
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed reductive decarboxylation of fatty acids for drop-in biofuel production†
Maria do S. B. da Silva ,
Jhudson G. L. de Araujo,
Júlia C. C. V. Bento,
Amanda M. de Azevedo,
Carlos R. O. Souto,
Aécia S. D. dos Anjos,
Aruzza M. M. de Araújo,
Djalma R. da Silva,
Fabrício G. Menezes,
Amanda D. Gondim and
Lívia N. Cavalcanti*
,
Jhudson G. L. de Araujo,
Júlia C. C. V. Bento,
Amanda M. de Azevedo,
Carlos R. O. Souto,
Aécia S. D. dos Anjos,
Aruzza M. M. de Araújo,
Djalma R. da Silva,
Fabrício G. Menezes,
Amanda D. Gondim and
Lívia N. Cavalcanti*
Federal University of Rio Grande do Norte, Institute of Chemistry, 59072-970, Natal, RN, Brazil. E-mail: livia.cavalcanti@ufrn.br
First published on 29th September 2022
Abstract
An operationally simple and highly selective method for the decarboxylation of fatty acids under remarkably mild conditions is described herein. The activation of the aliphatic carboxylic acids by esterification with N-hydroxyphthalimide (NHPI) enabled efficient deoxygenation to synthesize n-alkanes in up to 67% yield, employing inexpensive PMHS as a hydrogen source, NiCl2·6H2O, bipyridine, and zinc in THF. In contrast to the conventional thermo-catalytic approaches, this protocol does not require high temperature and high pressure of hydrogen gas to deoxygenate biomass-derived carboxylic acids, thus representing an attractive alternative for producing drop-in biofuels.
Introduction
Extensive consumption of fossil fuels to meet the global energy demand has intensified environmental concerns regarding the increase in emissions of CO2 and other greenhouse gases. In 2018, petroleum-based fuels were responsible for 94% of the energy use of the global transportation sector.1,2 In particular, the aviation industry, for example, accounts for approximately 2% of global CO2 emissions each year, which highlights the necessity of broadening the use of alternative low-carbon biofuels for preventing air pollution.3Vegetable oils are one of the most prominent types of renewable feedstock to produce biofuels such as biodiesel.4 However, as well as the crude oils, the fatty esters are still associated with poor fuel properties such as low heating value, high viscosity, and low oxidative stability.5,6 Additionally, the high oxygen content of this biofuel also results in poor cold flow properties, strongly limiting their use as blends with fossil fuels for diverse applications.7 Therefore, technical approaches for deoxygenating vegetable oils and their derived biofuels are important targets for increasing their compatibility with petroleum-based fuel infrastructure.
Several protocols have demonstrated the utility of vegetable oils for the production of biofuels through deoxygenation pathways.8–11 These reactions enable the conversion of biobased triglycerides, fatty acids, or esters into mixtures of renewable hydrocarbons that are able to either partially or completely replace fossil fuels as they are functionally equivalent to them.12 Remarkably, among the various types of suitable substrates for deoxygenation, fatty acids obtained upon hydrolysis of vegetable oils have met biofuel application more promisingly due to their low cost, availability, and carbon number lying within the range of fossil hydrocarbons.13,14
Depending on the biomass employed as feedstock, many technological routes such as hydroprocessing, catalytic hydrothermolysis, alcohol-to-jet, and gasification combined with the Fischer–Tropsch process are available for accomplishing the synthesis of renewable hydrocarbons.15,16 Particularly for the conversion of fatty acids and their derivatives, hydroprocessing has been a widely exploited approach17–19 (Scheme 1A). Although hydroprocessing has become increasingly relevant in petrochemical industry settings,20 it demands high temperatures (>250 °C) and high pressure (>10 bar) of hydrogen gas, which is usually obtained from non-renewable sources.21,22 Thermo-catalytic pyrolysis is a useful alternative that allows conversion without hydrogen. However, this process lacks selectivity, resulting in mixtures of products that require further upgrading to provide suitable biofuels.23–25 Hence, efficient deoxygenation methods that are operational in low temperature and ambient pressure under a hydrogen-free atmosphere are highly desirable.
