was obtained by dividing the average number of ligands determined from TGA analysis by the theoretical amount of surface Pd atoms based on the particle sizes determined by TEM analysis. The estimated molecular formula and ligand/surface Pd ratio is in accordance with the previously published work (∼Pd586(C12H25)142 and ∼0.52 ligand/surface Pd ratio) from our research group.36 TEM image in Fig. 1 (and S5†) showed that PdNPs are free of aggregations.
 | ||
| Fig. 1 TEM image of PdNPs synthesized via the thiosulfate synthetic method. The histogram of PdNPs was obtained by analysing several TEM images. | ||
Preparation of DSPC-PdNP LNAs
It has been shown that the hydrophobic metal nanoparticles of a similar or smaller size to the thickness of a phospholipid bilayer could be incorporated into liposomes via the thin film hydration method.40,41 The synthesized dodecanethiolate-capped PdNPs were embedded into liposome assemblies employing the same method. Chloroform was chosen as a solvent for homogeneous thin film formation considering its ability to dissolve both DSPC lipids and PdNPs in adequate concentrations. Dichloromethane was also investigated for the use as it dissolved PdNPs well, but ultimately was not chosen due to its modest ability to effectively solubilize phospholipids. The complex LNAs were formed from thin films by hydrating them to form liposomes, circular vesicles with a hydrophobic layer composed of phospholipids.DSPC-PdNP LNAs were characterized using 1H NMR, TEM, TGA, and dynamic light scattering (DLS). The 1H NMR spectra of DSPC liposomes showed chemical shifts above ∼2.0 ppm that represent hydrogens near the polar functional head groups such as the ammonium, phosphate, and acetate groups (Fig. S6†). By comparing both spectra of DSPC with and without PdNPs, the incorporation of the PdNPs with DSPC at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio was evident from the increase in –CH3 peak intensity at ∼0.8 ppm in the spectrum of DSPC-PdNPs. This change was much greater than the potential error normally associated with NMR integrations. The presence of stronger CH2 hydrogen signals at near ∼1.6–1.1 ppm also reflexed the presence of PdNPs with DSPC lipids.
:5 ratio was evident from the increase in –CH3 peak intensity at ∼0.8 ppm in the spectrum of DSPC-PdNPs. This change was much greater than the potential error normally associated with NMR integrations. The presence of stronger CH2 hydrogen signals at near ∼1.6–1.1 ppm also reflexed the presence of PdNPs with DSPC lipids.
TEM images of DSPC liposome and DSPC-PdNP LNAs are shown in Fig. 2. DSPC forms circular liposomal vesicles with diameter size within 20–70 nm without the presence of PdNPs (Fig. 2(a)). There appears to be no initial fusion of DSPC liposomal membranes and aggregation of vesicles based on this TEM image of fresh liposome samples. With the incorporation of PdNPs with a short heat treatment at 55 °C and a subsequent cooling, TEM images obtained from fresh samples showed the presence of many stand-alone liposomal vesicles of around ∼50 nm in size (Fig. 2(b)). The formation of LNAs was evidenced by the presence of spherical structures surrounded with a thin lipid bilayer. Darker PdNPs were clearly visible within or near the edge of vesicles indicating the embedding of the PdNPs into the bilayer of DSPC liposomes. A few PdNPs were also shown within the vesicles, but this is more likely due to the 3-D shape of the vesicles where the PdNPs are either shown embedded in the back or the front of the liposomes.42 Some PdNPs were observed to be outside of the liposomes, but they were mostly stabilized by small lipid bundles based on the presence of grey domains around or near PdNPs. The presence of joined or slightly fused LNAs was also clearly observed from TEM image. Application of intense stirring force was found to facilitate more embedding of the PdNPs, but at the same time these processes forced slight fusions and aggregations of liposomes which are clearly observed in TEM image shown in Fig. 2(c). PdNPs were mostly present within the bilayer of the LNAs, but a few PdNPs were exposed on the external surface suggesting a potential increase in hydrophobicity that would cause the aggregation of LNAs via the intermolecular hydrophobic interactions. Heat treatments of LNAs at ∼40 °C for 2 h in addition to additional drying and hydration of LNAs were found to facilitate the extensive fusion and aggregation of liposomes as shown in Fig. 2(d). It is necessary to clarify that TEM images were obtained in vitro after the removal of solvents from drop-casted samples to make vesicles stationary. Therefore, the drying process could have made the vesicles fuse more during the solvent evaporation considering the liposome's dynamic nature.
