
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel quinoxaline derivatives as dual EGFR and COX-2 inhibitors: synthesis, molecular docking and biological evaluation as potential anticancer and anti-inflammatory agents†
Eman A. Ahmeda,
Mamdouh F. A. Mohamed *b and
Omran A. Omrana
*b and
Omran A. Omrana
aDepartment of Chemistry, Faculty of Science, Sohag University, Sohag 82524, Egypt
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, 82524 Sohag, Egypt. E-mail: mamdouhfawzy3@yahoo.com; mamdouh.fawzi@pharm.sohag.edu.eg; Tel: (+20)-01018384461
First published on 5th September 2022
Abstract
Novel quinoxaline derivatives (2a–d, 3, 4a, 4b and 5–15) have been synthesized via the reaction of 4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide (1) with different aldehydes, ketones, diketones, ketoesters, as well as hydrazine, phenyl isothiocyanate, carbon disulphide. The synthesized products have been screened for their in vitro anticancer and COX inhibitory activities. Most of the synthesized compounds exhibited good anticancer and COX-2 inhibitory activities. MTT assay revealed that compounds 11 and 13 were the most potent and exhibited very strong anticancer activity against the three cancer cell lines with IC50 values ranging from 0.81 μM to 2.91 μM. Compounds 4a and 5 come next and displayed strong anticancer activity against the three cancer cell lines with IC50 values ranging from 3.21 μM to 4.54 μM. Mechanistically, compounds 4a and 13 were the most active and potently inhibited EGFR with IC50 = 0.3 and 0.4 μM, respectively. Compounds 11 and 5 come next with IC50 = 0.6 and 0.9 μM, respectively. Moreover, compounds 11 and 13 were the most potent as COX-2 inhibitors and displayed higher potency against COX-2 (IC50 = 0.62 and 0.46 μM, respectively) more than COX-1 (IC50 = 37.96 and 30.41 μM, respectively) with selectivity indexes (SI) of 61.23 and 66.11, respectively. Compounds 4a and 5 comes next with IC50 = 1.17 and 0.83 μM and SI of 24.61 and 48.58, respectively. Molecular docking studies into the catalytic binding pocket of both protein receptors, EGFR and COX-2, showed good correlation with the obtained biological results. Parameters of Lipinski's rule of five and Veber's standard were calculated and revealed that compounds 4a, 5, 11 and 13 had a reasonable drug-likeness with acceptable physicochemical properties. Therefore, based on the obtained biological results accompanied with the docking study and physicochemical parameters, it could be concluded that compounds 4a, 5, 11 and 13 could be used as promising orally absorbed dual anti-inflammatory agents via inhibition of COX-2 enzyme and anticancer candidates via inhibition of EGFR enzyme and could be used as a future template for further investigations.
1. Introduction
Cancer, with 7–10 million human mortalities annually worldwide, is still rising at an alarm rate as one of the most intractable diseases globally.1–3 It is a complex, heterogeneous, multigenic disease and is the leading cause of death preceded only by heart disease.4,5 There are many cancer hallmarks, tumor-promoting inflammation is now well-recognized as one of the cancer hallmarks. Moreover, both acute and chronic inflammatory processes have a significant influence on the development and growth of cancer.6–8 Recent evidence has expanded that inflammation is not only a vital component of tumor progression but it is also predisposing to the development of cancer and facilitates all stages of tumorigenesis.9 Till now, there are more than one hundred cancer types; each one needs unique diagnosis and treatment.2 Indeed, there are many effective anticancer drugs currently available on the market, such as the traditional anticancer chemotherapeutic agents that prevent cell division and replication of DNA. Unfortunately, most of these drugs lack selectivity and specificity, leading to issues such as the common severe adverse effects.2,10 Thus, there is an urgent necessity for the innovation and discovery of novel small molecules with potential to be effective as potent and selective anticancer agents which still represents a major challenge to medicinal chemists.2Currently, it is estimated that inflammatory reactions are responsible for up to 15–20% of cancer-related deaths.11 Additionally, administration of non-steroidal, anti-inflammatory drugs is connected with a lower risk of developing numerous tumors and decreased mortality further underlining the role of inflammation in neoplastic transformation.12 Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most commonly prescribed medication worldwide for the treatment of both acute and chronic inflammation,13 fever, pain14 and inflammation-related disorders.15 However, the long-term usage of non-selective NSAIDs has a number of undesirable side effects, including nephrotoxicity, hepatotoxicity, gastrointestinal irritation, bleeding and ulceration.16,17 Several methods have been reported for improving non-selective NSAIDs.18,19 One strategy is there placement of the acidic carboxylic functional groups in NSAIDs with alternative less acidic heterocyclic bioisosteres. Another strategy is the synthesis of selective COX-2 inhibitors as a new generation of NSAIDs, which would preserve the anti-inflammatory effectiveness while reducing gastric toxicity. Also, development of dual COX and LOX inhibitors with moderate selectivity towards COX-2 at micromole level.20 Recently, the more promising strategy through the development of NSAIDs/iNOS inhibitors for the treatment of inflammatory diseases by decreasing the over production of PGE2 and NO, respectively.17,21–23
Finally, Mantovani classified cancer-related inflammation into two pathways: (i) the intrinsic pathway, which is linked to genetic events that trigger inflammation and neoplastic transformation, and (ii) the extrinsic pathway, which explains inflammatory conditions that promote carcinogenesis. Based upon these observations, it has been proposed that cancer-related inflammation (CRI) might be considered as the “seventh hallmark” of cancer.24 Accordingly, the development of dual acting anti-inflammatory/anticancer candidates is urgently needed and represents a promising approach in treatment of both cancer and inflammatory diseases.
Over the years and due to their significant role in the drug design, development and discovery, nitrogen-based heterocycles have attracted considerable attention.25 Among the N-containing heterocyclic pharmacophores; quinoxaline moiety represents an important class of heterocyclic compounds due to their significant physiological properties and promising applications in medicinal chemistry.26 Notably, many approved drugs such as the antibiotics echinomycin, levomycin, and actinoleutin have quinoxaline as part of their structure.27 Moreover, quinoxaline moiety and its derivatives have received significant attention due to their wide spectrum of pharmaceutical and biological properties such as, among other insecticidal, antidepressant, antifungal, anticonvulsant, anthelmintic, anti-inflammatory, antiviral, antimalarial,antibacterial, antiprotozoal, and anticancer.26,28–30 CQS (4-amino-N-(5-chloroquinoxalin-2-yl) benzenesulfonamide, NSC 339004)31,32 and XK469 (2-[4-(7-chloroquinoxalin-2-yl)oxyphenoxy]propanoic acid, NSC 697887)33,34 are examples of potent anticancer quinoxalines. Moreover, these compounds have been widely used in technology as dyes, chemical switches, electroluminescent materials, photo-initiators, cavitands and organic semiconductors.26,28,29,35
On the other hand, hydrazones are found in numerous of the bioactive heterocyclic compounds that are of very important use because of their several biological and clinical medicinal applications.36,37 Hydrazone derivatives of heteroaromatic compounds have been proved to have, among other, antidepressant, antifungal, antimicrobial, antitubercular, anticonvulsant, analgesic, anti-inflammatory, antimalarial, antiplatelet, cardio protective and antiviral with anticancer activities being of significant interest.3,36,37
Motivated by the aforementioned facts and with the hope of yielding more potent, less toxic, selective, and effective dual acting anti-inflammatory/anticancer candidates, the main aim of this work is to synthesize a new series of quinoxaline-hydrazone derivatives as depicted in Fig. 1 and Scheme 1. Additionally, various substituents attached to the quinoxaline scaffold such as five membered rings (pyrazole and 1,3,4-oxadiazole), hydrazide or thiocarbohydrazide (Fig. 1 and Scheme 2) to investigate their anticancer activity as well as their EGFR, COX-1 and COX-2 inhibitory activities and study their structure activity relationship. Docking study, finally, will be carried out to investigate the binding interactions of the most potent compounds into the pocket of EGFR and COX-2 receptor proteins.