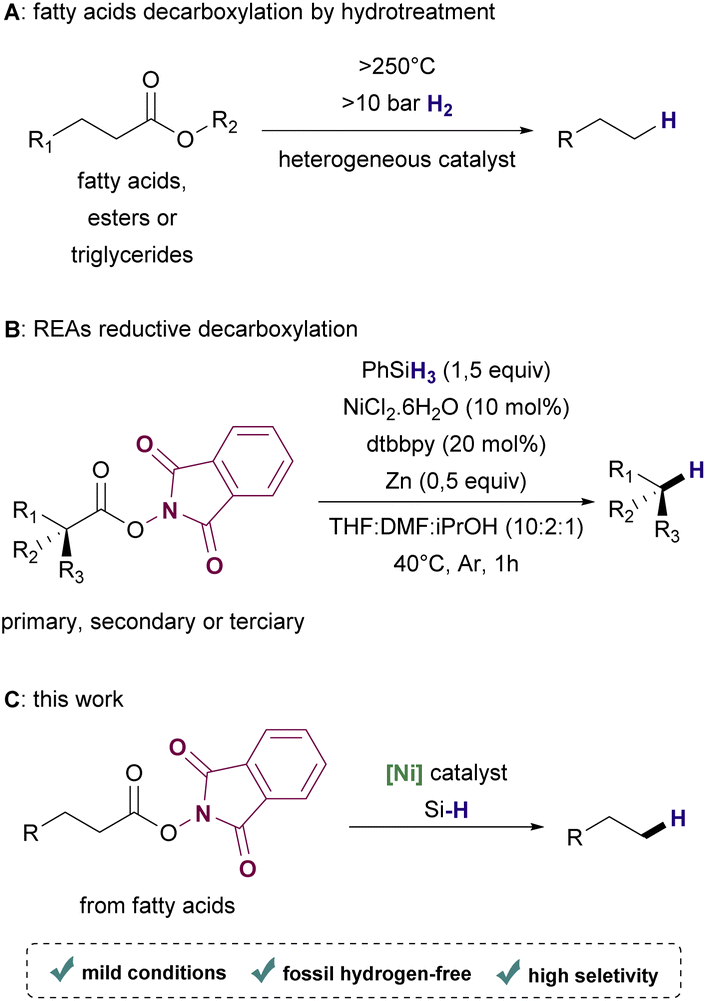 | ||
| Scheme 1 Reaction approaches for the decarboxylation of carboxylic acids. | ||
In the past few years, radical decarboxylation of redox-active esters (RAE) using metal catalysts under mild conditions has become a promising approach for forging C–C, C–X, and C–H bonds, introducing complexity in a variety of molecules with remarkable selectivity.26,27 Recently, Baran and coworkers reported the use of a Ni-based catalyst for the decarboxylation of RAEs in the presence of phenylsilane or Michael acceptors to afford hydrocarbons or coupling products respectively28 (Scheme 1B).
Although this protocol proved to be efficient to synthesize a variety of useful products from several examples of structurally diverse carboxylic acids, fatty acids were not included in the scope of the reductive method for hydrocarbon synthesis. Based on this important work, we envisioned the production of renewable hydrocarbons from bioderived fatty acids by reductive decarboxylation of RAEs under mild conditions using a Ni catalyst and a silane as the hydrogen source (Scheme 1C).
Results and discussion
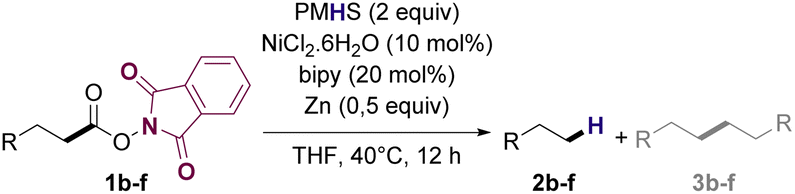
Based on previously reported protocols for the decarboxylation of RAE's,28 we started the optimization by employing NiCl2·6H2O as the catalyst, bipyridine as ligand, Zn as the reductive agent and inexpensive poly(methylhydrosiloxane) (PMHS) as the hydrogen source. PMHS has been previously reported as an efficient hydrogen source for other reductive decarboxylation processes29–32 and has a lower cost compared to other silanes already reported for similar transformations.33 It is a non-toxic, air and moisture stable reagent that can be easily obtained as by-product of silicon chemical industries.34,35 RAE 1a was selected as the model substrate to study the reductive deoxygenation since its corresponding fatty acid (lauric acid) is one of the major compounds in the composition of useful biofuel feedstocks such as coconut, licuri and babaçu oils.36–38Initial optimization studies revealed, after extensive solvent screening (see ESI for full details†), that THF was the ideal solvent for the transformation, resulting in the conversion of RAE into the desired hydrocarbon in 45% yield along with trace amounts of radical homocoupling byproduct (see Table ES1 in ESI†). Remarkably, the use of a mixture of THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF:iPrOH (10:2:1) (Table 1, entry 2), which was advantageous for the decarboxylation of a range of RAEs reported by Baran and coworkers,28 decreased the yield of 2a to 22%. Other Ni-based catalysts such as NiBr2 and Ni(OAc)2·4H2O (Table 1, entries 3 and 4) as well as other ligands such as dtbbpy and DPPB (Table 1, entries 5 and 6) also proved to be less effective to afford the decarboxylation product. Increasing the stoichiometry of both Ni and ligand diminished the yield of 2a (Table 1, entry 9).
