
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRaman mapping of piezoelectric poly(L-lactic acid) films for force sensors†
Ivan S. Babichuk‡
 ab,
Chubin Lin‡a,
Yuhui Qiua,
Huiyu Zhua,
Terry Tao Ye*c,
Zhaoli Gao*de and
Jian Yang*a
ab,
Chubin Lin‡a,
Yuhui Qiua,
Huiyu Zhua,
Terry Tao Ye*c,
Zhaoli Gao*de and
Jian Yang*a
aFaculty of Intelligent Manufacturing, Wuyi University, 529020 Jiangmen, P.R. China
bV. Lashkaryov Institute of Semiconductor Physics, NAS of Ukraine, 03680 Kyiv, Ukraine
cDepartment of Electrical and Electronic Engineering and University Key Laboratory of Advanced Wireless Communications of Guangdong Province, Southern University of Science and Technology, 518055, Shenzhen, P.R. China
dBiomedical Engineering Department, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, P.R. China
eCUHK Shenzhen Research Institute, Nanshan, 518060, Shenzhen, P.R. China
First published on 28th September 2022
Abstract
Poly-L-lactic acid (PLLA) is a synthetic, biocompatible, biodegradable polymer with good piezoelectric properties. The prepared PLLA films were annealed in the oven at 140 °C for 0 h, 3 h, 12 h, and 24 h, respectively. The influences of temperature treatment time on the optoelectronic properties of the PLLA films and piezoelectric sensors based on them were investigated. The morphology and crystal structure of the PLLA films obtained under various post-processing conditions were examined by scanning electron microscopy, X-ray diffraction, Raman spectroscopy, and ATR-FTIR spectroscopy. The micromechanical equipment for tension–compression measurements was built in the laboratory for the tested piezoelectric sensors. The analysis of the structure shows that the increase in the crystallite size of the PLLA film influences the growth of the piezoelectric signal of the sensors based on them. The vibrational analysis of the PLLA films confirmed their crystal structure. The improvement in the structure and the stretching of the dipole C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for the film obtained after 3 h treatment increased the piezoelectric properties of the PLLA films. The analysis of Raman mapping added information that the area of the ordered phase of the PLLA films depends on the time of temperature treatment. The maximum value of the piezoelectric signal was 0.98 mV for sensors prepared on films annealed for 3 h at a load of 20 N. For films without temperature annealing at the same load, the maximum value was 0.45 mV. Thus, efficient converters of mechanical energy into electrical energy were obtained, which opens new innovative perspectives for the creation of flexible pressure sensors based on PLLA.
O for the film obtained after 3 h treatment increased the piezoelectric properties of the PLLA films. The analysis of Raman mapping added information that the area of the ordered phase of the PLLA films depends on the time of temperature treatment. The maximum value of the piezoelectric signal was 0.98 mV for sensors prepared on films annealed for 3 h at a load of 20 N. For films without temperature annealing at the same load, the maximum value was 0.45 mV. Thus, efficient converters of mechanical energy into electrical energy were obtained, which opens new innovative perspectives for the creation of flexible pressure sensors based on PLLA.
Introduction
Flexible electronics are one of the important directions of the new generation of information technology. It has big application potential in the fields of tactile electronics, virtual reality, human–machine interaction, and health monitoring.1,2 In the last decade, interest in studying piezoelectric and triboelectric sensors with zero energy consumption has been constantly growing.3 Currently, active research is ongoing on materials and their properties to obtain sensors and actuators based on them.4,5 The development of new or the improvement of existing biocompatible piezoelectric materials will give the chance to apply them in the creation of small-scale devices for biomedical applications.6 Poly(lactide) (PLA) is one such promising green material because its raw material, lactic acid, can be efficiently produced by fermentation from renewable resources. In addition, owing to an asymmetric carbon atom, lactic acid has two optically active forms (called L- and D-lactic acid).7,8 This is possible due to the chiral nature of lactic acid. Poly-L-lactide (PLLA) is the product resulting from the polymerization of L, also known as L-lactide. The chemical properties of L- and D-lactide are the same while their steric structures are different,9,10 therefore, only the PLLA properties will be considered below.To date, three different crystalline modifications (α, β, and γ) of PLLA have been identified on changing the preparation conditions.11 The most common is the α-phase, which exhibits a 103 helical chain conformation and a pseudo-orthorhombic unit cell. It can be obtained easily via the melting or solution-phase crystallization methods. The β-phase, which has a left-handed 31 helical conformation and an orthorhombic or triclinic unit cell, can be produced by controlling the post-process drawing and annealing conditions.12 The γ-phase has recently been obtained through the epitaxial growth of PLLA on a hexamethylbenzene substrate.13 Although considerable progress has been made in elucidating the accurate crystalline structure of these crystal modifications of PLLA, some questions still remain unresolved. In particular, the crystalline structures of the most common α and β-phases are yet to be definitively assigned. The α form can coexist as the “pure” and the “distorted” 103 helix chain conformation owing to the interchain interactions between the CH3 groups.11 The crystal structure of the β form first was attributed to an orthorhombic unit cell with a = 1.031 nm, b = 1.821 nm, c = 0.900 nm, and a chain conformation with left-handed 31 helices.14 The other authors proposed an orthorhombic unit cell with two parallel chains.15 Further research has shown that the β phase is indeed a structure of three 31 helices in a trigonal unit cell of parameters a = b = 10.52 Å and c = 8.8 Å capable of accommodating the random up-down orientation of the neighboring chains.16,17 Currently, PLLA can be the pseudoorthorhombic α-structure (a = 10.6, b = 6.1, and c = 28.8 Å) that contains two chains in the unit cell, and for β-structure, an orthorhombic unit cell is proposed (a = 10.31, b = 18.21, and c = 9.00 Å) containing six chains.14,17 Obviously, further effort is required to address unresolved issues on the crystal structure of PLLA.
Other authors11,18 have showed that the crystallization temperature and molecular weight have a great effect on the IR spectral profiles of the PLLA samples annealed at different temperatures ranging from 80 to 140 °C. When PLLA crystallizes at a temperature over 120 °C, the band-splitting phenomena of CH3 and CO groups can be clearly observed, while for the samples crystallized at a temperature below 120 °C, relatively simple spectral profiles are obtained. The Raman and IR spectra of PLLA in the amorphous and semicrystalline states are shown in previous works.19,20 In these works, it was found that the splitting of the ν(CO) stretching and the δ(CCO) bend are characteristic of the crystalline phase and the presence of the two lines at 923 and 520 cm−1 indicates a 103 helical conformation. Another study20 showed a vibrational analysis, which provided a new understanding regarding the relative probability of a 103 or 31 helix for PLA.
PLLA has still some drawbacks about the physical properties compared with the other kinds of synthetic polymers such as polyethylene and polypropylene. For example, the Young's modulus of the bulk PLLA sample is 8 GPa and the tensioned film is easily broken at about 0.5 GPa.21 However, PLLA is one of the biodegradable and biocompatible synthetic polymers with piezoelectric properties that are safely degraded along the same metabolic pathway as lactic acid.22,23
The carbonyl groups present in the PLA structure induce polarity and it shows a d14 value of 10 pC N−1 without an additional poling step in its crystalline structure.24,25 In one study,26 it was reported for the first time that a PLLA polymer film can be used as a cantilever device for energy harvesting applications. In addition, even after being thermally treated at 140 °C for 24 h, this material was found to still show a similar piezoelectric coefficient of 9.57 pC N−1. Moreover, a 14.45 μW power output was obtained from the PLLA cantilever device for energy harvesting applications. Another practical application of PLLA was demonstrated27 where the authors reported the strong antibacterial effect of piezoelectric fabrics. Also, these authors have simulated the generating voltage of the yarn under tensile stress, considering filament slippage among adjacent filaments.28
The CO dipoles of α-PLLA are generally oriented in all the directions (360°) along the main chain of the helical structure, resulting in zero dipole moment. α-PLLA can be transformed to β-PLLA by stretching or drawing processes. The CO dipoles of β-PLLA are aligned along the backbone chain; thus, the total dipole moment is zero due to its helical structure. The shear piezoelectricity of PLLA or its piezoelectric-like behavior is of considerable interest to researchers.29 Shear piezoelectricity (d14) is induced along the primary axis of PLLA upon the application of shear stress as the CO dipoles in the helical backbone rotate, causing electrical polarization. In contrast, the β-crystalline phase of polyvinylidene fluoride (PVDF) can have a large net polarity because it has a linear molecular structure. PLLA-based piezoelectric materials generally have lower piezo-responses than PVDF or ceramic materials.30 This can be increased by adjusting the postprocessing treatment. Some previous studies31–33 have investigated the effects of drawing stress, temperature, and draw ratio on the crystalline phase transition in PLLA films containing highly oriented α-crystals. The report34 showed the analysis of the β form and proposes the microscopic mechanism of the stress-induced crystal phase transition between the α and β forms by focusing on the change of the upward and downward chain packing mode. Also, the piezoelectricity of PLLA can be enhanced by the application of the electric field in electrospinning processes.35 Thus, the question of how post-processing influences the optical and piezoelectric properties of this polymer is still open.