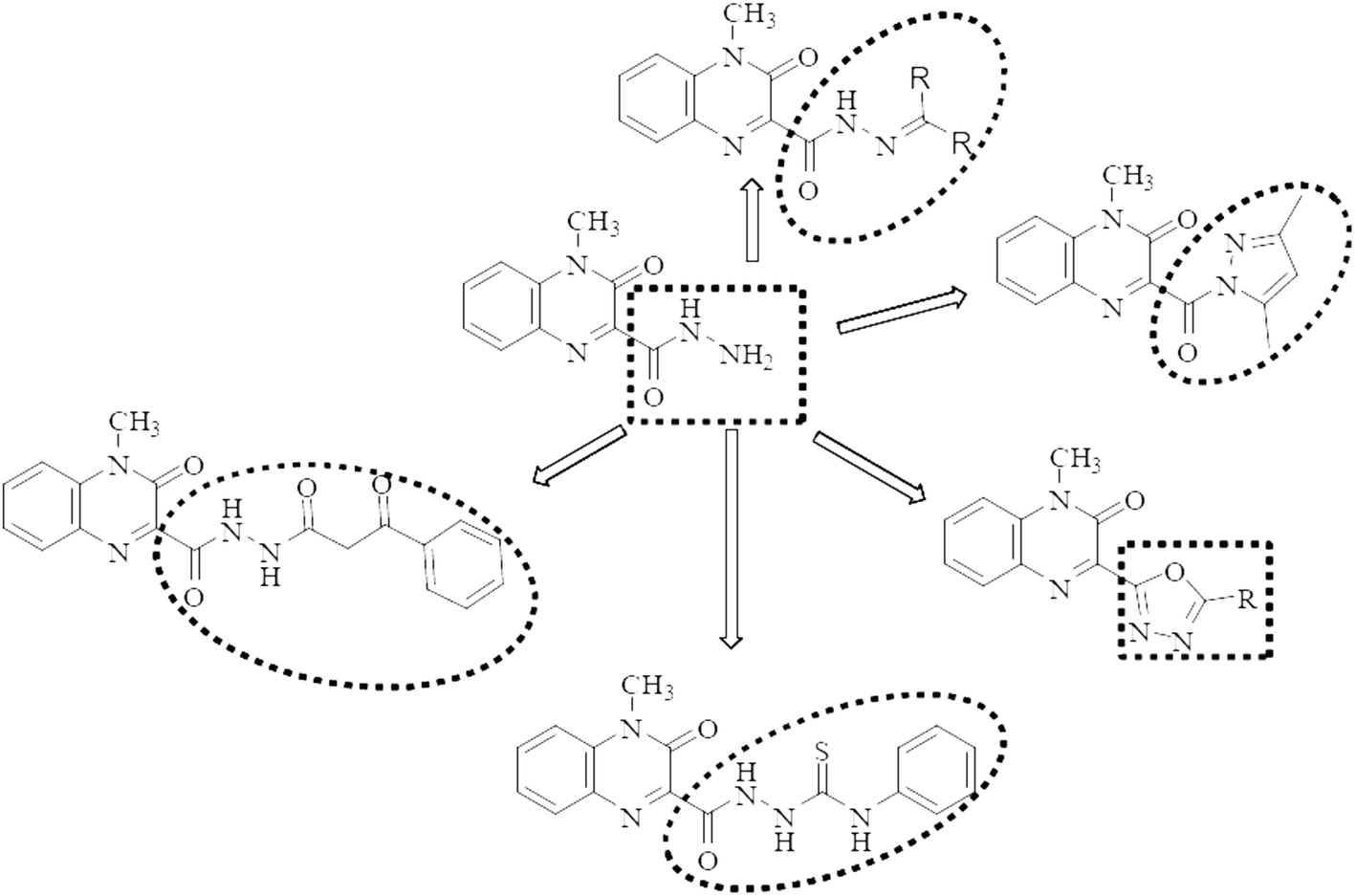 | ||
| Fig. 1 Rationale design of quinoxaline derivatives as anticancer/anti-inflammatory agents. | ||
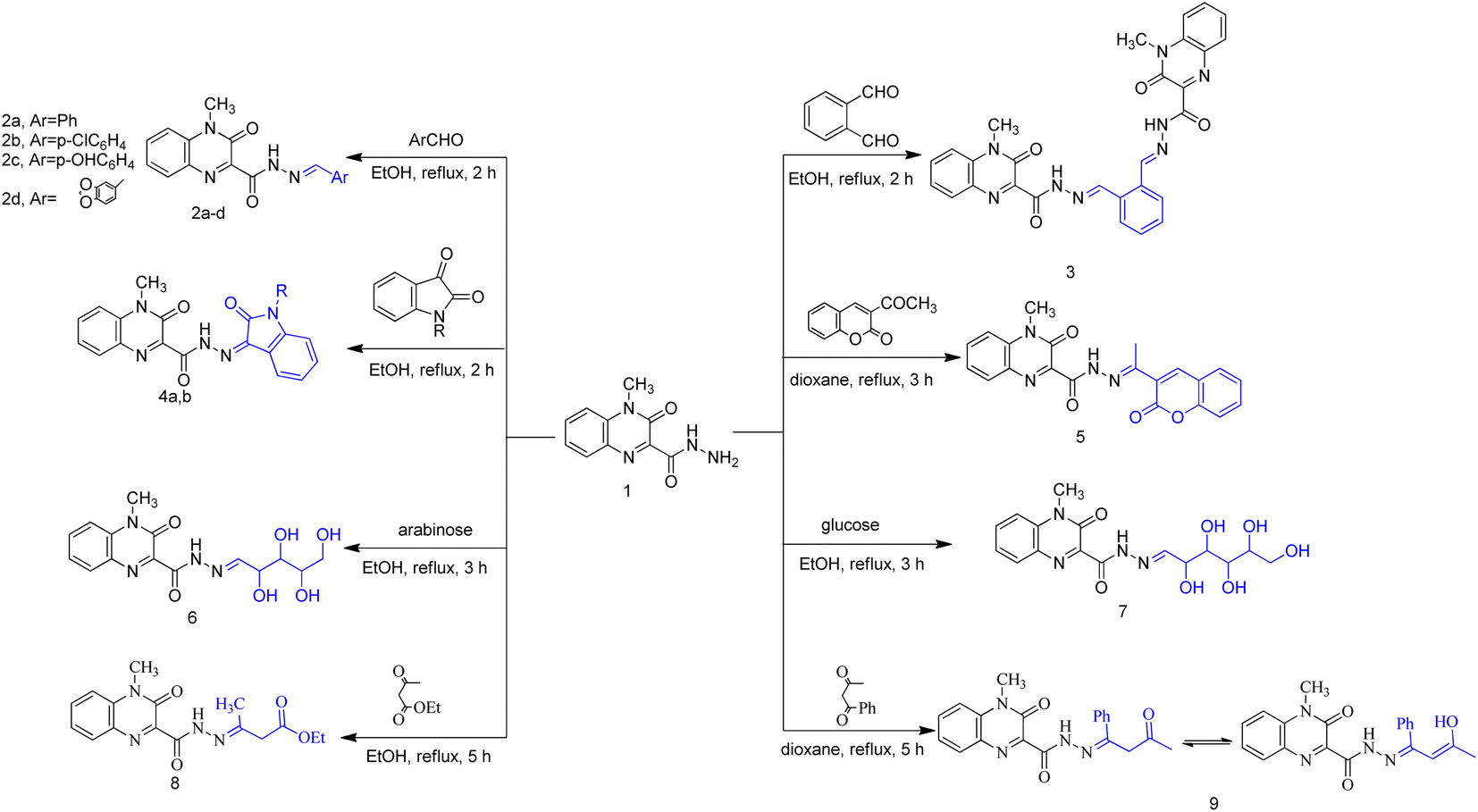 | ||
| Scheme 1 Synthesis of compounds 2–9. | ||
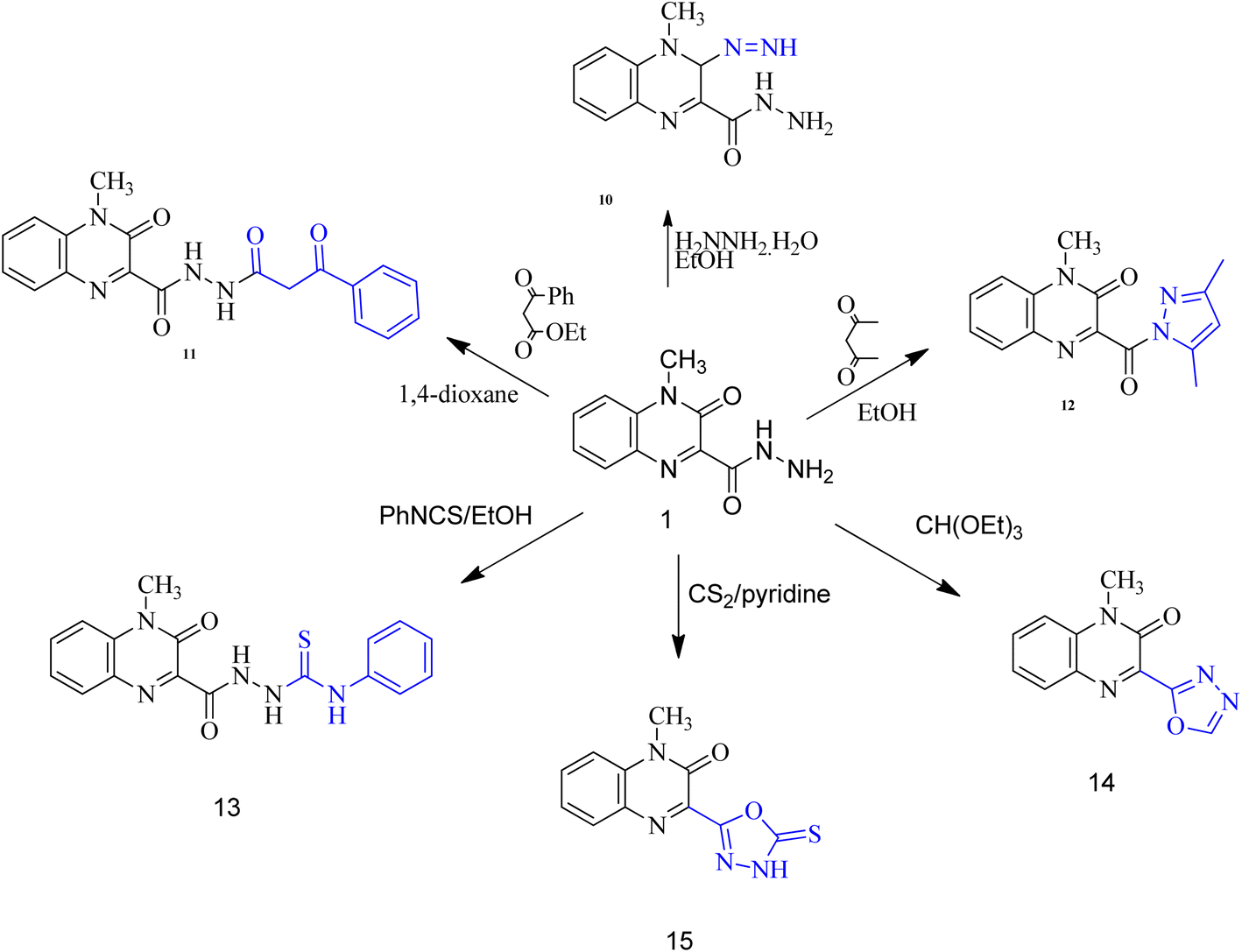 | ||
| Scheme 2 Synthesis of compounds 10–16. | ||
2. Results and discussion
2.1. Chemistry
Heterocycles, such as coumarin, oxindole among others, are highly valuable structures in medicinal chemistry and they represent ubiquitous fragments of several natural products, pharmaceuticals and designed bioactive drug candidates.38–40 Therefore, and in continuation of our work,41 we reported here synthesis of some novel molecules which have quinoxaline moiety coupled to N- or O-heterocyclic ring through acetyl/amide linkage as illustrated in Scheme 1 and 2. The 4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide (1) was prepared according to our reported method,41 via the treatment of ethyl 4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carboxylate with hydrazine hydrate. Carbohydrazide 1 was allowed to react with different aromatic aldehydes such as benzaldehyde, p-chlorobenzaldehyde, p-hydroxybenzaldehyde, and piperonlaldehyde to afford the corresponding N′-arylidene-4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide 2a–d, a mixture of keto- and enol-isomers of Schiff base was obtained in each case.The structure of compounds 2a–d was established on the basis of their spectral and analytical data. Their IR spectra showed the absence of absorption bands corresponding to NHNH2 groups and revealed a new absorption band at 3173–3111 cm−1 assignable to NH groups. Also, their 1H-NMR spectra showed the disappearance of the signal corresponding to NH2 group and exhibited a signal at δ 12.47–12.03 and 8.35–7.88 ppm due to presence of NH and azomethine protons, respectively, as well as increasing of aromatic protons. In addition, when compound 1 was subjected to react with dialdehyde such as o-phthaldehyde, the bis Schiff base N′,N′′-(1,2-phenylenebis(methan-1-yl-1-ylidene))bis(4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide) 3 were formed. The structure of compound 3 has been approved by disappearance of the absorption bands at 1700 cm−1 corresponding to formyl group. Also, 1H-NMR revealed that the ratio between aromatic and methyl protons is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in agreement with the desired structure. Compound 1 was then reacted with biologically active heterocyclic ketones such as isatin and acetyl coumarin. Thus, treatment with isatin and/or N-methyl isatin in boiling ethanol, gave 4-methyl-3-oxo-N′-(2-oxoindolin-3-ylidene)-3,4-dihydroquinoxaline-2-carbohydrazide 4a and/or 4-methyl-N′-(1-methyl-2-oxoindolin-3-ylidene)-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide 4b, respectively. Similarly, when acetyl coumarin was reacted with compound 1 in boiling dioxane, afforded 4-methyl-3-oxo-N′-(1-(2-oxo-2H-chromen-3-yl)ethylidene)-3,4-dihydroquinoxaline-2-carbohydrazide 5. IR spectra of compounds 4a, 4b and 5 showed new absorption bands at 1702 + 3 for the carbonyl of isatines or coumarin, in addition to an absorption band at 3438 corresponding to the new NH in the case of compound 4a. Also, 1H-NMR revealed a new signal at 10.93 due to the new NH in the case of compound 4a and new signals at 3.24 and 2.29 due to the new CH3 in the case of compounds 4b and 5, respectively. Condensation of compound 1 with two different aldoses, arabinose as an aldopentose and/or glucose as an aldohexose has been carried out to produce the polyhydroxyalkylidene quinoxaline carbohydrazide derivatives, 4-methyl-3-oxo-N′-(2,3,4,5-tetrahydroxypentylidene)-3,4-dihydroquinoxaline-2-carbohydrazide (6) and/or 4-methyl-3-oxo-N′-(2,3,4,5,6-pentahydroxyhexylidene)-3,4-dihydroquinoxaline-2-carbohydrazide (7), respectively. In the IR spectra, a series of absorption bands were observed at 3456, 3395, 3338, 3303 or at 3570, 3441, 3388, 3366, 3258 corresponding to hydroxy groups in the case of compounds 6 or 7, respectively.