:DMF:iPrOH (10:2:1) (Table 1, entry 2), which was advantageous for the decarboxylation of a range of RAEs reported by Baran and coworkers,28 decreased the yield of 2a to 22%. Other Ni-based catalysts such as NiBr2 and Ni(OAc)2·4H2O (Table 1, entries 3 and 4) as well as other ligands such as dtbbpy and DPPB (Table 1, entries 5 and 6) also proved to be less effective to afford the decarboxylation product. Increasing the stoichiometry of both Ni and ligand diminished the yield of 2a (Table 1, entry 9).
|
|||
|---|---|---|---|
| Entry | Modification from standard conditions | Yieldb (%) | |
| 2a | 3a | ||
| a Reactions were run on a 0.1 mmol scale.b Yield determined by GC-MS using hexadecane as internal standard. PMHS: poly(methylhydrosiloxane); bipy: 2,2-bipyridine; dtbbpy: 4,4-di-tert-butyl-2,2-dipyridyl; DPPB: 1,4-bis(diphenylphosphino)butane; PhSiH3: phenylsilane; TMDSO: 1,1,3,3-tetramethyldisiloxane. See ESI for additional details. | |||
| 1 | None | 45 | 2 |
| 2 | THF:DMF:iPrOH (10:2:1) |
22 | 0 |
| 3 | NiBr2 as catalyst | 19 | 0 |
| 4 | Ni(OAc)2·4H2O as catalyst | 10 | 2 |
| 5 | Dtbbpy as ligand | 28 | 0 |
| 6 | DPPB as ligand | 6 | 0 |
| 7 | PhSiH3 as hydrogen source | 28 | 26 |
| 8 | TMDSO as hydrogen source | 0 | 0 |
| 9 | 20 mol% NiCl2·6H2O + 40 mol% bipy | 6 | 0 |
| 10 | Mn as reducing agent | 0 | 0 |
| 11 | Addition 3 equiv. K2CO3 | 46 | 7 |
| 12 | Reaction concentration of 0.2 M (0.5 mL) | 36 | 0 |
| 13 | Reaction at 70 °C | 35 | 8 |
| 14 | No nickel or ligand | 0 | 0 |
| 15 | No PMHS | 0 | 0 |
| 16 | Methyl laureate or lauric acid instead of NHPI ester | 0 | 0 |
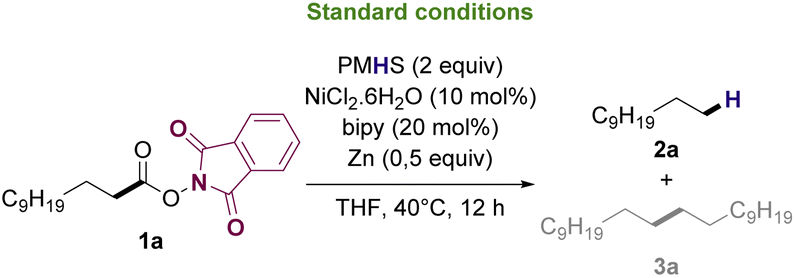
Replacing PMHS with different hydrogen sources commonly reported for this type of transformation39,40 significantly decreased the formation of the desired product under this set of conditions (Table 1, entries 7 and 8). Notably, other reductive agents such as Mn (Table 1, entry 10) completely inhibited the decarboxylation. The presence of a base such as K2CO3 as an additive (Table 1, entry 11) did not improve the yield of 2a, favoring radical homocoupling instead. Furthermore, increasing either reactants concentration or reaction temperature to 70 °C was also prejudicial to the formation of 2a (Table 1, entries 12 and 13). Control studies confirmed the necessity of Ni catalyst, ligand, and hydrogen source for the reaction to occur (Table 1, entries 14 and 15). Additionally, no product of decarboxylation was observed when using either the methyl laureate or lauric acid as substrates, proving that NHPI esters are crucial reactants to enable efficient reactivity (Table 1, entry 16).
After optimization experiments, the efficiency of the reductive deoxygenation of 1a using PMHS (2 equiv.), NiCl2·6H2O, bipyridine, and Zn in THF at 40 °C was confirmed by GC-FID with a calibration curve, which allowed to determine that 2a could be synthesized in 51% yield. It is noteworthy that, under the applied conditions, the reaction exclusively afforded hydrocarbons as products. Both undecane (n-C11) and docosane (n-C22) are valuable biofuel candidates since they have carbon number in the jet-fuel and diesel range, respectively. Furthermore, the N-phthalimide formed during the reaction work up, can be recovered and utilized to synthesize more N-hydroxyphthalimide41 that can be converted into 1a upon reaction with lauric acid.