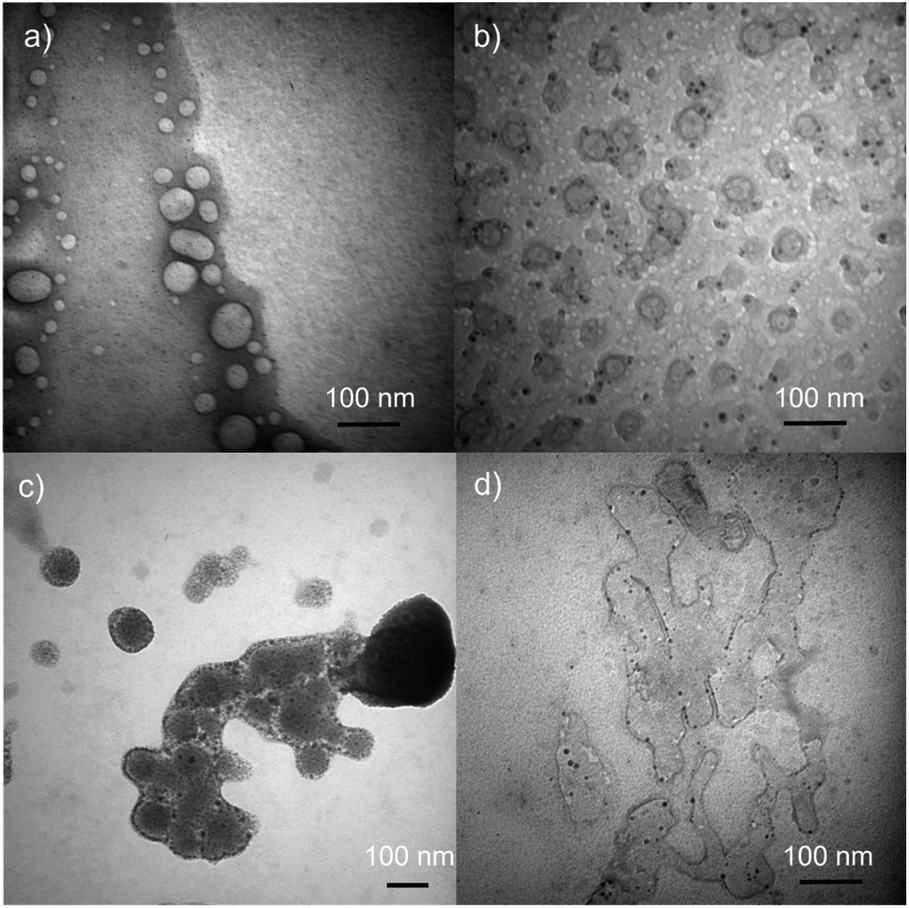 | ||
| Fig. 2 TEM Images of (a) DSPC liposomes, (b) DSPC-PdNP LNAs, (c) DSPC-PdNP LNAs after intense stirring, and (d) DSPC-PdNP LNAs after extended heating and drying. Size bars are 100 nm for all images. | ||
Size distributions of the DSPC liposomes and DSPC-PdNP LNAs in water obtained immediately after the preparations are depicted in DLS spectra shown in Fig. 3. For DSPC liposomes without PdNPs, the maximum intensity in the size distribution is 108.7 ± 13.2 nm. The range of the distribution appears to be between 80–170 nm in size. Within one week, however, the aggregation and fusion of vesicles could be further observed from DLS studies of DSPC liposomes (vide infra). Some vesicles shown in TEM image (Fig. 2(a)) are comparable in size to the DLS measurements as some vesicles are close to ∼100 nm. In contrast, measurements from DSPC-PdNP LNAs show the maximum peak distribution of 213.5 ± 25.9 nm. The results indicated that the incorporation of PdNPs to the bilayers of DSPC liposomal vesicles makes the vesicles less stable and causes them to aggregate or fuse, increasing the diameter size analyzed by the DLS. With the DSPC-PdNP LNAs constantly moving and bumping into each other in solution phase, the average size of destabilized liposomal vesicles could increase during the storage. It is important to mention that the TEM images from Fig. 2(c) and d of the irregularly shaped liposomal hybrids support the presence of liposomes with larger vesicle sizes observed in Fig. 3 for DSPC-PdNP LNAs. TEM and DSC characterization can complement each other but the results should still be interpretated with some uncertainties.
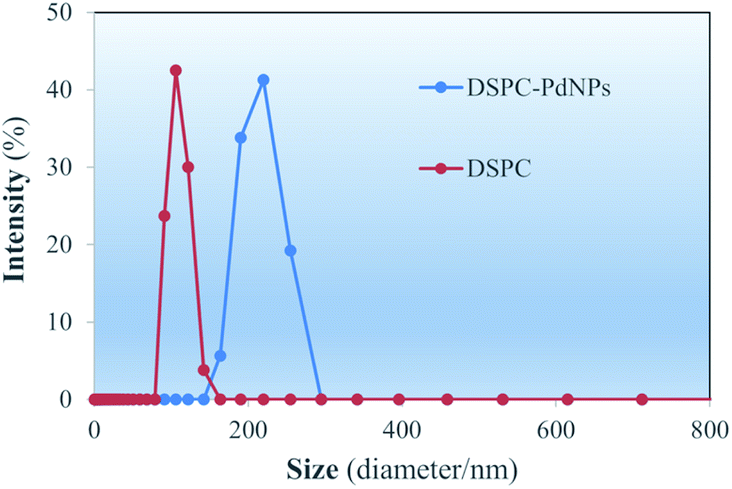 | ||
| Fig. 3 DLS size distributions of freshly prepared DSPC liposomes and DSPC-PdNP LNAs in water. | ||
The hydrophobic NPs used in this study are sufficiently small enough to be incorporated into liposomal formulations.42 Although the low NP concentrations used in liposomal drugs for magnetic resonance imaging or release of contents have not been deleterious to liposome stability,24 it was hypothesized the higher concentrations needed for effective NP catalysis might be deleterious to colloidal stability. The PdNPs are rigid and require deformation of the lipid bilayer to allow for their encapsulation. It was predicted the encapsulation of metal NP by the lipid bilayer would lead to further exposure of hydrophobic surfaces to the external aqueous solution.43 The hydrophobic surfaces could be derived from imperfect encapsulation and partial exposure of PdNPs.
Catalytic study of DSPC-PdNP LNAs
Previous work using octanethiolate-capped PdNPs with 1-pentene as a substrate in chloroform solvent under 1 atm hydrogen gas and at room temperature produced 2-pentene, the isomerization product, in ∼90% yield and pentane, the hydrogenation product, in ∼10% yield after 24 hours.28 The catalytic reaction of 1-octene using dodecanethiolate-capped PdNP in chloroform was examined under the same reaction condition. The result confirmed the unique selectivity of alkanethiolate-capped PdNPs toward the isomerization over the hydrogenation producing the isomerization product, 2-octene, in ∼90% yield (Scheme 2). In comparison, the heterogeneous reaction of 1-octene using dodecanethiolate-capped PdNP in water resulted in no reaction under the same reaction condition (Table S2†).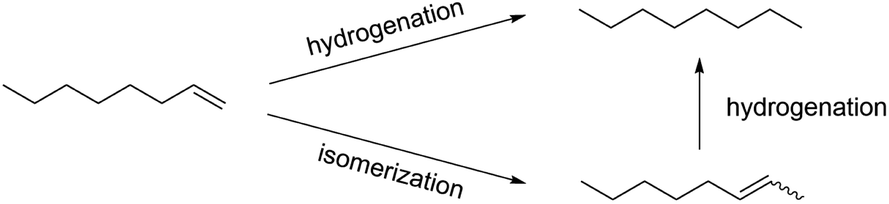 | ||
| Scheme 2 Catalytic reaction of 1-octene. | ||
For DSPC-PdNP LNAs, it was hypothesized that the permeability of the lipid bilayer should allow the embedded PdNP and H2 gas to form Pd–H species and allow the catalytic reactions to take place inside the liposomes. The catalytic activity of LNAs was studied under the same condition, an atmosphere pressure of H2 gas and room temperature, with a substrate loading of 20 mol1-octene molPd−1 with a catalyst loading of 2 mgPdNP in 10 mgDSPC. The reactions were independently attempted at various time periods ranging from 1 to 48 hours to obtain the kinetic information. Catalytic activity was evaluated based on the relative percentages of octane and the isomers of octene (E- and Z-2-octene and E-3-octene) present in the hydrophobic extract obtained from the post-catalysis colloidal dispersion of DSPC-PdNP LNAs. During the reactions, the colloidal dispersion was gently stirred and showed no obvious signs of aggregations.
