
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMn-doped Co3O4 for acid, neutral and alkaline electrocatalytic oxygen evolution reaction†
Ana Luisa Silva a,
Laura M. Estevescd,
Ludmila P. C. Silvac,
Vitor S. Ramosbe,
Fabio B. Passosc and
Nakédia M. F. Carvalho*a
a,
Laura M. Estevescd,
Ludmila P. C. Silvac,
Vitor S. Ramosbe,
Fabio B. Passosc and
Nakédia M. F. Carvalho*a
aUniversidade do Estado do Rio de Janeiro, Departamento de Química Geral e Inorgânica, Rio de Janeiro, RJ 20550-900, Brazil. E-mail: nakedia@uerj.com
bUniversidade do Estado do Rio de Janeiro, Departamento de Engenharia Mecânica, Rio de Janeiro, RJ 20940-903, Brazil
cUniversidade Federal Fluminense, Departamento de Engenharia Química e de Petróleo, Niterói, RJ 24210-240, Brazil. E-mail: ludmilapcs@id.uff.br; fabiopassos@id.uff.br
dLAQV-REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal. E-mail: lm.esteves@fct.unl.pt
eUniversidade Federal Do Rio de Janeiro, Instituto de Macromoléculas Professora Eloisa Mano, Rio de Janeiro, 21941-598, RJ, Brazil
First published on 21st September 2022
Abstract
This work reports the application of Mn-doped Co3O4 oxides in the electrocatalytic oxygen evolution reaction (OER). The materials were characterized by structural, morphological, and electrochemical techniques. The oxides with higher Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn molar ratio presented a lower electron transfer resistance, and consequently the most promising OER activities. Pure Co3O4 shows an overpotential at j = 10 mA cm−2 of 761, 490, and 240 mV, at pH 1, 7, and 14, respectively, and a high TOF of 1.01 × 10−1 s−1 at pH 14. Tafel slopes around 120 mV dec−1 at acidic pH and around 60 mV dec−1 at alkaline pH indicate different OER mechanisms. High stability for Co3O4 was achieved for up to 15 h in all pHs, and no change in the structure and morphology after the electrocatalysis was observed. The reported excellent OER activity of the Mn–Co oxides in a wide pH range is important to broaden the practical applicability in different electrolyte solutions.
:Mn molar ratio presented a lower electron transfer resistance, and consequently the most promising OER activities. Pure Co3O4 shows an overpotential at j = 10 mA cm−2 of 761, 490, and 240 mV, at pH 1, 7, and 14, respectively, and a high TOF of 1.01 × 10−1 s−1 at pH 14. Tafel slopes around 120 mV dec−1 at acidic pH and around 60 mV dec−1 at alkaline pH indicate different OER mechanisms. High stability for Co3O4 was achieved for up to 15 h in all pHs, and no change in the structure and morphology after the electrocatalysis was observed. The reported excellent OER activity of the Mn–Co oxides in a wide pH range is important to broaden the practical applicability in different electrolyte solutions.