With suitable conditions in hand, the generality of the decarboxylation method was evaluated. The scope of the fatty acids-derived RAE was examined by employing substrates with different carbon chain lengths. Reaction yield was determined by GC-FID using a calibration curve. The devised protocol proved to be effective for converting RAE 1b, which was synthesized from capric acid, into n-nonane in 45% yield, and octadecane in 2.5% yield (Table 2, entry 1). A better result was observed for the synthesis of n-tridecane (65% yield) after decarboxylation of 1c, which also resulted in the formation of the homocoupling product in a 4.9% yield (Table 2, entry 2). The reaction of the RAEs derived from palmitic and stearic acids delivered n-pentadecane in a 67% yield, and n-heptadecane in a 54% yield while the formation of the respective byproducts was diminished to less than 5% (Table 2, entries 3 and 4). Extending the scope to RAE 1f resulted in the complete suppression of 3f generation along with lower yields of 2f in relation to the other synthesized hydrocarbons. It is possible that the formation of 2f was limited by the reaction between the radicals formed upon decarboxylation and the unsaturation within the substrate, leading to a polymerization product that could not be detected by the employed technique.14
|
||||
|---|---|---|---|---|
| Entry | Feedstock | Principal product | Yieldb (%) | |
| 2a | 3a | |||
| a General decarboxylation reaction conditions: 0.1 mmol of redox-active ester, 0.2 mmol of PMHS, 0.01 mmol of NiCl2·6H2O, 0.02 mmol of bipyridine, 0.05 mmol of zinc in 1 mL of THF (0.1 M).b Yield determined by GC-FID using calibration curve. NPhth: N-phthalimide. | ||||
| 1 |  |
n-C9 2b | 45 | 2.5 |
| 2 |  |
n-C13 2c | 65 | 4.9 |
| 3 | 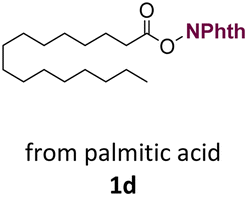 |
n-C15 2d | 67 | 4.2 |
| 4 | 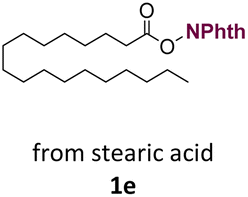 |
n-C17 2e | 54 | 1.7 |
| 5 | 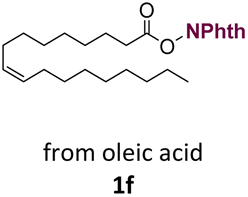 |
n-C17:1 2f | 33 | 0 |
Although moderate yields were determined for the n-alkanes, all reactions proceeded with more than 98% conversion, except for ester 1f, which afforded the respective product with an 85% conversion. These data indicate that the redox-active esters are likely to decompose under the reaction conditions, which is corroborated by the presence of fatty acids and phthalimide in crude reaction mixtures according to GC-FID analysis.
The optimized conditions were also employed to convert the fatty acids using a one-pot reaction approach. The decarboxylation protocol was applied to RAEs generated in situ to produce n-alkanes. Notably, the RAEs from lauric and palmitic acids (Table 3, entries 1 and 4) delivered the corresponding hydrocarbons in similar yields in relation to the protocol that starts from the isolated esters. For the other substrates, however, significantly lower yields were observed, especially in the decarboxylation of the RAE from oleic acid. Similar outcomes were already described for this type of transformation.28,42,43
|
||||
|---|---|---|---|---|
| Entry | Feedstock | Principal product | Yieldb (%) | |
| 2a | 3a | |||
| a Activation in situ conditions: fatty acid (0.1 mmol), N-hydroxyphthalimide (0.11 mmol), DIC (0.11 mmol) in DCM at rt, then 0.2 mmol of PMHS, 0.01 mmol of NiCl2·6H2O, 0.02 mmol of bipyridine, 0.05 mmol of Zn in 1 mL of THF (0.1 M).b Yield determined by GC-FID with calibration curve. NPhth: N-phthalimide. | ||||
| 1 |  |
n-C11 2a | 48 | 4.8 |
| 2 |  |
n-C9 2b | 28 | 1.2 |
| 3 |  |
n-C13 2c | 27 | <1 |
| 4 |  |
n-C15 2d | 51 | 2.3 |
| 5 |  |
n-C17 2e | 24 | 1.7 |
| 6 |  |
n-C17:1 2f | 10 | 0 |
Finally, the developed method was applied to a mixture of RAEs synthesized from saturated fatty acids. Fortunately, the materials could be converted into a mixture of hydrocarbons (C9–C17) within the diesel and jet-fuel ranges in 67% yield (Fig. 1). This result demonstrates the potential of the decarboxylation methodology to produce n-alkanes compatible with drop-in biofuels from more complex matrixes such as mixtures of fatty acids originating from vegetable oils or animal fat.