:1 in agreement with the desired structure. Compound 1 was then reacted with biologically active heterocyclic ketones such as isatin and acetyl coumarin. Thus, treatment with isatin and/or N-methyl isatin in boiling ethanol, gave 4-methyl-3-oxo-N′-(2-oxoindolin-3-ylidene)-3,4-dihydroquinoxaline-2-carbohydrazide 4a and/or 4-methyl-N′-(1-methyl-2-oxoindolin-3-ylidene)-3-oxo-3,4-dihydroquinoxaline-2-carbohydrazide 4b, respectively. Similarly, when acetyl coumarin was reacted with compound 1 in boiling dioxane, afforded 4-methyl-3-oxo-N′-(1-(2-oxo-2H-chromen-3-yl)ethylidene)-3,4-dihydroquinoxaline-2-carbohydrazide 5. IR spectra of compounds 4a, 4b and 5 showed new absorption bands at 1702 + 3 for the carbonyl of isatines or coumarin, in addition to an absorption band at 3438 corresponding to the new NH in the case of compound 4a. Also, 1H-NMR revealed a new signal at 10.93 due to the new NH in the case of compound 4a and new signals at 3.24 and 2.29 due to the new CH3 in the case of compounds 4b and 5, respectively. Condensation of compound 1 with two different aldoses, arabinose as an aldopentose and/or glucose as an aldohexose has been carried out to produce the polyhydroxyalkylidene quinoxaline carbohydrazide derivatives, 4-methyl-3-oxo-N′-(2,3,4,5-tetrahydroxypentylidene)-3,4-dihydroquinoxaline-2-carbohydrazide (6) and/or 4-methyl-3-oxo-N′-(2,3,4,5,6-pentahydroxyhexylidene)-3,4-dihydroquinoxaline-2-carbohydrazide (7), respectively. In the IR spectra, a series of absorption bands were observed at 3456, 3395, 3338, 3303 or at 3570, 3441, 3388, 3366, 3258 corresponding to hydroxy groups in the case of compounds 6 or 7, respectively.
When carbohydrazide 1 was reacted with ethyl acetoacetate and/or benzoyl acetone, open-chain products formed. Reaction with ethyl acetoacetate in boiling ethanol gave ethyl 3-(2-(4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbonyl)hydrazono)butanoate (8), the 1H NMR spectrum exhibited singlet signal at δ 11.6 ppm which was exchangeable with deuterium on addition of deuterium oxide and was assigned to N–H proton, new two signals were observed at 3.46 and 1.99 ppm for methylene and methyl protons, respectively, in addition to two sets of signals distinguish the presence of ethoxy group (OCH2CH3) at 4.16 and 1.26 ppm. The product was formed by nucleophilic addition of the amino group of 1 to the carbonyl acetyl group followed by elimination of water. Also, reaction of benzoylacetone with 1 in boiling dioxane, led to (Z)-4-methyl-3-oxo-N′-(3-oxo-1-phenylbutylidene)-3,4-dihydroquinoxaline-2-carbohydrazide (9). Compound 1 was allowed to react with hydrazine hydrate in boiling ethanol. The product of the reaction was proved as 3-diazenyl-4-methyl-3,4-dihydroquinoxaline-2-carbohydrazide (10) by spectral and analytical data. IR spectrum showed the presence of absorption bands corresponding to NH2; 2NH and CO groups, also, 1H-NMR revealed new four signals at 9.42, 4.43, 4.34 and 3.39 ppm due to 2NH; NH2 and CH protons.
Whereas, reaction of ethyl benzoylacetate with 1 in boiling dioxane, led to 4-methyl-3-oxo-N′-(3-oxo-3-phenylpropanoyl)-3,4-dihydroquinoxaline-2-carbohydrazide (11), the product was formed by nucleophilic addition of the amino group of 1 to the carbonyl ester group followed by elimination of ethanol molecule. On the other hand, upon heating the carbohydrazide 1 with acetylacetone in ethanol, the N-pyrazolo derivative was formed. Compound 12 showed in its 1H-NMR spectrum, new three singlet signals at 6.07, 2.72, 2.16 ppm due to 4-H pyrazole, two methyl protons at 5 and 3 positions of pyrazole moiety. In addition, the carbohydrazide 1 was allowed to react with phenyl isothiocyanate in boiling ethanol, to give 2-(4-methyl-3-oxo-3,4-dihydroquinoxaline-2-carbonyl)-N-phenylhydrazine-carbothioamide (13). Also, oxadiazole derivative 14 was formed from interaction of 1 with triethyl orthoformate. The 1H-NMR spectrum showed disappearance of signals related to NH and NH2 groups, and appearance of new signal due to CH proton of new oxadiazole ring. Finally, the interaction of 1 with carbon disulfide in boiling pyridine afforded 3-(5-mercapto-1,3,4-oxadiazol-2-yl)-1-methylquinoxalin-2(1H)-one (15). The spectral data of compound 15 are coincident with the suggested structure.
2.2. Biological evaluation
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as spacer, as in compound 12 with pyrazole moiety, or directly to the quinoxaline scaffold as in compound 14 (1,3,4-oxadiazole) and compound 15 (1,3,4-oxadiazole-2-thione) again we noticed a dramatic decrease in the anticancer activity against the three cancer cell lines.
O as spacer, as in compound 12 with pyrazole moiety, or directly to the quinoxaline scaffold as in compound 14 (1,3,4-oxadiazole) and compound 15 (1,3,4-oxadiazole-2-thione) again we noticed a dramatic decrease in the anticancer activity against the three cancer cell lines.
| MCF-7 | HepG2 | HCT-116 | |
|---|---|---|---|
| 2a | 82.85 ± 0.49 | 89.21 ± 0.63 | 69.25 ± 0.45 |
| 2b | 95.46 ± 1.25 | 90.76 ± 0.29 | 88.68 ± 0.52 |
| 2c | 69.72 ± 0.46 | 77.95 ± 0.56 | 61.92 ± 0.55 |
| 2d | 50.14 ± 0.46 | 56.95 ± 0.35 | 59.47 ± 0.78 |
| 3 | 46.52 ± 1.12 | 54.25 ± 0.60 | 63.79 ± 0.44 |
| 4a | 90.14 ± 0.76 | 91.82 ± 0.35 | 92.79 ± 0.15 |
| 4b | 61.47 ± 0.37 | 52.35 ± 0.74 | 59.46 ± 0.27 |
| 5 | 91.47 ± 0.25 | 90.79 ± 0.21 | 93.75 ± 0.29 |
| 6 | 10.82 ± 0.76 | 21.38 ± 0.52 | 41.79 ± 1.27 |
| 7 | 10.32 ± 0.48 | 26.37 ± 0.83 | 33.87 ± 1.27 |
| 8 | 95.96 ± 0.22 | 94.63 ± 0.35 | 96.42 ± 0.81 |
| 9 | 92.97 ± 0.54 | 96.83 ± 0.29 | 97.89 ± 0.69 |
| 10 | 49.76 ± 0.86 | 52.43 ± 0.69 | 86.63 ± 1.31 |
| 11 | 92.57 ± 0.43 | 92.78 ± 0.38 | 97.63 ± 0.16 |
| 12 | 33.47 ± 0.82 | 53.78 ± 1.38 | 49.22 ± 1.23 |
| 13 | 95.39 ± 0.27 | 96.75 ± 0.29 | 97.87 ± 0.19 |
| 14 | 29.27 ± 0.63 | 25.74 ± 1.67 | 33.34 ± 1.32 |
| 15 | 43.47 ± 0.63 | 59.47 ± 1.92 | 76.32 ± 1.22 |
The most potent compounds 2b, 4a, 5, 8, 9, 11 and 13 were further selected upon their 1st screening results for determination of their IC50 at 10-fold dilutions of five different concentrations (0.01, 0.1, 1, 10 and 100 μM) using doxorubicin as a reference drug. The obtained results, as shown in Table 2, revealed that the tested compounds showed variable results varying from very strong to moderate anticancer activity against the three used cancer cell lines. Among all, compounds 11 and 13 were the most potent and exhibited very strong anticancer activity against the three cancer cell lines with IC50 values ranging from 0.81 μM to 2.91 μM. Compounds 4a and 5 come next and displayed strong anticancer activity against the three cancer cell lines with IC50 values ranging from 3.21 μM to 4.54 μM. Compound 9 showed good anticancer activity with IC50 values 6.84, 5.54 and 8.75 μM against MCF-7, HepG2 and HCT-116, respectively. Finally, compounds 2b and 8 exhibited moderate anticancer activity against the three cancer lines with IC50 values more than 10 μM as illustrated in Table 2. From these results, it could be concluded that compounds 4a, 5, 11 and 13 could be considered as promising anticancer candidates.