In this work, we investigated the effects of temperature treatment time on the optoelectronic properties of PLLA films and piezoelectric sensors based on them for biomedical applications. The morphology and crystal structure of the PLLA films obtained under the various post-processing conditions were examined by scanning electron microscopy (SEM), X-ray diffraction (XRD), Raman, and ATR-FTIR spectroscopy. The piezoelectric characteristic of the PLLA sensors was determined by monitoring the output voltage and current with a force-tapping method.
Experimental
Preparation of PLLA samples
The ultrasonic bath to clean the measuring cylinder, glass pipette, slides, and other necessary vessels was utilized. After that, these dishes were washed twice in deionized water and placed in an oven (Binder ED 56, Germany) for drying at 70 °C for a few minutes.PLLA powder (MW = 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, polydispersity index ≤ 1, Sigma-Aldrich Corp., USA) was dissolved in dichloromethane solution and stirred at room temperature for 4 h to prepare 30 mg mL−1 PLLA solution. Then, a glass pipette was used to suck an equal amount of the PLLA solution, which was uniformly cast on four groups of glass slides (the size was 25 mm × 25 mm), and dried at room temperature for 24 h to obtain PLLA films. After that, the PLLA films was annealed in an oven (Binder ED 56, Germany) at 140 °C for 0 h, 3 h, 12 h, and 24 h. Finally, the films were removed from the glass slides using tweezers and placed in an Electronic Humidity Control Cabinet (Sirui HC-70, USA) for storage. The average thickness of the films was 120 ± 5 μm. A digital thickness gauge was used for this task (Shenzhen Yuan Hengtong Technology Co., Ltd, Shenzhen, China).
000, polydispersity index ≤ 1, Sigma-Aldrich Corp., USA) was dissolved in dichloromethane solution and stirred at room temperature for 4 h to prepare 30 mg mL−1 PLLA solution. Then, a glass pipette was used to suck an equal amount of the PLLA solution, which was uniformly cast on four groups of glass slides (the size was 25 mm × 25 mm), and dried at room temperature for 24 h to obtain PLLA films. After that, the PLLA films was annealed in an oven (Binder ED 56, Germany) at 140 °C for 0 h, 3 h, 12 h, and 24 h. Finally, the films were removed from the glass slides using tweezers and placed in an Electronic Humidity Control Cabinet (Sirui HC-70, USA) for storage. The average thickness of the films was 120 ± 5 μm. A digital thickness gauge was used for this task (Shenzhen Yuan Hengtong Technology Co., Ltd, Shenzhen, China).
Preparation of the sensor based on PLLA films
To obtain a patterned gold electrode on the surface of PLLA film, an ion sputtering instrument JYSC-2000 (Guangzhou Jingying Scientific Instrument Co., Ltd, Guangzhou, China) was used. A prepared mask plate was covered on the PLLA and gold was sputtered in this equipment during 200 s. The same was done on the other side. Then, conductive zebra paper was used to lead out the electrode and bridge it to the Flexible Printed Circuit line. Finally, the PLLA film was encapsulated with polyimide (PI) tape and put on polydimethylsiloxane (PDMS) film.Characterization
Structural characterization was carried out at room temperature by XRD and μ-Raman spectroscopy. XRD was carried out exploiting a PANalytical X'pert Pro MPD diffractometer with Cu-Kα-radiation (λ = 1.54056 Å).36A LabRAM HR Evolution (Horiba) and NOVA (ideaoptics/ideaoptics.com/, Shanghai, China) spectrometers were employed for Raman spectroscopy in the backscattering configuration with 785 nm and 532 nm excitation by Cobolt 08 NLD (Narrow Linewidth Diode) laser and Nd:YAG, respectively. Both laser modules were Diode-Pumped Solid-State (DPSS). A Gora-light system was used for Raman mapping with excitation at 532 nm. This confocal Raman imaging system included a highly sensitive spectrometer (NOVA, China), a high precision moving stage (Märzhäuser scanning stage with the TANGO controller, Germany), and a confocal microscope (Olympus BX-43, Japan). The scanned areas were 100 × 100 μm2 and 50 × 50 μm2 (lateral resolution of 5 and 2 μm, respectively). The laser radiation was focused on a sample with 50× and 20× objectives of a confocal Olympus microscope to a spot of ∼1 μm in diameter. The power density of laser irradiation did not exceed ∼106 W cm−2 to avoid changes in the film structure during the measurements. The spectra were registered at several points to prove film homogeneity.
The ATR-FTIR spectra of the PLLA films were recorded on a Nicolet-IS50 FT-IR spectrometer (ThermoFisher Scientific Inc. USA) within 4000–400 cm−1 at a resolution of 2 cm−1. Attenuated-total-reflectance (ATR) is a sampling mode that enhances the Fourier transform infrared (FTIR) spectroscopy signal obtained from the sample surfaces, increasing the sensitivity and allowing efficient measurements with minimal sample preparation. Similar to standard FTIR measurements, ATR-FTIR was used for the chemical qualification of samples.
The sample surface morphology was analyzed using a Zeiss Sigma 500 SEM (ZEISS International, Germany). SEM measurements were carried out at 10 kV operating voltage.
Piezoelectric test of the sensors
The PLLA sensor was fixed tightly on the compression disk of the micromechanical testing equipment, which was built in the laboratory (Fig. 1A). The indenter had a 1 mm diameter and was used to compress the PLLA sensor. The diagrams of the unit cell and piezoelectric effect of the flexible sensor based on PLLA are shown in Fig. 1B and C, respectively. The piezoelectric properties of the PLLA films at different loads (2, 5, 10, 15, 20 N) and impact of velocity (10, 20, 30, 40 mm s−1) were carried out with this mechanical test machine. The piezoelectric signal of the representative sensor at 10 N load is shown in Fig. 1D.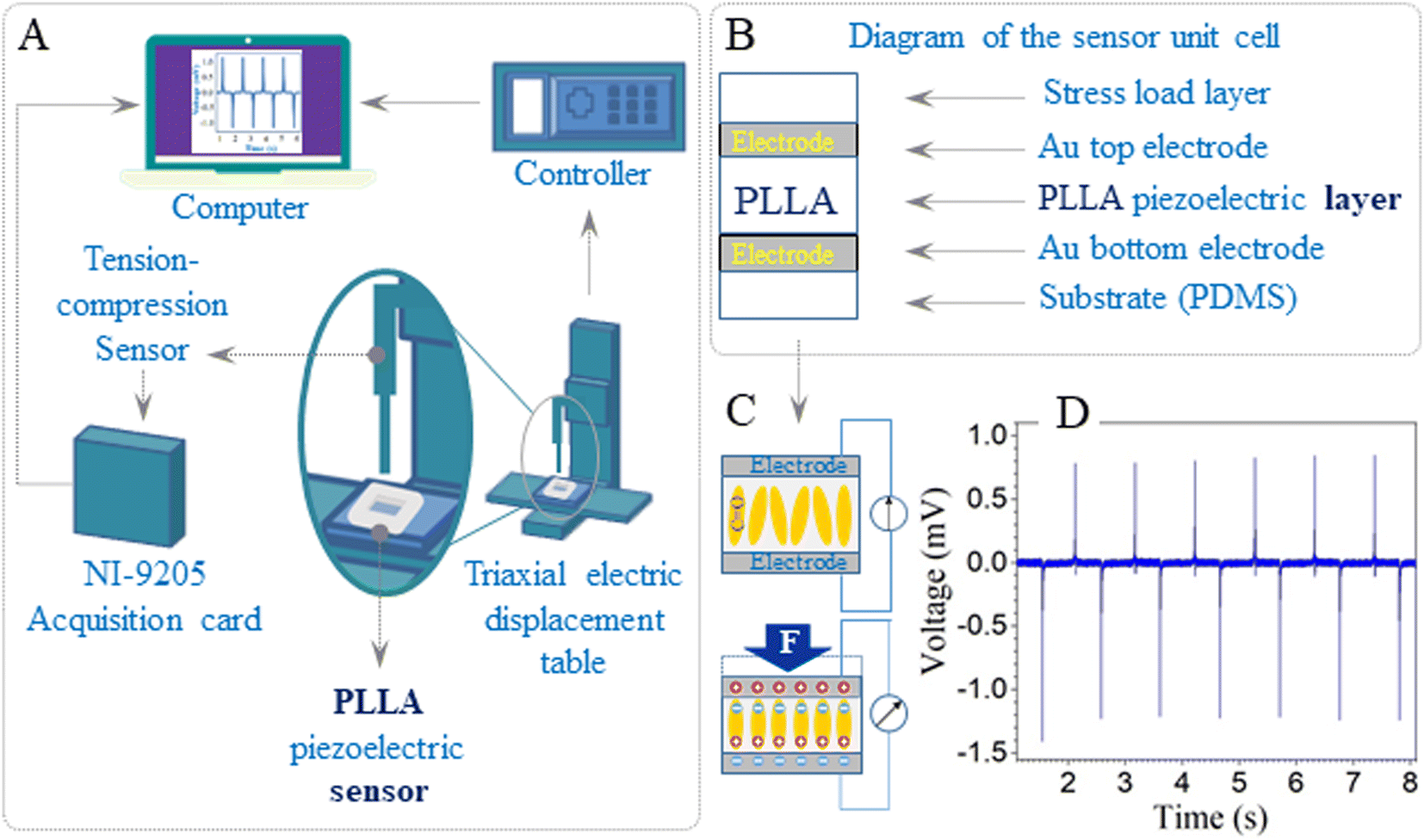 | ||
| Fig. 1 (A) The scheme of micromechanical testing equipment for tension–compression measurements. (B) Diagram of the unit cell of the PLLA sensor. (C) Diagram of the piezoelectric effect. (D) The piezoelectric signal of the representative sensor was based on the PLLA film. | ||