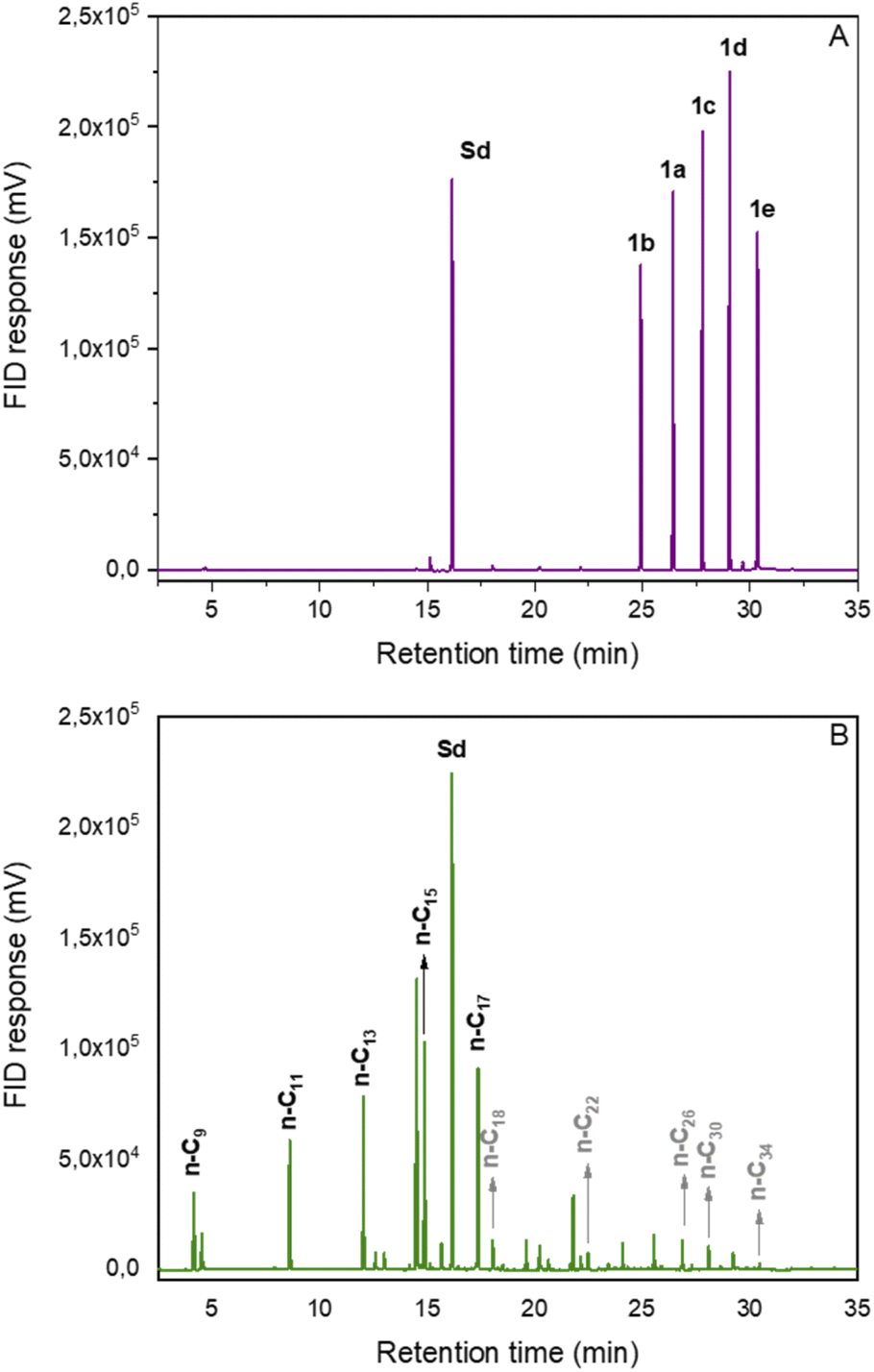 | ||
| Fig. 1 Chromatograms of the decarboxylation reaction of REAs mixture derived from fatty acids. (A) Initial reaction sample; (B) final reaction sample; Sd: internal standard. | ||
Based on these results, the proposed mechanism is analogous to previously reported Ni-catalyzed reductive decarboxylation of RAE (Scheme 2).27,44–46 On the basis of this mechanism the BiPy ligand is necessary to help stabilize the in situ formed Ni(I) catalyst II that undergoes transmetallation with PMHS to afford intermediate III. This complex would then donate an electron via SET to the RAE forming V and the radical anion IV which undergoes radical fragmentation (decarboxylation) yielding the primary alkyl radical VII and anionic phthalimide VI. Addition of radicals VII and VI to intermediate V gives intermediate VIII which after reductive elimination affords the desired alkane product and regenerates the catalyst.