The results suggested that the catalytic activity of PdNPs embedded in the bilayer of DSPC liposome was vastly different compared to that of hydrophobic PdNPs in CHCl3. While isomerization products (2-octene) were the major products (90%) formed in organic solvent for the catalytic reaction of 1-octene, the liposomal DSPC-PdNP LNAs produced the hydrogenation product as the only product at room temperature after 24 hours (Scheme 2). The kinetic studies shown in Fig. 4 revealed that the isomerization of 1-octene to 2-octene is the major pathway during the first few hours as the maximum percentage of 2-octene (∼<55%) occurred within the first 4 h of reaction. The isomerization of 1-octene to E-2-octene, more thermodynamically stable isomer, dominates over the isomerization of 1-octene to Z-2-octene and 3-octene. This is followed by the continued hydrogenation of all octene isomers to octane, the hydrogenation product. The percentage of hydrogenation products increased gradually to a maximum of 100% at 24 h. The hydrogenation reaction took over after the initial competition of isomerization reaction, which has the potentially lower activation energy. The formation of the σ bonded Pd–alkyl intermediate,30 the necessary intermediate for hydrogenation product, must have been promoted by the dynamic lipid chain interactions with hydrophobic substrates. The hydrophobic micellization of both substrates and H2 within hydrophobic regions in a more concentrated form44 should also have contributed to this activity change of PdNPs within DSPC liposome in water compared to that in organic solvents without lipid assemblies.
 | ||
| Fig. 4 Kinetic studies for the catalytic reactions of 1-octene to the isomerization and hydrogenation products by LNAs of DSPC-PdNP (1 h to 24 h) at room temperature. The standard deviations of each data point are less than ±5%. | ||
Effects of reaction temperature on the catalytic activity of DSPC-PdNP LNAs
Lipid bilayers composed of diacyl phosphatidylcholines exhibit temperature dependent chain dynamics.45–47 Although the exact temperatures at which a change in dynamics occurs are different depending on the structure of lipids, there is a general trend most phosphatidylcholines follow. At low temperatures, the acyl chains occupy an ordered gel phase characterized by restricted movement of the acyl chains and a narrowing of the phospholipid bilayer. At elevated temperatures, the acyl chains occupy a disordered liquid crystalline phase characterized by a widening of the phospholipid bilayer and free movement of the acyl chains. PC with sufficiently long acyl chains such as DSPC also have an intermediate phase called the rippled gel phase. DSPC is in the ripped gel phase at room temperature with a transition to liquid crystalline phase occurring at 26.3 ± 5.2 °C (Tg-to-l). This may explain the tendency of DSPC liposomes to form gels at room temperature. The melting transition from the liquid crystalline phase to amorphous phase takes place at 54.5 ± 1.5 °C (Tm).46–49The catalytic activity and selectivity of the DSPC-PdNP LNAs were examined at different temperatures in water. By using 1-octene as substrate, the catalytic activity of the LNAs was analyzed at 1, 3, 6, and/or 24 hours at 0, 22, 37, and 55 °C, which are water freezing point, room temperature (close to gel-to-liquid crystalline transition temperature (Tg-to-l) of DSPC), biological temperature, and melting transition temperature (Tm) of DSPC,46,49 to see the potential effect of the phase transition of the DSPC liposome bilayers. A summary of the results provided in Table 1 showed 100% conversion of 1-octene to the hydrogenation product, octane, for all conditions. A clear evidence of temperature effects on the catalytic activity of DSPC-PdNP was observed, when the temperature increases from 22 to 37 °C. The reactions completed at 0 and 22 °C exhibited a steady increase in the overall conversion as the reaction time progresses. However, at the biological body temperature (37 °C) and higher (55 °C), there is a significant increase in the conversion of the substrate at >98% within 1 h compared to the reactions completed at lower temperatures.
| Time (hr) | 0 °C | 22 °C | 37 °C | 55 °C |
|---|---|---|---|---|
| % | % | % | % | |
| 1 | 23.7 | 36.2 | 98.1 | 98.4 |
| 3 | 33.1 | 65.6 | — | — |
| 6 | 43.2 | 82.5 | — | — |
| 24 | 100 | 100 | 100 | 100 |
The results indicated that the dramatically increased catalytic activity of DSPC-PdNP LNAs is due to the gel-to-liquid crystalline transition of DSPC liposome bilayer rather than simple thermodynamic effects. The transition to the liquid crystalline phase has allowed free movement of the acyl chains and facile exposure of substrates to the catalytic surface. Since the incorporation of PdNPs within the bilayer of the DSPC could disrupt the packing of the DSPC bilayers and change the transition temperatures between different phases, differential scanning calorimetery (DSC) characterization of DSPC-PdNP LNAs was attempted. DSC spectrum in Fig. S6† indicated the intense broadening of transition temperature range while exhibiting a similar maximum Tm temperature compared to that of DSPC liposomes. This broadening of transition temperature indicated the heterogeneous nature of DSPC-PdNP LNAs resulted from their low colloidal stability and fusion problems. Therefore, it is also possible that the disordered liquid crystalline and amorphous phase of DSPC bilayers could co-exist at the temperature near 37 °C resulting in the improved catalytic activity of DSPC-PdNP LNAs at this temperature via the increased dynamic interactions between the fatty acid chain, the alkyl chain on the PdNPs, and the substrates.