| Compounds | MCF-7 | HepG2 | HCT-116 |
|---|---|---|---|
| 2b | 15.98 ± 0.06 | 12.41 ± 0.05 | 16.32 ± 0.24 |
| 4a | 4.42 ± 0.12 | 4.23 ± 0.09 | 4.54 ± 0.19 |
| 5 | 3.21 ± 0.10 | 3.62 ± 0.21 | 3.46 ± 0.15 |
| 8 | 10.50 ± 0.14 | 13.82 ± 0.06 | 12.97 ± 0.08 |
| 9 | 6.84 ± 0.07 | 5.54 ± 0.27 | 8.75 ± 0.25 |
| 11 | 2.91 ± 0.23 | 2.41 ± 0.07 | 2.38 ± 0.26 |
| 13 | 0.81 ± 0.13 | 0.96 ± 0.09 | 1.12 ± 0.19 |
| Doxorubicin | 0.90 ± 0.02 | 1.21 ± 0.08 | 0.51 ± 0.03 |
| Compounds | COX-1 | COX-2 | SI | EGFR |
|---|---|---|---|---|
| 2b | 4.42 ± 1.67 | 5.26 ± 0.49 | 0.84 | — |
| 4a | 28.79 ± 1.02 | 1.17 ± 0.15 | 24.61 | 0.3 ± 0.01 |
| 5 | 40.32 ± 3.35 | 0.83 ± 0.4 | 48.58 | 0.9 ± 0.01 |
| 8 | 1.62 ± 1.21 | 2.73 ± 0.04 | 0.59 | — |
| 9 | 38.72 ± 1.27 | 2.21 ± 0.14 | 17.52 | — |
| 11 | 37.96 ± 0.66 | 0.62 ± 0.07 | 61.23 | 0.6 ± 0.04 |
| 13 | 30.41 ± 1.63 | 0.46 ± 0.06 | 66.11 | 0.4 ± 0.02 |
| Erlotinib | — | — | — | 0.08 ± 0.03 |
| Indomethacin | 0.52 ± 0.02 | 0.84 ± 0.4 | 0.62 | — |
| Celecoxib | 29.49 ± 1.63 | 0.34 ± 0.06 | 86.74 | — |
3. Molecular docking studies
Docking studies have been carried out to elucidate the binding mode of compounds 13 and 11 as the most potent COX2 inhibitors and compounds 4a and 13 as the most potent EGFR inhibitors with the target COX2 (PDB ID: 3LN1)20,42 and EGFR (PDB ID: 1M17)43,44 enzymes using Discovery Studio software package. Firstly, the validation step was carried out via redocking of the ligand on both used crystal structures and the RMSD values were found to be less than 2 which proved the validity of the produced docking results.3.1. Molecular docking study of COX2 (PDB ID: 3LN1)
Analysis of the docking results of compound 13 revealed that it incorporated in the formation of six hydrogen bonds with Gln178, Leu338 (2 HB), Ser339 (2 HB) and Phe504 amino acid residues. In addition, compound 13 displayed several hydrophobic interactions such as Pi-Sigma, amide-pi shaped, alkyl and pi–alkyl interactions with Val335, Asp501, Ala502, Val509 and Ala513 amino acid residues as presented in Fig. 2.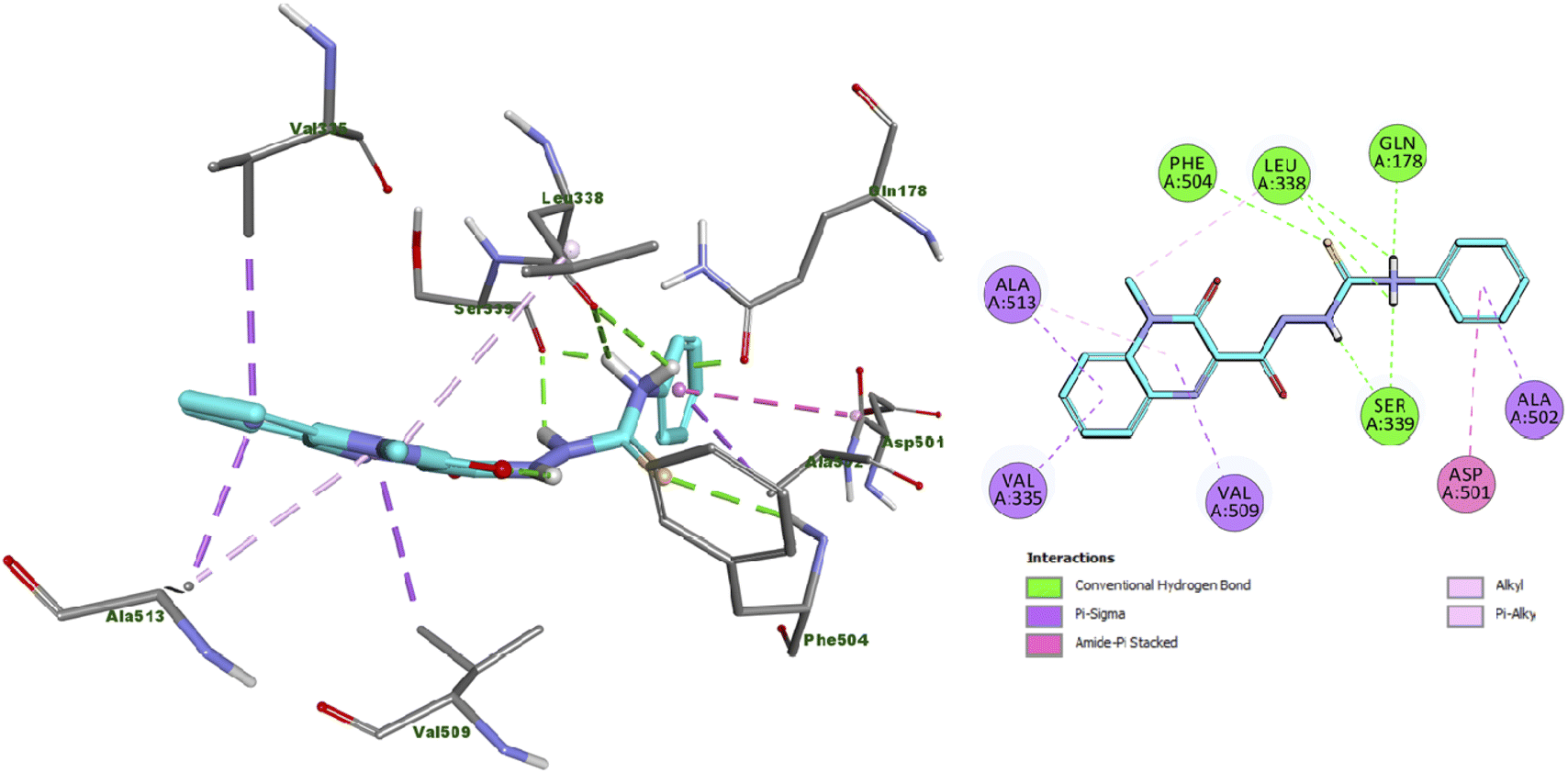 | ||
| Fig. 2 Docking and binding mode of 13 into the active site of COX2 (PDB ID: 3LN1) (A) 3D structure of 13 (cyan) (B) 2D structure of 13 (cyan). | ||
Moreover, the docking results of compound 11 (Fig. 3) showed that it engaged in three hydrogen bonds with Arg106, Arg499 and Ser516 amino acid residues. Additionally, it exhibited many hydrophobic interactions such as carbon hydrogen bond, pi-sigma, amide-pi stacked, alkyl and pi–alkyl interaction with Val335, Leu338, Tyr371, Ala502, Val509, Gly512 and Ala513 amino acid residues.