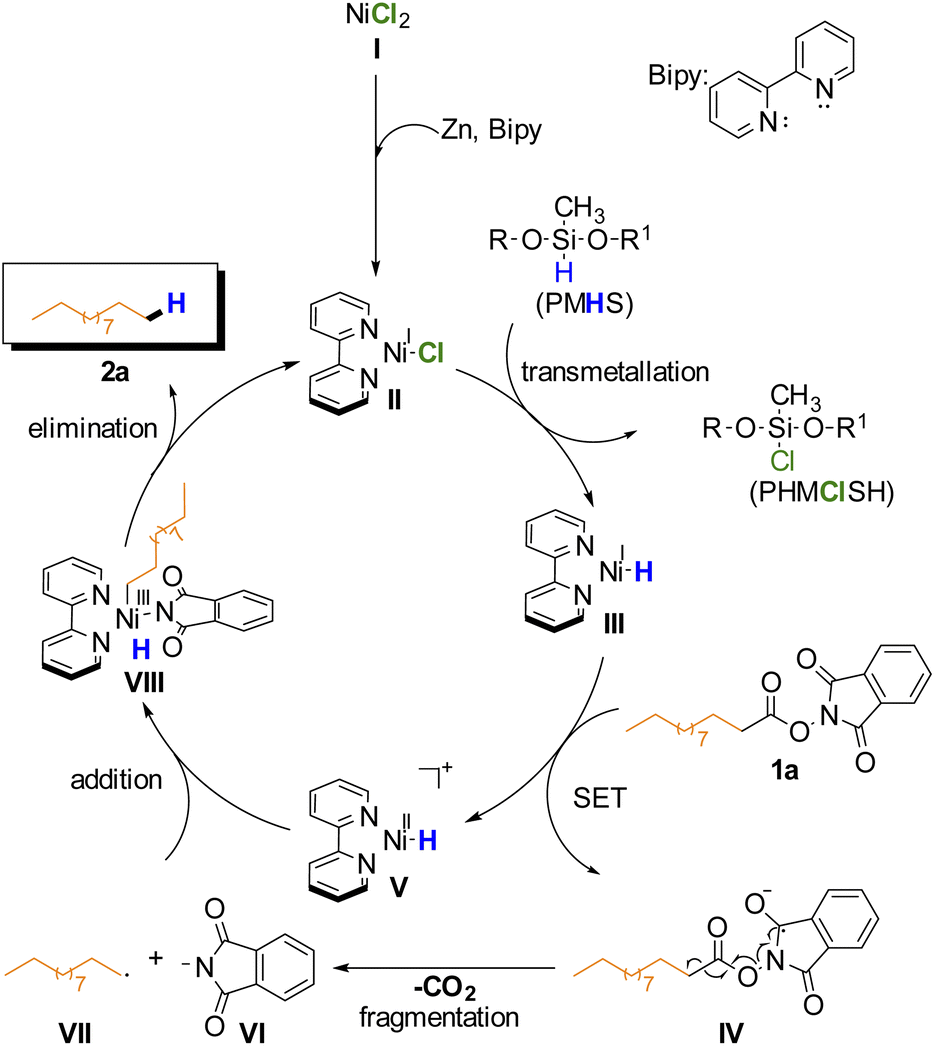 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusions
A method for the smooth conversion of redox-active esters derived from fatty acids to selectively produce renewable hydrocarbons was demonstrated. Prior activation of substrates enabled the process to be performed with a simple bench setup at 40 °C in ambient pressure. This chemistry was also efficient for the decarboxylation of fatty acids in a one-pot sequential protocol, avoiding the necessity of prior purification of the RAE formed in situ. Remarkably, a non-toxic, inexpensive silane proved to be effective, allowing the synthesis of n-alkanes under hydrogen-free conditions. To the best of our knowledge, no studies on applying Ni-catalysts combined with PMHS for decarboxylation of fatty acids to produce Cn−1 hydrocarbons with high selectivity under mild conditions have been reported. According to the results, hydrocarbons within the diesel and the jet-fuel ranges were synthesized at similar yields compared to conventional thermo-catalytic routes that apply harsh conditions for the deoxygenation of biomass.Author contributions
Silva MSB: methodology, formal analysis, investigation, writing – review & editing, Araujo JGL; Bento JCCV; Azevedo AM: methodology, formal analysis, Anjos ASD; Araújo AMM: methodology, formal analysis, Souto CRO; Menezes FG: writing – review & editing, data curation, validation, Silva DR; Gondim AD: conceptualization, supervision, Cavalcanti LN: conceptualization, data curation, validation, review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by Programa de Formação de Recursos Humanos of ANP (PRH 37/ANP). Authors thank Programa de Pós-graduação em Química (PPGQ-UFRN) e PRH37/ANP for M. S. B. da Silva fellowship. Authos also thank the Analytical Center at Institute of Chemistry (CA-IQ-UFRN) and Núcleo de Processamento Primário e Reuso de Água Produzida e Resíduos of UFRN (Nupprar/UFRN) for providing GC/MS, GC/FID and NMR analysis.Notes and references
- International Energy Agency, Annual Review, 2020 Search PubMed.
- World Health Organization, COP24 Special Report, 2018 Search PubMed.
- J. Yang, Z. Xin, Q. He, K. Corscadden and H. Niu, An overview on performance characteristics of bio-jet fuels, Fuel, 2019, 237, 916 CrossRef CAS.
- G. Cruz, A. V. S. Silva, J. B. S. Silva, R. N. Caldeiras and M. E. P. Souza, Biofuels from oilseed fruits using different thermochemical processes: opportunities and challenges, Biofuels, Bioprod. Biorefin., 2020, 14, 696 CrossRef CAS.
- S. L. Douvartzides, N. D. Charisiou, K. N. Papageridis and M. A. Goula, Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines, Energies, 2019, 12, 809 CrossRef CAS.
- R. Loe, E. Santillan-Jimenez and M. Crocker Upgrading of lipids to fuel-like hydrocarbons and terminal olefins via decarbonylation/decarboxylation, in Chemical Catalysts for Biomass Upgrading, Wiley-VCH, 2020, p. 497 Search PubMed.
- E. Santillan-Jimenez and M. Crocker, Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation, J. Chem. Technol. Biotechnol., 2012, 87, 1041 CrossRef CAS.
