
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFundamental understanding of the size and surface modification effects on r1, the relaxivity of Prussian blue nanocube@m-SiO2: a novel targeted chemo-photodynamic theranostic agent to treat colon cancer†
Panchanan Sahooab,
Sudip Kundua,
Shubham Royc,
S. K. Sharmad,
Jiten Ghoshe,
Snehasis Mishraa,
Abhishek Mukherjee *b and
Chandan Kumar Ghosh*a
*b and
Chandan Kumar Ghosh*a
aSchool of Materials Science and Nanotechnology, Jadavpur University, Kolkata-700032, India. E-mail: chandan.kghosh@jadavpuruniversity.in
bAgricultural and Ecological Research Unit, Biological Science Division, Indian Statistical Institute, Giridih, Jharkhand, India. E-mail: abhi.mukh@yahoo.com
cDepartment of Physics, Jadavpur University, Kolkata-700032, India
dEko X-Ray & Imaging Institute, 54, Jawaharlal Nehru Road, Kolkata-700071, India
eXRD and SEM Units, Materials Characterization and Instrumentation Division, CSIR-Central Glass and Ceramic Research Institute, India
First published on 30th August 2022
Abstract
A targeted multimodal strategy on a single nanoplatform is attractive in the field of nanotheranostics for the complete ablation of cancer. Herein, we have designed mesoporous silica (m-SiO2)-coated Prussian blue nanocubes (PBNCs), functionalized with hyaluronic acid (HA) to construct a multifunctional PBNC@m-SiO2@HA nanoplatform that exhibited good biocompatibility, excellent photodynamic activity, and in vitro T1-weighted magnetic resonance imaging ability (r1 ∼ 3.91 mM−1 s−1). After loading doxorubicin into the as-prepared PBNC@m-SiO2@HA, the developed PBNC@m-SiO2@HA@DOX displayed excellent pH-responsive drug release characteristics. Upon irradiation with 808 nm (1.0 W cm−2) laser light, PBNC@m-SiO2@HA@DOX exhibited synergistic photodynamic and chemotherapeutic efficacy (∼78% in 20 minutes) for human colorectal carcinoma (HCT 116) cell line compared to solo photodynamic or chemotherapy. Herein, the chemo-photodynamic therapeutic process was found to follow the apoptotic pathway via ROS-mediated mitochondrion-dependent DNA damage with a very low cellular uptake of PBNC@m-SiO2@HA@DOX for the human embryonic kidney (HEK 293) cell line, illustrating its safety. Hence, it may be stated that the developed nanoplatform can be a potential theranostic agent for future applications. Most interestingly, we have noted variation in r1 at each step of the functionalization along with size variation that has been the first time modelled on the basis of the Solomon–Bloembergen–Morgan theory considering changes in the defect crystal structure, correlation time, water diffusion rate, etc., due to varied interactions between PBNC and water molecules.
Introduction
Coordination polymer nanostructures are one of the emerging types of inorganic nanostructures where metal ions are linked together, forming a cyanide-bridged infinite network. They have gained increasing interest in recent times due to their ability to combine multiple activities within the multifunctional nano-object, which can be ascribed to their molecular characteristics.1,2 One of the promising candidates in this domain is the Prussian blue (PB), a face-centered cubic (fcc) structure-based metal–organic framework (MOF).3 Its easy tuneable porosity and tailorable structures impart PB with a wide range of prospective applications such as energy storage, hydrogen storage, electrochromic, and photomagnetic devices. Apart from the pure phase, due to high ligand field stabilization energy (LFSE) in comparison with iron compounds such as Fe3O4. PB forms a thermodynamically stable mixed-valence phase with the generic formula A1−xFeIII[FeII(CN)6]1−x/4/Δx/4, where A and Δ represent alkali ion and hexacyanoferrate vacancies, respectively, which facilitate easy functionalization on PB and widen the applicational prospects.4 Alkali-free PB is also known as insoluble PB, containing water molecules with an increased amount of Fe3+ leading to charge balance.5 PB also exhibits strong potentiality in biomedical applications such as the drug delivery system and contrast agents (CAs) for different diagnostics purposes including X-ray computed and positron emission tomography, optical biomarkers, ultrasound imaging, and NIR-assisted therapeutic activities such as photothermal ablation and photodynamic therapy.6–8 Among various diagnostics modalities, special thrusts are generally given to design PB as CAs for magnetic resonance imaging (MRI) since it has the advantage of non-ionizing type radiation, high sensitivity to afford non-destructive details of soft tissues and functional information on lesions, limitless penetration depth and high spatial resolution (∼100 μm) in comparison with other imaging modalities.9,10Herein, it may be stated that MRI, an example of the preferred non-invasive imaging technique, is commonly adopted in a modern clinical study to diagnose the internal body structure with high spatial resolution and exquisite anatomical contrast efficiency, and utilizes the basic principle of nuclear magnetic resonance (NMR) where a magnetic field gradient is applied to encode the signal in all three spatial directions. NMR signals are generally weighed by the density of H2O molecules and the longitudinal (T1) and/or transverse (T2) relaxation times of the tissues and efficiency of CAs depends on two parameters, namely longitudinal (r1) and transverse (r2) relaxation of protons. For 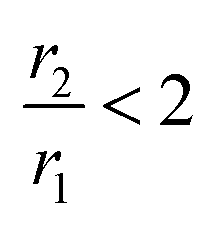 , CA is positive and it is called T1-relaxing CA, which gives a brighter spot of the diagnostic area. Mainly, mononuclear Gd3+ complexes with r1 ∼ 3–5 mM−1 s−1 at 20 MHz and 298 K are used T1 CAs. In other cases,
, CA is positive and it is called T1-relaxing CA, which gives a brighter spot of the diagnostic area. Mainly, mononuclear Gd3+ complexes with r1 ∼ 3–5 mM−1 s−1 at 20 MHz and 298 K are used T1 CAs. In other cases, 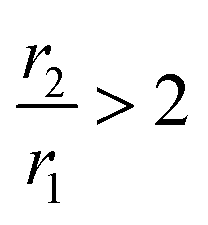 gives a dark signal, the CA is said to be negative and is defined as T2-relaxing CA that mostly involves superparamagnetic iron oxide-based nanoparticles.11 In this context, it may be stated that mostly Gd3+ based chelating agents, namely DOTAREM® and MAGNEVIST®, are nowadays used at a dose of 10 to 20 mL of 0.5 mM solution depending on the body weight; besides, it also has serious side effects such as gadolinium toxicity and nephrogenic systemic fibrosis.12,13 Herein, the current research effort focuses on improving the effectiveness of CA in order to be able to reduce the dose of administration of CA with very mild side effects.
gives a dark signal, the CA is said to be negative and is defined as T2-relaxing CA that mostly involves superparamagnetic iron oxide-based nanoparticles.11 In this context, it may be stated that mostly Gd3+ based chelating agents, namely DOTAREM® and MAGNEVIST®, are nowadays used at a dose of 10 to 20 mL of 0.5 mM solution depending on the body weight; besides, it also has serious side effects such as gadolinium toxicity and nephrogenic systemic fibrosis.12,13 Herein, the current research effort focuses on improving the effectiveness of CA in order to be able to reduce the dose of administration of CA with very mild side effects.
Concerning, PB, which is being used as the sole U.S. Food and Drug Administration approved T1-weighted CA for MRI has the advantage of a high signal-to-noise ratio, fast scanning, bright field image, etc. in comparison with T2-weighted CAs.6 Despite several prospects, the practical utility of pure PB nanostructures is limited due to their instability, particularly under acidic pH conditions in the neighborhood of cancer cells or tumor microenvironment; hence, they are often stabilized within various matrixes such as polymers, biopolymers, alumina, amorphous and mesoporous silica or by using stabilizing ligands in the solution. It is well-reported that these protocols for the stabilization of PB nanostructures fall into different matrixes or in organic phase colloids and only a few methods allow the preparation of PB nanostructures in aqueous solutions, facilitating the scope of using PB nanostructures in several bioanalytical and biomedical applications.6,7,14,15 Herein, several reports exist for other materials e.g. Gd2O3 and Fe3O4 to illustrate that various functionalizations alter the contrast efficiency of the agent, as well as the size and shape of their nanostructure form, play an important role. Though many theoretical and experimental efforts were previously given to examine the influence of these parameters in order to increase contrast efficiency, no such reports, except a few, exist to indicate the influence of the surface functionalities, shape, and size of PB nanostructures on MRI contrast efficiency.16,17 As an example, PB nanoparticles (∼13 nm), stabilized with citric acid and conjugated with 5-(amino-acetomido) fluorescein dye, show modest r1 ∼ 0.079 mM−1 s−1, while Gd3+, Mn2+ doping increase r1 by at least 1–2 orders of magnitude.18,19 In this context, Guari et al. have demonstrated that Gd3+ doped PB nanoparticles, coated with chitosan, exhibit r1, which is two times higher than that for clinically approved paramagnetic Gd3+ chelate complexes.20 Hence, in this study, our primary objective was to investigate the role of the size, surface functional groups, and defects of PB nanostructures on the r1-relaxation parameter. In this context, it may be mentioned that functionalized nanoparticles are further modified with RGD peptides, hyaluronic acid, etc. for easy localization on an over-expressed receptor on the cancer cell and chemotherapeutic drugs are loaded to increase the therapeutic efficiency.6,16,21 To the best of our knowledge, no report exists to indicate the role of target specific agent or drug in the efficiency of CAs in MRI; hence in this study we have also emphasized understanding their influence on r1-parameter using hyaluronic acid (HA) and doxorubicin (DOX) as the target-specific agents and chemotherapeutic drugs, respectively. Currently, the experimental parameters influencing r1-relaxation are expressed in terms of the change of internal time correlation factors such as rotational correlation time, residence time, diffusion time, and electron correlation time; however, they are very hard to be analysed in the case of complicated nanoparticle-based CAs containing multiple paramagnetic sites, coordinated with water molecules. In this study, we have developed an intuitive model according to the Solomon–Bloembergen–Morgan theory to explain the variation of r1-relaxation parameter in each functionalization step.19 In this study, we developed an intuitive model according to Solomon–Bloembergen–Morgan theory to explain the influence of structural defects, different modifications such as m-SiO2 coating, HA functionalization, and DOX loading on r1 relaxivity values in differently sized PBNC nanostructures. Finally, we evaluated the theranostic performances of our synthesized PB nanostructures, coated with mesoporous SiO2 (m-SiO2) and functionalized with HA, followed by DOX loading on the colon carcinoma cell line (HCT 116). Several reports demonstrated that CD44 expression in the colon cancer tissue was substantially higher than that in normal mucosa.22 On the basis of this background, HA functionalization was executed, which provided the target-specific cellular uptake to the HCT 116 cell line. Moreover, the functionalization of HA not only increases CD44 receptor-based target specificity but also makes the nanoplatform very much stable for the biological environment.7 Apart from stabilization, herein, m-SiO2 makes HA functionalization easy and its porous structure facilitates DOX loading. In this context, we have compared the localization of our synthesized nanostructures using the human embryonic kidney cell line (HEK 293) as a reference.
Experimental section
Chemicals and cell lines
K4[Fe(CN)6]·3H2O, tetraethyl orthosilicate (TEOS), N-cetyl-N,N,N-trimethyl ammonium bromide (CTAB), citric acid anhydrous, hydrochloric acid (35 wt%) were brought from Merck. Absolute ethanol, acetone, and concentrated ammonia aqueous solution (25 wt%) were purchased from Merck, Amplura. FeCl3·6H2O, doxorubicin hydrochloride, N-(3-dimethylaminopropyl)-N/-ethylcarbodiimide hydrochloride (EDC), 1,3-diphenylisobenzofuran (DPBF) were purchased from Sigma-Aldrich. Hyaluronic acid (HA) was obtained from TCI chemicals. Cell culture medium components viz. Dulbecco's modified eagle medium (DMEM), penicillin–streptomycin–neomycin (PSN) antibiotic cocktail, fetal bovine serum (FBS), ethylenediaminetetraacetic acid (EDTA), and trypsin were obtained from Gibco, USA. Other compulsory fine and raw chemicals were obtained from SRL, India, and Sigma-Aldrich, USA. Human colorectal carcinoma (HCT 116) and human embryonic kidney 293 (HEK 293) cell lines were purchased from the National Centre for Cell Sciences (NCCS), India. Antibodies were procured from cell signalling technology (CST), and eBioscience, USA. Other reagents were obtained from the best existing commercial sources and all the chemicals were used without further purification.Synthesis of insoluble Prussian blue nanocubes
Citric acid-capped insoluble Prussian blue nanocubes (PBNCs) were synthesized by a little-modified reflux technique using a single anionic source.6,23 Briefly, solution A and solution B were prepared separately by dissolving 5.40 mg of FeCl3·6H2O and 8.45 mg of K4Fe(CN)6·3H2O in 30 mL of DI water, respectively, taken into three neck flasks, followed by the addition of 392.00 mg of citric acid at 60 °C. Maintaining the same temperature, solution A was added dropwise to solution B under vigorous stirring and a bright blue dispersed solution appeared after 1.0 h of stirring. After completing the reaction, the mixture solution was cooled to room temperature and the dispersed solution was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm for 15 minutes. After washing with acetone three times, the final product in the powder form was obtained after drying at 60 °C under vacuum conditions. In order to obtain larger-sized PBNCs, another experiment was performed by maintaining the mixing reaction temperature at 80 °C. Two PBNCs, prepared at two different temperatures, were named PBNC60 and PBNC80.
500 rpm for 15 minutes. After washing with acetone three times, the final product in the powder form was obtained after drying at 60 °C under vacuum conditions. In order to obtain larger-sized PBNCs, another experiment was performed by maintaining the mixing reaction temperature at 80 °C. Two PBNCs, prepared at two different temperatures, were named PBNC60 and PBNC80.
Synthesis of mesoporous silica-coated PBNC
Mesoporous silica (m-SiO2) coated PBNCs (PBNC@m-SiO2) were synthesized using a CTAB-directed sol–gel method.24 Typically, the as-prepared PBNCs (25.00 mg), DI water (1 mL), 25% NH3 (0.5 mL), and TEOS (13 μL) were mixed with 10 mL ethanol and were stirred continuously for 5 minutes at 30 °C. Then, CTAB (0.06 g) and DI water (7.5 mL) were added to the above solution. After 12 h of continuous stirring, 25% NH3 (140 μL) and TEOS (100 μL) were added with stirring for another 2 h. Then, the particles were centrifuged at 12500 rpm for 15 minutes and were washed with ethanol three times. To remove CTAB, the particles were extracted in an ethanol solution containing 10 mL water and 1 mL HCl (35%) for 4.5 h. Finally, the mixture was centrifuged again, washed with ethanol thrice, and dried at 45 °C under vacuum for further use. Two different-sized PBNCs, coated with m-SiO2, were named PBNC60@m-SiO2 and PBNC80@m-SiO2, respectively.