 | ||
| Fig. 3 Docking and binding mode of 11 into the active site of COX2 (PDB ID: 3LN1) (A) 3D structure of 11 (yellow) (B) 2D structure of 11 (yellow). | ||
3.2. Molecular docking study of EGFR (PDB ID: 1M17)
The docking results of compound 4a (Fig. 4) showed that it formed three hydrogen bonds with Met769 and Asp831 (two HBs). Additionally, compound 4a presented many hydrophobic interactions such as carbon hydrogen bond, pi-donor hydrogen bond, pi-sigma, alkyl and pi–alkyl interactions with Leu694, Val702, Lys721, Pro770 and Thr830 amino acid residues.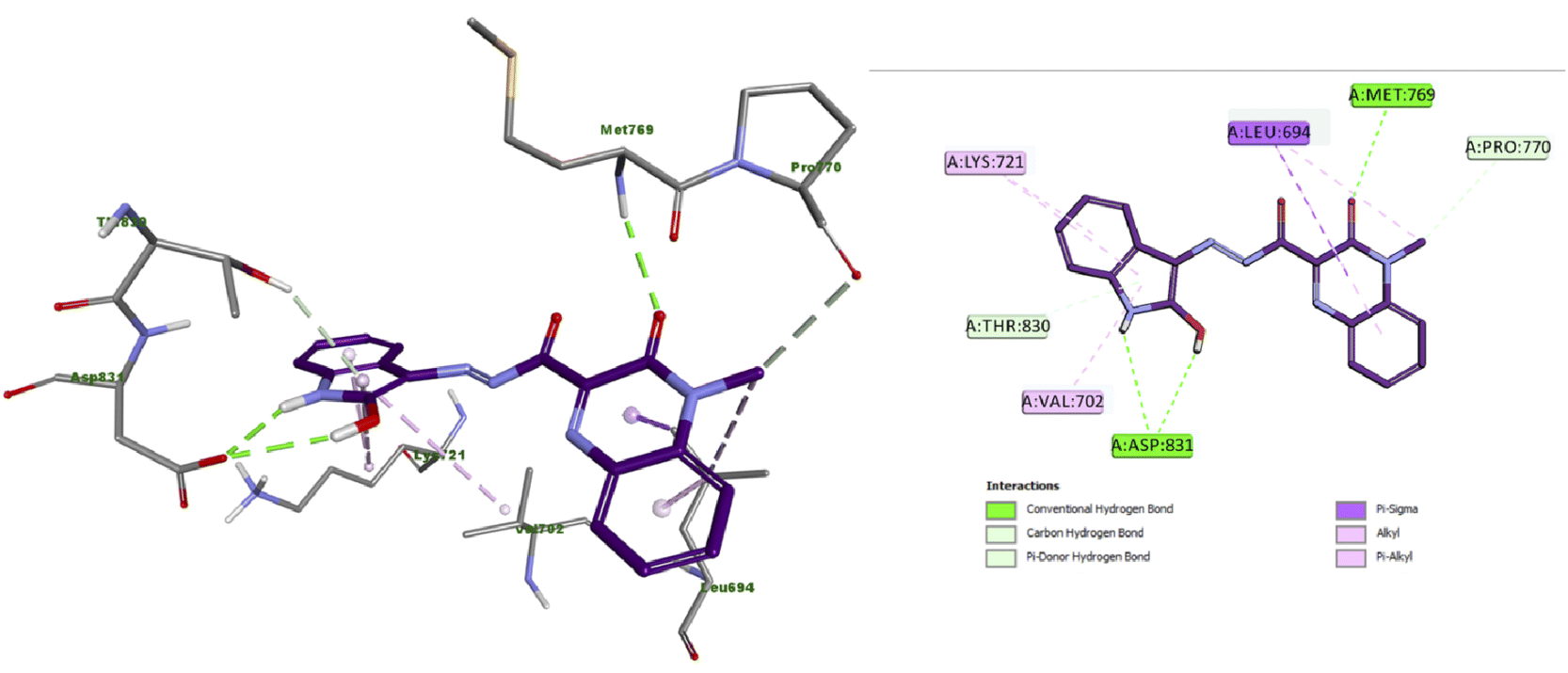 | ||
| Fig. 4 Docking and binding mode of 4a into the active site of EGFR (PDB ID: 1M17) (A) 3D structure of 4a (violet) (B) 2D structure of 4a (violet). | ||
Furthermore, the docking results of compound 13 displayed that it engaged in the formation of three hydrogen bonds with Met769 (two HB) and Pro770 amino acid residues. Also, compound 13 engaged in the formation of numerous hydrophobic interactions as pi-sigma, alkyl and pi–alkyl interactions with Leu694, Val702, Val721, Lys721 and Leu820 amino acid residues as illustrated in Fig. 5.
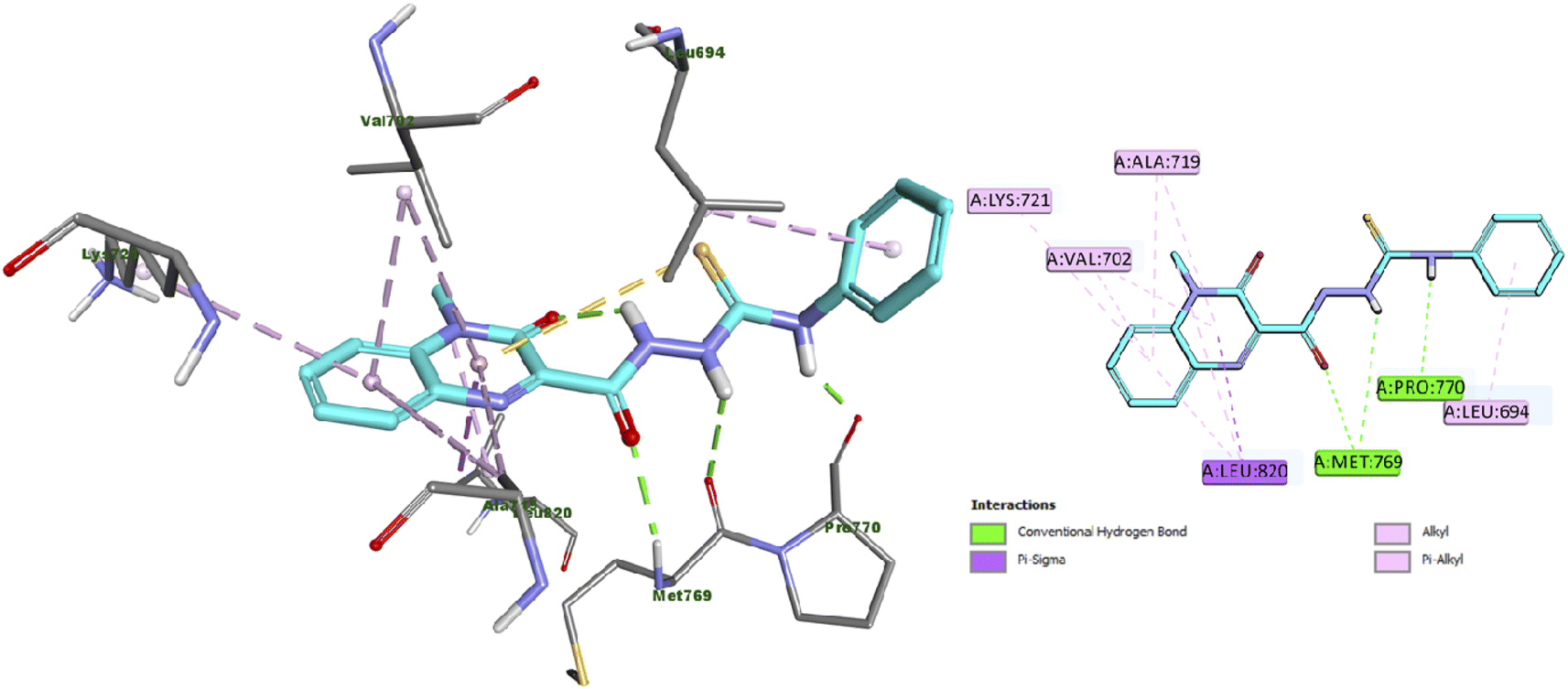 | ||
| Fig. 5 Docking and binding mode of 13 into the active site of EGFR (PDB ID: 1M17) (A) 3D structure of 13 (cyan) (B) 2D structure of 13 (cyan). | ||
Collectively, the docking results were in good agreement with the biological screening results suggesting that compounds 4a, 5, 11 and 13 are promising dual anticancer candidates via inhibition of EGFR-TK enzyme and anti-inflammatory agents via inhibition of COX-2 enzyme.
4. Calculations of Lipinski's rule and other in silico parameters
Oral bioavailability is a crucial factor in the development of therapeutically bioactive candidates.45,46 Thus, Lipinski formulated his rule (Rule of Five) for prediction of oral bioavailability as well as drug likeness.47 This rule depends on using some descriptors as molecular weight, lopP (partition coefficient), hydrogen bond acceptors and donors.48 Later, Veber added extra parameters for drug bioavailability such as nRB (number of rotatable bonds) and TPSA (topological polar surface area).49 Therefore, prediction of Lipinski rule of five and Veber's standards for the most potent quinoxaline derivatives 4a, 5, 11 and 13 were performed via the Pre-ADMET on line server. As shown in Table 4, all the estimated quinoxaline derivatives have no violation and in full accordance to Lipinski's rule and Veber's standards.| Comp. | MWc | Log Pd | HBAe | HBDf | nRBg | nVsh | TPSAi | % ABSj |
|---|---|---|---|---|---|---|---|---|
| a Reference values of Lipinski.b Reference values of Veber.c MW, molecular weight.d LogP, lipophilicity (O/W).e HBA, number of hydrogen bond acceptors.f HBD, number of hydrogen bond donors.g nRB, number of rotatable bonds.h nVs, number of Lipinski rule violations.i TPSA, topological polar surface area (PSA) (Å2).j % ABS, percentage of oral absorption. | ||||||||
| Lipinskia | ≤500 | ≤5 | ≤10 | ≤5 | — | ≤1 | — | — |
| Veberb | — | — | — | — | ≤10 | — | ≤140 | — |
| 4a | 347.33 | 1.88 | 8 | 2 | 2 | 0 | 109.22 | 71.13 |
| 5 | 388.38 | 2.57 | 8 | 1 | 3 | 0 | 106.57 | 72.23 |
| 11 | 364.36 | 1.18 | 8 | 2 | 5 | 0 | 110.16 | 70.99 |
| 13 | 353.41 | 1.69 | 7 | 3 | 5 | 0 | 88.05 | 78.62 |
The molecular weight of all predicted quinoxaline derivatives was less than 500, also, they presented LogP values in the range of 1.18 to 2.57 (LogP < 5) and the number of hydrogen bond acceptors and donors in all derivatives were in the acceptable range (HBA < 10 and HBD < 5), which indicated that all estimated quinoxaline derivatives met all criteria of Lipiski's rule of five. Regarding Veber's standards, the number of rotatable bonds were less than 10 and the topological polar surface areas (TPSA) were found in the range of 88.05 to 110.16 Å2 (<140 Å2), which accorded to Veber's rule. The percentage of oral absorption (% ABS) values of all estimated quinoxaline derivatives ranged from 70.99% to 78.62%, indicating that these derivatives would have acceptable molecular flexibilities and accordingly good membrane permeability and good oral bioavailability. Based on these results, we could conclude that hybrids 4a, 5, 11 and 13 can be served as good orally-absorbed dual acting antitumor and anti-inflammatory candidates and these properties can be improved by further modifications and SAR studies.