Results and discussion
XRD: crystal structure characterization
The crystal structure of the PLLA films was analyzed using XRD (Fig. 2). The dominant diffraction peaks positioned at 2θ ∼ 16.65° ± 0.05° (200/110 plane) and ∼19.05° ± 0.05° (203/111 plane) indicate the orientation of the PLLA structure.18 Other small diffraction peaks located at 2θ ∼ 12.4° (004/103), 14.7° (010), 20.7° (204), 22.3° (015), 23.9° (016), and 24.95° ± 0.05° (206), labeled in Fig. 2, also correspond to the PLLA structure that belongs to an orthorhombic system, according to the ICDD 00-064-1624 database. Based on the experimental data and appropriate structural model, the calculations of reflection relative intensities were performed, and the parameters of the elementary cell were refined using the Rietveld method37 and FullProf Suite.38 The lattice parameters a, b, and c refined for the studied PLLA films have been calculated (Table 1). It is known that PLLA has three kinds of crystal forms (i.e., α, β, and γ forms). The most common polymorph, α form, has a 103 helical chain conformation and is obtained by the crystallization from the melt state, the same as our experimental conditions. The obtained parameters are in agreement with the literature for α-form crystals structure.14,21,39 The analysis of data in Table 1 also allows for the estimation of treatment time effects on the unit cell structure. It is seen that during treatment (3 and 12 h), the films undergo a slight reduction in the lattice parameters a, b, and c relative to the initial PLLA film. At the same time, the shift in the diffraction peak 200/110 to higher angles, the decrease in its FWHM, and the increase in intensity give reasons to claim an increase in the quality of the crystal structure of the films that were annealed for 3 and 12 h. Subsequent annealing during 24 h showed a slight increase in the unit cell parameters a and b but not c, relative to the parameters for the film annealed during 12 h. The decrease in the intensity and position of the peak 200/110 relative to the film with annealing of 12 h was also observed. It can be noted that annealing improves the structure of the films but only up to a certain time.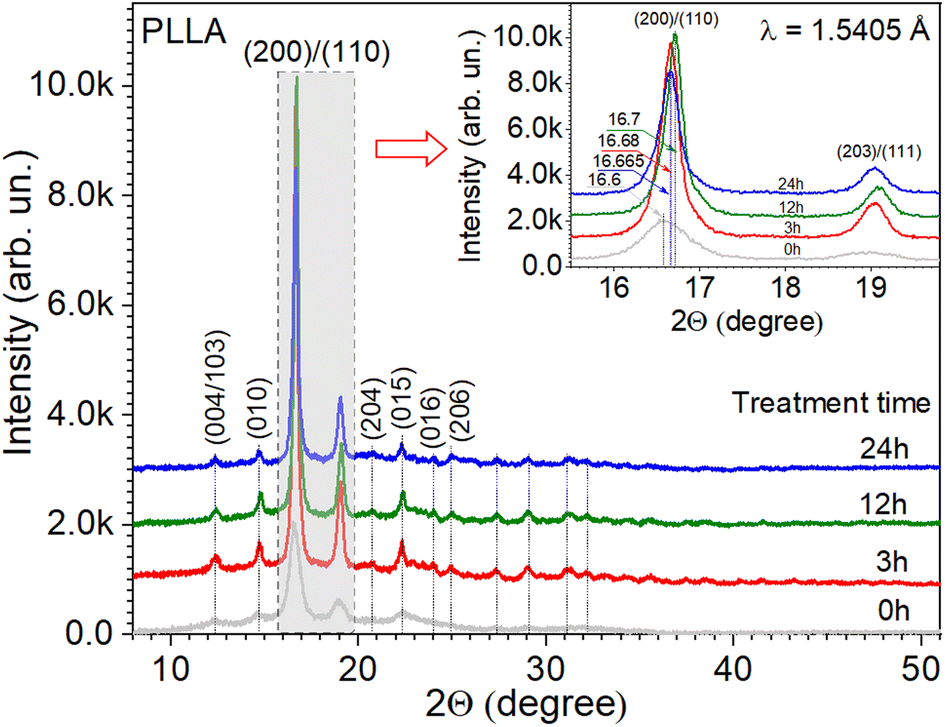 | ||
| Fig. 2 X-ray diffraction patterns of PLLA at varied treatment times. (Inset) The detailed range of dominant diffraction peaks. | ||
| Sample (treatment time) | a, Å | b, Å | c, Å | Peak position (200) & (110) (°) | FWHM (°) | Height (arb. un.) | Crystallite size, D (nm) | Degree of the crystallite orientation, χ (%) |
|---|---|---|---|---|---|---|---|---|
| 0 h | 10.68 ± 0.01 | 6.18 ± 0.01 | 28.89 ± 0.02 | 16.60 ± 0.05 | 0.67 ± 0.02 | 1886 ± 2 | 12.3 ± 0.1 | 0.996 ± 0.001 |
| 3 h | 10.64 ± 0.02 | 6.14 ± 0.02 | 28.84 ± 0.01 | 16.68 ± 0.03 | 0.26 ± 0.01 | 8435 ± 5 | 31.4 ± 0.2 | 0.998 ± 0.001 |
| 12 h | 10.61 ± 0.01 | 6.13 ± 0.01 | 28.81 ± 0.01 | 16.70 ± 0.05 | 0.22 ± 0.01 | 8054 ± 5 | 35.9 ± 0.2 | 0.999 ± 0.001 |
| 24 h | 10.64 ± 0.02 | 6.15 ± 0.01 | 28.76 ± 0.03 | 16.66 ± 0.01 | 0.22 ± 0.01 | 5514 ± 3 | 30.6 ± 0.2 | 0.998 ± 0.001 |
The next important parameters for the PLLA films are crystallite size and orientation, which influence their piezoelectric properties. The crystallite size (also termed domain size) Dhkl and the observed profiles for the (hkl) planes in PLLA film were calculated through the application of Scherrer's equation.
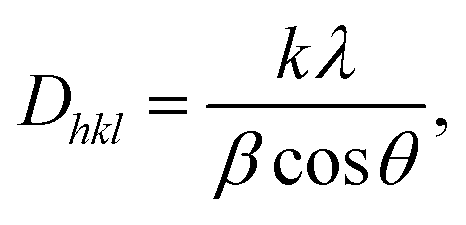 | (1) |
The degree of the crystallite orientation χ was defined by eqn (2).
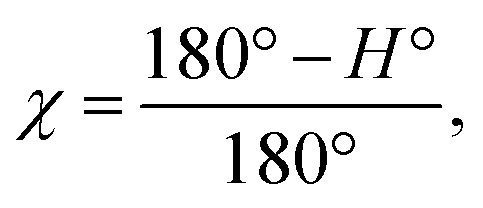 | (2) |
The analysis of the diffraction patterns in Fig. 2 and the calculated data in Table 1 give grounds to summarize that the temperature treatment time has a significant effect on the crystal structure of the PLLA films. It can be noted that at an annealing time of 12 h in this series, there is a maximum intensity of the diffraction peaks at a relatively minimum FWHM. Also, the maximum size of the crystallite was found at the same treatment time. To samples with annealing times of 3 and 24 h at temperature 140 °C, the similar results are observed, which gives grounds to argue about the possibility of using a shorter treatment time.
Surface morphology of the PLLA films
The morphology of the PLLA films at varied treatment times is presented in Fig. 3, and as can be seen, is strongly dependent on the annealing time. It should be noted that all samples had an opaque milky color, which is because the solid (surface morphology)/air interface reflects in the visible radiation and even infrared in the range of 2000–4000 cm−1, causing a slope in the spectrum baseline.43 The infrared properties of the PLLA films will be described in more detail below. Another important factor that affects color is cavities. These cavities are almost missing for the pristine sample (Fig. 3A) and were well manifested in the sample obtained at annealing times of 3 and 12 h (Fig. 3B and C, respectively). A feature of the pristine sample has areas with a spiral shape and diameter of about 45 μm. It has been suggested that these spiral areas became centers of spherical granule formation during annealing. These granules with an average diameter of 30–50 μm are formed after 3 and 12 h of temperature treatment. After 12 h of annealing, their diameter decreases by 10 μm. The further increase in the annealing time led to the fusion of these granules and the loss of a relatively regular round shape (Fig. 3D). | ||
| Fig. 3 SEM surface images of PLLA at varied temperature treatment times: (A) pristine 0 h; (B) 3 h; (C) 12 h; and (D) 24 h. | ||
Annealing was demonstrated to be effective for the improvement of the surface morphology due to the increased crystallinity and more perfect crystals. Under these conditions, after 3 and 12 h treatment at 140 °C, spherical granules with a diameter between 30 and 50 μm were formed. These samples showed good transmittance and flexibility, which facilitated the preparation of devices based on them.