Functionalization of PBNC@m-SiO2 with hyaluronic acid
The m-SiO2 coated PBNC nanostructures were first functionalized with –NH2.25 Briefly, 20.0 mg of the as-coated samples were dispersed in 40.0 mL ethanol, subsequently 2.4 mL of APETS was added and was left for 30.0 h with continuous stirring at 80 °C under reflux conditions. Then, the mixture was centrifuged and was washed with ethanol 3 times for further use (ascertained as solution C). Secondly, an EDC linker was used to link between –NH2 and –COOH of the HA. Briefly, 30.0 mg EDC was added to 22.0 mL PBS buffer (pH 7.4) solution containing 12.0 mg HA, followed by continuous stirring for 4.0 h at room temperature. Then, solution C was added dropwise into the HA-containing solution. Furthermore, 24.0 h of continuous stirring was conducted after which the particles were collected by centrifugation, followed by washing with a PBS buffer 3 times. Finally, the collected particles were designated as PBNC60@m-SiO2@HA and PBNC80@m-SiO2@HA according to PBNC60@m-SiO2 and PBNC80@m-SiO2 as precursors, respectively.Characterization
The crystallography and phase information on the synthesized PBNCs were acquired using powder X-ray diffraction recorded on an ULTIMA IV X-ray diffractometer (Rigaku) operating at 40 kV and 40 mA using CuKα radiation. Microstructures of the as-prepared samples were investigated by field emission scanning electron microscopy (FESEM, S-4800, Hitachi, Japan) and high-resolution transmission electron microscopy (HRTEM, JEM-2100, JEOL, Japan). Thermal gravimetric analysis (TGA), absorption characteristics (200–900 nm), surface charge, and surface functionalizations were determined using a DTA-TGA instrument (SHIMADZU, DTG-60H), UV-vis-NIR spectrophotometer (PerkinElmer Instruments, Waltham, MA), Nano-ZetaSizer (Brookhaven Instruments, Holtsville, NY) and Fourier transform infrared spectrometer (FTIR, PerkinElmer, Spectrum-2000), respectively. The Quantachrome, NOVA 1000e system was adopted to capture the nitrogen adsorption/desorption isotherms on which Barret–Joyner–Halenda (BHJ) and Brunauer–Emmett–Teller (BET) analyses were performed to determine the surface area, pore diameter and pore volume of the as-prepared nanostructures.NIR triggered extracellular singlet oxygen (1O2) generation
To examine 1O2 generation, we chose the DPBF probe trapping method by monitoring the absorption peak of DPBF at 410 nm through UV-vis spectroscopy. The generation of the 1O2 oxidised DPBF leads to a decrease in absorption. For this, 3 mL of 100 μg mL−1 PBNC60@m-SiO2@HA alcoholic solution was mixed with 1 mL of 68 μg mL−1 DPBF alcoholic solution in each vial under irradiation of 808 nm (1.0 W cm−2) NIR light in a time-dependent manner. Only the DPBF solution or the mixed solution of PBNC60@m-SiO2@HA and DPBF under the dark was taken as a control.7DOX loading and releasing profile
DOX, an anticancer drug, was loaded in the pores of mesoporous silica on PBNC60@m-SiO2@HA to improve the anticancer activity. Briefly, 2 mg DOX containing 1.2 mL of PBS solution was dropwise added with 178 μg mL−1 of 4 mL PBNC60@m-SiO2@HA solution under stirring, where the concentration of the as-prepared solution was measured using UV-vis-NIR spectroscopy by monitoring the absorption of PBNC60. Then, the mixed solution was left overnight with continuous stirring at 300 rpm under dark. After that PBNC60@m-SiO2@HA@DOX nanoplatforms were collected by centrifugation and washing with PBS. The collected PBNC60@m-SiO2@HA@DOX was redispersed in PBS buffer for future use and the supernatant was also collected for the measurement of DOX loading efficiency through UV-vis spectroscopy by monitoring absorption at 480 nm. DOX loading efficiency was measured using the following formula,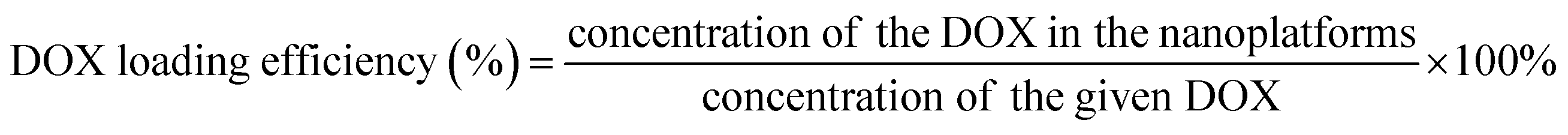
To investigate the release behaviour of DOX from PBNC60@m-SiO2@HA@DOX, we dispersed 1.0 mL of the above DOX-loaded nanoplatform in 4.0 mL PBS buffer (pH 5 and pH 7.4) separately and poured it in a dialysis bag (MWCO 12 kDa).16 After that the dialysis bag was immersed in a beaker containing 50.0 mL of the fresh buffer with slow stirring. Then, at a predetermined time interval 1.0 mL of fresh buffer was replaced after collecting 1.0 mL of DOX-loaded buffer from the beaker to maintain the concentration. Finally, the release profile was measured by UV-vis spectroscopy by monitoring at 480 nm.
Cell culture
Briefly, cells were cultured in DMEM containing 10% fetal bovine serum (FBS) and 1% antibiotic cocktail at 37 °C under humidified conditions at constant 5% CO2. After 75–80% confluency, cells were harvested with trypsin (0.25%), and EDTA (0.52 mM) in phosphate-buffered saline (PBS), plated at a necessary density to allow them to re-equilibrate before the experimentation.26In vitro T1-weighted cellular MRI contrast measurement
For cellular MR imaging, ∼6 × 103 numbers of HCT 116 cells were incubated in a 96 well plate. Cells were treated with PBNC60@m-SiO2@HA@DOX under six different concentrations of PBNC (0.025, 0.05, 0.1, 0.2, 0.4, 0.6 mM) and incubated for 24 h. Then, cells were washed with PBS three times and fixed using paraformaldehyde. To avoid air susceptibility 100 mL of 2% low-melting agarose was added to each well. Then, the plate was kept at 4 °C to solidify the cell suspensions. MR contrast measurements of both bare PBNC along with each step of modification were also evaluated at different concentrations (0.025–0.6 mM) in 96 well plates under the dispersion in PBS. MRI was performed by keeping the samples under a 3T clinical MRI scanner (Siemens MAGNETOM Verio), using a pre-fabricated sample holder. MR phantom images were obtained by applying a spin-echo multi-section pulse sequence. In order to determine the T1 relaxation of the sample, coronal images were acquired under the following constant parameters: magnetic field (B), 3T; inversion time (TI), 1100 ms; echo time (TE), 9.6 ms; repetition time (TR), 2000 ms; matrix size, 256 mm × 256 mm; field of view (FOV), 150 mm × 150 mm; slice thickness, 3 mm. The MRI signal intensity (SI) and visualization of the phantom images were performed using the standard software provided by the manufacturer.The longitudinal relaxivity value (r1) at a fixed magnetic field (3T) and inversion time (1100 ms) for all samples was extracted from the plot of signal intensity versus the concentration of the contrast agent (CA) based on the following equation
| SI = A − Pe−(TI×r1)C |
Cell viability assay
MTT [(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay was carried out to screen the cytotoxic effects of our synthesized nanoplatform in HEK 293 and HCT 116 cell lines with concentration and 808 nm NIR laser exposure time-dependent manner.28 Cells at the required density (2 × 106 cells per well) were seeded in each well of the 96 well plates and treated under different concentrations (0–70 μg mL−1) of PBNC60@m-SiO2@HA@DOX and the individuals PBNC60, PBNC60@m-SiO2 and PBNC60@m-SiO2@HA. Soon after the treatment, plates were kept in an incubator for 24 h at 37 °C in a humidified CO2-rich condition (5%). After the completion of the incubation period, cells were thoroughly washed with PBS followed by the addition of MTT solution (4 mg mL−1) and kept in an incubator for 4 h. The absorbance of the DMSO-solubilized intracellular formazan salt was recorded at 595 nm using an ELISA reader (Emax, Molecular Device, USA). In all cases, the samples were sonicated before treatment in a cell line to obtain the homogenized mixtures. The final reported biological data represent the average obtained after repeating the experiments in triplicate.In vitro photodynamic therapy (PDT)
On the basis of preliminary screening experimentation, 21 μg mL−1 of PBNC60@m-SiO2@HA@DOX was chosen as the concentration for further experimentation to check whether they exhibited any photosensitizing effect within the cells. Hence, treated HCT 116 and HEK 293 cells were exposed under 808 nm irradiation in a time-dependent manner (0, 10, 20, 30, and 40 min) following the MTT assay to determine the photodynamic and chemo-dynamic efficacy.Quantification of apoptosis using Annexin V-FITC kit
Apoptosis was examined through the use of an Annexin V-FITC apoptosis detection kit (Calbiochem, CA, USA).29,30 After treatment with PB60@m-SiO2@HA@DOX as per the IC50 value, HCT 116 cells were irradiated for 0, 10, and 20 min, and the cells were washed and stained with Annexin V-FITC and propidium iodide (PI) in accordance with the manufacturer's instructions. The percentages of apoptotic (early and late), necrotic along with viable cells were evaluated by flow cytometry (BD LSRFortessa, San Jose, CA, USA). For each sample analysis, 106 cells were taken.Determination of intracellular ROS (iROS)
Mitochondria are the primary source of the amplification of the reactive oxygen species (ROS) production in mammalian cells and it play a significant role in the stimulation of apoptosis in a variety of cells.31 To determine the iROS, we irradiated 21 μg mL−1 of PB60@m-SiO2@HA@DOX-treated HCT 116 cells in a time-dependent manner, after that the HCT 116 cells were incubated with 10 μM H2DCFH-DA (2′,7′-dichlorofluorescein diacetate) at 37 °C for 25 min before the analysis using the flow cytometer (BD LSRFortessa, San Jose, CA, USA). The increment of DCF fluorescence directly reflects the generated ROS inside cells, which were represented as the mean fluorescence intensity of DCF where untreated cells were considered a control. For each sample analysis, 106 cells were taken.Confocal microscopy
Briefly, after the treatment with 21 μg mL−1 of PB60@m-SiO2@HA@DOX, HCT 116 cells were irradiated in a time-dependent manner and incubated for 24 h. Then, the coverslips containing HCT 116 cells were washed twice for 10 min each in 0.01 M PBS and incubated for 1 h in blocking the solution containing 2% normal bovine serum, and 0.3% Triton X-100 in PBS. After blocking, the slides were incubated overnight at 4 °C with the proper primary antibody (Gamma H2AX, PARP, Cytochrome c, and SOD). Alexa Fluor 555 tagged Gamma H2AX was used as the primary tagged antibody and in the case of SOD and cytochrome c; FITC tagged anti-rabbit secondary was used. Secondary antibodies were diluted, 1:100, in blocking solution and incubated for 2 h. The slides were then counterstained with 6-diamidino-2-phenylindole (DAPI) for 10 min and mounted with the Prolong anti-fade reagent (Molecular Probe, Eugene, OR, USA). The fluorescence intensity of doxorubicin was also measured using a confocal laser scanning microscope (FV 10i, Olympus, Japan).32 Further, the fluorescence intensity of doxorubicin was also observed in the HEK 293 cell line where phalloidin 488 and DAPI were used as a counter-stainer.