1. Introduction
In recent years, much research has been done to remedy the environmental damage caused by the combustion of fossil fuels. The development of efficient technologies for the conversion and storage of energy using renewable sources has been intensified, especially those related to the use of solar energy.1 However, some factors hamper the implementation of a solar-based generation system, such as the intermittence of sunlight and the high cost of silicon cells. These barriers make even more urgent the search for new materials to play the different functions in an artificial leaf device, such as light-harvesting, electron conduction, and hydrogen/oxygen catalytic generation from the water splitting reaction.2,3 Hydrogen has been claimed as the most promising clean fuel, with high energy output, which could be elegantly produced by the electrochemical water splitting without generating carbon in the process.4,5Water splitting is the chemical reaction in which water is broken down into O2 and H2 (eqn (1)), constituted by the oxygen evolution reaction (OER) (eqn (2) and (3)) and the hydrogen evolution reaction (HER). However, water splitting is an uphill process with a large overpotential, mainly because of the thermodynamically and kinetically unfavorable OER that is the bottleneck of the process.5 Thus, the development of efficient, stable, low-cost, and environmentally benign OER electrocatalysts is essential to diminish the reaction overpotential and makes the water splitting feasible for practical applications.6
| 2H2O → O2 + 2H2 | (1) |
| OER (pH 0): 2H2O → O2 + 4H+ + 4e− Eanode0 = −1.23 V vs. NHE | (2) |
| OER (pH 14): 4OH− → O2 + 2H2O + 4e− Eanode0 = −0.40 V vs. NHE | (3) |
Nanoscience is highlighted as one of the most attractive and promising areas for the technological development of this century. Materials at the nanoscale have become very popular in several scientific areas because of their unique physical and chemical properties, with a substantial increase in solar energy storage and transformation. High surface area and consequently more exposure of the active sites are important features for catalytic activity and has accelerated the pursuit of new materials for their use in water splitting.5,7 Nano-oxides based on transition metals have been efficiently employed as catalysts for OER.8–11 First-row transition elements are of particular importance due to their high Earth-abundance, highly oxidized redox couples as Co3+/Co4+ and Mn3+/Mn4+, and robustness in harsh chemical conditions. However, they still present limitations as lower catalytic rates and poor electrical conductivities, compared to the highly active, high cost and scarce, noble metal oxides RuO2 and IrO2.12
Cobalt oxides (CoOx) present high catalytic activity for OER in alkaline solution, especially the Co3O4 spinel that has been widely investigated as an attractive anode material with high activity, corrosion resistance in alkaline electrolytes, excellent redox property originating from its Co2+/Co3+ mixed valence nature.1,3,5,6,13–15 Furthermore, the development of acid-stable OER electrocatalysts is still a challenge.5 The electrolyte pH has a significant impact on the choice of the electrocatalyst, for instance, for the application of proton exchange membrane (PEM) electrolyzer for hydrogen production at acid pH, active and resistant catalysts are necessary.16,17 CoOx has shown higher OER electrocatalytic activity and low stability in alkaline solution, but has been poorly investigated in acid and neutral pH.
A strategy that has gained popularity is the combination of different elements to tune the activity through synergistic effects. Mn–Co mixed oxides have been prepared by several methods and have shown improved electrocatalytic activity in relation to the respective pure oxides. The spinel MnCo2O4 prepared with citric acid and ethylene glycol,17,18 or oxalic acid,19 at different calcination temperatures; or MnxCo3−xO4 prepared via a sol–gel EDTA–citric acid–ethylene glycol method at different Mn:Co molar ratio,20 were tested in alkaline electrolyte with low overpotentials at j = 10 mA cm−2 around 350 mV. Physical mixture of α-MnO2 and Co3O4 also showed improved OER electroactivity in alkaline solution.21 Mixed Mn-promoted mesoporous Co3O4 reported OER electrocatalytic performance comparable to the precious metals in alkaline media.6
The main goal of this work is to prepare active and stable electrocatalysts for OER in a wide range of pH. To address this question, Mn-doped Co3O4 in different Mn:Co molar ratios were prepared to improve the physical-chemical, electrochemical and catalytic properties by the synergistic effect between Mn and Co, and to reach low overpotential and high stability. The oxides were characterized by powder X-ray diffraction (XRD) and Rietveld refinement, Raman and X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), field emission scanning electron microscopy (FESEM), and atomic force microscopy (AFM). Electrochemical characterization was carried out by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The materials were tested in the electrocatalytic OER in acidic, neutral, and alkaline pH electrolytes. Low overpotentials, high TOF, and low Tafel slopes were obtained in all conditions. The materials showed high stability and preservation of structure and morphology after OER at all pHs. It is known that Mn–Co bimetallic oxides are efficient for OER, but in general, these were tested in alkaline electrolytes. This work reports promising results in acidic and neutral pH as well as in alkaline conditions.