Vibrational analysis of PLLA films
A molecule composed of n-atoms has 3n degrees of freedom, six of which are translations and rotations of the molecule itself. This leaves 3n − 6 degrees of vibrational freedom (3n − 5 if the molecule is linear).Vibrational modes are often given descriptive names, such as stretching, bending, scissoring, rocking, and twisting. According to group theory, there are 27 fundamental vibrations for PLA. The active modes of PLA are classified as 27A + 27B + 27E1 + 27E2, with the A and E1 species being IR and Raman active, whereas the B and E2 species are only Raman active.19 Since 103 and 31 helices are also similar in structure, for simplicity, our analysis used a 103 helix. In addition, the 31 helix has 9 atoms for each chemical repeat unit. Three such units with a phase angle difference of 120° form a translational repeat unit. The optical activity of PLA has 25A modes and 26 degenerate E modes.20
To clarify the structural differences in the PLLA samples annealed at different times, their behaviors are further investigated by Raman and ATR-FTIR spectroscopy. These two techniques are non-destructive and complementary. In addition, a cross-investigation was performed on two different Raman spectrometers with different laser wavelengths to better reconcile the obtained data. It is known that in this way the peak positions remain unchanged. Only the intensity of the individual peaks can change due to resonant excitation conditions. As can be seen in Fig. 4A and B, our result coincides with our expectations and changes in the intensity of the peaks shown in Fig. 4C and D.
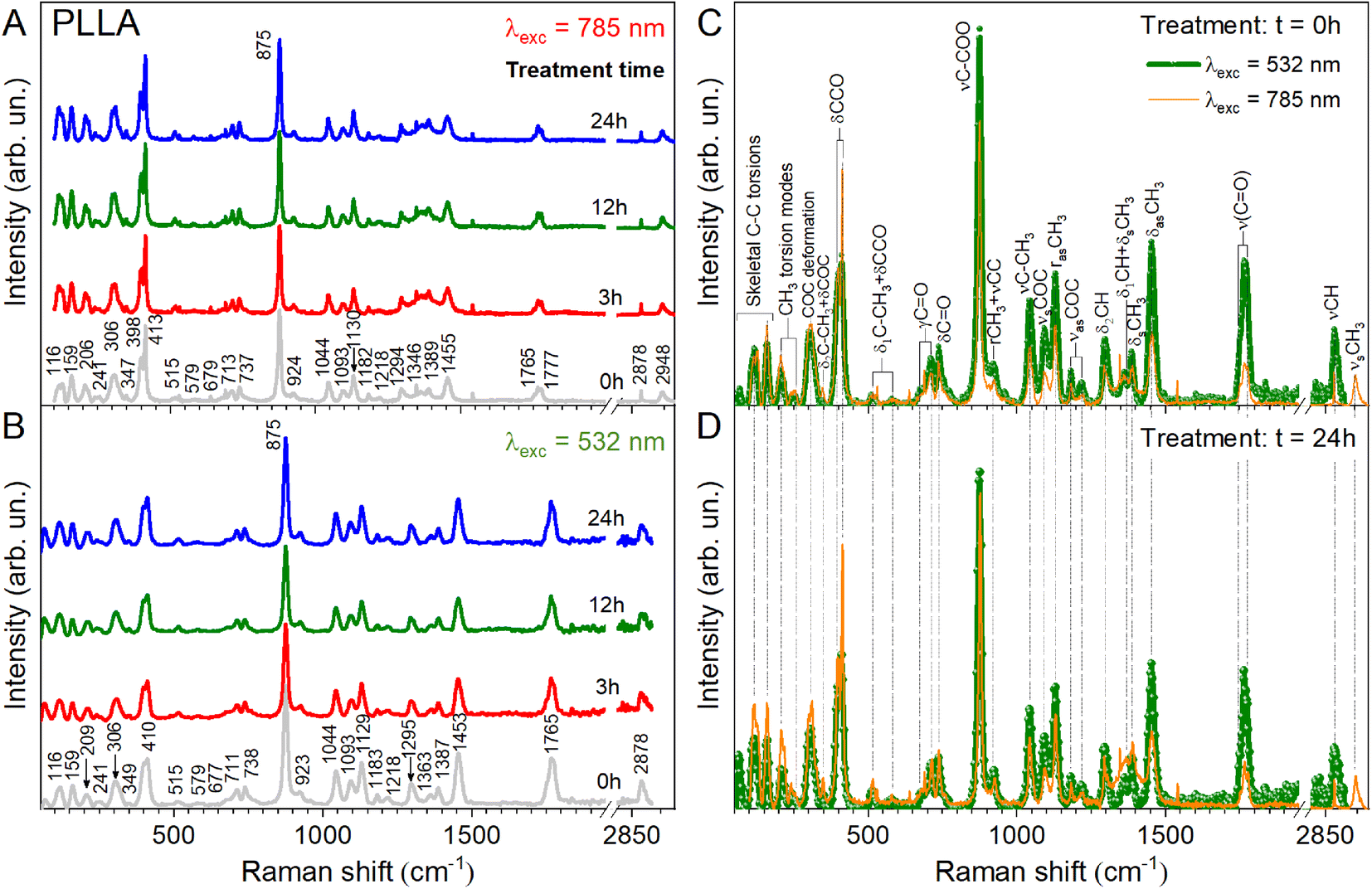 | ||
| Fig. 4 Raman spectra of PLLA films at varied temperature treatment times with different laser radiation (A) λ = 785 nm, (B) λ = 532 nm. Comparison of the spectra: (C) the pristine film at different laser excitation and (D) the film after temperature treatment for 24 h. | ||
The Raman and ATR-FTIR spectra for PLLA at varied temperature treatment times are shown in Fig. 4 and 5. Molecular vibrational assignments are given in Table S1 (ESI).† All observed experimentally significant shifts correlate with known vibrational assignments.19
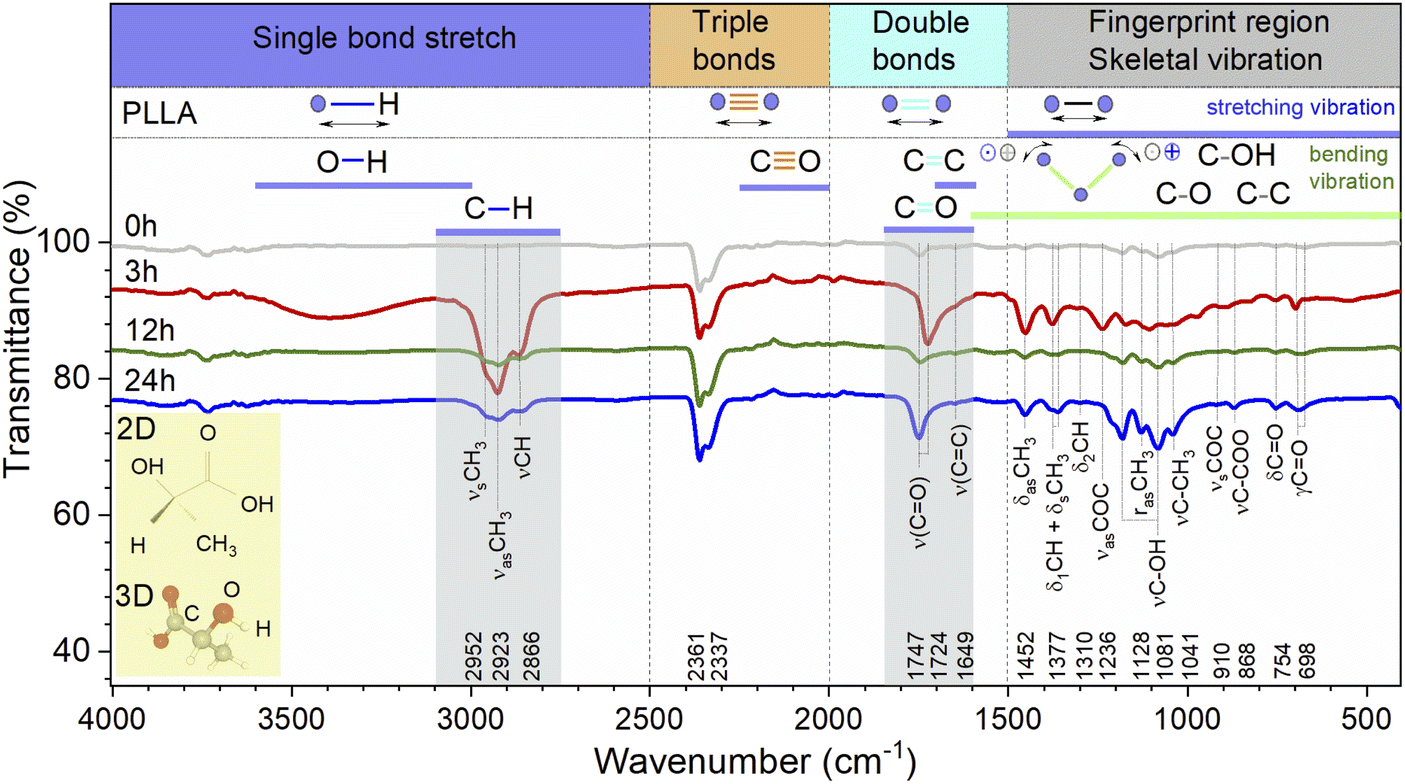 | ||
| Fig. 5 ATR-FTIR spectra of PLLA films at varied temperature treatment times. The blue-coloured sections refer to stretching vibrations and the green-coloured band encompasses bending vibrations. (Inset) 2D and 3D diagrams of the PLLA molecule. | ||
The general regions of the ATR-FTIR in which various kinds of vibrational bands are observed are outlined in Fig. 5. Note that the blue-colored sections refer to stretching vibrations and the green-colored band below encompasses bending vibrations. Transmittance bands in the 4000 to 1450 cm−1 region are usually due to stretching vibrations of diatomic units, and this is sometimes called the group frequency region. The complexity of the infrared spectra in the 1450 to 600 cm−1 region makes it difficult to assign all the absorption bands, and because of the unique patterns found there, it is often called the fingerprint region.