Caspase-3 and caspase-9 activity assays
HCT 116 cells were irradiated under 808 nm NIR light after treatment with 21 μg mL−1 solution of PB60@m-SiO2@HA@DOX. Caspase-3 and caspase-9 activities were quantified with commercially available caspase-3 and caspase-9 colorimetric assay kits (BioVision Research Products, Mountain View, CA), respectively. Caspase activities were detected at 405 nm using a spectrophotometer on an ELISA reader.33Statistical analysis
Data are presented as mean ± SEM. Statistical significance and differences among the groups were assessed via one-way analysis of variance (ANOVA) using OriginPro 8.0 software (San Diego, CA, USA). ‘p-value of 0.05 was considered significant.Results and discussions
Synthesis and characterization
The preparation, size-dependent contrast efficiency in MRI diagnosis, and chemo-photodynamic therapeutic activity of PBNC@m-SiO2@HA@DOX nanoplatform are schematically shown in Scheme 1. Herein, m-SiO2 was coated onto PBNC using the CTAB-assisted sol–gel technique with subsequent functionalization with HA for stabilization and target specificity against CD44 receptors, which are commonly overexpressed within the human colorectal carcinoma (HCT 116) cell line. After loading with DOX, PBNC@m-SiO2@HA@DOX nanoplatforms were examined for combined photodynamic and chemotherapy. Herein, it may be stated that the CTAB coating plays the role of secondary surfactant making PBNCs hydrophilic, which as a consequence acts as seeds for the coating of m-SiO2.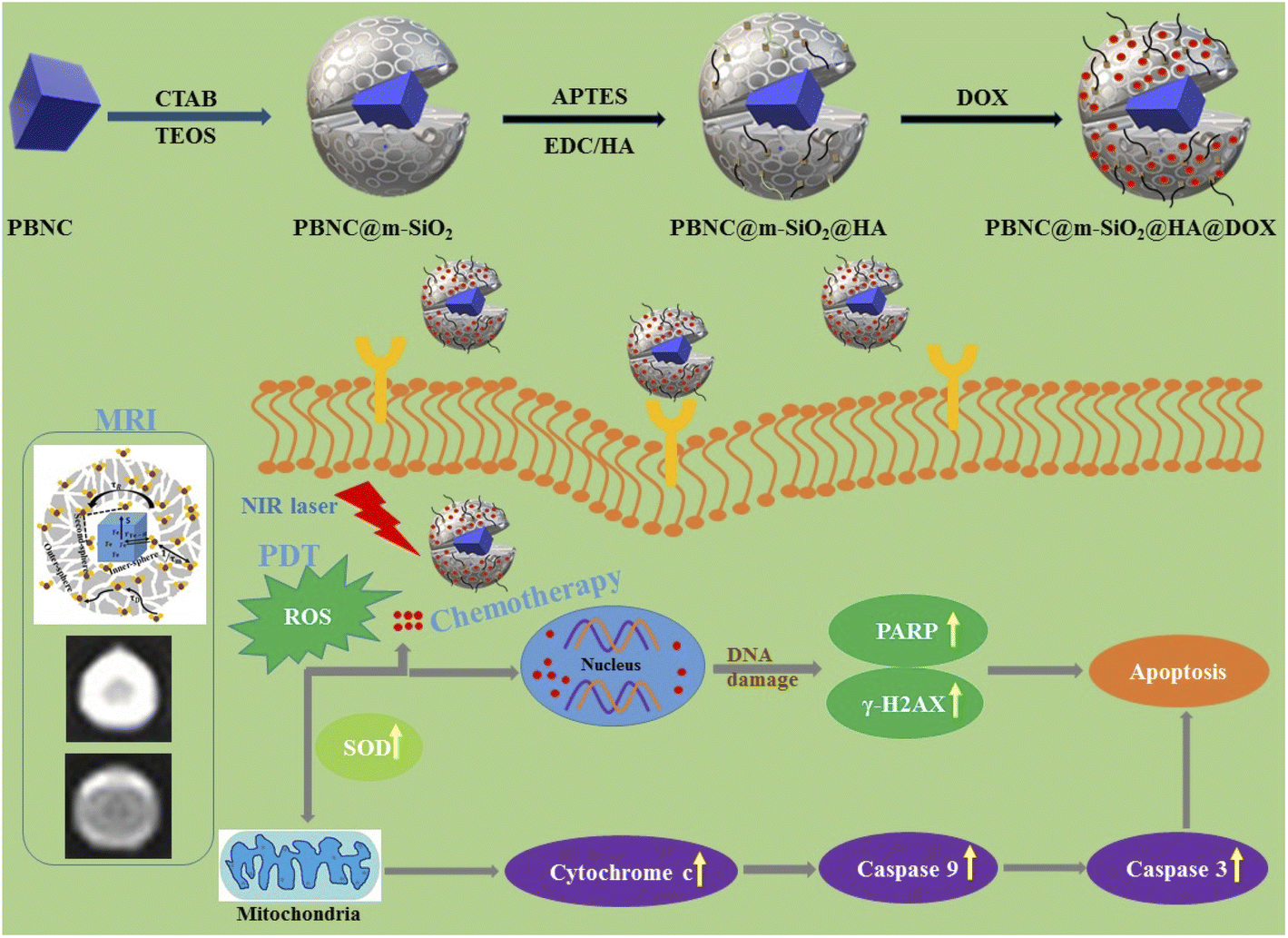 | ||
| Scheme 1 Demonstration of synthesis of PBNC@m-SiO2@HA@DOX nanoplatforms, size-dependent MRI contrast efficiency, and cell death through targeted chemo-photodynamic therapy upon irradiation with the 808 NIR laser. | ||
XRD patterns of the samples (shown in Fig. 1(a) and (b)) consist of eleven peaks, measured at 2θ = 17.50, 24.82, 35.42, 39.74, 43.80, 50.96, 54.28, 57.46, 66.37, 69.29, 77.65°, which can be readily indexed to Bragg's reflections from the (200), (220), (400), (420), (422), (440), (600), (620), (640), (642) and (121) planes, respectively, corresponding to face centred cubic phase of PBNC (JCPDS card no. 73-0687). The absence of any other peak confirms the phase purity of the samples. It has been studied previously that bare PBNCs having Pm3m space group grow with [Fe2+(CN)6]4− vacancy sites, replaced with H2O molecules. They are either bonded directly with Fe3+, known as coordinated H2O molecules, or bonded with coordinated H2O molecules, referred to as zeolitic-like H2O molecules. As both of them are highly dependent on the synthesis condition and have a significant influence on several physical properties of PBNCs, hence we have qualitatively determined coordinated and zeolitic H2O molecules from Rietveld analysis using Maud software according to the method prescribed by Samain et al.34 Herein, it is being considered that the ordered vacancies can be described by the parameter κ representing occupancy probability of Fe2+ at the 1b site corresponding to [Fe2+(CN)6]4− at the centre of the unit cell, with κ = 1 illustrating a completely ordered structure. Particularly, κ = 3/4 illustrates that 25% of [Fe2+(CN)6]4− sites remain unoccupied. After rigorous refinements, we obtained the occupancy of atoms at different sites as well as lattice parameters, bond lengths, unit cell volume, crystallite sizes, etc. as listed in Tables 1 and 2, while a schematic representation of the unit cell is presented in Fig. 1(c). It is being noted that the particle size and strain increase from PBNC60 to PBNC80, while lattice parameters, and unit cell volumes are decreased. In this context, it may be stated that an increase in the particle size can be assigned to the effect of higher synthesis temperature while an increasing strain is attributed to a decrease in the lattice parameters. A decrease in Fe3+ (3c)–O (6f, 12h) and an increase in the Fe3+ (3c)–C (6e) bond length has been noted in PBNC80 with respect to PBNC60. It was also noticed that the occupancy of Fe2+ at the 1b site remains unchanged; as well as the occupancy of the coordinated H2O at the 6f site changes hardly. In contrast, the occupancy of the zeolitic H2O at the 8g site is found to be less in PBNC80 in comparison with PBNC60. Herein, we assigned the reduced zeolitic H2O with the lattice parameter.
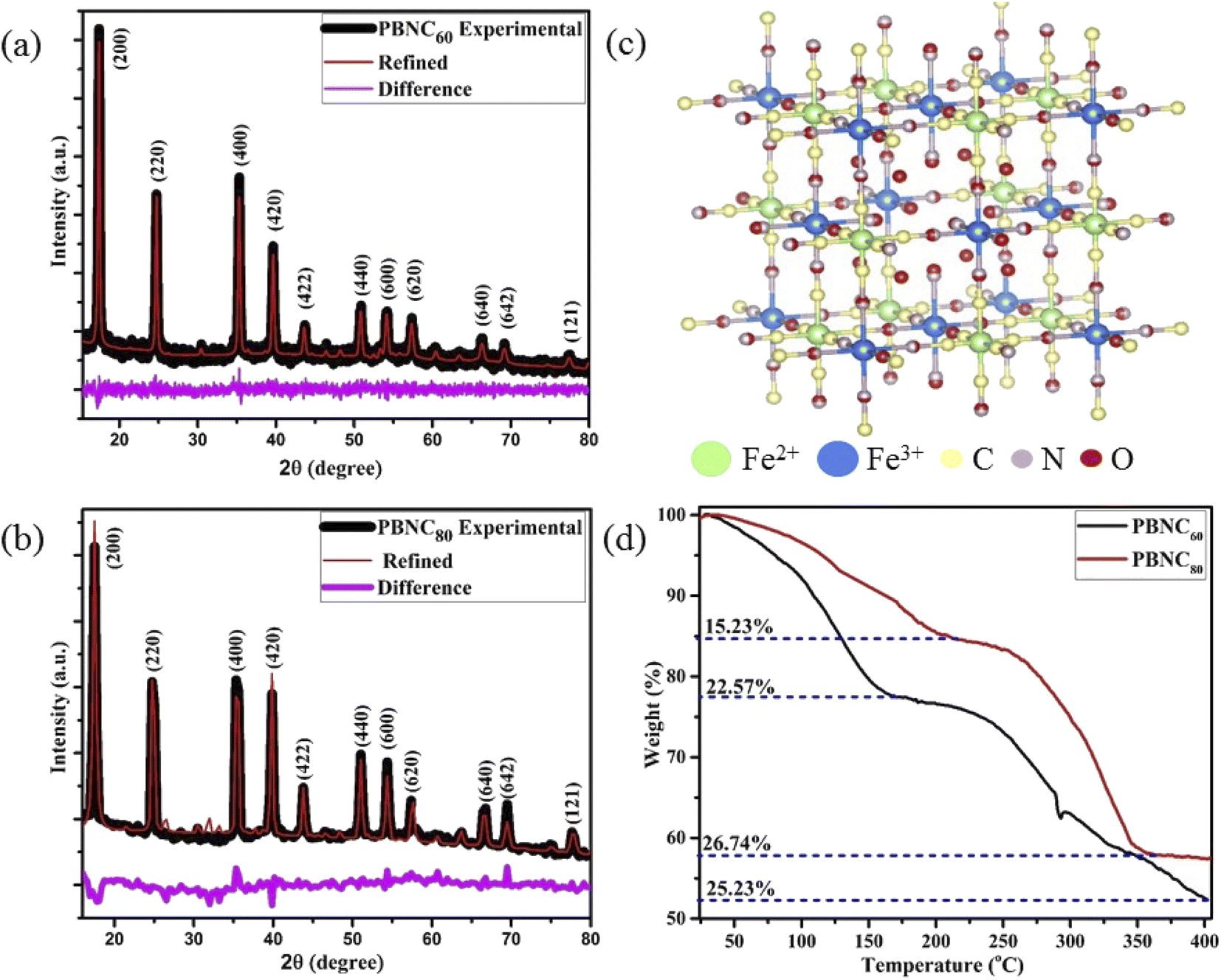 | ||
| Fig. 1 Rietveld refinement of XRD patterns with difference between the experimental and refined patterns of (a) PBNC60 with Rp = 18.68%, Rwp = 15.71%, χ2 = 1.19, (b) PBNC80 with Rp = 10.42%, Rwp = 12.34%, χ2 = 0.84. (c) Unit cell diagram of PBNC60. (d) TGA plots of both PBNC60 and PBNC80. | ||
| Atom | Site | PBNC60 | PBNC80 | ||
|---|---|---|---|---|---|
| Position (x) | Occupancy | Position (x) | Occupancy | ||
| Fe3+ | 1a | 0.0 | 1.0 | 0.0 | 1.0 |
| 3c | 0.0 | 1.0 | 0.0 | 1.0 | |
| Fe2+ | 1b | 0.50486345(9) | 0.6592583(21) | 0.5001262(13) | 0.6588522(9) |
| 3d | 0.5002564(13) | 0.7803208(17) | 0.4998143(19) | 0.80423933(18) | |
| N | 6e | 0.20095892(5) | 0.7803186(23) | 0.20295562(14) | 0.78014165(23) |
| 6f | 0.20777132(6) | 0.6590573(8) | 0.19910882(12) | 0.65888876(12) | |
| 12h | 0.3086982(18) | 0.7803359(19) | 0.3042289(9) | 0.7801389(28) | |
| C | 6e | 0.31160247(2) | 0.7803157(12) | 0.28431782(17) | 0.7801409(11) |
| 6f | 0.2108135(8) | 0.6592680(10) | 0.4952381(11) | 0.6588363(19) | |
| 12h | 0.19350913(21) | 0.7803082(13) | 0.19597772(21) | 0.78011984(26) | |
| O | 6e | 0.20546116(6) | 0.2196236(24) | 0.20186207(25) | 0.21956125(18) |
| 6f | 0.31103197(8) | 0.3418713(15) | 0.25275186(16) | 0.4129339(14) | |
| 12h | 0.21343586(11) | 0.2196520(17) | 0.3001274(8) | 0.21956046(13) | |
| O | 8g | 0.28311816(7) | 0.9769835(23) | 0.28644013(9) | 0.89194755(29) |
| Sample | PBNC60 | PBNC80 | |
|---|---|---|---|
| Lattice parameter (a in Å) | 10.16460 | 10.124104 | |
| Unit cell volume (Å3) | 1050.192443 | 1037.684371 | |
| Crystallite sizes (nm) | 67.9 | 96.4 | |
| Lattice strain | 0.0016 | 0.0026 | |
| Bond length (Å) | Fe3+ (3c)–N (12h) | 1.9445(16) | 1.9799(14) |
| Fe3+ (3c)–N (6f) | 2.1119(17) | 2.0153(14) | |
| Fe3+ (1a)–N (6e) | 2.0426(17) | 2.0540(15) | |
| Fe2+ (3d)–C (6e) | 1.9149(16) | 2.1817(16) | |
| Fe2+ (3d)–C (12h) | 1.9669(16) | 1.9833(14) | |
| Fe3+ (3c)–O (6f) | 3.1615(3) | 2.5584(18) | |