2. Experimental
2.1. Reagents and chemicals
The chemicals and reagents were used as received: Co(NO3)2·6H2O ≥ 98.0%, Mn(NO3)2·4H2O ≥ 97.0%, NaOH ≥ 97.0%, NH2CONH2 (urea) 99.5% and Na2SO4 ≥ 99.0% from Sigma-Aldrich; ethyl alcohol > 99.0% and isopropyl alcohol > 99.0% from Tedia; and HNO3 ≥ 65% from Merck. 5% Nafion 117 solution and fluoride tin oxide (FTO) glass plates with 13 Ω per square surface resistivity were purchased from Sigma-Aldrich. FTO glass plates were previously cut into slides of 1 cm × 3.5 cm (W × H). Before deposition, the FTO slides were first sonicated in soap water, then in ethanol and acetone for 10 minutes, and finally rinsed with deionized water before use. All solutions were prepared by direct dissolution of suitable reagents in distilled water.2.2. Synthesis of the oxides
Pure and Mn-doped Co3O4 were synthesized by a hydrothermal methodology in presence of urea, in different molar ratios of Mn:Co (0:1, 1:4, 1:2, and 1:1) named respectively Co3O4, Mn@Co3O4-1, Mn@Co3O4-2, and Mn@Co3O4-3. A typical procedure is exemplified for Mn@Co3O4-2, where a mixture of 6.04 g (20 mmol) of Co(NO3)2·6H2O, 2.48 g (10 mmol) of Mn(CH3CO2)2·4H2O, and 1.25 g (20 mmol) of CO(NH2)2 were dissolved in 60 mL of distilled water under continuous stirring and heating at 60 °C. After that, the resulting solution was transferred to a Teflon-lined stainless-steel autoclave for hydrothermal reaction at 90 °C for 15 h. The products were cooled down to room temperature, filtered out, and washed thoroughly with water and ethanol. Finally, the solid was dried at 60 °C for 1 h in an oven and then calcined at 500 °C with a heating rate of 2 °C min−1 in a furnace.
2.3. Characterization methods
X-ray diffraction analysis of the powder catalysts was performed with a Rigaku Miniflex II X-ray diffractometer (Rigaku®, Japan) using monochromatic Cu-Kα radiation (λ = 1.540 Å). The XRD patterns of the catalysts were recorded in a range of 2θ from 5 to 80°, with a pitch of 0.05° and 1.0 seconds per pitch. The diffractograms were refined using the Rietveld method to obtain the composition of the phases. The Rietveld refinement calculation was carried out using the program TOPAS academic V5.0, based on the fundamental parameters approach, using the instrumental parameters with background correction. The data were refined, if necessary, with the following parameters: unit cell dimensions; sample height displacement; zero-shift; weight fraction (scaling); preferred orientation; atomic species/substitutions; atomic coordinates; site occupancies; thermal displacement parameters; crystallite size, and lattice strain.The textural properties of the samples were evaluated by N2 physisorption at −196.15 °C using an ASAP 2020 plus (Micromeritics, Norcross, GA, USA) over a relative pressure (P/P0) range from about 0 to 0.995. Prior to the measurements, all samples were degassed at 350 °C for 12 h. The specific surface area (SBET) was determined by Brunauer–Emmett–Teller (BET) method using adsorption data in the P/P0 ranging from 0.05 to 0.30. The average pore diameter (dp) was calculated by the BJH method (dp = 4Vp/SBET, where Vp is the specific pore volume). The total pore volume was calculated by converting the amount of N2 adsorbed at a P/P0 of 0.995 to the volume of liquid adsorbate.
The catalysts films were characterized by Raman spectroscopy, using an Alpha 300 system (Witec®, Germany). Raman experiments were performed at 25 °C using a ×100 objective lens and a Nd:YAG green laser with 532 nm excitation wavelength and with a small entrance (1000 ct) to avoid the decomposition of possible organic compounds in the samples during the analyses.
XPS analyses of the powder catalysts were carried out in Escalab 250Xi spectrometer (ThermoScientific) using a monochromatic Al Kα (hν = 1486.6 eV) X-ray source. The powdered samples were mounted on a double-sided adhesive carbon tape. The pressure was kept around 10−8 mbar inside the analysis chamber. For the survey spectra, the analyzer was operated with a pass energy of 100 eV and an energy step of 1 eV. Though, for the high-resolution spectra, these parameters corresponded to 25 eV and 0.05 eV, respectively. The peak positions of all spectra were corrected using the C 1s peak at 284.8 eV as a reference and the charging effect was minimized with the help of a flood gun.