- B. P. Pattanaik and R. D. Misra, Effect of reaction pathway and operating parameters on the deoxygenation of vegetable oils to produce diesel range hydrocarbon fuels: A review, Renewable Sustainable Energy Rev., 2017, 73, 545 CrossRef CAS.
- V. Rathore, B. L. Newalkar and R. P. Badoni, Processing of vegetable oil for biofuel production through conventional and non-conventional routes, Energy Sustainable Dev., 2016, 31, 24 CrossRef CAS.
- T. P. Oliveira, M. F. V. Santos, A. C. M. Batista, A. M. M. Araújo, M. M. Conceição, V. J. Fernandes Jr and A. D. Gondim, CaO–TiO2 bimetallic mixed oxide applied to the production of biodiesel from cotton oil (Gossypium hisutum): monitoring of the procedure by TGA, J. Therm. Anal. Calorim., 2021, 146, 2667 CrossRef CAS.
- K. S. Castro, L. F. M. Costa, V. J. Fernandes Jr, R. O. Lima, A. M. M. Araújo, M. C. Sant'Anna, N. A. Santos and A. D. Gondim, Catalytic pyrolysis (Ni/Al-MCM-41) of palm (Elaeisguineensis) oil to obtain renewable hydrocarbons, RSC Adv., 2021, 11, 555 RSC.
- S. Karatzos, S. Dyk, J. D. McMillan and J. Saddler, Drop-in biofuel production via conventional (lipid/fatty acid) and advanced (biomass) routes. Part I, Biofuels, Bioprod. Biorefin., 2017, 11, 344 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S. Chang, J. Haveren, K. P. Jong, J. H. Bitter and D. S. Es, Reaction Pathways for the Deoxygenation of Vegetable Oils and Related Model Compounds, ChemSusChem, 2013, 6, 1576 CrossRef CAS PubMed.
- Z. Huang, Z. Zhao, C. Zhang, J. Lu, H. Liu, N. Luo, J. Zhang and F. Wang, Enhanced photocatalytic alkane production from fatty acid decarboxylation via inhibition of radical oligomerization, Nat. Catal., 2020, 3, 170 CrossRef CAS.
- H. Wei, W. Liu, X. Chen, Q. Yang, J. Li and H. Chen, Renewable bio-jet fuel production for aviation: A review, Fuel, 2019, 254, 115599 CrossRef CAS.
- C. Gutiérrez-Antonio, F. I. Gómez-Castro, J. A. Lira-Flores and S. Hernández, A review on the production processes of renewable jet fuel, Renewable Sustainable Energy Rev., 2017, 79, 709 CrossRef.
- M. Ammen, M. T. Azizan, S. Yusup, A. Ramli and M. Yasir, Catalytic hydrodeoxygenation of triglycerides: An approach to clean diesel fuel production, Renewable Sustainable Energy Rev., 2017, 80, 1072 CrossRef.
- R. Chen and W. Wang, The production of renewable aviation fuel from waste cooking oil. Part I: Bio-alkane conversion through hydro-processing of oil, Renewable energy, 2019, 135, 819 CrossRef CAS.
- M. Zula, M. Grilc and B. Likozar, Hydrocracking, hydrogenation and hydro-deoxygenation of fatty acids, esters and glycerides: Mechanisms, kinetics and transport phenomena, Chem. Eng. J., 2022, 444, 136564 CrossRef CAS.
- A. Vonortas and N. Papayannakos, Comparative analysis of biodiesel versus green diesel, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 3 CAS.
- K. A. Rogers and Y. Zheng, Selective Deoxygenation of Biomass-Derived Bio-oils within Hydrogen-Modest Environments: A Review and New Insights, ChemSusChem, 2016, 9, 1750 CrossRef CAS.
- M. C. Vásquez, E. E. Silva and E. F. Castillo, Hydrotreatment of vegetable oils: A review of the technologies and its developments for jet biofuel production, Biomass Bioenergy, 2017, 105, 197 CrossRef.
- S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, Trasforming triglycerides and fatty acids into biofuels, ChemSusChem, 2009, 2, 1109 CrossRef CAS PubMed.
- M. Zhang, Y. Hu, H. Wang, H. Li, X. Han, Y. Zeng and C. C. Xu, A review of bio-oil upgrading by catalytic hydrotreatment: Advances, challenges, and prospects, Mol. Catal., 2021, 504, 111438 CrossRef CAS.
- Y. Sorunmu, P. Billen and S. Spatari, A review of thermochemical upgrading of pyrolysis bio-oil: Techno-economic analysis, life cycle assessment, and technology readiness, GCB Bioenergy, 2019, 12, 4 CrossRef.
- H. Chen, Y. Liu and X. Liao, Recent progress in radical decarboxylative functionalizations enabled by transition-metal (Ni, Cu, Fe, Co or Cr) catalysis, Synthesis, 2020, 52, 1 CrossRef.