5. Structure activity relationship
Study of the structure activity relationship (SAR), as illustrated in Fig. 6, showed that, presence of hydrazone or hydrazide group is essential for both anticancer and COX-2 inhibitory activities. Moreover, polar hyrazones or polar groups are not favored and results in decrease or abolishment of the anticancer or COX-2 inhibitory activities. Additionally, the presence of oxygen at position 2 is essential for activity if it is replaced with other group, as in compound 10, results in significant decrease in both the potency and broadness. Ongoing throughout the results, it is obvious that the hydrazide obtained from the reaction of compound 1 with ethylacetoacetate (8), benzoylacetone (9), showed high activity; when R1 = phenyl and R2 = methyl (9) > R1 = methyl and R2 = ethoxy (8). Also, introducing the coumarin moiety, significantly improved both the anticancer and COX-2 inhibitory activities (5). Shifting to isatin (4a) enhanced the activity as anticancer and COX-2 inhibitor, while replacement with N-methyl isatin (4b) resulted in decrease in the anticancer activity. N-benzylidene derivatives(2a–d)are still active, in particular, inpresence of lipophilic electron withdrawing group as Cl (2b) is the most potent while substitution with polar donating groups such as OH or dioxolyl group (2c and 2d, respectively) led to significant decrease in the activity. The order of activity is as follows; Cl > H > OH > dioxolyl. On contrary, hydrazides obtained from polar aldehydes such as arabinose or glucoseled to a dramatic decrease in anticancer activity. Additionally, masking the hydrazide group via incorporation of five membered rings remarkably decreased the anticancer activity (12, 14 and 15). Finally, substitution with ethyl benzoyl acetate (11), phenyl isothiocyanate (13), seems to be the optimum for anticancer and COX-2 inhibitory activities.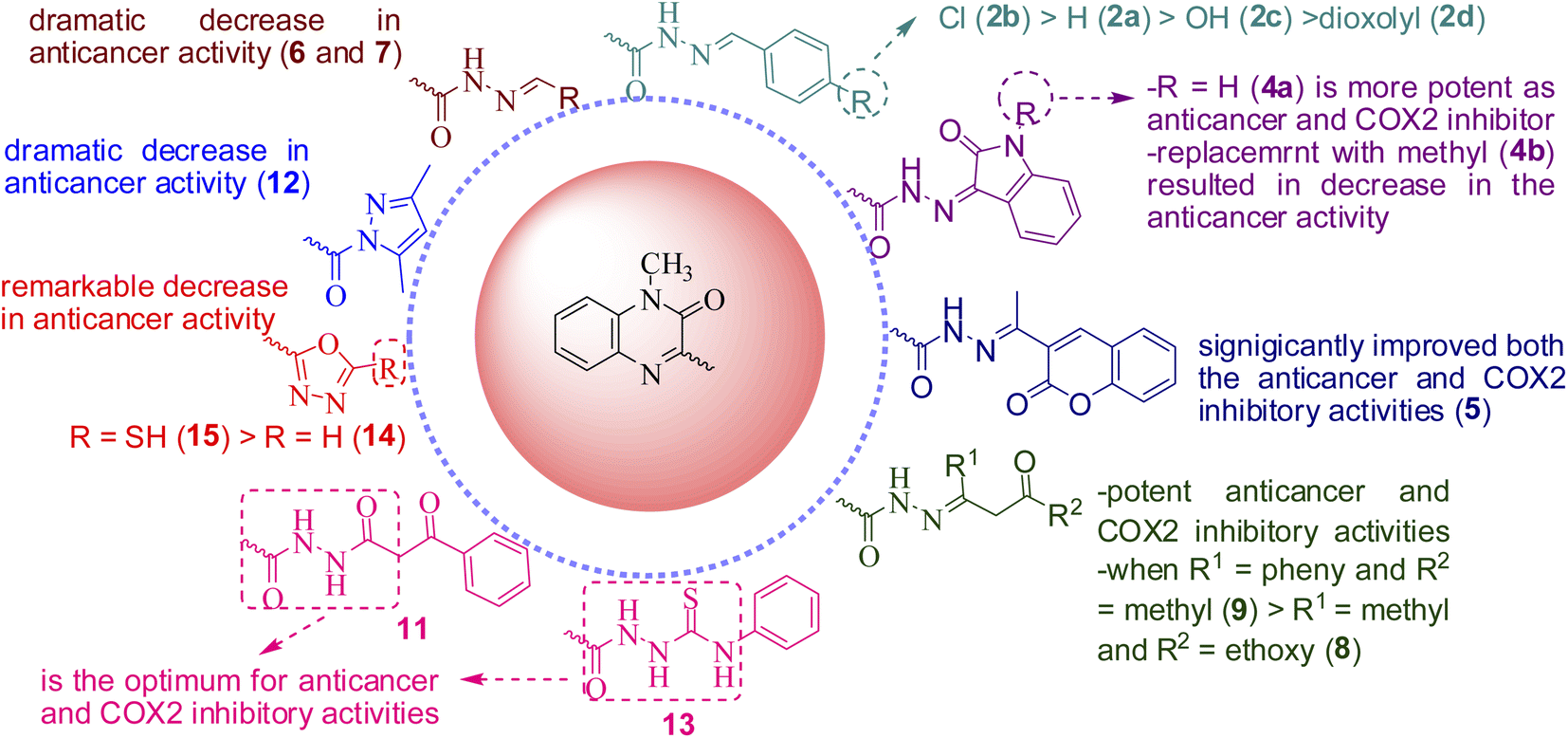 | ||
| Fig. 6 Structure activity relationship (SAR) for the newly synthesized quinoxaline derivatives. | ||
6. Conclusion
In summary, 18 novel quinoxaline derivatives have been synthesized and evaluated for their anticancer and COX inhibitory activities. Most of the tested compounds displayed good anticancer and COX-2 inhibitory activities. In particular, quinoxaline derivatives 11 and 13 exhibited potent anticancer activity against the three cancer cell lines with IC50 values ranging from 0.81 μM to 2.91 μM. Also, compounds 4a and 5 showed strong anticancer activity against the three-cancer cell line with IC50 values ranging from 3.21 μM to 4.54 μM. These derivatives strongly inhibited EGFR with IC50 values in the range of 0.3–0.9 μM. Regarding COX enzyme inhibition, compounds 4a, 5, 11 and 13 were more potent and selective against COX-2 than COX-1, among them, compounds 11 and 13 were the most potent as COX-2 inhibitors and showed higher potency against COX-2 (IC50 = 0.62 and 0.46 μM, respectively) more than COX-1 (IC50 = 37.96 and 30.41 μM, respectively) with selectivity indexes (SI) of 61.23 and 66.11, respectively. Compound 5 comes next with IC50 = 0.83 μM and SI of 48.58. Finally, compounds 4a were the least active with IC50 values equal to 1.17 μM and SI of 24.61. The molecular docking into the catalytic binding pocket of the both protein receptors; EGFR (PDB ID: 1M17) and COX-2 (PDB ID: 3LN1) strongly correlated with the biological results. The calculated parameters of Lipinski's rule of five were and Veber's standards revealed that compounds 4a, 5, 11 and 13 had a reasonable drug-likeness with acceptable physicochemical properties. Based on the obtained results of biological investigation as well as molecular docking study and physiochemical parameters, it could be concluded that quinoxalinehydrazide derivatives, particularly, 4a, 5, 11 and 13 are promising scaffold for innovation and discovery of new bioactive candidates. Moreover, compounds 4a, 5, 11 and 13 might be used as promising orally absorbed dual anticancer candidates via inhibition of EGFR enzyme and anti-inflammatory agents via inhibition of COX-2 enzyme and entitled to be used as a future template for further modifications and more SAR study.7. Experimental
7.1. Chemistry
For details; see ESI File.†O), 1640 (CO) cm−1; 1H NMR (DMSO-d6) δ: 12.47 (s, 1H, NH disappeared on addition of D2O), 8.35 (s, 1H, CHSchiffbase), 8.02–7.58 (m, 3H, Harom.), 7.57–7.18 (m, 6H, Harom.), 3.74 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 166.9, 159.4, 154.5, 154.0, 152.5, 149.9, 147.9, 145.3, 134.1, 133.9, 133.9, 133.7, 133.0, 132.3, 132.1, 131.5, 131.1, 130.6, 130.1, 128.8, 128.7, 127.8, 127.0, 124.6, 124.0, 114.9, 114.8, 29.6, 29.1. Anal. calcd for C17H14N4O2 (306.32): C, 66.66; H, 4.61; N, 18.29; O, 10.45, found: C, 66.62; H, 4.65; N, 18.33.O) cm−1; 1H NMR (DMSO-d6) δ: 12.27 (s, 1H, NH disappeared on addition of D2O), 8.06 (s, 1H, CHSchiffbase), 7.98–7.68 (dd, 1H, Harom.), 7.83–7.63 (m, 3H, Harom.), 7.58–7.54 (d, 1H, Harom.), 7.53–7.43 (m, 1H, Harom.), 7.39 (s, 2H, Harom.),3.71 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 166.9, 160.3, 154.5, 153.3, 152.5, 150.6, 148.0, 144.2, 135.3, 134.9, 134.2, 133.8, 133.3, 132.9, 132.8, 132.0, 132.0, 131.9, 130.5, 130.1, 129.4, 129.4, 129.4, 128.7, 124.6, 124.4, 115.7, 115.6, 29.6, 29.4. Anal. calcd for C17H13ClN4O2 (340.76): C, 59.92; H, 3.85; Cl, 10.40; N, 16.44. Found: C, 59.88; H, 3.82; Cl, 10.43; N, 16.47.O) cm−1; 1H NMR (DMSO-d6) δ: 12.03 (s, 1H, NH disappeared on addition of D2O), 10.09–9.87 (d, 1H, OH), 7.96–7.94 (m, 1H, Harom.), 7.90–7.88 (d, 1H,CH Schiffbase), 7.80–7.70 (m, 2H, Harom.), 7.68–7.58 (d, 1H, Harom.), 7.54–7.46 (m, 1H, Harom.), 7.22–7.16 (d, 1H, Harom.), 6.90–6.85 (d, 1H, Harom.), 6.73–6.69 (d, 1H, Harom.), 3.71 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 166.5, 160.2, 159.9, 159.7, 154.8, 153.4, 152.5, 150.7, 149.6, 145.7, 134.1, 133.8, 132.8, 132.0, 131.9, 131.9, 130.5, 130.0, 129.7, 128.9, 125.3, 125.0, 124.6, 124.4, 116.2, 116.1, 115.6, 115.6, 29.6, 29.4. Anal. calcd for C17H14N4O3 (322.32): C, 63.35; H, 4.38; N, 17.38. Found: C, 63.35; H, 4.38; N, 17.38.O) cm−1; 1H NMR (DMSO-d6) δ: 12.10 (s, 1H, NH disappeared on addition of D2O), 8.25–6.85 (m, 8H, CH + Harom.), 6.00 (s, 2H, CH2), 3.69 (s, 3H, N–CH3). Anal. calcd for C18H14N4O4 (350.33): C, 61.71; H, 4.03; N, 15.99. Found: C, 61.75; H, 3.99; N, 15.96.O), 1660 (CO) cm−1; 1H NMR (DMSO-d6) δ: 12.43–12.18 (dd, 2H, 2NH disappeared on addition of D2O), 8.86–7.28 (m, 14H, CH + Harom.), 3.71 (s, 