In the case of the PLLA film annealed for 3 h, the evidence for a C–H⋯OC hydrogen bond is observed above 3000 cm−1 (3100–3600 cm−1 in the ATR-FTIR spectrum). Under this experimental condition, the distance between the H atom and the O atom was probably sufficient in the 103 helical structure of the α crystal of PLLA for the C–H⋯OC hydrogen bond to be generated. The medium Raman (Fig. 4) and strong ATR-FTIR (Fig. 5) bands observed at ∼2950 ± 2, 2923 ± 1, and 2878 (2864–2868 cm−1 IR) cm−1 were assigned to νsCH3, νasCH3, and νCH modes, respectively, and were related to the A and E1 modes. The strong intensity band at 2923 ± 1 cm−1 is due to the asymmetric vibration of the methyl group. A similar increase in the intensity for the band at 2878 cm−1 was observed for the film treatment time during 3 h (Fig. 4B). Also, a noticeable feature in Raman spectra was a variation in the intensities of the PLLA characteristic peaks. For example, the intensity of other peaks (Fig. 4B) decreases at treatment times from 3 to 12 h and increases from 12 to 24 h. This behavior of the spectra was associated with increased scattering due to the presence of pores between the spherical granules in the films mentioned above and shown in Fig. 3. At the same time, for example, the FWHM for the 875 cm−1 band differed by only 1 cm−1 from the pristine sample, and was 16 and 15 cm−1 for the pristine and all annealed samples, respectively.
Herein, we need to understand how annealing time influences the CO band since the piezoelectric properties are dependent on this dipole in the PLLA film. It is known that at an annealing temperature of 140 °C, the spectral band-splitting phenomena of the CO group can be clearly observed. The CO stretching vibration modes are very sensitive to the structural changes of PLLA. In our work, the Raman spectra of all the films showed splitting of bands at 1777, 1765, and 1749 cm−1, which were assigned to the A, B, E1, and E2 modes of α-form helical PLLA.18,19 The spectral changes indicate that there are some changes in the crystal structure of PLLA in the treatment processes. The bands associated with CO bond stretching are usually very strong in IR spectroscopy because a large change in the dipole takes place in that mode. The CO bands have one of the strongest intensities in the whole FTIR spectral region of PLLA. There is particular interest toward this band because the CO dipoles in the helical backbone rotate upon the application of stress and cause the electrical polarization of PLLA. It should be noted that the FTIR spectra shows significant shift of this band in the low-frequency region from 1747 to 1724 cm−1 and an increase in the intensity for the film annealed for 3 h. In general, the intensity increased after treatment for all the treatment films. This allows to claim the stretching of the dipole CO and an improvement in the structure of the films, which will probably increase the influences on the piezoelectric properties of PLLA.
The weak shoulder (Fig. 5) at about 1649 cm−1 corresponds to the stretching vibrations ν(CC). The asymmetric deformation (δasCH3) appeared near 1453 ± 2 cm−1 as an intense band in the Raman and FTIR spectra. No significant shift was found in the spectrum of the pristine film with respect to the annealed samples. The δCH3 symmetric bending mode was split in the Raman spectrum at 1389 and 1346 cm−1. The δ1CH band component coupled with the δCH3 mode was found at 1378 ± 1 and 1361 cm−1 in the FTIR spectra and at 1365 cm−1 in the Raman spectra. The weak intensity bands at 1310 and 1299 cm−1 (FTIR) were mainly assigned to the δ2CH modes, appearing as triplet medium intensity bands at 1315, 1303, and 1297 cm−1 for pristine film and 1312, 1303, and 1295 cm−1 for treatment films in the Raman spectra. The bands at 1218, 1182 cm−1 (Raman), and 1236 cm−1 (FTIR) were assigned to the C–O–C stretching modes of ester groups as asymmetric bands. In the FTIR spectra for pristine and 12 and 24 h treated films, the ν(C–OH) modes were found at 1205 and 1081 cm−1. Also, one weak band at 1171 cm−1 in the FTIR spectrum for film annealed for 3 h was found and assigned to the δCH3 deformation band. Two bands appear near 1130 ± 1 and 1128 cm−1 (1120 cm−1 for film treatment during 3 h tensile stress) in the Raman and FTIR spectra, respectively. They are found in lactic acid at the same wavenumbers and are assigned to rasCH3 asymmetric rocking.19
The band at about 1093 cm−1 (Raman) is ascribed to the symmetric vibrations of the νsCOC mode. At the same time, the band at about 1059 cm−1 (FTIR-for film annealed for 3 h) was assigned to the νCOC modes. The bands at about 1044 (Raman) and 1041 cm−1 (1036 cm−1, film annealed for 3 h-FTIR) were assigned to the νC–CH3 stretching mode.
The weak absorption bands at 974, 891, and 827 cm−1 (FTIR) were assigned to flat deformations of C–H of in-plane –CC–H for film treated for 3 h. The band at 924 cm−1 was assumed to be one of the characteristic vibrations of the helical backbone mixed with the CH3 rocking modes. The presence of the two lines at 924 and 515 cm−1 indicates a 103 helical conformation. One of the strong intensity bands in Raman spectra at 875 cm−1 was assigned to the νC–COO stretching mode of the repeat unit, as shown in the course of L-lactic acid polycondensation.19
The bands observed at 737 cm−1 (Raman) and at 754 (FTIR) were mainly due to the δC–O in-plane bending. The 713 and 679 cm−1 Raman bands and the 715 and 698 cm−1 absorption bands were assigned to γC–O out-of-plane bending. The modes that appeared near 579 and 515 cm−1 (Raman) were mainly assigned to the δ1C–CH3 + δCCO band. Generally, this band was not observed in the spectrum of amorphous PLLA.19 The 413–398 cm−1 Raman doublet of strong intensity was associated with δCCO deformation, observed in all films as a strong and asymmetric band. The δ2C–CH3 and δsC–CH3 bands located at 347 and 306–305 cm−1 are weak and medium-intensity Raman bands, respectively. The weaker and medium bands at 241 and 208 cm−1 correspond to CH3 torsion modes, and the 159 and 116 cm−1 bands correspond to skeletal C–C torsions.
Thus, after the vibrational analysis of the PLLA films, their crystal structure and some features of the spectra were confirmed. In particular, the O–H stretching vibration band was relatively increased for the film annealed for 3 h when compared to pristine PLLA. The band-splitting phenomena of CH3 in the spectra was clearly observed. Also, it was found that the splitting of the ν(CO) stretching and the δ(CCO) band is characteristic of all the films and corresponds to the crystalline PLLA phase. The improvement in the structure and the stretching of the dipole CO for the film obtained after 3 h treatment can increase the piezoelectric properties of PLLA films.
Further investigations were performed by analyzing the PLLA samples by Raman mapping technique. The purpose of this analysis was to determine if there were microstructural differences in the films due to the variation in their annealing times on large areas. Fig. 6 shows the Raman mapping of a representative PLLA film with characteristic bands described above. Mapping was performed on an area of 50 × 50 μm2 with a lateral resolution of 2 μm. High-quality mapping was provided by a moving stage (Märzhäuser scanning stage) and a confocal microscope (Olympus) with a 20× lens. A laser with a wavelength of 532 nm was used. The measurement time at one point lasted 5 s (during this time, laser irradiation did not influence the film). The total measurement time was 52 min. Fig. 6A shows the optical photo and the mapping area on it (Fig. 6B). The area in the optical photo (Fig. 6B) corresponds to the Raman mapping of all the bands (Fig. 6C–U). This allowed getting a picture of the distribution of the type and quality of chemical bonds. Moreover, in our case, the distribution of intensity can identify the quality crystal structure of the film. The Raman mapping of skeletal CC torsions (116 and 159 cm−1), CH3 torsion (208 and 241 cm−1), δsC–CH3 (306 cm−1), and δ(CCO) (408 cm−1) bands showed high intensity in the areas of formation spherical granules and low intensity between them, which correlates with the morphology of the films. The same result was observed for the most intense band at 875 cm−1 (νC–COO) that corresponds to the spectrum in Fig. 6V. Based on this, it can be argued that in the places of granule formation, we have a crystal structure of the PLLA film and some distortion of the structure between them. Regarding other bands, it can be seen that their intensity is several times lower (intensity scale in Fig. 6U) with correspondingly lower quality of crystallization (fewer of these bonds in this sample). It is worth noting the bands at 710, 735, 1043, 1090, and 1128 cm−1, for which a clear contrast is still visible at the boundary of the spherical granules. This also confirms their high crystallinity.