- S. Parida, T. Mandal, S. Das, S. K. Hota, S. D. Sarkar and S. Muraka, Single electron transfer-induced redox processes involving N-(Acyloxy)phthalimides, ACS Catal., 2021, 11, 1640 CrossRef CAS.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Nickel-catalyzed Barton decarboxylation and Giese reactions: A practical take on classic transforms, Angew. Chem., Int. Ed., 2017, 56, 260 CrossRef CAS PubMed.
- A. Volkov, K. P. J. Gustafson, C. Tai, O. Verho, J. Bäckvall and H. Adolfsson, Mild deoxygenation of aromatic ketones and aldehydesover Pd/C using polymethylhydrosiloxane as the reducing agent, Angew. Chem., Int. Ed., 2015, 54, 5122 CrossRef CAS PubMed.
- H. Yue, L. Guo, S. Lee, X. Liu and M. Rueping, Selective reductive removal of ester and amide groups from arenes and heteroarenes through nickel-catalyzed C–O and C–N bond activation, Angew. Chem., Int. Ed., 2017, 56, 3972 CrossRef CAS PubMed.
- X. Li, R. Shang, M. Fu and Y. Fu, Conversion of biomass-derived fatty acids and derivatives into hydrocarbons using a metal-free hydrodeoxygenation process, Green Chem., 2015, 17, 2790 RSC.
- X. Lu, B. Xiao, L. Liu and Y. Fu, Formation of C(sp3)–C(sp3) Bonds through Nickel-Catalyzed Decarboxylative Olefin Hydroalkylation Reactions, Chem.–Eur. J., 2016, 22, 11161 CrossRef CAS PubMed.
- W. Jianxin and F. Mingchen, Visible light induced Barton decarboxylation free of radical initiators, J. Univ. Sci. Technol. China, 2020, 50, 1403 Search PubMed.
- N. J. Lawrence, M. D. Drew and S. Bushell, Polymethylhydrosiloxane: A versatile reducing agent for organic synthesis, J. Chem. Soc., Perkin Trans. 1, 1999, 23, 3381 RSC.
- B. Pachaly and J. Weis, The Direct Process to Methylchlorosilanes: Reflections on Chemistry and Process Technology, in Organosilicon Chemistry Set: From Molecules to Materials, ed. N. Auner and J. Weis, Wiley, 2005, p. 478 Search PubMed.
- Y. Sugami, E. Minami and S. Saka, Hydrocarbon production from coconut oil by hydrolysis coupled with hydrogenation and subsequent decarboxylation, Fuel, 2017, 197, 272 CrossRef CAS.
- P. H. M. Araújo, A. S. Maia, A. M. T. M. Cordeiro, A. D. Gondim and N. A. Santos, Catalytic deoxygenation of the oil and biodiesel of Licuri (Syagrus coronata) to obtain n-alkanes with chains in the range of biojet fuels, ACS Omega, 2019, 4, 15849 CrossRef PubMed.
- L. C. Bauer, L. S. Santos, K. A. Sampaio, S. P. B. Ferrão, R. C. I. Fontan, L. A. Minim, C. M. Veloso and R. C. F. Bonomo, Physicochemical and thermal characterization of babassu oils (Orbignya phalerata Mart.) obtained by different extraction methods, Food Res. Int., 2020, 137, 109474 CrossRef CAS PubMed.
- C. Liu, Z. Qin, C. Ji, X. Hong and M. Szostak, Highly-chemoselective step-down reduction of carboxylic acids to aromatic hydrocarbons: via palladium catalysis, Chem. Sci., 2019, 10, 5736 RSC.
- A. Cook, H. MacLean, P. Onge and S. G. Newman, Nickel-catalyzed reductive deoxygenation of diverse C–O Bond-bearing functional groups, ACS Catal., 2021, 11, 13337 CrossRef CAS.
- C. Einhorn, J. Einhorn and C. Marcadal-Abbadi, Mild and convenient one pot synthesis of N-hydroximides from N-unsubstituted imides, Synth. Commun., 2001, 31, 741 CrossRef CAS.
- T. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Quaternary Centers by Nickel-Catalyzed Cross-Coupling of Tertiary Carboxylic Acids and (Hetero)Aryl Zinc Reagents, Angew. Chem., Int. Ed., 2019, 58, 2454 CrossRef CAS PubMed.
- J. Wang, T. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids, Angew. Chem., Int. Ed., 2016, 55, 9676 CrossRef CAS PubMed.
- H. Chen, L. Hu, W. Ji, L. Yao and X. Liao, Nickel-Catalyzed Decarboxylative Alkylation of Aryl Iodides with Anhydrides, ACS Catal., 2018, 8, 10479 CrossRef CAS.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, Practical Ni-Catalyzed Aryl-Alkyl Cross-Coupling of Secondary Redox-Active Esters, J. Am. Chem. Soc., 2016, 138, 2174 CrossRef CAS PubMed.
- J. Wang, H. Lundberg, S. Asai and P. S. Baran, Kinetically guided radical-based synthesis of C(sp3)–C(sp3) linkages on DNA, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04057c |
| This journal is © The Royal Society of Chemistry 2022 |