TGA of the powder catalysts were carried out on a TG 209 F1 Iris (Netzsch®, Germany), by placing approximately 10 mg of each sample into alumina crucibles and heating them from 50 to 1000 °C at a heating rate of 10 °C min−1, under 50 mL min−1 Helium gas flow.
FESEM analyses of the catalyst films were carried out on a SEM-FEG JEOL JSM-7100 F microscope (JEOL®, Japan) operated at 15 kV acceleration voltage, at the working distance of 3.7 mm at up to 150k× magnitude. AFM images of the catalyst films were obtained on a Park NX10 (Park®, Korea). Experiments were conducted using a 2.8 N m−1 Pt–Ir probe in intermittent contact (topography, phase contrast, and electrical images) were acquired in air by a single pass scanning at room temperature and humidity between 2 and 5%. Kelvin force and capacitance coupling measurements were conducted in parallel by applying an electric AC signal at 10 kHz to the metal-coated cantilever. The electrical potential of the sample is deduced by the DC potential applied to the cantilever to nullify the AC signal at 10 kHz. Furthermore, the second harmonic of the AC signal (35 kHz), which is shown to be proportional to the capacitance gradient (∂C/∂z), or capacitance coupling, of the tip to the sample, was monitored. The images were generated with scans covering an area of about 5 μm × 5 μm, containing 512 × 512 pixels at maximum resolution. The Gwyddion 2.54 software package was used to analyze the AFM images.
2.4. Electrochemical performance of the films
All electrochemical experiments were performed in triplicate, using an Autolab PGSTAT302N potentiostat/galvanostat (Metrohm®, Switzerland), controlled by the NOVA® software (Metrohm®). The electrochemical system consisted of a 30 mL three-electrode cell. An Ag|AgCl electrode (3 mol L−1 KCl) was used as the reference electrode, a platinum bar as the counter electrode, and the FTO glass plates modified with the synthesized oxides used as the working electrode (WE). Experiments of CV and EIS were conducted at room temperature inside a homemade Faraday's cage, using different electrolytic solutions, 0.1 mol L−1 HNO3 at pH 1, 0.1 mol L−1 Na2SO4 at pH 7, and 0.1 mol L−1 NaOH at pH 13.Thin films of the synthesized mixed oxides were prepared by drop casting on FTO glass plates, which consists of dripping a 20 μL aliquot of a suspension containing 2.0 mg of the oxides prepared in 200 μL of isopropyl alcohol (10 mg mL−1) over a delimited area of 1 cm2 of the FTO substrate. The total catalyst loading was 0.2 mg cm−2.
CV measurements were carried out at 50 mV s−1, and automatic iR (ohmic drop) compensation was employed, where the electrode and solution resistance were determined by EIS. EIS measurements were conducted using a frequency range of 105 Hz to 0.01 Hz and voltage amplitude of 0.01 VRMS AC amplitude, at 1.1 V vs. RHE, no iR compensation was applied during these measurements. Charge transfer resistance (Rct) and solution electrochemical resistance (Rs) were calculated by electrochemical fits based on a Randles equivalent circuit model employing constant phase (CPE) and Warburg (W) elements.
2.5. OER electrocatalytic performance of the films
OER electrocatalytic tests were conducted in two different modified WE:FTO or Pt RDE (rotating disk electrode from Metrohm). Experiments using a 3.0 mm diameter Pt RDE were carried out at 1600 rpm according to a benchmarking protocol previously developed to evaluate and compare the performance of electrocatalysts for OER process.22,23 The tests were carried out at different electrolytic solutions, 0.1 mol L−1 HNO3 at pH 1; 0.1 mol L−1 Na2SO4 at pH 7; and 1.0 mol L−1 NaOH at pH 14.