| Fe3+ (3c)–O (12h) | 2.3029(19) | 2.0214(14) | |
| Fe3+ (1a)–O (6e) | 2.0884(17) | 2.0428(15) | |
| O (6f)–O (8g) | 3.2891(4) | 3.0721(3) | |
In order to gain more insight into the variation of the coordinated and zeolitic H2O, we carried out TG analyses (revealed in Fig. 1(d)), which correspond to two regions of weight loss, assigned to the removal of zeolitic and coordinated H2O molecules, respectively. Careful calculations revealed approximately 25% loss coordinated H2O molecules from both samples, whereas ∼22 and 15% zeolitic H2O molecule losses were noted from the samples PBNC60 and PBNC80, respectively, indicating that TG analysis highly corroborates the Rietveld refinement. The morphological investigations by FESEM (shown in Fig. 2(a) and (b)) and subsequent histogram (shown in the inset of Fig. 2(a) and (b)) illustrate that PBNC60 are monodispersed and cubic in shape with a very smooth surface with average size 50–70 nm, while the size of PBNC80 varies widely from 150 to 280 nm retaining the cubic shape. TEM image of one representative bare sample (shown in Fig. 2(c)) further confirms the cubic shape of the as-prepared samples. DLS data (shown in Fig. S1 of ESI†) shows that the diameter of the bare PBNCs is higher than that obtained from FESEM and TEM due to the hydration layer within the solution and DLS size is calculated based on the spherical model considering this layer. More specifically, m-SiO2 and HA are well-known for their high hydration capacity, thus the high rise in the size of PBNC after each step, i.e., m-SiO2 coating and HA functionalization corroborate the phenomenon of successful coating and functionalization as well as their stability.35
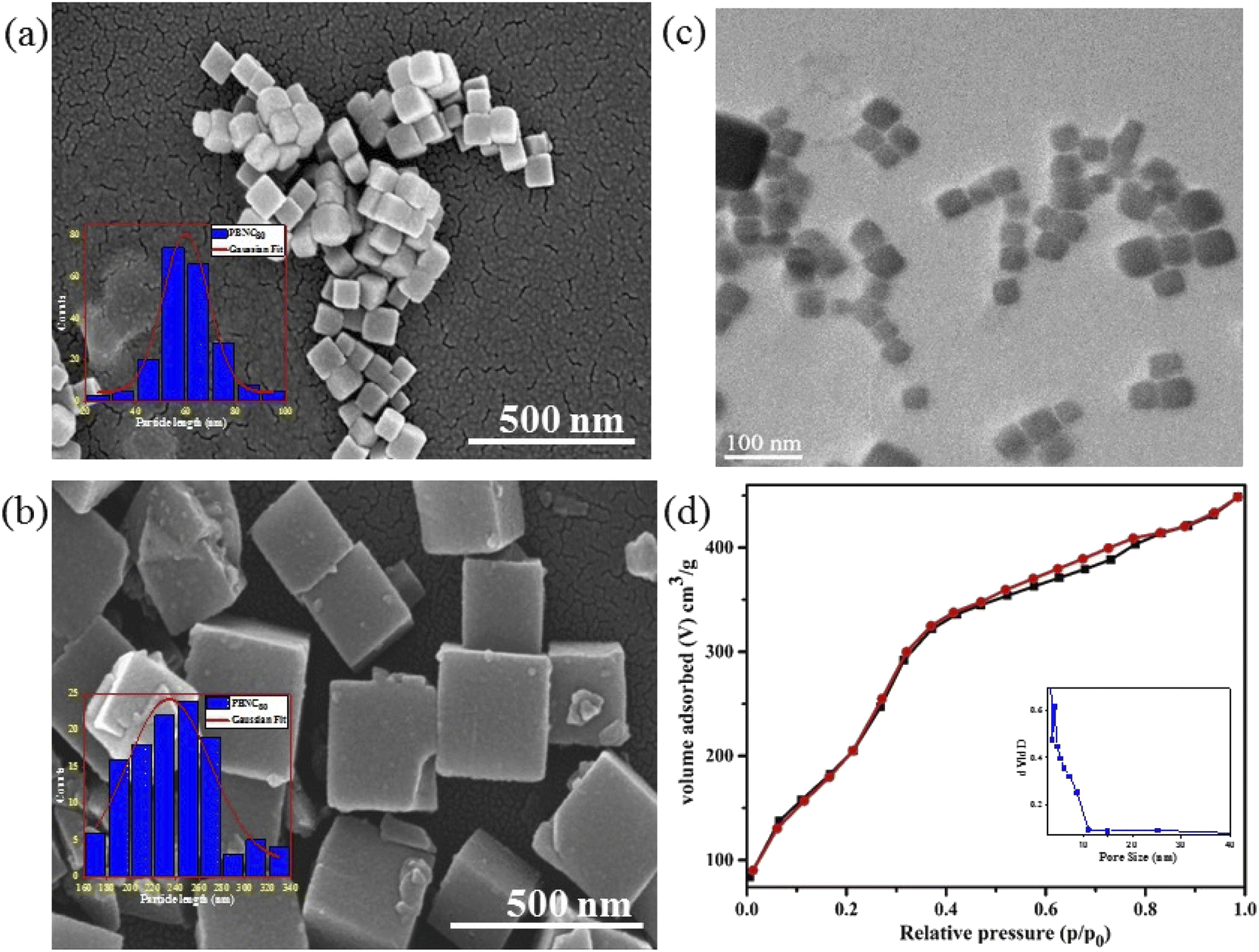 | ||
| Fig. 2 FESEM image with histogram plot in the inset of (a) PBNC60 and (b) PBNC80. (c) TEM image of PBNC60. (d) N2 adsorption–desorption isotherms with pore size distribution in the inset of PBNC60@m-SiO2. | ||
The nitrogen adsorption–desorption isotherms of one typical m-SiO2 coated PBNCs sample (shown in Fig. 2(d)) exhibits a typical IV curve, illustrating the mesoporous structure, while the BET surface area was found to be 938.2 m2 g−1 and the simultaneous presence of a large pore volume ∼0.29 cm3 g−1 and average pore size ∼3.2 nm. In this context, it may be stated that the as-pure PBNCs exhibit no N2 adsorption and we had coated m-SiO2 maintaining similar experimental conditions, thus the other m-SiO2 coated sample most likely had a similar surface area, pore volume, and pore diameter.14 In this context, it may be stated that FTIR and zeta potential analysis was performed to validate m-SiO2 coating and functionalization with HA, followed by DOX loading. From Fig. 3(a), the zeta potentials of bare PBNC60 and PBNC60@m-SiO2 were measured to be −25.7 and −21.7 mV, respectively, while the modification with APTES and HA reduced the zeta potential to −20.1 mV. Such systematic variation of zeta potential indicates successful coating of m-SiO2 and functionalization of HA onto the surface of PBNC.36 As the zeta potential of PBNC@m-SiO2@HA was negative, while that of –NH2 contained DOX was positive, it may be stated that DOX will be loaded onto the surface of PBNC@m-SiO2@HA by forming a bond through electrostatic attraction. The formation of the above-mentioned composite was further confirmed by FTIR analysis. As displayed in Fig. 3(b), representative FTIR spectra of PBNC60, PBNC60@m-SiO2, PBNC60@m-SiO2@HA, and PBNC60@m-SiO2@HA@DOX show strong C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration at 2072 cm−1 and a broad band in between 3200 and 3675 cm−1 due to the –OH from the surface adsorbed H2O, which plays an active role in adsorption via hydrogen bonding.
N stretching vibration at 2072 cm−1 and a broad band in between 3200 and 3675 cm−1 due to the –OH from the surface adsorbed H2O, which plays an active role in adsorption via hydrogen bonding.
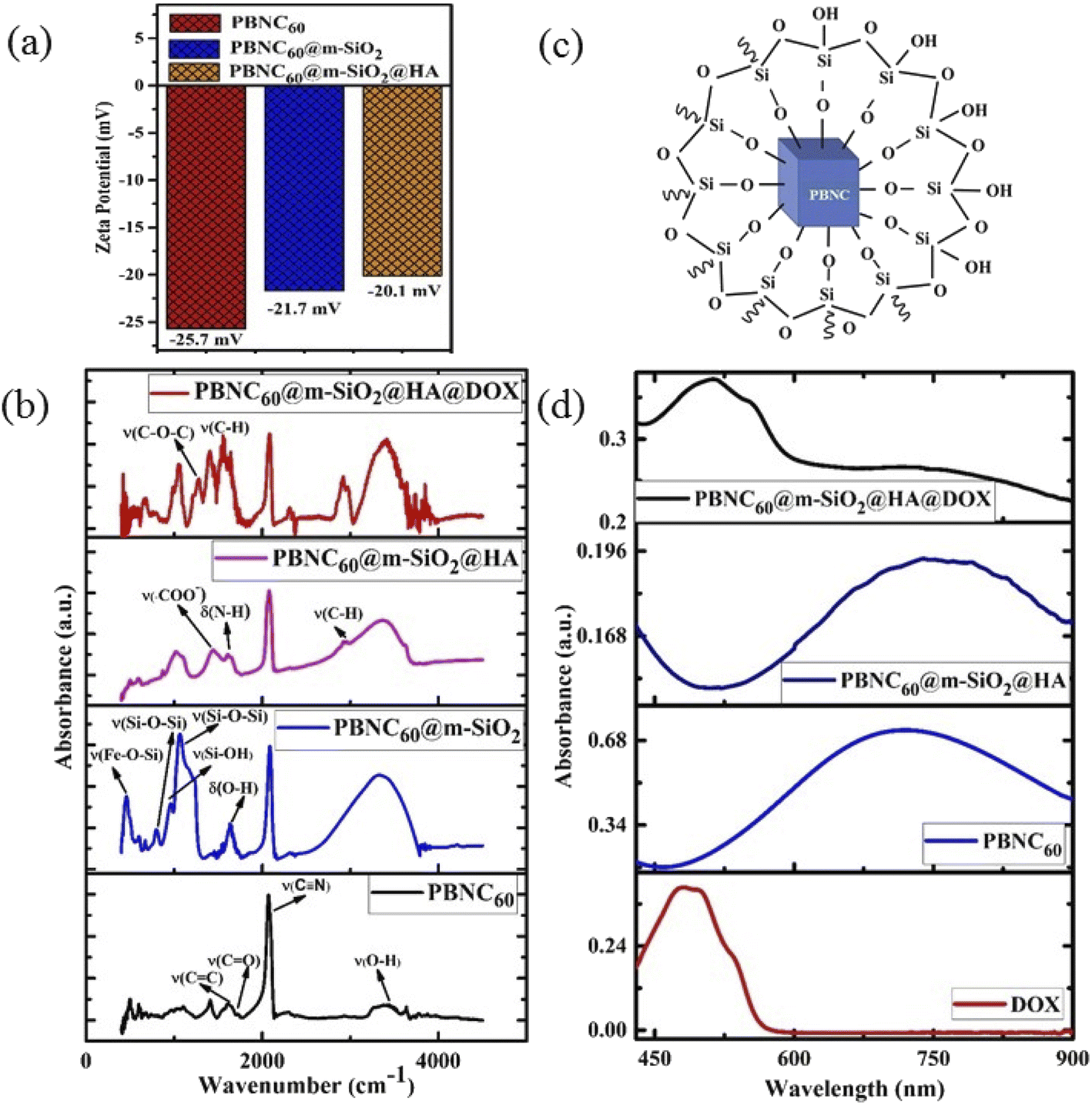 | ||
| Fig. 3 (a) Zeta potential of PBNC60, PBNC60@m-SiO2, and PBNC60@m-SiO2@HA. (b) FTIR spectra of PBNC60 along with each step of modification. (c) Design of m-SiO2 coated PBNC. (d) UV-vis-NIR spectroscopy of pure DOX, PBNC60, PBNC60@m-SiO2@HA, and PBNC60@m-SiO2@HA@DOX. | ||
In addition, PBNC60 exhibits absorption peaks at 1721 and 1610 cm−1, assigned to CO and CC stretching vibrations of citric acid that acts as an electrostatic stabilizer of PBNC60.37 PBNC60@m-SiO2 illustrates two absorption peaks at 1063 and 798 cm−1 corresponding to asymmetric stretching of ν(Si–O–Si) and symmetric stretching of ν(Si–O–Si), respectively, testifying the formation of the Si–O–Si framework, while the peak at 462 cm−1 is ascribed to ν(Fe–O–Si) indicating the formation of the chemical bond between PBNC60 and m-SiO2 and the considerable reduction of the intensity corresponding to C–N stretching with respect to OH indicates the presence of m-SiO2 (schematically shown in Fig. 3(c)) framework around PBNC60.38 Herein, the peak at 1637 cm−1 originates from δ(O–H) corresponding to OH present at the surface of m-SiO2 that facilitates easy functionalization with APTES. PBNC60@m-SiO2@HA shows very strong absorption peaks at 1443 and 1611 cm−1 due to ν(COO−) and δ(N–H), respectively, of HA. Briefly, δ(N–H) originates from the amidation reaction between the COOH bond of HA and –NH2 group of APTES functionalized m-SiO2. Furthermore, PBNC60@m-SiO2@HA@DOX displays the absorption peaks at 1572 and 1282 cm−1, assigned to ν(C–H) of the aromatic ring and asymmetric ν(C–O–C) of DOX, respectively, confirming the successful loading of DOX onto PBNC60@m-SiO2@HA through the electrostatic attraction.
The optical absorption spectrum as shown in Fig. 3(d) has the absorption maxima at 714 nm for sample PBNC60, while that of sample PBNC80 gets red-shifted at 852 nm due to the higher particle size (shown in Fig. S2 of ESI†). In addition, a wider full width at half maxima (FWHM) of the absorption spectra indicates non-uniformity in the size of PBNC80 supporting FESEM studies. Free DOX displays a broad absorption band from 420 nm to 560 nm, centred at 480 nm, hence the absorption band between 420 and 900 nm with an absorption peak at 510 nm is assigned to DOX within PBNC@m-SiO2@HA@DOX, illustrating successful loading of DOX onto PBNC@m-SiO2@HA, hence the phenomenon well corroborates with the FTIR studies.