6H, 2N–CH3). Anal. calcd for C18H14N4O3 (334.33): C, 64.66; H, 4.22; N, 16.76. Found: C, 64.62; H, 4.19; N, 16.81.O) cm−1; 1H NMR (DMSO-d6) δ: 13.21 (s, 1H, NH disappeared on addition of D2O), 10.93 (s, 1H, NH disappeared on addition of D2O), 8.26–8.19 (d, 1H, Harom.), 8.12–8.08 (d, 1H, Harom.), 7.92–7.87 (t, 1H, Harom.), 7.82–7.78 (d, 1H, Harom.), 7.62–7.56 (t, 1H, Harom.), 7.51–7.45 (t, 1H, Harom.), 7.21–7.16 (t, 1H, Harom.), 7.00–6.96 (d, 1H, Harom.), 3.83 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 166, 160, 159, 155, 144, 143, 134.1, 133.8, 131.5, 125.3, 125.1, 123.0, 122.6, 115.9, 115.7, 115.5, 111.6, 30.3. Anal. calcd for C18H13N5O3 (347.33): C, 62.24; H, 3.77; N, 20.16. Found: C, 62.21; H, 3.81; N, 20.13.O) cm−1; 1H NMR (DMSO-d6) δ: 13.26 (s, 1H, NH disappeared on addition of D2O), 8.28–7.15 (m, 8H, Harom.), 3.83 (s, 3H, N–CH3), 3.24 (s, 3H, N–CH3). Anal. calcd for C19H15N5O3 (361.35): C, 63.15; H, 4.18; N, 19.38. Found: C, 63.11; H, 4.21; N, 19.41.O) cm−1; 1H NMR (DMSO-d6) δ: 12.01 (s, 1H, NH disappeared on addition of D2O), 8.34–7.29 (m, 9H, CH + Harom.), 3.75 (s, 3H, N–CH3 quinoxaline), 2.29 (s, 3H, CH3). Anal. calcd for C21H16N4O4 (388.38): C, 64.94; H, 4.15; N, 14.43. Found: C, 64.91; H, 4.18; N, 14.47.O) cm−1; 1H NMR (DMSO-d6) δ: 10.41 (s, 1H, NH exchanged with D2O), 7.96–7.92 (d, 1H, Harom.), 7.80–7.74 (t, 1H, Harom.), 7.70–7.65 (d, 1H, Harom.), 7.51–7.45 (t, 1H, Harom.), 6.15–6.10 (t, 1H, OH), 6.08–6.04 (d, 1H, CH), 5.04–5.03 (d, 1H, OH), 4.96–4.93 (d, 1H, OH), 4.57–4.56 (d, 1H, OH), 4.46–4.43 (d, 1H, CH), 3.96–3.91 (t, 1H, CH), 3.80–3.76 (d, 1H, CH), 3.69 (s, 3H, NCH3), 3.55–3.49 (m, 2H, CH2); 13C NMR (DMSO-d6) δ: 166.6, 162.2, 155.3, 153.6, 149.9, 134.0, 132.6, 131.8, 130.6, 124.5, 115.6, 93.2, 91.3, 69.7, 68.0, 67.8, 63.1, 29.6. Anal. calcd for C15H18N4O6 (350.33): C, 51.43; H, 5.18; N, 15.99. Found: C, 51.39; H, 5.21; N, 16.02.IR (KBr) ν: 3570 (OH), 3441 (OH), 3388 (OH), 3366 (OH), 3258 (OH), 3169 (NH), 3070 (CH-aromatic), 2918, 2850 (CH-aliphatic), 1704 (CO), 1649 (CO) cm−1; 1H NMR (DMSO-d6) δ: 10.33 (s, 1H, NH exchanged with D2O), 7.93–7.91 (d, 1H, Harom.), 7.81–7.74 (t, 1H, Harom.), 7.69–7.58 (d, 1H, Harom.), 7.50–7.40 (t, 1H, Harom.), 6.09–6.05 (t, 1H, OH), 5.63–5.59 (d, 1H, CH), 5.11–5.08 (t, 1H, OH), 5.02–4.98 (d, 1H, OH), 4.95–4.92 (d, 1H, OH), 4.45–4.43 (t, 1H, OH), 3.92–3.88 (dd, 1H, CH), 3.68 (s, 3H, NCH3), 3.51–2.84 (m, 5H, Haliph). Anal. calcd for C16H20N4O7 (380.35): C, 50.52; H, 5.30; N, 14.73. Found: C, 50.48; H, 5.35; N, 14.77.
O), 1631 (CO) cm−1; 1H NMR (DMSO-d6) δ: 11.06 (s, 1H, NH disappeared on addition of D2O), 7.99–7.95 (d, 1H, Harom.), 7.83–7.77 (t, 1H, Harom.), 7.54–7.48 (t, 1H, Harom.), 4.16–4.11 (q, 2H, CH2), 3.72 (s, 3H, N–CH3), 3.46 (s, 2H, CH2CO), 1.99 (s, 3H, CH3CN), 1.27–1.22 (t, 3H, CH3CH2). 13C NMR (DMSO-d6) δ: 169.9, 169.2, 167.5, 159.7, 155.2, 154.2, 153.2, 152.3, 149.7, 149.3, 134.1, 134.0, 133.7, 132.9, 132.3, 131.9, 131.7, 130.7, 129.9, 124.7, 124.2, 115.6, 115.5, 61.0, 60.4, 44.3, 44.2, 29.8, 29.3, 17.2, 17.0, 14.5, 14.4, 14.1. Anal. calcd for C16H18N4O4 (330.34): C, 58.17; H, 5.49; N, 16.96. Found: C, 58.21; H, 5.53; N, 17.01.O), 1626 (CO) cm−1; 1H NMR (DMSO-d6) δ: 12.73 (s, 1H, NH exchanged with D2O), 11.43 (s, 1H, OH exchanged with D2O), 8.00–7.96 (d, 1H, Harom.), 7.95–7.91 (d, 1H, Harom.), 7.84–7.79 (t, 1H, Harom.), 7.74–7.70 (d, 1H, Harom.), 7.63–7.56 (t, 1H, Harom.), 7.54–7.46 (t, 3H, Harom.), 7.44–7.39 (t, 1H, Harom.), 6.07 (s, 1H, CH), 3.73 (s, 3H, NCH3), 2.18 (s, 3H, CH3). Anal. calcd for C20H18N4O3 (362.38): C, 66.29; H, 5.01; N, 15.46. Found: C, 66.33; H, 4.97; N, 15.49.O) cm−1; 1H NMR (DMSO-d6) δ: 9.42 (s, 1H, NH disappeared on addition of D2O), 7.25–6.96 (dd, 2H, Harom.), 6.82–6.77 (t, 1H, Harom.), 6.70–6.66 (d, 1H, Harom.), 4.43 (s, 1H, CH), 4.34 (s, 2H, NH2 disappeared on addition of D2O), 3.39 (s, 1H, NH disappeared on addition of D2O), 2.79 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 166.0, 163.8, 136.6, 135.1, 129.0, 123.6, 118.4, 114.5, 111.2, 66.0, 35.5, 29.0. Anal. calcd for C10H12N6O (232.24): C, 51.72; H, 5.21; N, 36.19. Found: C, 51.68; H, 5.24; N, 36.23.O), 1633 (CO) cm−1; 1H NMR (DMSO-d6) δ: 11.39 (s, 1H, NH disappeared on addition of D2O), 11.05 (s, 1H, NH disappeared on addition of D2O), 8.06–7.47 (m, 9H, Harom.), 4.1 (s, 2H, CH2), 3.70 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 194.5, 169.9, 168.9, 164.3, 160.3, 153.6, 148.9, 136.5, 134.1, 133.0, 132.0, 130.7, 129.3, 129.2, 128.9, 125.8, 124.7, 115.7, 45.3, 29.7. Anal. calcd for C19H16N4O4 (364.35): C, 62.63; H, 4.43; N, 15.38. Found: C, 62.66; H, 4.39; N, 15.42.O), 1648 (CO) cm−1; 1H NMR (CDCl3) δ: 7.99–7.94 (d, 1H, Harom.), 7.68–7.64 (t, 1H, Harom.), 7.46–7.40 (t, 1H, Harom.), 7.30 (s, 1H, Harom.), 6.07 (s, 1H, Hpyrazole), 3.77 (s, 3H, N–CH3), 2.72 (s, 3H, CH3), 2.16 (s, 3H, CH3). 13C NMR (CDCl3) δ: 159.6, 149.5, 148.3, 148.1, 139.6, 129.1, 127.6, 126.9, 126.2, 119.3, 109.2, 107.2, 24.3, 9.2, 9.1. Anal. calcd for C15H14N4O2 (282.30): C, 63.82; H, 5.00; N, 19.85. Found: C, 63.86; H, 4.96; N, 19.89.O), 1631 (CO) cm−1. 1H NMR (DMSO-d6) δ: 11.02 (br, 1H, NH disappeared on addition of D2O), 10.20 (br, 1H, NH disappeared on addition of D2O), 9.81 (br, 1H, NH disappeared on addition of D2O), 8.00–7.98 (d, 1H, Harom.), 7.85–7.81 (t, 1H, Harom.), 7.77–7.73 (d, 1H, Harom.), 7.67–7.63 (d, 2H, Harom.), 7.58–7.52 (t, 1H, Harom.), 7.42–7.36 (t, 2H, Harom.), 7.23–7.17 (t, 1H, Harom.), 3.76 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 154.3, 139.3, 133.9, 133.2, 132.3, 130.8, 128.8, 125.0, 115.9, 30.0. Anal. calcd for C17H15N5O2S (353.40): C, 57.78; H, 4.28; N, 19.82; S, 9.07. Found: C, 57.82; H, 4.24; N, 19.78; S, 9.12.O) cm−1. 1H NMR (DMSO-d6) δ: 9.52 (s, 1H, Hoxadiazole), 7.99–7.41 (m, 4H, Harom.), 3.72 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 161.0, 155.5, 152.4, 141.3, 134.7, 133.7, 132.2, 131.0, 124.6, 115.7, 29.9. Anal. calcd for C11H8N4O2 (228.21): C, 57.89; H, 3.53; N, 24.55. Found: C, 57.93; H, 3.50; N, 24.51.IR (KBr) ν: 3233 (NH), 3046 (CH-aromatic), 2917, 2847 (CH-aliphatic), 1645 (CO) cm−1. 1H NMR (DMSO-d6) δ: 14.98 (br, 1H, NH disappeared on addition of D2O), 7.99–7.97 (d, 1H, Harom.), 7.82–7.77 (t, 1H, Harom.), 7.68–7.66 (d, 1H, Harom.), 7.52–7.47 (t, 1H, Harom.), 3.70 (s, 3H, N–CH3). 13C NMR (DMSO-d6) δ: 157.4, 151.7, 139.1, 134.5, 133.7, 132.1, 130.9, 124.7, 115.6, 29.8. Anal. calcd for C11H8N4O2S (260.27): C, 50.76; H, 3.10; N, 21.53; O, 12.29; S, 12.32. Found: C, 50.80; H, 3.14; N, 21.49; S, 12.33.