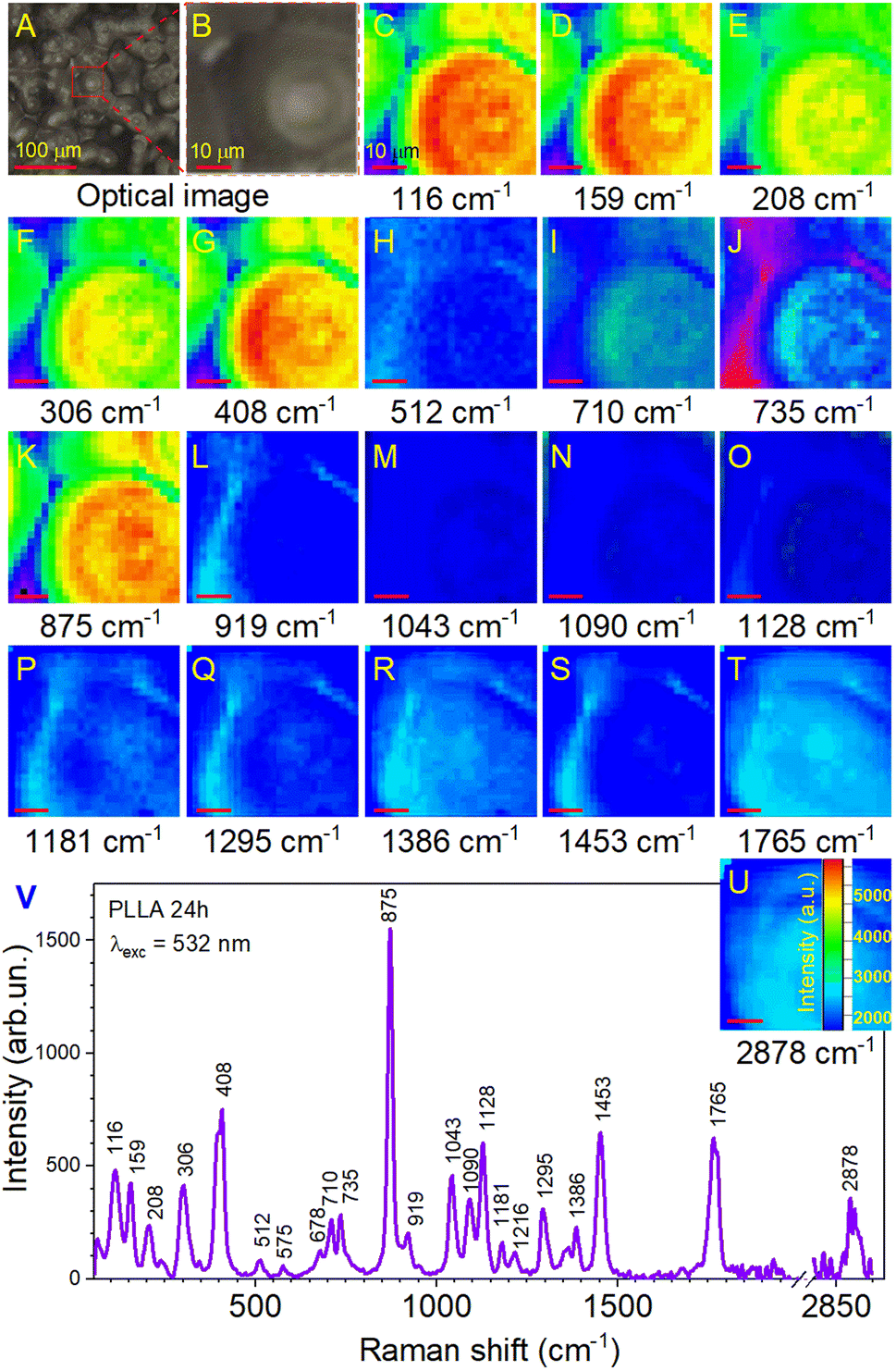 | ||
| Fig. 6 (A) The optical photo of the surface PLLA film. (B) The mapping area on this surface. (C–U) The Raman mapping of all yjr bands corresponds to the area in the optical photo. (V) Raman spectrum of the PLLA film obtained after annealing for 24 h. | ||
The reflection of the influence of the annealing time of the films on the Raman mappings is shown in Fig. 7 for the most intense band at 875 cm−1. In this case, the study area was doubled to 100 × 100 μm2 with a resolution of 5 μm. The measurement time at one point lasted 10 s, and the total measurement time was 67 min. These were minimally sufficient conditions to ensure the effective evaluation of film uniformity in our study. Fig. 7A shows that the pristine PLLA film had a uniform distribution of intensity over a large area, which corresponds to the good quality of the synthesized sample. For the PLLA film annealed for 3 h, an increase in the intensity is seen in half of the investigation area, which gives a ground to talk about improving the crystal structure and crystallinity of the film (Fig. 7B). The film obtained after 12 h of annealing shows that high-intensity areas still have a significant value, but they are divided into separate sections (Fig. 7C). It has been suggested that this is due to a change in the size of the spherical granules. Remember that the maximum intensity was on the granules of the films during mapping, and those granules for this film decreased by 10 microns compared to the 3 h-annealed film.
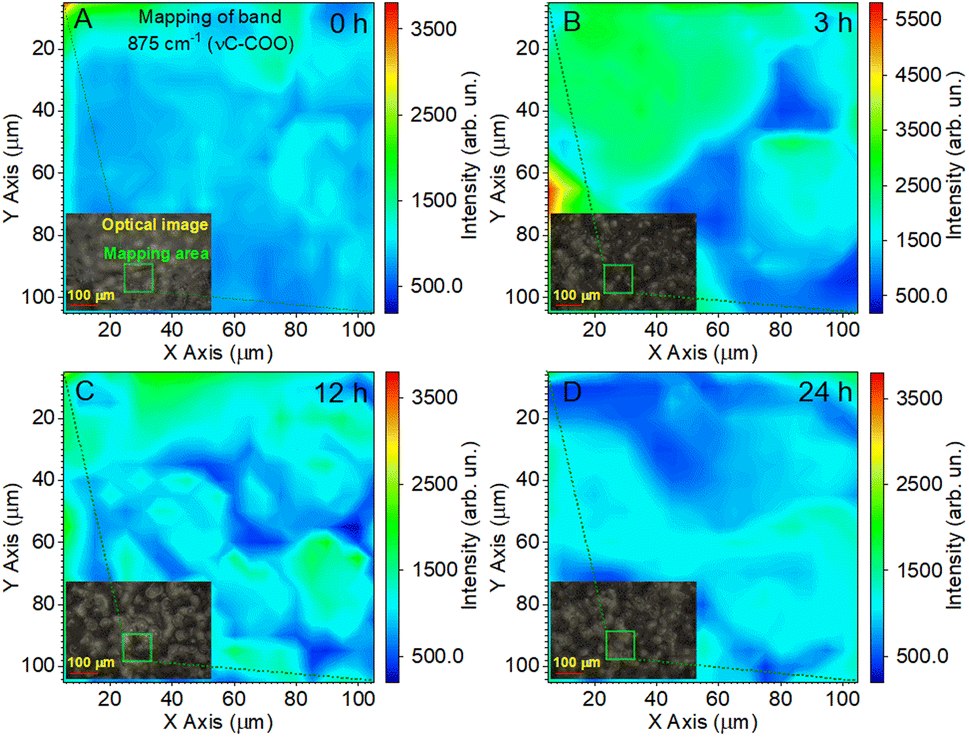 | ||
| Fig. 7 Raman mapping of the 875 cm−1 band of the PLLA films at varied temperature treatment times: (A) pristine 0 h; (B) 3 h; (C) 12 h; and (D) 24 h. | ||
As for the PLLA film obtained after 24 h of annealing (Fig. 7D), it was characterized by a slight decrease in the intensity relative to the film annealed for 12 h, but, at the same time, slightly increased the homogeneity of the film.
Thus, the analysis of Raman mapping further added information that the area of the ordered phase of PLLA films depends on the time of the temperature treatment.44 Temperature treatment times from 3 to 24 h (T = 140 °C) allows for the improvement of the uniformity of films. The uniform area can be important in enhancing the piezoelectric properties of PLLA films. The next section will consider how our assumptions about the change in the position of the bands in the vibration of the CO dipole correlate with the piezoelectric properties of the films.
Piezoelectric characterization of sensors
The PLLA exhibits shear piezoelectricity due to the electrical polarity present in the carbon–oxygen (CO) double-bond branching off from the polymer backbone chain. Due to a low dielectric constant, PLLA could performed the same energy conversion as efficiently as the common piezoelectric polymer PVDF.30 Knowing this feature, after the preparation of PLLA films and their characterization, the prototypes of pressure sensors based on them were developed to study the piezoelectric properties.
The influence of the load at a constant impact of velocity (40 mm s−1) and the effect of the change of the impact of velocity at a constant load (10 newtons) on the value of the piezoelectric signal are shown in Fig. 8A and B. The analysis of the relationship of the piezoelectric signal with optical and structural parameters is shown in Fig. 8C. It can be noted that increasing the load and the impact of velocity contribute to the growth of the piezoelectric signal. Another important factor is the temperature treatment time, which has a significant effect on this value.