Substrate scope on the catalytic activity of DSPC-PdNP LNAs
To further investigate the structural scope of substrates, the catalytic reactions of 2-octene isomers were attempted. For the catalytic reaction of 1-octene, the kinetic studies indicated the isomerization of 1-octene to E-2-octene and Z-2-octene are initially the main pathway before the hydrogenation of octene isomers takes place. It was projected that the hydrogenation of internal alkenes, both E-2-octene and Z-2-octene, would likely take place within 24 h, but the kinetics of hydrogenation for two substrates could be different. The catalysis results showed that the catalytic reaction of Z-2-octene resulted in complete hydrogenation to octane within 24 h reaction (Fig. 5). Due to the greater thermodynamic stability of the E-isomer, the Z-isomer substrate was converted to the E-isomers while they also transform into octane directly at the similar rate during the initial stages of reactions (<6 h). Formation of 3-octene, a product not seen formed in other reactions of the PdNPs in organic solvent, was also observed within 10%. Once the yields of E-isomer and 3-octene reached 40% and 10%, respectively, they were consumed while a sharp increase in the formation of octane product occurred. The complete hydrogenation of octene isomers to octane after the isomerization indicated that the hydrogenation of internal alkenes takes place well when the concentration of E-isomers is relatively low.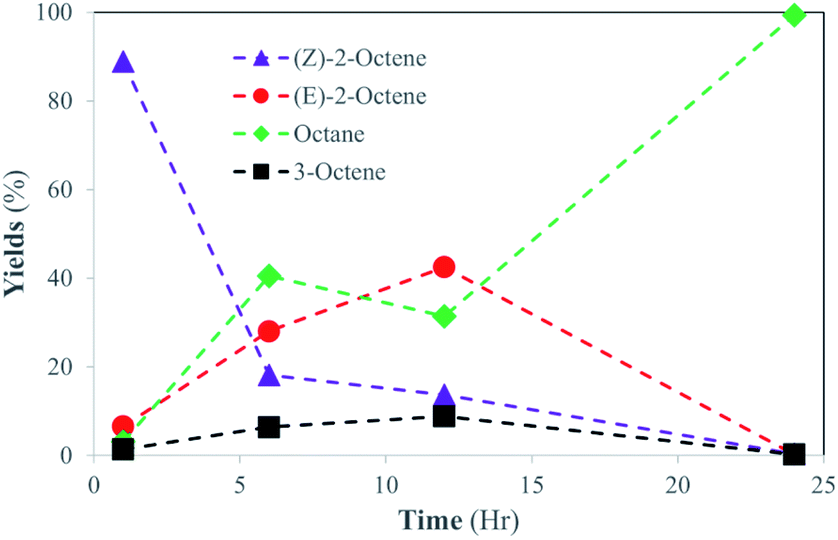 | ||
| Fig. 5 Catalytic Conversion of Z-2-octene into isomerization and hydrogenation products at room temperature. The standard deviations of each data point are less than ±8%. The large deviations in some data points are because some kinetics data points veered off from the overall results as outliers. This inconsistency was caused by running reactions independently from one another to preserve the PdNP:DSPC:Substrate ratio. | ||
In comparision, the catalytic reaction of E-2-octene in Fig. 6 showed that there is a logarithmic trend in the consumption of substrate and formation of hydrogenation product. The reaction did not go to completion as octane yield was at ∼55% while substrate consumption nearly stopped at ∼70% after 24 h. The results indicated that the hydrogenation of E-isomer is more difficult compared to that of Z-isomer implicating the impact of steric interactions between E-2-octene with alkanethiolate ligands on PdNPs. Formation of the other isomers remained within 10–15% with minimal formation of the Z-2-octene isomer. This observation is attributed to the greater thermodynamic stability of the E-isomer compared to that of the Z-isomer. The hydrogenation yield increased only slightly even after the 48 hours reaction (data not shown). It is proposed that the steric interaction of E-2-octene isomer would make the formation of the di–σ bonded Pd–alkyl intermediate needed for octane formation more difficult within liposome bilayer. The steric interaction and space saturation by E-isomer inside the bilayer also cause the diffusion of E-2-octene to PdNP more difficult to take place.
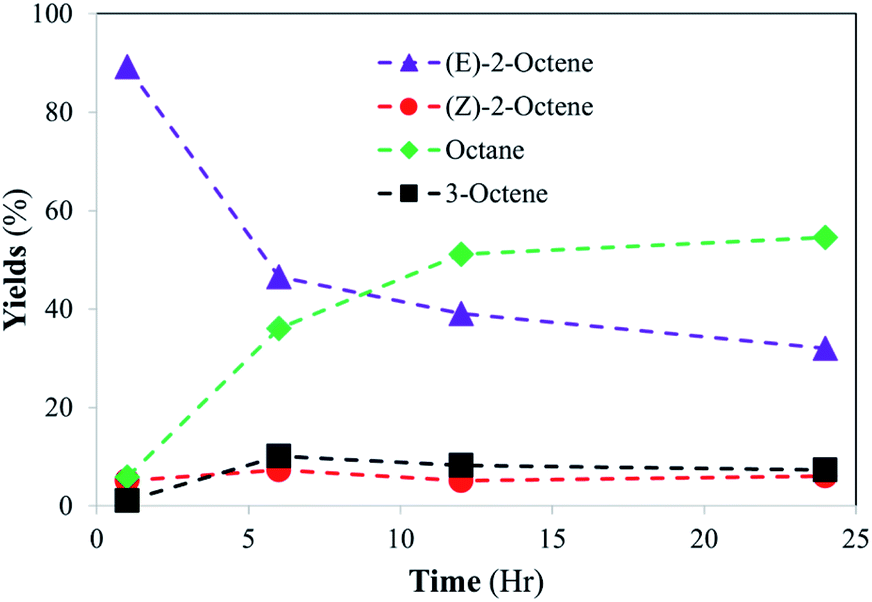 | ||
| Fig. 6 Catalytic conversion of E-2-octene into isomerization and hydrogenation products at room temperature. The standard deviations of each data point are less than ±5%. | ||
Effects of lipid moderator (cholesterol) on the stability of DSPC liposomes