7.2. Biological evaluation
8. Docking methodology
For docking analysis, Discovery Studio 2.5 software (Accelrys Inc., San Diego, CA, USA) was used. Fully automated docking tool using “Dock ligands (CDOCKER)” protocol running on Intel (R) core (TM) i32370 CPU@2.4 GHz 2.4 GHz, RAM Memory 2 GB under the Windows 7.0 system. The 3.5 ÅA 3D crystal structures of EGFR (PDB ID: 1M17)43,44 and COX-2 (PDB ID: 3LN1)20,42 were retrieved from protein data bank. See ESI File.†9. In silico prediction of physicochemical properties and pharmacokinetic profile
For prediction of Lipinski's rule (rule of five), molecular property prediction and pre-ADMET estimation; the free accesses to websites https://www.molsoft.com/servers.html and https://preadmet.bmdrc.kr/ were used.Conflicts of interest
There are no conflicts to declare.References
- M. F. Mohamed and G. E.-D. A. Abuo-Rahma, RSC Adv., 2020, 10, 31139–31155 RSC.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and I. H. Eissa, Bioorg. Chem., 2021, 114, 105105 CrossRef CAS PubMed.
- F. A. Rodrigues, S. Bomfim Ida, B. C. Cavalcanti, Ó. Pessoa Cdo, J. L. Wardell, S. M. Wardell, A. C. Pinheiro, C. R. Kaiser, T. C. Nogueira, J. N. Low, L. R. Gomes and M. V. de Souza, Bioorg. Med. Chem. Lett., 2014, 24, 934–939 CrossRef CAS PubMed.
- A. K. A. Bass, E.-S. M. Nageeb, M. S. El-Zoghbi, M. F. A. Mohamed, M. Badr and G. E.-D. A. Abuo-Rahma, Bioorg. Chem., 2022, 119, 105564 CrossRef CAS PubMed.
- A. K. A. Bass, M. S. El-Zoghbi, E.-S. M. Nageeb, M. F. A. Mohamed, M. Badr and G. E.-D. A. Abuo-Rahma, Eur. J. Med. Chem., 2021, 209, 112904 CrossRef CAS PubMed.
- I. Piotrowski, K. Kulcenty and W. Suchorska, Rep. Pract. Oncol. Radiother., 2020, 25, 422 CrossRef PubMed.
- F. R. Greten and S. I. Grivennikov, Immunity, 2019, 51, 27–41 CrossRef CAS PubMed.
- R. Medzhitov, Cell, 2010, 140, 771–776 CrossRef CAS PubMed.
- L. Wu, S. Saxena, M. Awaji and R. K. Singh, Cancers, 2019, 11, 564 CrossRef CAS PubMed.
- T. S. Ibrahim, A. M. Malebari and M. F. A. Mohamed, Pharmaceuticals, 2021, 14, 1177 CrossRef CAS PubMed.
- A. Mantovani, P. Allavena, A. Sica and F. Balkwill, Nature, 2008, 454, 436–444 CrossRef CAS PubMed.
- P. Igor, K. Katarzyna and S. Wiktoria, Rep. Pract. Oncol. Radiother., 2020, 35(3), 422–427 Search PubMed.
- B. L. Sng and S. A. Schug, Ann. Acad. Med. Singapore, 2009, 38, 960–966 Search PubMed.
- X. Y. Sun, C. Hu, X. Q. Deng, C. X. Wei, Z. G. Sun and Z. S. Quan, Eur. J. Med. Chem., 2010, 45, 4807–4812 CrossRef CAS PubMed.
- R. Eccles, J. Clin. Pharm. Ther., 2006, 31, 309–319 CrossRef CAS PubMed.
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, M. Abdel-Aziz, K. N. Dalby and T. S. Kaoud, Bioorg. Chem., 2017, 75, 242–259 CrossRef CAS PubMed.
- M. F. A. Mohamed, A. A. Marzouk, A. Nafady, D. A. El-Gamal, R. M. Allam, G. E.-D. A. Abuo-Rahma, H. I. El Subbagh and A. H. Moustafa, Bioorg. Chem., 2020, 105, 104439 CrossRef CAS PubMed.
- H. Kakuta, X. Zheng, H. Oda, S. Harada, Y. Sugimoto, K. Sasaki and A. Tai, J. Med. Chem., 2008, 51, 2400–2411 CrossRef CAS PubMed.
- M. G. Perrone, D. D. Lofrumento, P. Vitale, F. De Nuccio, V. La Pesa, A. Panella, R. Calvello, A. Cianciulli, M. A. Panaro and A. Scilimati, Pharmacology, 2015, 95, 22–28 CrossRef CAS PubMed.
- B. G. M. Youssif, M. F. A. Mohamed, M. M. Al-Sanea, A. H. Moustafa, A. A. Abdelhamid and H. A. M. Gomaa, Bioorg. Chem., 2019, 85, 577–584 CrossRef CAS PubMed.
- K. M. Lim, J. Y. Lee, S. M. Lee, O. N. Bae, J. Y. Noh, E. J. Kim, S. M. Chung and J. H. Chung, Br. J. Pharmacol., 2009, 156, 328–337 CrossRef CAS PubMed.
- L. Ma, C. Xie, Y. Ma, J. Liu, M. Xiang, X. Ye, H. Zheng, Z. Chen, Q. Xu, T. Chen, J. Chen, J. Yang, N. Qiu, G. Wang, X. Liang, A. Peng, S. Yang, Y. Wei and L. Chen, J. Med. Chem., 2011, 54, 2060–2068 CrossRef CAS PubMed.
- A. H. Abdelazeem, S. A. Abdelatef, M. T. El-Saadi, H. A. Omar, S. I. Khan, C. R. McCurdy and S. M. El-Moghazy, Eur. J. Pharm. Sci., 2014, 62, 197–211 CrossRef CAS PubMed.
- F. Colotta, P. Allavena, A. Sica, C. Garlanda and A. Mantovani, Carcinogenesis, 2009, 30, 1073–1081 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, Molecules, 2021, 26, 4742 CrossRef CAS PubMed.
- S. A. Khan, K. Saleem and Z. Khan, Eur. J. Med. Chem., 2007, 42, 103–108 CrossRef CAS PubMed.
- S. K. Suthar, N. S. Chundawat, G. Pal Singh, J. M. Padrón and Y. Kunwar Jhala, Eur. J. Med. Chem. Rep., 2022, 100040, DOI:10.1016/j.ejmcr.2022.100040.
- J. A. Pereira, A. M. Pessoa, M. N. D. S. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- H. Gao, E. F. Yamasaki, K. K. Chan, L. L. Shen and R. M. Snapka, Cancer Res., 2000, 60, 5937–5940 CAS.
- T. Uehara, Y. Minoshima, K. Sagane, N. H. Sugi, K. O. Mitsuhashi, N. Yamamoto, H. Kamiyama, K. Takahashi, Y. Kotake, M. Uesugi, A. Yokoi, A. Inoue, T. Yoshida, M. Mabuchi, A. Tanaka and T. Owa, Nat. Chem. Biol., 2017, 13, 675–680 CrossRef CAS PubMed.
- H. Gao, K. C. Huang, E. F. Yamasaki, K. K. Chan, L. Chohan and R. M. Snapka, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12168–12173 CrossRef CAS PubMed.
- S. D. Undevia, F. Innocenti, J. Ramirez, L. House, A. A. Desai, L. A. Skoog, D. A. Singh, T. Karrison, H. L. Kindler and M. J. Ratain, Eur. J. Cancer, 2008, 44, 1684–1692 CrossRef CAS PubMed.
- M. Liu, Y. Wang, W.-z. Wangyang, F. Liu, Y.-l. Cui, Y.-s. Duan, M. Wang, S.-z. Liu and C.-h. Rui, J. Agric. Food Chem., 2010, 58, 6858–6863 CrossRef CAS PubMed.
- S. Rollas and S. G. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS PubMed.
- G. Verma, A. Marella, M. Shaquiquzzaman, M. Akhtar, M. R. Ali and M. M. Alam, J. Pharm. BioAllied Sci., 2014, 6, 69–80 CrossRef PubMed.
- N. Radhoff and A. Studer, Chem. Sci., 2022, 13, 3875–3879 RSC.
- I. Khan, S. Zaib and A. Ibrar, Org. Chem. Front., 2020, 7, 3734–3791 RSC.
- Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed.
- E. A. Ahmed, M. F. A. Mohamed, A. Omran and H. Salah, Synth. Commun., 2020, 50, 2924–2940 CrossRef CAS.
- N. A. Qandeel, A. K. El-Damasy, M. H. Sharawy, S. M. Bayomi and N. S. El-Gohary, Bioorg. Chem., 2020, 102, 103890 CrossRef CAS PubMed.
- T. S. Ibrahim, A. M. Malebari and M. F. A. Mohamed, Pharmaceuticals, 2021, 14, 1177 CrossRef CAS PubMed.
- H. A. Abou-Zied, B. G. M. Youssif, M. F. A. Mohamed, A. M. Hayallah and M. Abdel-Aziz, Bioorg. Chem., 2019, 89, 102997 CrossRef CAS PubMed.
- B. J. Aungst, J. Pharm. Sci., 2017, 106, 921–929 CrossRef CAS PubMed.
- M. F. Mohamed, A. A. Marzouk, A. Nafady, D. A. El-Gamal, R. M. Allam, G. A. Abuo-Rahma, H. I. El Subbagh and A. H. Moustafa, Bioorg. Chem., 2020, 104439 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- M. A. Bakht, M. S. Yar, S. G. Abdel-Hamid, S. I. Al Qasoumi and A. Samad, Eur. J. Med. Chem., 2010, 45, 5862–5869 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- M. J. Uddin, P. N. P. Rao and E. E. Knaus, Bioorg. Med. Chem., 2004, 12, 5929–5940 CrossRef CAS PubMed.
- M. A. A. El-Sayed, N. I. Abdel-Aziz, A. A. M. Abdel-Aziz, A. S. El-Azab, Y. A. Asiri and K. E. H. ElTahir, Bioorg. Med. Chem., 2011, 19, 3416–3424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04498f |
| This journal is © The Royal Society of Chemistry 2022 |