MRI studies and phantom images in aqueous medium: size, coating, and functionalization-dependent on relaxivity
Performance of T1-weighted contrast agents, i.e., shortening of the relaxation rate of protons in the presence of the unit concentration of paramagnetic agents (in mM−1 s−1), defined by their relaxivity (r1), of bare as well as functionalized PBNCs were herein explored on a 3.0 T analyzer with pure PBS as the control. As shown in Fig. S3 of ESI† and Table 3, pristine PBNC60 and PBNC80 exhibit r1 relaxivity ∼5.14 and 3.95 mM−1 s−1, respectively, whereas r1 value of PBNC60@m-SiO2 and PBNC80@m-SiO2 raises up to 5.65 and 4.19 mM−1 s−1, respectively. Consecutively, PBNC60@m-SiO2@HA and PBNC80@m-SiO2@HA show r1 ∼ 5.30 and 4.14 mM−1 s−1, respectively, while PBNC60@m-SiO2@HA@DOX and PBNC80@m-SiO2@HA@DOX exhibit r1 ∼ 3.76 and 2.93 mM−1 s−1, respectively.| Sample | PBNC60 | PBNC80 |
|---|---|---|
| r1 (mM−1 s−1) | r1 (mM−1 s−1) | |
| Bare PBNC | 5.14 | 3.95 |
| PBNC@m-SiO2 | 5.65 | 4.19 |
| PBNC@m-SiO2@HA | 5.30 | 4.14 |
| PBNC@m-SiO2@HA@DOX | 3.76 | 2.93 |
Importantly, r1 of PBNC60@m-SiO2@HA@DOX was found to be larger than that of clinically used Gd-DPTA (∼3.1 mM−1 s−1).39 Possible explanations for the higher r1 of PBNC60 relative to PBNC80 is as follows: considering PBNC as macromolecules, r1 consists of contributions from the inner sphere (rIS1), second sphere (rSS1) and outer sphere (rOS1) H2O molecules and can be written as r1 = rIS1 + rSS1 + rOS1.19,24 Briefly, rIS1 depends on the interaction between Fe and directly coordinated H2O molecules, while rSS1 involving the interaction of Fe with nearby zeolitic H2O molecules. According to the theory of Solomon–Bloembergen–Morgan, rIS1 can be written as shown in eqn (1):
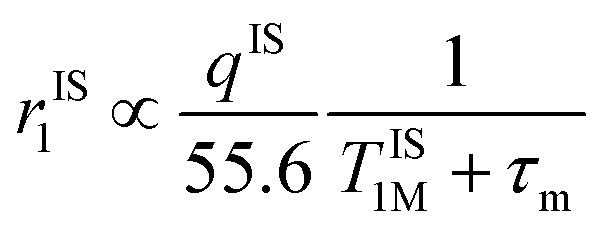 | (1) |
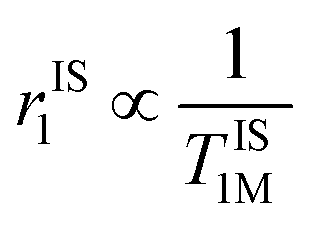 . Herein, it is to be mentioned that TIS1M consists of two contributions, namely, scalar (TSC1) and magnetic dipole–dipole (TDD1) due to the electronic and nuclear interactions between water proton and PBNCs, respectively, and may be written as the following eqn (2):
. Herein, it is to be mentioned that TIS1M consists of two contributions, namely, scalar (TSC1) and magnetic dipole–dipole (TDD1) due to the electronic and nuclear interactions between water proton and PBNCs, respectively, and may be written as the following eqn (2):
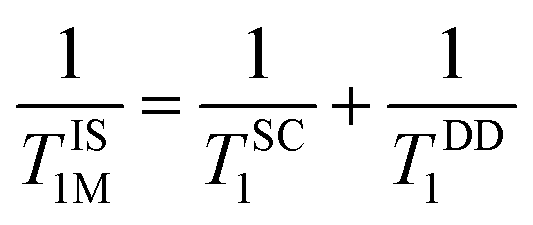 | (2) |
At magnetic field ≥0.25T, TSC1 appears to be insignificant, indicating 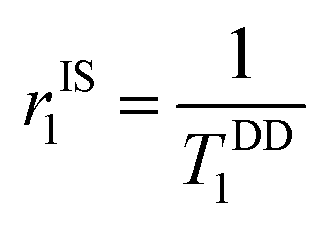 , whereas at the higher field (∼3.0T) TDD1 can be given by the expression
, whereas at the higher field (∼3.0T) TDD1 can be given by the expression 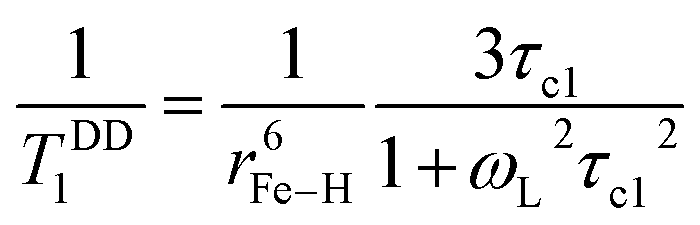 , where ωL and rFe–H represent the Larmor frequency and Fe–H distance, respectively, while τc1 is defined as the time constant of the fluctuating magnetic dipole that consists of two contributions as shown below (eqn (3)):
, where ωL and rFe–H represent the Larmor frequency and Fe–H distance, respectively, while τc1 is defined as the time constant of the fluctuating magnetic dipole that consists of two contributions as shown below (eqn (3)):
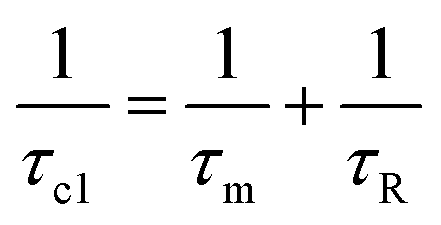 | (3) |
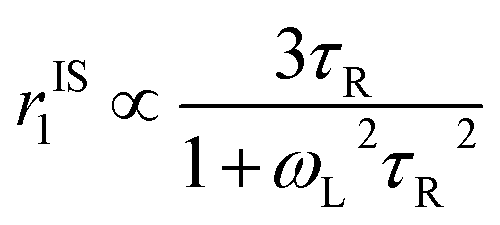 . Since our clinical MRI operates at 127.74 MHz frequency; hence neglecting ωL2τR2 in the denominator, we have to write rIS1 ∝ τR. Now according to Debye–Stokes relation,
. Since our clinical MRI operates at 127.74 MHz frequency; hence neglecting ωL2τR2 in the denominator, we have to write rIS1 ∝ τR. Now according to Debye–Stokes relation, 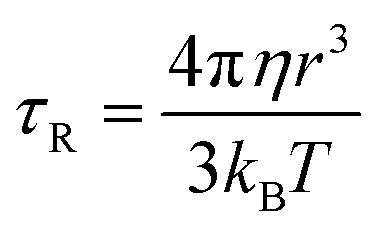 , where, η, kB, and T represent the viscosity of the medium,41 Boltzmann constant, and absolute temperature, respectively, indicating that PBNC80 should exhibit higher r1, which is not in consequence of our experimental observation. Thus, it may be conceived that no significant variation in r1 between PBNC60 and PBNC80 originates from rIS1.42 In this context, we next considered rSS1 contribution to r1, which is given by eqn (4):
, where, η, kB, and T represent the viscosity of the medium,41 Boltzmann constant, and absolute temperature, respectively, indicating that PBNC80 should exhibit higher r1, which is not in consequence of our experimental observation. Thus, it may be conceived that no significant variation in r1 between PBNC60 and PBNC80 originates from rIS1.42 In this context, we next considered rSS1 contribution to r1, which is given by eqn (4):
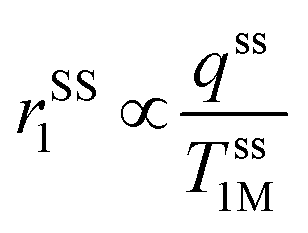 | (4) |
In comparison with bare nanostructures, PBNC@m-SiO2 samples show higher r1 that may be attributed to the synergistic effect of the enhanced τR by virtue of the Fe–O–Si bond formation between PBNC and rigid m-SiO2 and increased proton number of hydroxyls at the surface of m-SiO2 and H2O molecules, entrapped at the mesopores of SiO2.43 Noticeably, higher fractional change of relaxivity 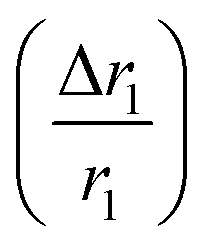 for sample PBNC60 may be explained as follows: considering zero wastage of TEOS, a simple calculation on the basis of conservation of mass illustrate that thickness (l) of m-SiO2 layer on PBNCs is proportional to the size of PBNCs, i.e., ‘l’ is higher for PBNC80. As is known, the surface-to-volume ratio (S/V) becomes higher as the size of the nanoparticle goes down, thus higher
for sample PBNC60 may be explained as follows: considering zero wastage of TEOS, a simple calculation on the basis of conservation of mass illustrate that thickness (l) of m-SiO2 layer on PBNCs is proportional to the size of PBNCs, i.e., ‘l’ is higher for PBNC80. As is known, the surface-to-volume ratio (S/V) becomes higher as the size of the nanoparticle goes down, thus higher 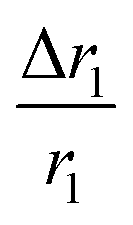 ratio may be attributed to the S/V ratio. Briefly, entrapped H2O molecules experience the local field generated by Fe2+ and/or Fe3+ ions residing at the surface of PBNCs, hence the interaction would be more for H2O molecules in the vicinity of PBNC's surface and would drastically decrease with increasing distance from the surface of PBNCs.44 Hence, the interaction between Fe2+ and/or Fe3+ at the surface and entrapped H2O molecules within m-SiO2 is more in PBNC60 giving higher
ratio may be attributed to the S/V ratio. Briefly, entrapped H2O molecules experience the local field generated by Fe2+ and/or Fe3+ ions residing at the surface of PBNCs, hence the interaction would be more for H2O molecules in the vicinity of PBNC's surface and would drastically decrease with increasing distance from the surface of PBNCs.44 Hence, the interaction between Fe2+ and/or Fe3+ at the surface and entrapped H2O molecules within m-SiO2 is more in PBNC60 giving higher 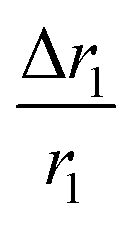 in comparison with PBNC80, which is schematically represented in Fig. 4(a and b).45 Therefore, we may simply assume that change in the longitudinal relaxation of the H2O proton due to functionalization is proportional to the S/V ratio of the nanostructure.
in comparison with PBNC80, which is schematically represented in Fig. 4(a and b).45 Therefore, we may simply assume that change in the longitudinal relaxation of the H2O proton due to functionalization is proportional to the S/V ratio of the nanostructure.
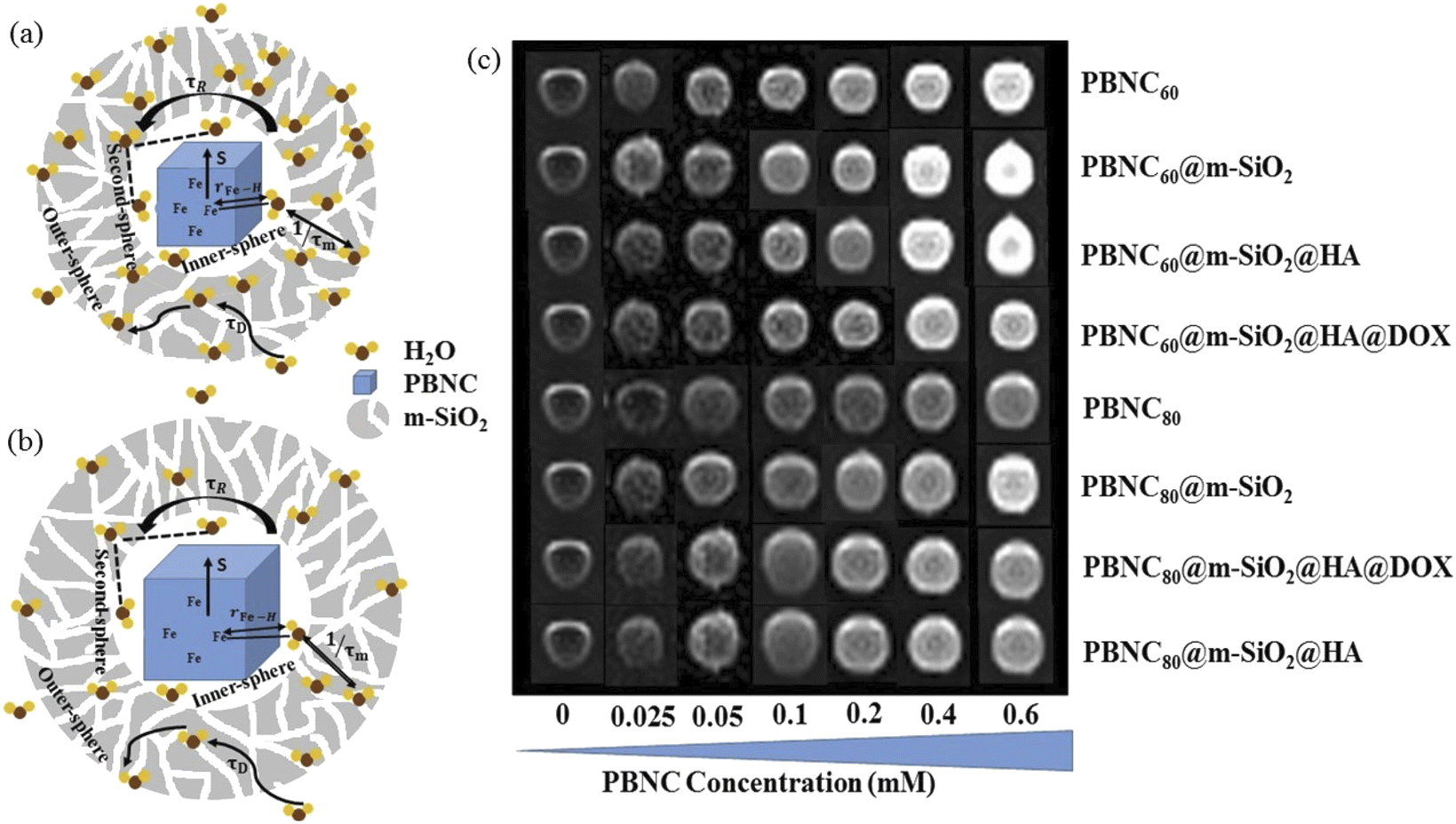 | ||
| Fig. 4 Schematic representation for the fractional change in r1 relaxivity due to m-SiO2 on (a) PBNC60; (b) PBNC80. (c) T1-weighted MR Phantom images of both PBNC along with each step of modification in PBS at different concentrations. | ||
HA functionalization further reduces r1 with reference to PBNC@m-SiO2 counterpart and it may be explained on the basis of relaxivity contribution from the outer sphere according to the following expression:
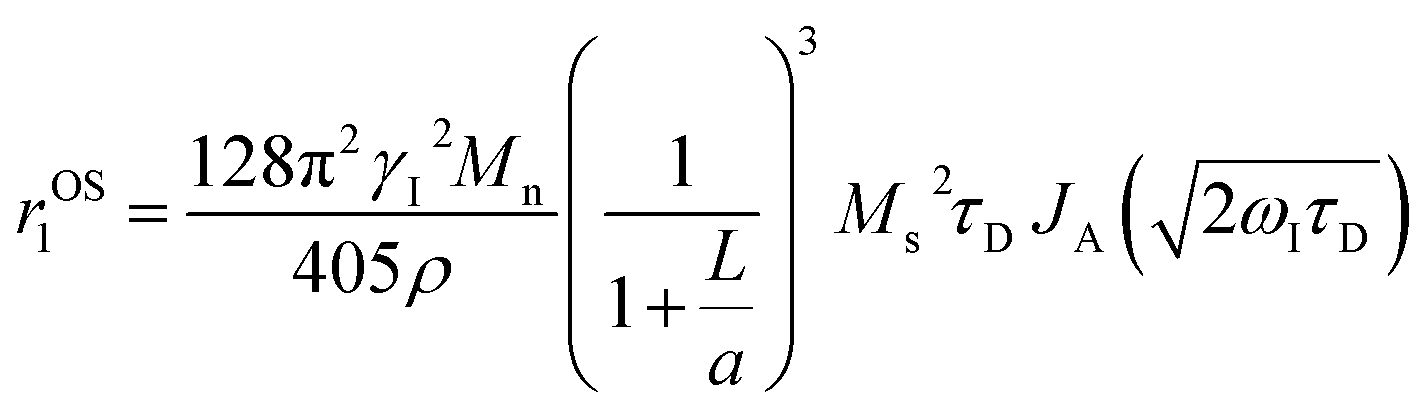 | (5) |
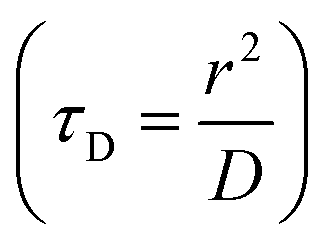 , where D and ‘r’ represent the diffusion coefficient of H2O molecules and effective radius of the particles, respectively, which is the sum of the actual radius (r) and thickness (L) of the impermeable surface coating i.e. r = a + L. Herein, we assume that HA functionalization forms an impermeable surface, which limits the diffusion of H2O molecules; hence, according to eqn (5) we attribute the reduced r1 of PBNC@m-SiO2@HA with respect to PB@m-SiO2 with the impermeable surface to HA. In this context, we calculated the ratio
, where D and ‘r’ represent the diffusion coefficient of H2O molecules and effective radius of the particles, respectively, which is the sum of the actual radius (r) and thickness (L) of the impermeable surface coating i.e. r = a + L. Herein, we assume that HA functionalization forms an impermeable surface, which limits the diffusion of H2O molecules; hence, according to eqn (5) we attribute the reduced r1 of PBNC@m-SiO2@HA with respect to PB@m-SiO2 with the impermeable surface to HA. In this context, we calculated the ratio 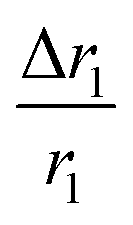 as ∼−0.06 and −0.01 for PBNC60@m-SiO2@HA and PBNC80@m-SiO2@HA, respectively, and the variation can be understood as follows: from eqn (5), we have found
as ∼−0.06 and −0.01 for PBNC60@m-SiO2@HA and PBNC80@m-SiO2@HA, respectively, and the variation can be understood as follows: from eqn (5), we have found 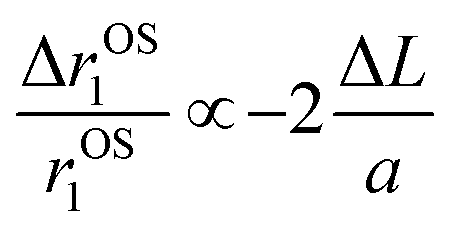 , indicating that for thick HA, the reduction is more. Therefore, it may be concluded that PBNC60 containing thicker HA corresponds to more reduction in
, indicating that for thick HA, the reduction is more. Therefore, it may be concluded that PBNC60 containing thicker HA corresponds to more reduction in 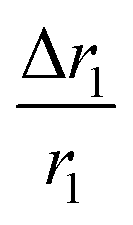 with reference to PBNC80, i.e., the surface functionalization effect is said to be more prominent for PBNC60. A further reduction of r1 in DOX-loaded samples is again assigned to the presence of an impermeable DOX layer onto the surface of PB@m-SiO2@HA. In this context,
with reference to PBNC80, i.e., the surface functionalization effect is said to be more prominent for PBNC60. A further reduction of r1 in DOX-loaded samples is again assigned to the presence of an impermeable DOX layer onto the surface of PB@m-SiO2@HA. In this context, 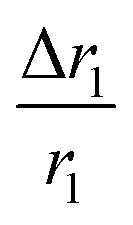 ∼−0.29 for both samples indicates identical DOX loading on both samples, i.e., the DOX loading appears to be independent of the size of the nanoparticles. Furthermore, the variation of r1 relaxivity value due to different functionalization for both PBNC was pictured (shown in Fig. 4(c)) through T1-weighted MRI Phantom images in PBS. The brightening effect with increasing concentration was observed and the results suggest that PBNC60 is more efficient as an MRI CA than PBNC80. Besides this, the images revealed that both PBNC60@m-SiO2@HA and PBNC60@m-SiO2@HA@DOX might be used as an MRI CA with minimal concentration for prolonged detection of cancer.