Liposome stability is related to the presence of packing defects leading to exposure of hydrophobic surfaces to the outer aqueous environment.50,51 At room temperature, liposomal formulations in the form of modulators, spacers, and ionogens have been used for the stabilization of PC liposomes.52–54 In this study cholesterol was chosen as a bilayer modulator due to its prevalence in the membranes of mammalian cells and its well documented effects on the physical properties of lipid bilayers.55–57 The inclusion of various amounts of cholesterol into liposome formulations was investigated to determine the effect cholesterol concentration has and the optimal amount able to be included in the liposomal formulations.The stability of liposomes prepared in this study was investigated using DLS (Fig. 7 and Table 2), which shows the effect of cholesterol content on the intensity weighted particle size distributions of aged (∼one week) DSPC-based liposome formulations. DLS measurements showed similar D values of liposomes with 9% chol (10.0.1 – mass (in mg) of DSPC, PdNP, and cholesterol, respectively, used per 1 mL of each formulation) to that of pure DSPC liposomes (10.0.0) but with a lower intensity of light scattered by the smallest liposomes, suggesting an increase in aggregation potential. Liposomes prepared with 17%, 23%, and 29% cholesterol (10.0.2, 10.0.3, and 10.0.4) showed a much narrower size distribution supporting its purported use as a modulator by intercalating into hydrophobic defect sites. At 33% chol (10.0.5) the formation of larger particles was observed again. Concentrations that high are known to form cholesterol monohydrate crystals and could be the reason larger particles are observed.57
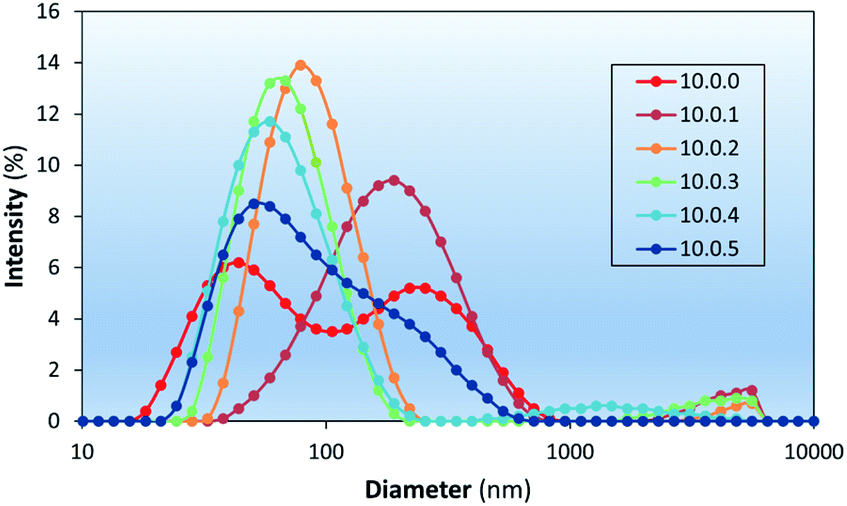 | ||
| Fig. 7 The effects of cholesterol on the colloidal stability of DSPC liposomes (aged ∼ one week). Labels represent the mass (in mg) of DSPC, PdNP, and cholesterol (e.g., 10.0.2) used per 1 mL of each formulation. | ||
| Formulation | D10 | D50 | D90 |
|---|---|---|---|
| 10.0.0 | 29 | 93 | 348 |
| 10.0.1 | 93 | 175 | 408 |
| 10.0.2 | 53 | 89 | 155 |
| 10.0.3 | 39 | 64 | 120 |
| 10.0.4 | 34 | 60 | 130 |
| 10.0.5 | 35 | 73 | 228 |
| 10.2.0 | 125 | 441 | 1027 |
| 10.2.2 | 73 | 171 | 408 |
The effects of dodecanethiolate-capped PdNP on the colloidal stability of LNAs composed of 83% DSPC:17% chol (10.0.2) and 72% DSPC:14% PdNP:14% chol (10.2.2) are investigated as shown in Fig. 8. The colloidal LNAs made from hydrating the film of lipids and PdNP showed that stability of LNAs decreased with the addition of PdNP even in the presence of cholesterol. Furthermore, the results also showed that the trend of cholesterol increasing colloidal stability of DSPC liposomes is also true for corresponding LNAs based on the D value shown in Table 2 (10.2.0 vs. 10.2.2). Further research into the stabilizing effects of lipid composition on LNAs could assess whether the changes in colloidal stability are due to modification of the phase behaviour, a morphological change, or another mechanism.
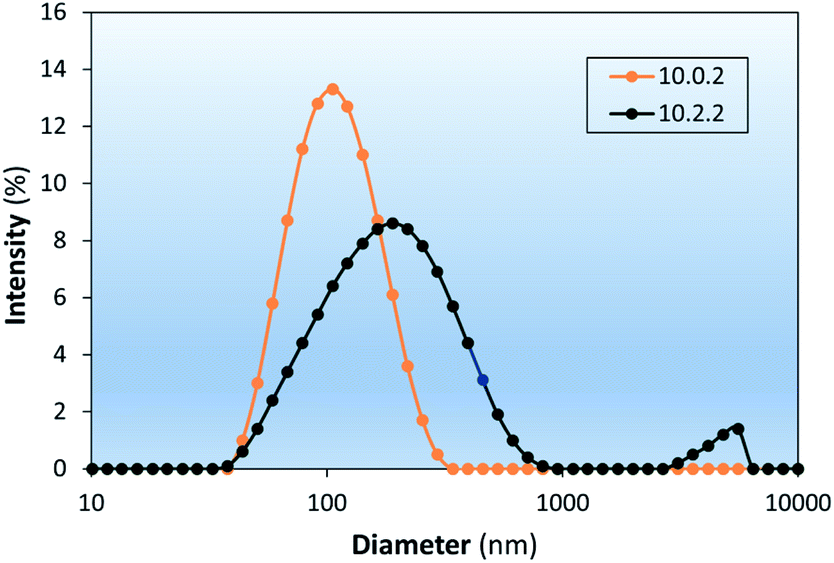 | ||
| Fig. 8 The colloidal stability of DSPC:chol liposomes is also decreased with the addition of PdNP, but to a lesser extent than pure DSPC counterparts. LNAs contained 30% cholesterol. Labels represent the mass (in mg) of DSPC, PdNP, and cholesterol (e.g., 10.0.2) used per 1 mL of each formulation. | ||
Effects of lipid modulator (cholesterol) on the catalytic activity of DSPC-PdNP LNAs
Modifying the lipid composition to give the LNAs greater colloidal stability may lead to unintended consequences to the effectiveness of these materials as catalysts. It is also potentially plausible that stabilizing liposomes with modulators such as cholesterol could aid diffusion of substrates to active sites of the catalyst within the lipid bilayer by modifying bilayer phase behavior. Cholesterol is a component of most eukaryotic cellular membranes and plays a role in bilayer protein folding.56 It strongly interacts with the polar termini of phospholipids thereby helping vesicles resist aggregation.58 The high cholesterol content in biological membranes makes the Lo phase of importance for modeling the