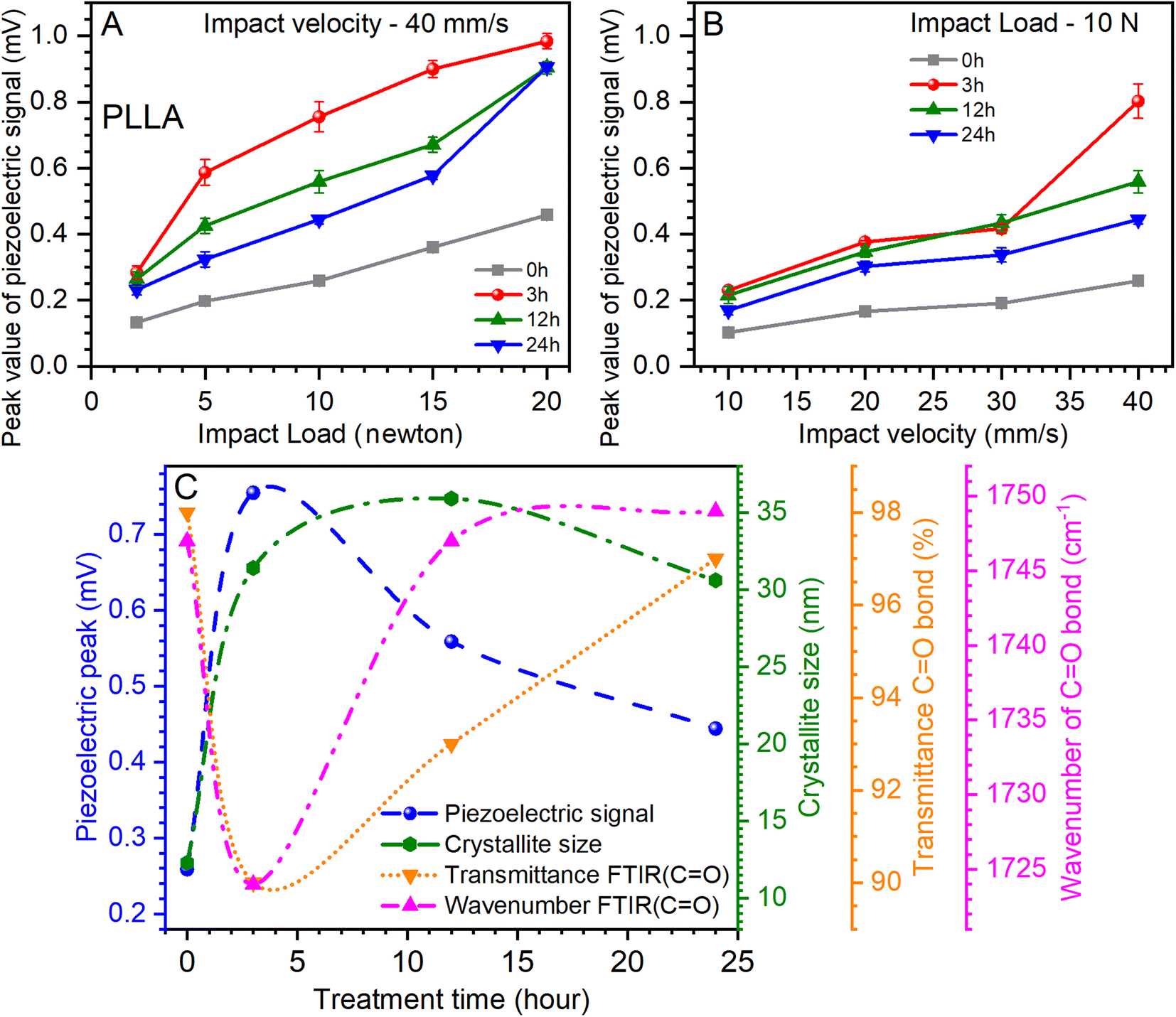 | ||
| Fig. 8 (A) The influence of the different loads on the piezoelectric properties of PLLA at constant impact of velocity (40 mm s−1). (B) The impact of the different velocities on the piezoelectric signal of PLLA at continual load (10 N). (C) Relationship between optical, structural, and piezoelectric parameters. The piezoelectric signal was obtained from (A) at a load of 10 N and an impact velocity of 40 mm s−1. Transmission and band position for the CO bond were obtained from the ATR-FTIR measurements. | ||
Fig. 8A shows that the maximum value of the piezoelectric signal was 0.98 mV for sensors prepared on films annealed for 3 h at a load of 20 N.45 For films without temperature annealing at the same load, the maximum value was 0.45 mV. Next, a relative decrease in the piezoelectric signal was seen for sensors prepared on annealed films at 12 and 24 h at 9 mV, respectively (Fig. 8A). Similar dynamics are inherent at increasing the impact of velocity (from 10 to 40 mm s−1) at a constant load, which also shows an increase in the value of the piezoelectric signal (Fig. 8B). Thus, for the initial film (0 h), this value increased from 0.1 to 0.25 mV, for films annealed at 3 h from 0.22 to 0.8 mV, for 12 h-annealed films from 0.21 to 0.55 mV, and for 24 h-annealed films from 0.16 to 0.44 mV. Thus, it can be seen again that for sensors prepared on films that have been annealed at 3 h, the value of the piezoelectric signal was the maximum. It should be noted that the annealing of the films can be most effective from 3 to 12 h, as the values of the piezoelectric signal are relatively maximum for sensors prepared on the basis of these films.
Analyzing the piezoelectric signal at a load of 10 N and an impact velocity of 40 mm s−1 (Fig. 8A), optical (Fig. 5), and structural (Table 1) parameters at varied temperature treatment times, some patterns can be found. A parametric equation for a modified Bezier curve was used for curve fitting. As can be seen from Fig. 8C, this ensured that the curves were accurately constructed based on the experimental data. The first thing that can be seen immediately is the rapid growth of the piezoelectric signal of the PLLA sensors and the increase in the crystallite size of the film domains (Fig. 8C). This suggests a direct relationship between these two parameters. Regarding the optical parameters, Fig. 8C shows the values of transmission and band position for the CO bond, which are obtained from ATR-FTIR measurements (Fig. 5). Recall that the piezoelectric changes in the PLLA film are directly related to this bond. We note the inverse dependence of the optical parameters on the piezoelectric signal when the temperature treatment time changes. In particular, the piezoelectric signal increases as the transmission decreases, and a shift of the band to the low-frequency region of the spectrum is seen. As we expected, the stretching of the dipole CO associated with band shift and an improvement in the structure of the films increase the piezoelectric properties of PLLA. Therefore, it can be argued that the first hours of annealing have a fast effect on the properties of PLLA films, which is reflected in the analysis in Fig. 8C. This confirms the effectiveness of the use of temperature treatment time to improve the piezoelectric properties of the sensors based on PLLA. Further detailed investigation of such films with a temperature treatment time from 1 to 12 h may reveal additional properties of this material, and by creating multilayer PLLA sensors, one can achieve an even higher conversion efficiency of mechanical energy to electricity, similar to that of ceramic materials. All this will facilitate the faster application of such sensors in various fields of electronics.
Conclusions
The PLLA films and piezoelectric sensors based on them at varied temperature treatment times were investigated. The morphology and crystal structure of the PLLA films obtained under various post-processing conditions were examined by SEM, XRD, Raman, and ATR-FTIR spectroscopy. The micromechanical equipment for tension–compression measurements was built in the laboratory for the tested piezoelectric sensors.The annealing time was demonstrated to be effective for the improvement of the morphology and structure due to the increased crystallite size. After 3 h treatment at 140 °C, the crystallite size increased to 31.4 nm, while for the initial film, this value was 12.3 nm. The increase in the crystallite size of the PLLA film influences the growth of the piezoelectric signal of sensors based on them. The maximum value of the piezoelectric signal was 0.98 mV for sensors prepared on films annealed for 3 h at a load of 20 N. For films without temperature annealing at the same load, the maximum value was 0.45 mV. The analysis of Raman mapping gave the information that the area of the ordered phase of the PLLA films depends on the time of temperature treatment. It was shown that a larger area of the ordered PLLA phase was intrinsic to the films annealed from 3 to 12 h. Thus, all the above give grounds to claim that high-quality films have been obtained as efficient converters of mechanical energy into electricity, which opens new innovative perspectives for the creation of pressure sensors based on PLLA.
Author contributions
Ivan S. Babichuk, Lin Chubin: conceptualization, methodology. Yuhui Qiu, Huiyu Zhu: visualization, investigation. Ivan S. Babichuk, Chubin Lin, Yuhui Qiu and Huiyu Zhu: formal analysis, writing-original draft preparation. Terry Tao Ye, Zhaoli Gao: project administration, writing-reviewing and editing. Jian Yang: supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their sincere thanks to Dr Dawei Wen and Dr Chengqun Xu (Wuyi University, Jiangmen, China) for the helpful XRD and SEM measurements. This research was supported by Key-Area Research and Development Program of Guangdong Province [2020B0101030002], and in part by the United Program of National Natural Science Foundation of China with Shenzhen [U1613212] and Innovation Projects of Department of Education of Guangdong Province [2018KQNCX264]. The authors also thank the support from Wuyi University-Hongkong-Macao Joint Research Funds [2019WGALH19].References
- N. Matsuhisa, X. Chen, Z. Bao and T. Someya, Chem. Soc. Rev., 2019, 48, 2946–2966, 10.1039/c8cs00814k.