∼−0.29 for both samples indicates identical DOX loading on both samples, i.e., the DOX loading appears to be independent of the size of the nanoparticles. Furthermore, the variation of r1 relaxivity value due to different functionalization for both PBNC was pictured (shown in Fig. 4(c)) through T1-weighted MRI Phantom images in PBS. The brightening effect with increasing concentration was observed and the results suggest that PBNC60 is more efficient as an MRI CA than PBNC80. Besides this, the images revealed that both PBNC60@m-SiO2@HA and PBNC60@m-SiO2@HA@DOX might be used as an MRI CA with minimal concentration for prolonged detection of cancer.
In vitro cellular theranostic measurement
Motivated by higher r1 in comparison with commercially available Gd-based CA for MRI, we employed PBNC60@m-SiO2@HA for in vitro theranostic investigations against the HCT 116 cell line. Prior to in vitro studies, we checked the stability, singlet oxygen (1O2) generation, DOX loading capacity, and release kinetics of PBNC60@m-SiO2@HA. Herein, the generation of 1O2, believed to be the major cytotoxic agent giving photodynamic therapy (PDT) was investigated through a DPBF-fading experiment. Fig. 5(a) shows the decrease in absorbance intensity at 410 nm as a function of exposure time that illustrates 1O2 generation during irradiation with 808 nm laser light, hence the observation clearly revealed the potential of PBNC60@m-SiO2@HA for PDT. Very high DOX loading efficiency (∼94.43%) is attributed to the highly porous character of SiO2 and attractive electrostatic interaction between the charged Si–O− groups of m-SiO2 and DOX. Drug release profiles, carried out at PBS buffer solutions of different pH, are shown in Fig. 5(b). It is noted from the figure that only 19% of the drug gets released within 22 h at normal PBS buffer solution (pH ∼ 7.4), while only 50% drug is released over 22 h and 77% over 70 h at pH ∼ 5, which is the pH of the most cancerous microenvironment. Herein, the ultraslow release is attributed to hydrophobic–hydrophobic interaction between m-SiO2 and DOX, while acidic pH makes DOX hydrophilic and facilitates the release process.47 PBNC60@m-SiO2@HA as checked by monitoring the absorbance peak of PBNC (shown in Fig. S4 of ESI†) is highly stable at normal pH, while 55% gets degraded after 7 days at pH ∼ 5, indicating that our synthesized nanoplatform may be a potential candidate for synergistic theranostic applications.48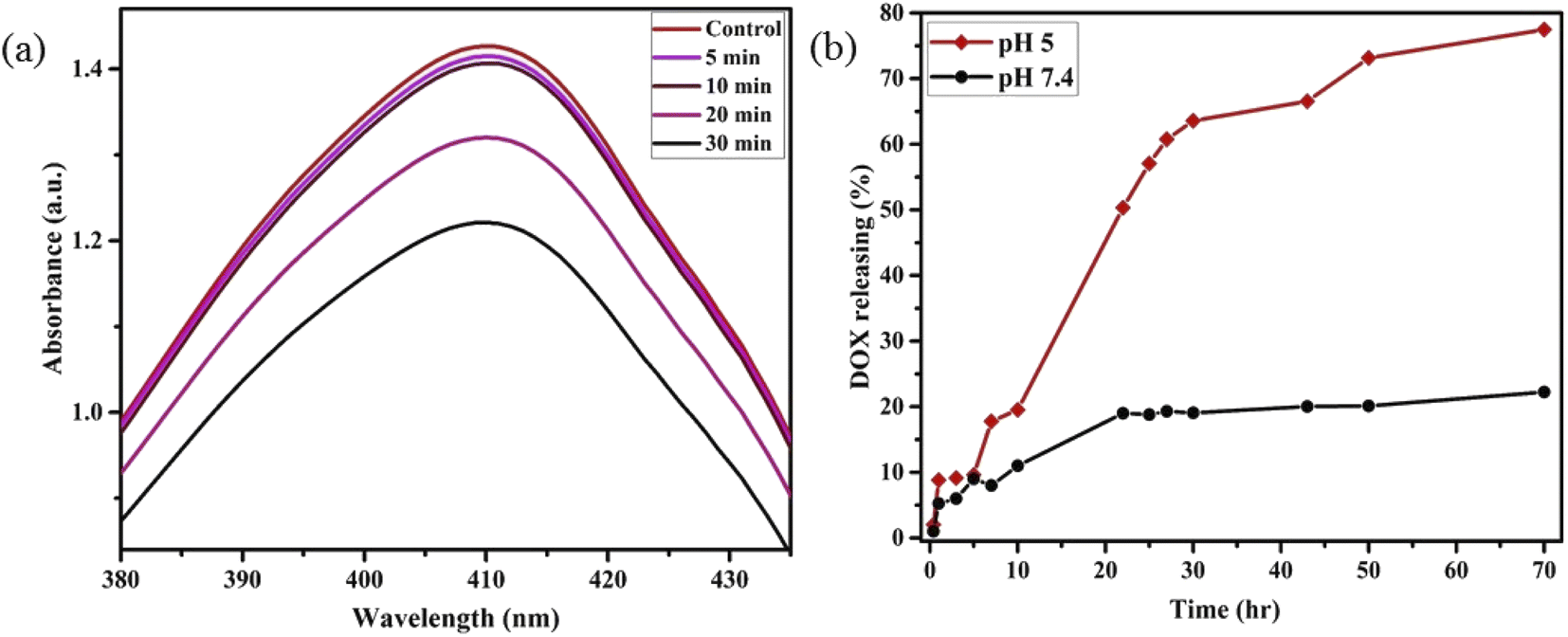 | ||
| Fig. 5 (a) UV-vis spectra for time-dependent degradation of DPBF by PBNC60@m-SiO2@HA under NIR laser irradiation. (b) DOX-release plot of PBNC60@m-SiO2@HA@DOX in different pH of PBS buffer without NIR irradiation. | ||
In vitro cellular toxicity measurement (shown in Fig. 6(a)) has been performed on HCT 116 cells using MTT assay, revealing that PBNC60, PBNC60@m-SiO2, and PBNC60@m-SiO2@HA exhibit limited toxicity of up to 70 μg mL−1 concentration, while approximately 91.05% cell death was observed only in the presence of 40 μg mL−1 PBNC60@m-SiO2@HA@DOX, indicating its cytotoxic efficacy as a chemotherapeutic agent. Herein, the very low IC50 value (∼21 μg mL−1) of PBNC60@m-SiO2@HA@DOX might indicate high uptake due to the presence of an abundance of HA receptors on HCT 116, which was further confirmed by red fluorescence emission from DOX in the confocal laser scanning microscopic (CLSM) images (shown in Fig. 6(b)), taken with light irradiation after 24 h incubation. Thus, the HA-functionalized nanoplatform is selectively delivered into the cancerous site (HCT 116) via its enhanced permeability and retention (EPR) effect. As HA has a strong affinity for the overexpressed CD44 receptor, therefore receptor-mediated active-targeting mechanism plays a major role in comparison to passive targeting.7,49 In order to eliminate the interference of conjugated DOX during the photodynamic ablation efficiency, we have compared MTT assay results with/without 808 nm laser irradiation for various times (10, 20, 30, and 40 minutes) (shown in Fig. 6(c)). The result demonstrates increasing cell death with an increase of irradiation time, reaching ∼78% at 20 minutes and the phenomenon suggests high PDT efficiency of the synthesized nanoplatform. We have observed red fluorescence emission from the nuclear region that indicates DOX binding on the nucleus and it well corroborates with previous reports.50 Importantly, we did not notice any significant variation in DOX fluorescence with exposure time, however, an upregulated expression of SOD at different irradiation times indicates more generation of enzymatic antioxidants to neutralize oxidative stress that confirms the PDT effect of the nanoplatform. Moreover, the DCFH-DA staining method was employed here to examine iROS production for actual research on the phototoxic efficacy of PBNC60@m-SiO2@HA@DOX. As expected, quantified by the DCF fluorescein intensity (shown in Fig. 6(d)) on flow cytometry assay (FACS), PBNC60@m-SiO2@HA@DOX leads to the most remarkable iROS production under laser irradiation compared with that without irradiation. In addition, the figure clearly reveals an increase in DCF fluorescein intensity with increasing light irradiation, illustrating prolonged iROS, which is advantageous for photodynamic therapy. Hence, the MTT and FACS results indicate that PBNC60@m-SiO2@HA@DOX-induced chemo-photodynamic results are more striking than chemo or photodynamic treatment alone.
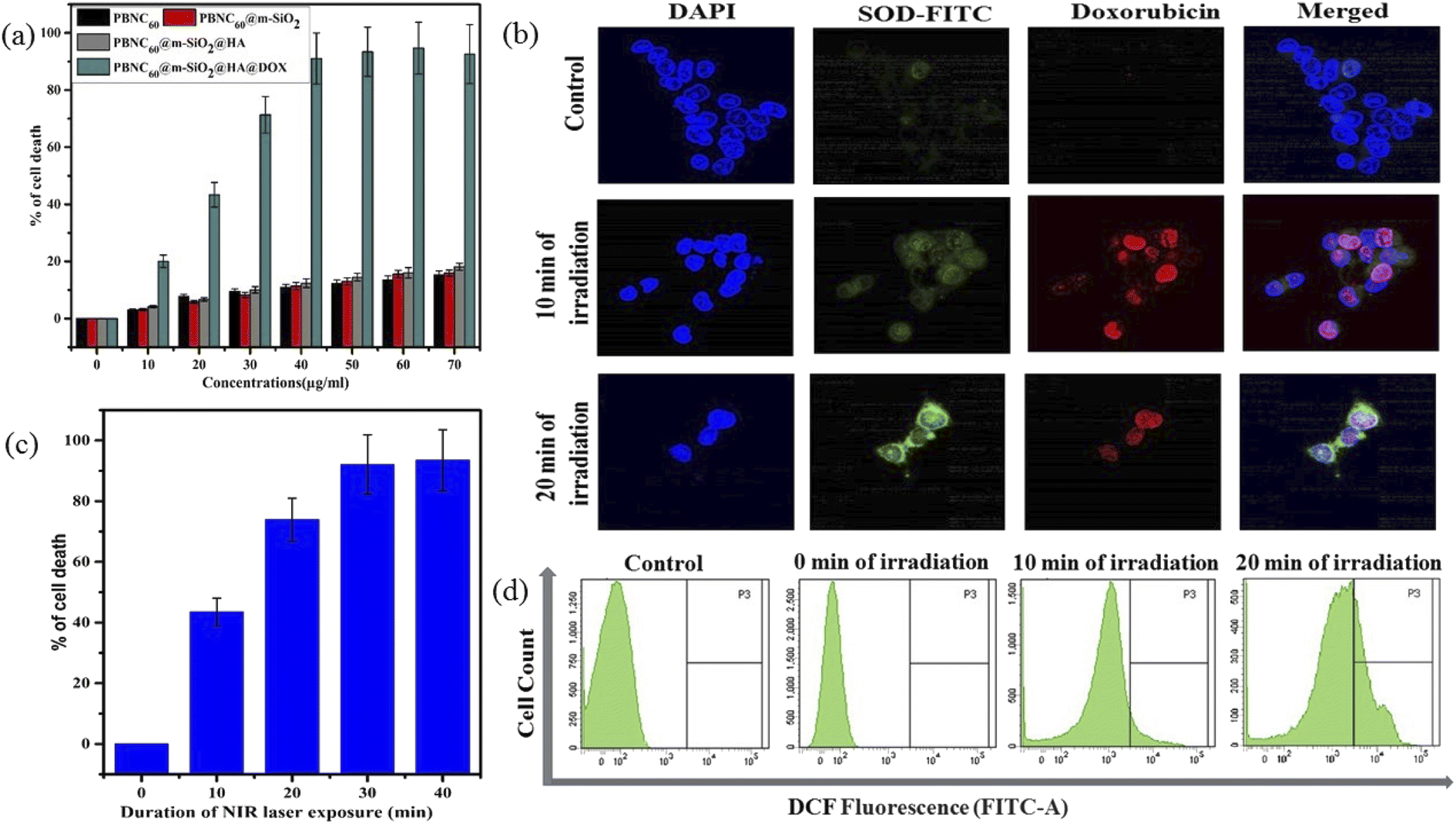 | ||
| Fig. 6 (a) Concentration-dependent MTT assay plot after 24 h incubation with PBNC60 along with each step of modification in HCT 116 cell line. (b) Expression of SOD and internalization of DOX in PBNC60@mSiO2@HA@DOX (21 μg mL−1) treated NIR irradiated (10 and 20 min) HCT 116 cell line; DAPI as a nuclear stainer. (c) Time-dependent cell death plot and (d) determination of iROS generation of HCT 116 cells after 24 h incubation with PBNC60@mSiO2@HA@DOX (21 μg mL−1) with laser irradiation (808 nm, 1 W cm−2). | ||
In general, the success of any therapeutic approach significantly depends on the early response of the cancer cell to the therapeutic process, where cell death mechanisms such as apoptosis (programmed cell death) and necrosis (accidental cell death) play a crucial role.51 Presently, chemo-photodynamic-induced cell death mechanism using PBNC60@m-SiO2@HA@DOX was assessed by FACS measurement using the Annexin V-FITC/PI kit. Prior to the measurement, as shown in Fig. 7(a), cell viability was found to be 99.7%, which was used as a control. After 24 h incubation with the DOX-containing nanoplatform (21 μg mL−1) under dark, cell viability is decreased to 83.9% along with 2.5% early/late apoptotic and 11.3% necrotic populations. After 10 minutes of light irradiation, cell viability reduces to 22.3% with enhanced early apoptotic (28.9%), late apoptotic (30.4%), and necrotic (18.4%) population. Further increase of the irradiation time (20 minutes) reduces cell viability to 0.1% along with an increase of necrosis to apoptosis ratio (31.3% late apoptotic and 68.6% necrotic populations). Importantly, a very drastic reduction of apoptotic percentage only after 20 minutes of irradiation indicates high PDT activity of PBNC60@m-SiO2@HA@DOX in comparison with sole chemotherapeutic activity.