effects of catalytic activity that may arise when LNAs are applied to biological systems. Bilayers containing DSPC with high cholesterol loadings could have regions of or are entirely in a liquid ordered (Lo) phase. The addition of cholesterol into vesicle formulations, for example, has made them more resistant to mechanical stresses and less permeable to small hydrophilic molecules and ions.55Comparison studies to determine the effect of lipid composition on catalytic activity were undertaken using DSPC-PdNP (10.2.0) and DSPC-PdNP-chol (10.2.2, 72% DSPC:14% PdNP:14% chol) LNAs having the better colloidal stability determined by DLS. In contrast to the DSPC-PdNP LNAs, the DSPC-PdNP-chol LNAs were not able to completely hydrogenate 1-octene within 24 h. After 3 h a maximum of 25% isomerization was achieved and remained constant up to 24 h as shown in Fig. 9. 1-Octene slowly decreased to 5% left at 24 h reaction time indicating continuous hydrogenation. This result suggested that the catalytic activity of PdNP within the lipid bilayers of DSPC-PdNP-chol LNAs is reduced compared to those within DSPC-PdNP LNAs. The use of cholesterol as a bilayer modulator appeared to have a slight negative effect on the catalytic activity of LNAs. It is possible that there may be the competition for space around the NP surface between cholesterol and 1-octene. The presence of cholesterol might also inhibit the diffusion of octene isomers to the catalytic sites on PdNPs. The result suggested that the use of alternative stabilizers such as phosphatidylglycerol or PEGylated lipids instead of lipid bilayer modulator may be a more effective way to enhance colloidal stability while limiting the detrimental effects on catalysis. PEGylation of vesicles is a common modification used in drug design and can be done by using PEGylated PE as one of the lipid components.59 In contrast to PEGylation, the inclusion of cationic lipids is known to encourage an interaction with cells via transfection while also preventing aggregation during storage.60
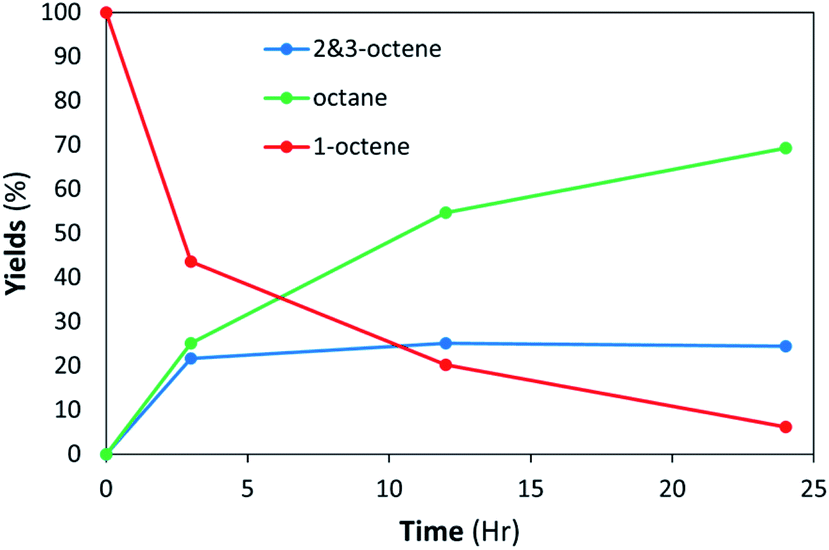 | ||
| Fig. 9 LNAs of DSPC-PdNP and cholesterol (10.2.2) are unable to complete hydrogenation within 24 h at room temperature. The standard deviations of each data point are less than ±5%. | ||
Experimental
Synthesis of palladium nanoparticles using the thiosulfate method
Dodecanethiolate-capped Pd nanoparticles were synthesized via the thiosulfate method we have previously reported.36 K2PdCl4 (0.4 mmol) was dissolved in 12 mL of water. TOAB (2.0 mmol) was dissolved in 25 mL of toluene. The two solutions were mixed for 15 min. This step allows the phase transfer of PdCl42− into the toluene layer. The aqueous layer was separated and disposed of. Additional amount of TOAB (2.0 mmol) was added to the mixture. Sodium S-dodecyl thiosulfate ligand (0.8 mmol) was dissolved in 10 mL of 25% methanol and added to the reaction flask. The reaction mixture was stirred for 15 min. NaBH4 (8.0 mmol) was dissolved in 7 mL of water and added to the stirring (350 rpm) reaction mixture over 10 s. The aqueous layer was separated and discarded after 3 h of stirring. Toluene was removed by rotary evaporation. Crude PdNPs were washed with ethanol, methanol, and acetone several times and dried under vacuum conditions for extended time.Lipid-PdNP thin film
Stock solutions of each component were prepared in chloroform immediately prior to use and then combined as necessary (Table S1†). For DSPC-PdNP film, 10 mg of DSPC and 2 mg of PdNP (5:1 weight ratio) were added to a 50 mL RBF. A 16 mL of chloroform was then added to the 50 mL flask and stirred for 10 min with a magnetic stirring bar. Organic solvent was dried under the stream of N2 gas until a dark thin dry film was observed. Sample was further dried in a vacuum oven overnight.