- T. R. Ray, J. Choi, A. J. Bandodkar, S. Krishnan, P. Gutruf, L. Tian, R. Ghaffari and J. A. Rogers, Chem. Rev., 2019, 119, 5461–5533, DOI:10.1021/acs.chemrev.8b00573.
- M. Zhu, T. He and C. Lee, Appl. Phys. Rev., 2020, 7, 031305, DOI:10.1063/5.0016485.
- J. Li, Y. Long, F. Yang and X. Wang, Curr. Opin. Solid State Mater. Sci., 2020, 24, 100806, DOI:10.1016/j.cossms.2020.100806.
- M. T. Chorsi, E. J. Curry, H. T. Chorsi, R. Das, J. Baroody, P. K. Purohit, H. Ilies and T. D. Nguyen, Adv. Mater., 2019, 31, 1802084, DOI:10.1002/adma.201802084.
- D.-M. Shin, S. W. Hong and Y.-H. Hwang, Nanomaterials, 2020, 10, 123, DOI:10.3390/nano10010123.
- B. Gupta, N. Revagade and J. Hilborn, Prog. Polym. Sci., 2007, 32, 455–482, DOI:10.1016/j.progpolymsci.2007.01.005.
- J. Yang, F. Liu, L. Yang and S. Li, Eur. Polym. J., 2010, 46, 783–791, DOI:10.1016/j.eurpolymj.2009.12.017.
- M. Brzeziński and T. Biela, Stereocomplexed Polylactides, in Encyclopedia of Polymeric Nanomaterials, ed., S. Kobayashi and K. Müllen, Springer-Verlag Berlin, Heidelberg, 1 edn, 2015, pp. 1–10, DOI:10.1007/978-3-642-36199-9_394-1.
- J. Yang, F. Liu, S. Tu, Y. Chen, X. Luo, Z. Lu, J. Wei and S. Li, J. Biomed. Mater. Res., Part A, 2010, 94, 396–407, DOI:10.1002/jbm.a.32677.
- J. Zhang, Y. Duan, H. Sato, H. Tsuji, I. Noda, S. Yan and Y. Ozaki, Macromolecules, 2005, 38, 8012–8021, DOI:10.1021/ma051232r.
- H. J. Oh, D.-K. Kim, Y. C. Choi, S.-J. Lim, J. B. Jeong, J. H. Ko, W.-G. Hahm, S.-W. Kim, Y. Lee, H. Kim and B. J. Yeang, Sci. Rep., 2020, 10, 16339, DOI:10.1038/s41598-020-73261-3.
- L. Cartier, T. Okihara, Y. Ikada, H. Tsuji, J. Puiggali and B. Lotz, Polymer, 2000, 41, 8909–8919, DOI:10.1016/s0032-3861(00)00234-2.
- W. Hoogsteen, A. R. Postema, A. J. Pennings, G. Ten Brinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642, DOI:10.1021/ma00204a041.
- D. Brizzolara, H.-J. Cantow, K. Diederichs, E. Keller and A. J. Domb, Macromolecules, 1996, 29, 191–197, DOI:10.1021/ma951144e.
- L. Jiang, T. Shen, P. Xu, X. Zhao, X. Li, W. Dong, P. Ma and M. Chen, e-Polym., 2016, 16, 1–13, DOI:10.1515/epoly-2015-0179.
- J. Puiggali, Y. Ikada, H. Tsuji, L. Cartier, T. Okihara and B. Lotz, Polymer, 2000, 41, 8921–8930, DOI:10.1016/s0032-3861(00)00235-4.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905, DOI:10.1021/ma071258d.
- G. Kister, G. Cassanas, M. Vert, B. Pauvert and A. Térol, J. Raman Spectrosc., 1995, 26, 307–311, DOI:10.1002/jrs.1250260409.
- S. Kang, S. L. Hsu, H. D. Stidham, P. B. Smith, M. A. Leugers and X. Yang, Macromolecules, 2001, 34, 4542–4548, DOI:10.1021/ma0016026.
- K. Wasanasuk, K. Tashiro, M. Hanesaka, T. Ohhara, K. Kurihara, R. Kuroki, T. Tamada, T. Ozeki and T. Kanamoto, Macromolecules, 2011, 44, 6441–6452, DOI:10.1021/ma2006624.
- E. J. Curry, K. Ke, M. T. Chorsi, K. S. Wrobel, A. N. Miller, A. Patel, I. Kim, J. Feng, L. Yue, Q. Wu, C.-L. Kuo, K. W. H. Lo, C. T. Laurencin, H. Ilies, P. K. Purohit and T. D. Nguyen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 909–914, DOI:10.1073/pnas.1710874115.
- R. Fitzgerald, L. M. Bass, D. J. Goldberg, M. H. Graivier and Z. P. Lorenc, Aesthetic Surg. J., 2018, 38, S13–S17, DOI:10.1093/asj/sjy012.
- J. Morvan, E. Buyuktanir, J. L. West and A. Jákli, Appl. Phys. Lett., 2012, 100, 063901, DOI:10.1063/1.3683482.
- S. Mishra, L. Unnikrishnan, S. K. Nayak and S. Mohanty, Macromol. Mater. Eng., 2019, 304, 1800463, DOI:10.1002/mame.201800463.
- C. Zhao, J. Zhang, Z. L. Wang and K. Ren, Adv. Sustainable Syst., 2017, 1, 1700068, DOI:10.1002/adsu.201700068.
- M. Ando, S. Takeshima, Y. Ishiura, K. Ando and O. Onishi, Jpn. J. Appl. Phys., 2017, 56, 10PG01, DOI:10.7567/jjap.56.10pg01.
- M. Ando, D. Tamakura, T. Inoue, K. Takumi, T. Yamanaga, R. Todo, K. Hosoya and O. Onishi, Jpn. J. Appl. Phys., 2019, 58, SLLD09, DOI:10.7567/1347-4065/ab3b1b.
- J. Zhu, L. Jia and R. Huang, J. Mater. Sci.: Mater. Electron., 2017, 28, 12080–12085, DOI:10.1007/s10854-017-7020-5.
- K. Kapat, Q. T. H. Shubhra, M. Zhou and S. Leeuwenburgh, Adv. Funct. Mater., 2020, 30, 1909045, DOI:10.1002/adfm.201909045.
- R. Xu, J. Xie and C. Lei, RSC Adv., 2017, 7, 39914–39921, 10.1039/c7ra05422j.
- K. Takahashi, D. Sawai, T. Yokoyama, T. Kanamoto and S.-H. Hyon, Polymer, 2004, 45, 4969–4976, DOI:10.1016/j.polymer.2004.03.108.
- K. Tashiro, N. Kouno, H. Wang and H. Tsuji, Macromolecules, 2017, 50, 8048–8065, DOI:10.1021/acs.macromol.7b01468.
- H. Wang, J. Zhang and K. Tashiro, Macromolecules, 2017, 50, 3285–3300, DOI:10.1021/acs.macromol.7b00272.
- S. J. Lee, A. P. Arun and K. J. Kim, Mater. Lett., 2015, 148, 58–62, DOI:10.1016/j.matlet.2015.02.038.
- H. Liu, W. Zhang, H. Liang, Q. Zeng, J. Shi and D. Wen, J. Mater. Chem. C, 2021, 9, 1622–1631, 10.1039/d0tc05379a.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71, DOI:10.1107/s0021889869006558.
- C. Frontera and J. Rodriguez-Carvajal, Phys. B, 2003, 335, 219–222, DOI:10.1016/S0921-4526(03)00241-2.
- K. Wasanasuk and K. Tashiro, Polymer, 2011, 52, 6097–6109, DOI:10.1016/j.polymer.2011.10.046.
- I. S. Babichuk, M. O. Semenenko, S. Golovynskyi, R. Caballero, O. I. Datsenko, I. V. Babichuk, J. Li, G. Xu, R. Qiu, C. Huang, R. Hu, I. Golovynska, V. Ganus, B. Li, J. Qu and M. Leon, Sol. Energy Mater. Sol. Cells, 2019, 200, 109915, DOI:10.1016/j.solmat.2019.109915.
- S. Lee, M. Kimoto, M. Tanaka, H. Tsuji and T. Nishino, Polymer, 2018, 138, 124–131, DOI:10.1016/j.polymer.2018.01.051.
- I. Navarro-Baena, A. Marcos-Fernandez, J. M. Kenny and L. Peponi, J. Appl. Crystallogr., 2014, 47, 1948–1957, DOI:10.1107/s1600576714022468.
- R. Gregorio, J. Appl. Polym. Sci., 2006, 100, 3272–3279, DOI:10.1002/app.23137.
- I. S. Babichuk, M. O. Semenenko, R. Caballero, O. I. Datsenko, S. Golovynskyi, R. Qiu, C. Huang, R. Hu, I. V. Babichuk, R. R. Ziniuk, M. Stetsenko, O. A. Kapush, J. Yang, B. Li, J. Qu and M. Leon, Sol. Energy, 2020, 205, 154–160, DOI:10.1016/j.solener.2020.05.043.
- D. M. Correia, L. C. Fernandes, B. Cruz, G. Botelho, V. de Zea Bermudez and S. Lanceros-Méndez, Polymers, 2020, 12, 1187, DOI:10.3390/polym12051187.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04241j |
| ‡ I. S. Babichuk and C. Lin contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2022 |