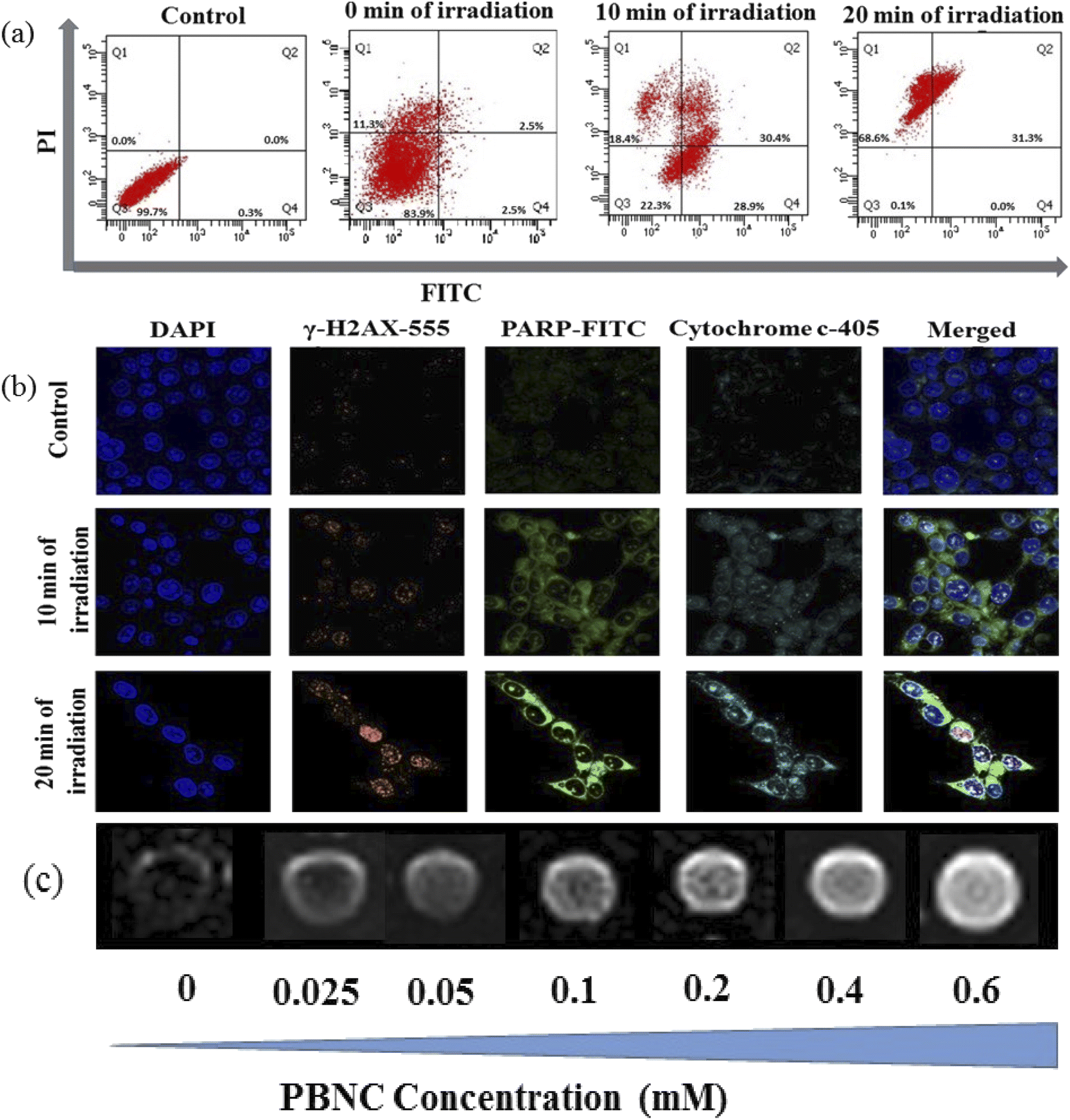 | ||
| Fig. 7 (a) Quantification of apoptosis; (b) expression of γ-H2AX, PARP, and cytochrome c through CLSM images of HCT 116 cells with PBNC60@m-SiO2@HA@DOX (21 μg mL−1) incubated with 24 h under time-dependent laser irradiation; DAPI as a nuclear stainer. (c) In vitro T1-weighted MR Phantom images of PBNC60@m-SiO2@HA@DOX incubated for 24 h at different concentrations of PBNC60. | ||
Apoptosis is mostly initiated by intracellular (mitochondrial disruption) or extracellular (death receptor activation) stimuli, called intrinsic and extrinsic pathways, whereas extreme physicochemical stress results in necrosis.52 Apoptosis in tissues is an asynchronous characteristic that leads to signalling pathways from the damage site of the cell onto the central leading to the final execution phase of the apoptosis where different hydrolytic enzymes are activated by a family of caspases based catalyses.53 Among them, caspase 3 and caspase 9 play the most pivotal roles in the early apoptotic process.54 In PDT-treated cells, both caspases cause several biochemical changes, including phosphatidylserine (PS), phosphatidylethanolamine (PE), plasma membrane depolarization, cell external membrane leaflet, cytoplasm acidification and DNA fragmentation, depending on the particular cell line, type of PDT agent and its subcellular location, overall dose, etc. To determine the apoptotic pathways of PBNC60@m-SiO2@HA@DOX-mediated chemo-photodynamic therapy, we performed a series of CLSM-based biochemical assays. It can be noticed from Fig. S5 in ESI† that light irradiation enhances the expressions for caspase 3 and caspase 9 in comparison to that without light, taken as a control, hence the results demonstrate prominent 1O2 stress-induced biochemical changes in PS, PE, etc. Herein, it may be stated that an altered level of pro/anti-apoptotic proteins triggers the release of cytochrome c (cyt-c) to the cytosol from the outer mitochondrial membrane, hence confirming whether mitochondria are involved in the apoptosis or not, we have checked cyt-c release using the confocal microscope. In this context, confocal microscopic data in Fig. 7(b) shows an increased expression of cyt-c release, indicating that the apoptosis is initiated from mitochondria. In any cell, mitochondrial superoxide dismutase (SOD) plays an important role to protect from oxidative damage by scavenging 1O2, however, excessive 1O2 depletes anti-oxidant defenses and disrupts antioxidant/prooxidant balance, which leads to DNA damage.55 Presently, we have observed increased expression of DNA damage (shown in Fig. 7(b)) with increasing irradiation time, while PBNC60@m-SiO2@HA@DOX-incubated sample without light irradiation was considered as a control. The DNA damage involves double-strand breaks (DSB), which may be induced directly or indirectly due to extensive iROS generation. In the literature, limited studies exist on the illustration of PDT-induced DSB, particularly in the presence of 808 nm NIR laser.56 We examined the phenomenon by assessing the expression of phosphorylation of H2AX, which is a well-considered marker of DSBs. Briefly, H2AX, one of the key variants of the H2A protein family, consists of the histone octamer of nucleosomes and is phosphorylated during the DNA damage due to any kind of exogenous and endogenous reasons and forms gamma-H2AX (γ-H2AX). It can be seen from Fig. 7(b) that the number of γ-H2AX foci increases with increasing irradiation time, hence the study demonstrates that the apoptotic is associated with DSBs. Herein, it is worth mentioning that poly(-adenosine diphosphate (ADP)-ribose) polymerase (PARP), an abundant nuclear protein that plays a pivotal role as a first responder to maintain genomic integrity and DNA repair, is being considered an indicator for apoptotic induction, while its inhibition enhances therapeutic efficiency.57 Recently, we have noticed that PARP cleavage is enhanced with increasing irradiation time in the presence of 21 μg mL−1 of PBNC60@m-SiO2@HA@DOX, while very insignificant PARP expression was noticed in the control. This result confirms that the PDT efficacy of PBNC60@m-SiO2@HA@DOX is due to the ROS-mediated mitochondrion-dependent DNA damage pathway. Moreover, our developed nanoplatform exhibits higher NIR laser-induced chemo-photodynamic therapeutic efficacy against HCT 116 cancer in comparison with other nanoparticles e.g. Fe3O4, Au, or upconversion nanoparticles.58–60
Furthermore, in vitro cellular T1 weighted MRI was also carried out to confirm the efficiency of PBNC60@m-SiO2@HA@DOX as an MRI CA against HCT 116 cells. The concentration-dependent brightening effect was visualised through phantom images, as shown in Fig. 7(c) and r1 relaxivity was measured to be 3.91 mM−1 s−1 (as shown in Fig. S6 of ESI†), which was well matched the previously measured one in solution. Hence, therapeutics and MR measurements attributed the potential of PBNC60@m-SiO2@HA@DOX as a theranostic CA against HCT 116 cell line with optimal concentration. Herein, MTT assay (shown in Fig. 8(a) and (b)) revealed very low death of the normal cell (HEK 293), taken as a control, which might be attributed to poor uptake of PBNC60@m-SiO2@HA@DOX due to the absence of HA receptor and was further confirmed from the confocal microscopic image (shown in Fig. 8(c)) from low red fluorescence from DOX. Thus, the study reveals very high selectivity of the nanoplatform for normal and HCT 116 cells, as well it is expected to be accumulated within HCT 116 cells via enhanced permeability and the retention (EPR) effect, and hence the present study illustrates a very high HCT 116 cell-killing efficiency of PBNC60@m-SiO2@HA@DOX.