Hydration of thin film
The film was hydrated with 1 mL diluted (10x) PBS (pH = 7.4) solution followed by three rounds of sonication and vortex for 1 min each round. The heterogeneous mixture was heated to 55 °C for 30 min and gently stirred while cooling for 2 h to ensure embedding of the PdNP into the bilayer and a DSPC liposome bilayer phase transition to prevent leakage of PdNPs. The sample was then allowed to sit overnight before being transferred to refrigerator. The formed DSPC-PdNP hybrid assemblies were characterized via 1H NMR after solvent removal, which proved the incorporation of the PdNPs with DSPC (Fig. S6†).Catalytic reactions
Catalytic assays were performed at room temperature (∼22 °C), 0, 37, and 55 °C in 50 mL round bottom flasks using 40 μL (0.25 mmol) of substrate and 1 mL lipid dispersion containing 2 mg PdNP [5 mol% PdNPs; 0.0125 mmol Pd] in 10x diluted phosphate buffered saline (PBS). The reaction flask was capped with a rubber septum and magnetic stir bar, purged with H2 gas for 10 min (the flask is charged with ∼10 mmol H2 gas). Reactions were done at various time periods (1, 3, 4, 6, 12, 24 and/or 48 hours). Reaction products were extracted by disrupting the aqueous lipid dispersions by adding 0.5 mL of CH2Cl2 and slightly vortexing the 2-layered mixture. Purification of organic products from PdNPs and lipids was done by filtration through a Pasteur pipette silica gel column chromatography using CH2Cl2 as eluent. The isolated products were analysed via GC/MS.Conclusions
Hybrid catalytic assemblies composed of DSPC phospholipid vesicles embedded with the hydrophobic PdNPs were prepared in water. The catalysis studies showed that the hydrophobic micellization effect and the dynamic lipid bilayer–substrate interactions enhance the catalytic activity of PdNPs embedded in liposomal assemblies compared to that of PdNPs in organic solvent. When comparing the catalysis at varying temperatures, such as ripped gel to liquid crystalline transition temperature (Tg-to-l), biological temperature, and melting phase transition temperature (Tm) of DSPC, it was indeed evident that there is an increase in the kinetics of the 1-octene hydrogenation as the temperature approaches the ripped gel to liquid crystalline transition temperature of the DSPC. It was also shown that the colloidal stability of LNAs was influenced by the incorporation of PdNPs and cholesterol to the bilayers of liposomes. PdNPs were confirmed to lower the colloidal stability of DSPC liposomes. Cholesterol was confirmed to be an effective stabilizer for both DSPC liposome and DSPC-PdNP LNAs but at the cost of catalytic activity. Overall, the liposomal DSPC-PdNP hybrid system has shown a great potential in its robust catalytic capabilities in transforming organic substrates in an aqueous environment. However, the preparation of hybrid assemblies with increased colloidal stability is necessary to solve the problem of occasional inconsistent results. Future research into structural properties of these novel composite materials may manifest such results and allow for a more targeted approach when it comes to engineering them for specific purposes.Author contributions
D. O. and N. P. conducted the experimental work (N. P. studied the effects of cholesterols and D. O investigated all others). D. O. wrote the original draft with N. P. adding his contributions. Y.-S. S. supervised the project, provided resources, and edited the draft. All authors have given approval to the last version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1954659) and CSULB for the financial supports. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.Notes and references
- R. Raliya, T. S. Chadha, K. Haddad and P. Biswas, Curr. Pharm. Des., 2016, 22, 2481–2490 CrossRef CAS PubMed
.
- E. Aznar, E. Climent, L. Mondragon, F. Sancenón and R. Martínez-Máñez, Functionalized Mesoporous Materials with Gate-Like Scaffoldings for Controlled Delivery, Polymers in Regenerative Medicine: Biomedical Applications from Nano-to Macro-Structures, 2015, pp. 337–366 Search PubMed
- E. Che, Y. Gao, L. Wan, Y. Zhang, N. Han, J. Bai, J. Li, Z. Sha and S. Wang, Microporous Mesoporous Mater., 2015, 204, 226–234 CrossRef CAS
- S. Bajpai, S. K. Tiwary, M. Sonker, A. Joshi, V. Gupta, Y. Kumar, N. Shreyash and S. Biswas, ACS Appl. Nano Mater., 2021, 4, 6441–6470 CrossRef CAS
- C. Chung, Y. K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC
- M. R. K. Ali, Y. Wu, D. Ghosh, B. H. Do, K. Chen, M. R. Dawson, N. Fang, T. A. Sulchek and M. A. El-Sayed, ACS Nano, 2017, 11, 3716–3726 CrossRef CAS PubMed
- F. S. Rosarin and S. Mirunalini, J. Bioanal. Biomed., 2011, 3, 85–91 Search PubMed
- J. Dolai, K. Mandal and N. R. Jana, ACS Appl. Nano Mater., 2021, 4, 6471–6496 CrossRef CAS
- S. Singha, K. Shao, K. K. Ellestad, Y. Yang and P. Santamaria, ACS Nano, 2018, 12, 10621–10635 CrossRef CAS PubMed
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed
- Q. Mu, G. Jiang, L. Chen, H. Zhou, D. Fourches, A. Tropsha and B. Yan, Chem. Rev., 2014, 114, 7740–7781 CrossRef CAS PubMed
- K. M. Vargas and Y.-S. Shon, J. Mater. Chem. B, 2019, 7, 695–708 RSC
- W. Gao, D. Vecchio, J. Li, J. Zhu, Q. Zhang, V. Fu, J. Li, S. Thamphiwatana, D. Lu and L. Zhang, ACS Nano, 2014, 8, 2900–2907 CrossRef CAS PubMed
- L. Calucci, A. Grillone, E. R. Riva, V. Mattoli, G. Ciofani and C. Forte, J. Phys. Chem. C, 2017, 121, 823–829 CrossRef CAS
- N. Kanwa, A. Patnaik, S. K. De, M. Ahamed and A. Chakraborty, Langmuir, 2019, 35, 1008–1020 CrossRef CAS PubMed
- P. Thanasekaran, C.-H. Chu, S.-B. Wang, K.-Y. Chen, H.-D. Gao, M. M. Lee, S.-S. Sun, J.-P. Li, J.-Y. Chen, J.-K. Chen, Y.-H. Chang and H.-M. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 84–95 CrossRef CAS PubMed
- A. Luchini and G. Vitiello, Front. Chem., 2019, 7, 343 CrossRef CAS PubMed
- S. Chakraborty, A. Abbasi, G. D. Bothun, M. Nagao and C. L. Kitchens, Langmuir, 2018, 34, 13416–13425 CrossRef CAS PubMed
- M. R. Preiss, A. Hart, C. Kitchens and G. D. Bothun, J. Phys. Chem. B, 2017, 121, 5040–5047 CrossRef CAS PubMed
- E. Porret, L. Sancey, A. Martin-Serrano, M. I. Montañez, R. Seeman, A. Yahia-Ammar, H. Okuno, F. Gomez, A. Ariza, N. Hildebrandt, J.-B. Fleury, J.-L. Coll and X. L. Guével, Chem. Mater., 2017, 29, 7497–7506 CrossRef CAS
- S.-H. Park, S.-G. Oh, J.-Y. Mun and S.-S. Han, Colloids Surf., B, 2005, 44, 117–122 CrossRef CAS PubMed
- S.-H. Park, S.-G. Oh, J.-Y. Mun and S.-S. Han, Colloids Surf., B, 2006, 48, 112–118 CrossRef CAS PubMed
- E. Amstad, J. Kohlbrecher, E. Müller, T. Schweizer, M. Textor and E. Reimhult, Nano Lett., 2011, 11, 1664–1670 CrossRef CAS PubMed
- Q.-Y. Bao, N. Zhang, D.-D. Geng, J.-W. Xue, M. Merritt, C. Zhang and Y. Ding, Int. J. Pharm., 2014, 477, 408–415 CrossRef CAS PubMed
- E. Sadeghmoghaddam, H. Gu and Y.-S. Shon, ACS Catal., 2012, 2, 1838–1845 CrossRef CAS PubMed
- D. J. Gavia, J. Koeppen, E. Sadeghmoghaddam and Y.-S. Shon, RSC Adv., 2013, 3, 13642–13645 RSC
- J. S. Zhu and Y.-S. Shon, Nanoscale, 2015, 7, 17786–17790 RSC
- K. A. San and Y.-S. Shon, Nanomaterials, 2018, 8, 346 CrossRef PubMed
- K. M. Vargas, K. A. San and Y.-S. Shon, ACS Appl. Nano Mater., 2019, 2, 7188–7196 CrossRef CAS PubMed
- P. Tieu, V. Nguyen and Y.-S. Shon, Front. Chem., 2020, 8, 599 CrossRef CAS PubMed
- C. Wang, Y. Zhang and Y. Dong, Acc. Chem. Res., 2021, 54, 4283–4293 CrossRef CAS PubMed
- U. Elia, S. Ramishetti, R. Rosenfeld, N. Dammes, E. Bar-Haim, G. S. Naidu, E. Makdasi, Y. Yahalom-Ronen, H. Tamir, N. Paran, O. Cohen and D. Peer, ACS Nano, 2021, 15, 9627–9637 CrossRef PubMed
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Acc. Chem. Res., 2022, 55, 2–12 CrossRef CAS PubMed
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed
- D. Gavia and Y.-S. Shon, Langmuir, 2012, 28, 14502–14508 CrossRef CAS PubMed
- T.-A. Chen and Y.-S. Shon, Catal. Sci. Technol., 2017, 7, 4823–4829 RSC
- T.-A. Chen and Y.-S. Shon, Catalysts, 2018, 8, 428 CrossRef PubMed
- M. A. Mahdaly, J. S. Zhu, V. Nguyen and Y.-S. Shon, ACS Omega, 2019, 4, 20819–20828 CrossRef CAS PubMed
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS PubMed
- M. R. Rasch, E. Rossinyol, J. L. Hueso, B. W. Goodfellow, J. Arbiol and B. A. Korgel, Nano Lett., 2010, 10, 3733–3739 CrossRef CAS PubMed
- H. Sharma and E. E. Dormidontova, ACS Nano, 2017, 11, 3651–3661 CrossRef CAS PubMed
- G. V. White II, Y. Chen, J. Order-Hanna, G. D. Bothun and C. L. Kitchens, ACS Nano, 2021, 6, 4678–4685 CrossRef PubMed
- D. J. Gavia, M. S. Maung and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2013, 5, 12432–12440 CrossRef CAS PubMed
- J. Bodosa, S. S. Iyer and A. Srivastava, J. Membr. Biol., 2020, 253, 551–562 CrossRef CAS PubMed
- R. Koynova and M. Caffrey, Biochim. Biophys. Acta, 1998, 1376, 91–145 CrossRef CAS
- S. Matviykiv, H. Deyhle, J. Kohlbrecher, F. Neuhaus, A. Zumbuehl and B. Müller, Langmuir, 2019, 35, 11210–11216 CrossRef CAS PubMed
- C. Naumann, T. Brumm and T. M. Bayerl, Biophys. J., 1992, 63, 1314–1319 CrossRef CAS PubMed
- W. Chen, F. Duša, J. Witos, S.-K. Ruokonen and S. K. Wiedmer, Sci. Rep., 2018, 8, 14815 CrossRef PubMed
- D. J. A. Crommelin, J. Pharm. Sci., 1984, 73, 1559–1563 CrossRef CAS PubMed
- M. N. Jones, Curr. Opin. Colloid Interface Sci., 1996, 1, 91–100 CrossRef CAS
- J. N. Israelachvili, S. Marčelja and R. G. Horn, Q. Rev. Biophys., 1980, 13, 121–200 CrossRef CAS PubMed
- M. B. Sankaram and T. E. Thompson, Biochemistry, 1990, 29, 10670–10675 CrossRef CAS PubMed
- G. Cevc, J. Controlled Release, 2012, 160, 135–146 CrossRef CAS PubMed
- E. Sparr, L. Hallin, N. Markova and H. Wennerström, Biophys. J., 2002, 83, 2015–2025 CrossRef CAS PubMed
- J. Oates, B. Faust, H. Attrill, P. Harding, M. Orwick and A. Watts, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2228–2233 CrossRef CAS PubMed
- T. M. Konyakhina, J. Wu, J. D. Mastroianni, F. Heberle and G. W. Feigenson, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2204–2214 CrossRef CAS PubMed
- M. Bloom, E. Evans and O. G. Mouritsen, Q. Rev. Biophys., 1991, 24, 293–397 CrossRef CAS PubMed
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed
- S. A. Walker and J. A. Zasadzinski, Langmuir, 1997, 13, 5076–5081 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials; instrumentation; synthesis and characterization of sodium S-dodecanethiosulfate ligand; characterization of Pd nanoparticles synthesized by alkyl thiosulfate method; characterization of lipid and lipid-Pd nanoparticles assemblies; summary of catalytic reaction results. 1H NMR data and FT-IR spectrum of sodium S-dodecanethiosulfate ligand. UV-vis spectrum, 1H NMR spectrum, TGA, and an additional TEM image of dodecanethiolate-capped PdNPs. 1H NMR spectra of DSPC and DSPC-PdNP LNAs. DSC result of DSPC-PdNP LNAs. See https://doi.org/10.1039/d2ra03974e |
| This journal is © The Royal Society of Chemistry 2022 |