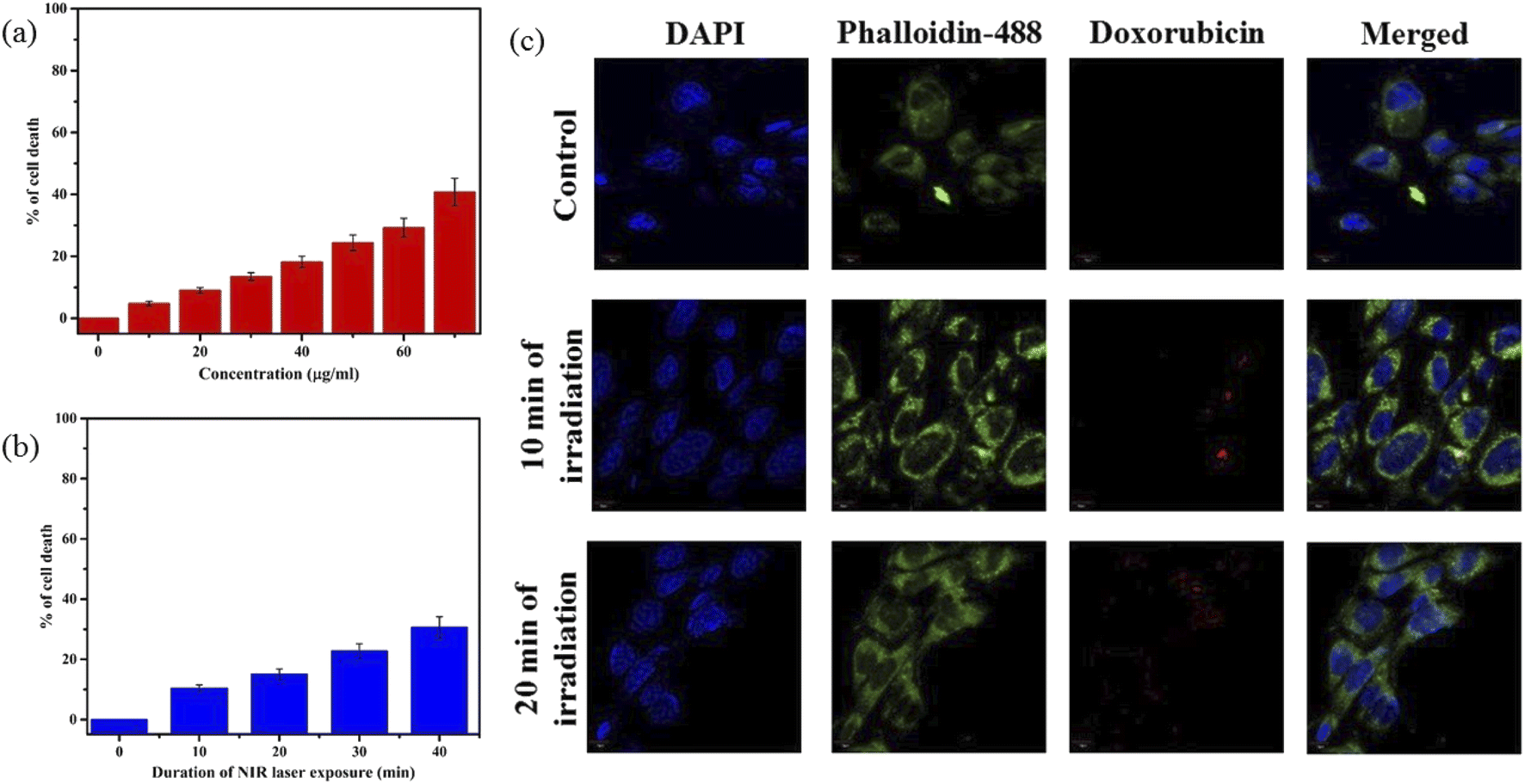 | ||
| Fig. 8 (a) concentration and (b) laser irradiation time dependant MTT assay plot of HEK 293 cells after 24 h incubated with PBNC60@m-SiO2@HA@DOX. (c) Expression of DOX through CLSM images of HEK 293 cells with PBNC60@m-SiO2@HA@DOX (21 μg mL−1) incubated with 24 h under time-dependent laser irradiation; DAPI and phalloidin-448 as a nuclear and cytoskeleton stainer. | ||
Conclusions
In summary, we have reported the synthesis of HCT 116 cell-specific, NIR (808 nm) triggering, a highly dispersed, coordination polymer-based nanoplatform PBNC60@m-SiO2@HA by coating PBNC60 with m-SiO2 and modification with hyaluronic acid as the next-generation theranostic nanoplatform for image-guided combined photodynamic and chemotherapy. PBNC60@m-SiO2@HA, which consists of amphiphilic micelles for conjugating DOX showed good biocompatibility, ultrahigh DOX loading as well as pH-responsive DOX-release efficiency. Besides this, the nanoplatform acts as in vitro T1-weighted MR agent having r1 value greater than r1 of commercially available Gd-based MRI contrast agent. Moreover, a high rate of cell (HCT 116) death was achieved over a lower dose of PBNC60@m-SiO2@HA@DOX due to the mutual effect of the laser (808 nm) and DOX, followed by iROS-mediated mitochondrion-dependent DNA damage pathway. Therefore, the multimodal theranostic nanoplatform demonstrates fascinating approaches and prospects against colon cancer for future perspectives.More interestingly, our detailed study on higher r1 value for smaller-sized PBNC is assigned to the contribution of the second sphere zeolitic water molecules leading to crystal vacancy. Furthermore, variation of r1 value at each functionalization step illustrates that m-SiO2 coating on PBNC increases r1 due to the quantum confinement effect, while HA and DOX addition lowers r1. Fractional changes in r1 are ascribed to size-dependent correlation time and diffusion of water molecules where the thickness of m-SiO2 plays a significant role.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
P. S. acknowledges the Department of Science & Technology, Government of India for financial support through the DST-INSPIRE program (Grant No. DST/INSPIRE Fellowship/2018/194) while S. K. shows gratitude to Council of Scientific & Industrial Research, Government of India for awarding him JRF. We are thankful to Dr. Sumita Kundu and Dr. S. Paul of Eko X-Ray & Imaging Institute, Kolkata for their kind help in the execution of the MRI experiment.Notes and references
- L. Catala and T. Mallah, Coord. Chem. Rev., 2017, 346, 32–61 CrossRef CAS.
- J. A. Webb, Y. C. Ou, S. Faley, E. P. Paul, J. P. Hittinger, C. C. Cutright, E. C. Lin, L. M. Bellan and R. Bardhan, ACS Omega, 2017, 2, 3583–3594 CrossRef CAS PubMed.
- L. Su, Y. Xiong, H. Yang, P. Zhang and F. Ye, J. Mater. Chem. B, 2016, 4, 128–134 RSC.
- K. R. Dunbar and R. A. Heintz, Prog. Inorg. Chem., 2007, 45, 283–391 Search PubMed.
- F. Shiba, U. Mameuda, S. Tatejima and Y. Okawa, RSC Adv., 2019, 9, 34589–34594 RSC.
- Y. Dou, X. Li, W. Yang, Y. Gou, M. Wu, X. Li, X. Zhang and J. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 1263–1272 CrossRef CAS PubMed.
- B. Zhou, B. P. Jiang, W. Sun, F. M. Wei, Y. He, H. Liang and X. C. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 18036–18049 CrossRef CAS PubMed.
- J. Long, Y. Gauri, C. Guérin and J. Larionova, Dalton Trans., 2016, 45, 17581–17587 RSC.
- M. Shokouhimehr, E. S. Soehnlen, J. Hao, M. Griswold, C. Flask, X. Fan, J. P. Basilion, S. Basu and S. D. Huang, J. Mater. Chem., 2010, 20, 5251–5259 RSC.
- M. Shokouhimehr, E. S. Soehnlen, A. Khitrin, S. Basu and S. D. Huang, Inorg. Chem. Commun., 2010, 13, 58–61 CrossRef CAS.
- M. Perrier, S. Kenouche, J. Long, K. Thangavel, J. Larionova, C. Goze-Bac, A. Lascialfari, M. Mariani, N. Baril, C. Guercin and B. Donnadieu, Inorg. Chem., 2013, 52, 13402–13414 CrossRef CAS PubMed.
- P. Mathieu, M. Chalet, M. M. Clain, L. Teulon, E. Benoist, N. Leygue, C. Picard, S. Boutry, S. Laurent, D. Stanicki and C. Hénoumont, New J. Chem., 2020, 44, 18031–18047 RSC.
- M. López-Pedrouso, J. M. Lorenzo, Z. Varela, J. Á. Fernández and D. Franco, Antioxidants, 2022, 11, 369 CrossRef PubMed.
- Y. Y. Su, Z. Teng, H. Yao, S. J. Wang, Y. Tian, Y. L. Zhang, W. F. Liu, W. Tian, L. J. Zheng, N. Lu and Q. Q. Ni, ACS Appl. Mater. Interfaces, 2016, 17038–17046 CrossRef CAS PubMed.
- X. Lin, Y. Cao, J. Li, D. Zheng, S. Lan, Y. Xue, F. Yu, M. Wu and X. Zhu, Biomater. Sci., 2019, 7, 2996–3006 RSC.
- K. Feng, J. Zheng, H. Dong, Z. Li, N. Gu, M. Ma and Y. Zhang, ACS Appl. Nano Mater., 2021, 4, 5176–5186 CrossRef CAS.
- Z. Zhou, L. Yang, J. Gao and X. Chen, Adv. Mater., 2019, 31, 1804567–1804598 CrossRef PubMed.
- W. Zhu, K. Liu, X. Sun, X. Wang, Y. Li, L. Cheng and Z. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 11575–11582 CrossRef CAS PubMed.
- M. F. Dumont, H. A. Hoffman, P. R. Yoon, L. S. Conklin, S. R. Saha, J. Paglione, R. W. Sze and R. Fernandes, Bioconjugate Chem., 2014, 25, 129–137 CrossRef CAS PubMed.
- Y. Gauri, J. Larionova, M. Corti, A. Lascialfari, M. Marinone, G. Poletti, K. Molvinger and C. Guérin, Dalton Trans., 2008, 28, 3658–3660 RSC.
- M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3–43 CrossRef CAS PubMed.
- L. H. Zhao, Q. L. Lin, J. Wei, Y. L. Huai, K. J. Wang and H. Y. Yan, Int. J. Clin. Exp. Pathol., 2015, 8, 692 CAS.
- L. Cheng, H. Gong, W. Zhu, J. Liu, X. Wang, G. Liu and Z. Liu, Biomaterials, 2014, 35, 9844–9852 CrossRef CAS PubMed.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- S. Mishra, K. Manna, U. Kayal, M. Saha, S. Chatterjee, D. Chandra, M. Hara, S. Datta, A. Bhaumik and K. D. Saha, RSC Adv., 2020, 10, 23148–23164 RSC.
- S. M. Yu, S. H. Choi, S. S. Kim, E. H. Goo, S. Y. Ji and B. Y. Choe, Curr. Appl. Phys., 2013, 13, 857–863 CrossRef.
- R. Nandi, S. Mishra, T. K. Maji, K. Manna, P. Kar, S. Banerjee, S. Dutta, S. K. Sharma, P. Lemmens, K. D. Saha and S. K. Pal, J. Mater. Chem. B, 2017, 5, 3927–3939 RSC.
- P. Bhanja, S. Mishra, K. Manna, A. Mallick, K. D. Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2017, 9, 31411–31423 CrossRef CAS PubMed.
- Y. Su, S. Lu, P. Gao, M. Zheng and Z. Xie, Mater. Chem. Front., 2019, 3, 1747–1753 RSC.
- D. L. Sai, J. Lee, D. L. Nguyen and Y. P. Kim, Exp. Mol. Med., 2021, 53, 495–504 CrossRef CAS PubMed.
- A. Alam, S. Mishra, A. Hassan, R. Bera, S. Dutta and K. D. Saha, ACS Omega, 2020, 5, 4250–4260 CrossRef CAS PubMed.
- M. Brentnall, L. Rodriguez-Menocal, R. L. De Guevara, E. Capero and L. H. Boise, BMC Cell Biol., 2013, 14, 1–9 CrossRef PubMed.
- L. Samain, F. Grandjean, G. J. Long, P. Martinetto, P. Bordet and D. Strivay, J. Phys. Chem. C, 2013, 117, 9693–9712 CrossRef CAS.
- S. Amorim, A. Martins, N. M. Neves, R. L. Reis and A. R. Pires, J. Mater. Chem. B, 2014, 2, 6939–6946 RSC.
- H. Xu, Z. Wang, Y. Li, Y. Gou, H. Zhou, Y. Li, F. Wu, L. Zhang, X. Yang, B. Lu and Z. Huang, RSC Adv., 2016, 6, 40427–40435 RSC.
- X. Hou, H. Xu, L. Pan, Y. Tian, X. Zhang, L. Ma, Y. Li and J. Zhao, RSC Adv., 2015, 5, 103760–103766 RSC.
- R. K. Sharma, S. Dutta and S. Sharma, New J. Chem., 2015, 40, 2089–2101 RSC.
- Y. Miao, H. Zhang, J. Cai, Y. Chen, H. Ma, S. Zhang, J. B. Yi, X. Liu, B. H. Bay, Y. Guo and X. Zhou, Nano Lett., 2021, 21, 1115–1123 CrossRef CAS PubMed.
- Q. Liu, S. Chen, J. Chen and J. Du, Macromolecules, 2015, 48, 739–749 CrossRef CAS.
- C. Y. Chen, H. Yang, W. Tang, X. Cui, W. Wang, X. Chen, Y. Yuan and A. Hu, J. Mater. Chem. B, 2013, 1, 5443–5449 RSC.
- H. Zhang, L. Li, X. L. Liu, J. Jiao, C. T. Ng, J. B. Yi, Y. E. Luo, B. H. Bay, L. Y. Zhao, M. L. Peng and N. Gu, ACS Nano, 2017, 11, 3614–3631 CrossRef CAS PubMed.
- K. Ni, Z. Zhao, Z. Zhang, L. Yang, L. Wang, H. Ai and J. Gao, Nanoscale, 2016, 8, 3768–3774 RSC.
- L. Zhang, Y. Wang, Y. Tang, Z. Jiao, C. Xie, H. Zhang, P. Gu, X. Wei, G. Y. Yang, H. Gu and C. Zhang, Nanoscale, 2013, 5, 4506–4516 RSC.
- L. Zhou, W. Yin, W. Ren, Z. Gu, W. Li, S. Jin, L. Yan, G. Tian, Z. Hu and Y. Zhao, New J. Chem., 2012, 36, 2599–2606 RSC.
- X. Zhang, B. Blasiak, A. J. Marenco, S. Trudel, B. Tomanek and F. C. van Veggel, Chem. Mater., 2016, 28, 3060–3072 CrossRef CAS.
- J. Liu, X. Hu, S. Jin, X. J. Liang and X. Ma, J. Mater. Chem. B, 2022, 10, 384–395 RSC.
- B. Zargar and A. Hatamine, Anal. Methods, 2014, 6, 5951–5956 RSC.
- M. B. Zakaria, A. A. Belik, C. H. Liu, H. Y. Hsieh, Y. T. Liao, V. Malgras, Y. Yamauchi and K. C. W. Wu, Chem.–Asian J., 2015, 10, 1457–1462 CrossRef CAS PubMed.
- P. Gao, S. Liu, Y. Su, M. Zheng and Z. Xie, Bioconjugate Chem., 2020, 31, 646–655 CrossRef CAS PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- L. Portt, G. Norman, C. Clapp, M. Greenwood and M. T. Greenwood, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 238–259 CrossRef CAS PubMed.
- N. L. Oleinick, R. L. Morris and I. Belichenko, Photochem. Photobiol. Sci., 2002, 1, 1–21 CrossRef CAS PubMed.
- D. Zhang, Q. Jin, Y. Ni and J. Zhang, J. Drug Targeting, 2020, 28, 904–912 CrossRef CAS PubMed.
- B. L. Tan, M. E. Norhaizan, W. P. P. Liew and H. S. Rahman, Front. Pharmacol., 2018, 9, 1162 CrossRef PubMed.
- X. Ji, L. Ge, C. Liu, Z. Tang, Y. Xiao, W. Chen, Z. Lei, W. Gao, S. Blake, D. De, B. Shi, X. Zeng, N. Kong, X. Zhang and W. Tao, Nat. Commun., 2021, 12, 1–17 CrossRef PubMed.
- F. U. Rahman, A. Ali, R. Guo, Y. C. Zhang, H. Wang, Z. T. Li and D. W. Zhang, Dalton Trans., 2015, 44, 2166–2175 RSC.
- A. Maximenko, J. Depciuch, N. Łopuszyńska, M. Stec, Ż. Światkowska-Warkocka, V. Bayev, P. M. Zieliński, J. Baran, J. Fedotova, W. P. Węglarz and M. Parlinska-Wojtan, RSC Adv., 2020, 10, 26508–26520 RSC.
- H. S. Kim and D. Y. Lee, Polymers, 2018, 10, 961 CrossRef PubMed.
- S. Y. Lee, R. Lee, E. Kim, S. Lee and Y. I. Park, Front. Bioeng. Biotechnol., 2020, 275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03995h |
| This journal is © The Royal Society of Chemistry 2022 |