The preparation of the modified WE films by drop cast followed a modified protocol from the literature.24 Stock suspensions of the catalysts were prepared by adding 80.0 mg of the oxide in a mixture containing 3.98 mL of deionized water, 1.0 mL of 2-propanol, and 0.02 mL of 5% Nafion 117 solution, afterward the suspension (16.0 mg mL−1) was sonicated for 10 min to form an ink. 4.0 μL of the oxide ink was pipetted onto the 3.0 mm diameter Pt disc electrode or 20.0 μL of the oxide ink was pipetted onto 1 cm2 delimited surface of a FTO glass. Finally, the prepared films were dried in a vacuum desiccator for 10 min. The entire process was carried out at room temperature. The catalyst loading was 0.90 mg cm−2 for RDE and 0.32 mg cm−2 for FTO.
Electrocatalytic activity for OER was assessed by linear sweep voltammetry (LSV) from 0.7 to 1.7 V vs. RHE, under a scan rate of 5 mV s−1, with a potential step of 5 mV. All electrochemical data were treated to correct the ohmic resistance (iR) to correctly characterize the behaviours of the catalysts in relation to the currents measured in each potential.
Potentials were converted to the RHE scale by the following relation (eqn (4)),
| ERHE = EAg|AgCl + 0.210 V + 0.059 V × pH | (4) |
The overpotentials (η) for the OER were calculated according to eqn (5).
| η = ERHE − 1.23 V | (5) |
The turnover frequency (TOF) was calculated at the measured current density j (A cm−2) at η = 0.45 V by eqn (6):
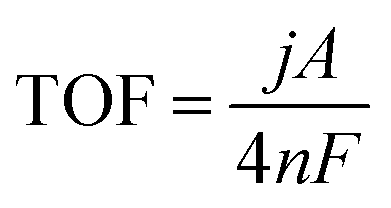 | (6) |
485C mol−1).25
Electrocatalytic long-term stability was tested for Co3O4 by chronopotentiometry at a constant density current of 10 mA (j = 10 mA cm−2 for FTO, j = 141 mA cm−2 for RDE), under the same conditions of the OER tests, but with different times according to the electrode: FTO over 2 h and RDE over 15 h.
3. Results and discussion
3.1. Structural characterization of the mixed oxides
Pure and Mn-doped Co3O4 were prepared by hydrothermal synthesis in presence of urea, starting from different proportions of Mn(II) and Co(II) precursors. Fig. 1A shows the X-ray diffraction patterns of the prepared catalysts. Rietveld refinement (Fig. S1 and Table S1†) of Co3O4 showed a single phase of the face-centered-cubic structure typical of the spinel, with space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227) (JCPDS 73-1701).26–28 XRD patterns of Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3 revealed Co3O4 as the major phase with 97.0, 82.8 and 46.8wt%, respectively. The minor phase for Mn@Co3O4-1 was the spinel MnCo2O4 with 3.0wt%, for Mn@Co3O4-2 was CoMnO3 (JCPDS: 75-2090) with 17.2 wt%, and for Mn@Co3O4-3 a mixture of phases of different Mn valences, such as Mn3O4 at Hausmannite structure (Mn3O4: JCPDS 80-0382) with 12.1 wt%, in addition to the phases CoMnO3 with 36.6 wt%, and the spinel MnCo2O4 with 4.5 wt%. The main phases and respective proportions for each material are summarized in Table 1. A similar mixture of MnOx and CoOx phases was also described for Mn–Co–O catalysts with different Mn/Co molar ratios.17,29
m (227) (JCPDS 73-1701).26–28 XRD patterns of Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3 revealed Co3O4 as the major phase with 97.0, 82.8 and 46.8wt%, respectively. The minor phase for Mn@Co3O4-1 was the spinel MnCo2O4 with 3.0wt%, for Mn@Co3O4-2 was CoMnO3 (JCPDS: 75-2090) with 17.2 wt%, and for Mn@Co3O4-3 a mixture of phases of different Mn valences, such as Mn3O4 at Hausmannite structure (Mn3O4: JCPDS 80-0382) with 12.1 wt%, in addition to the phases CoMnO3 with 36.6 wt%, and the spinel MnCo2O4 with 4.5 wt%. The main phases and respective proportions for each material are summarized in Table 1. A similar mixture of MnOx and CoOx phases was also described for Mn–Co–O catalysts with different Mn/Co molar ratios.17,29
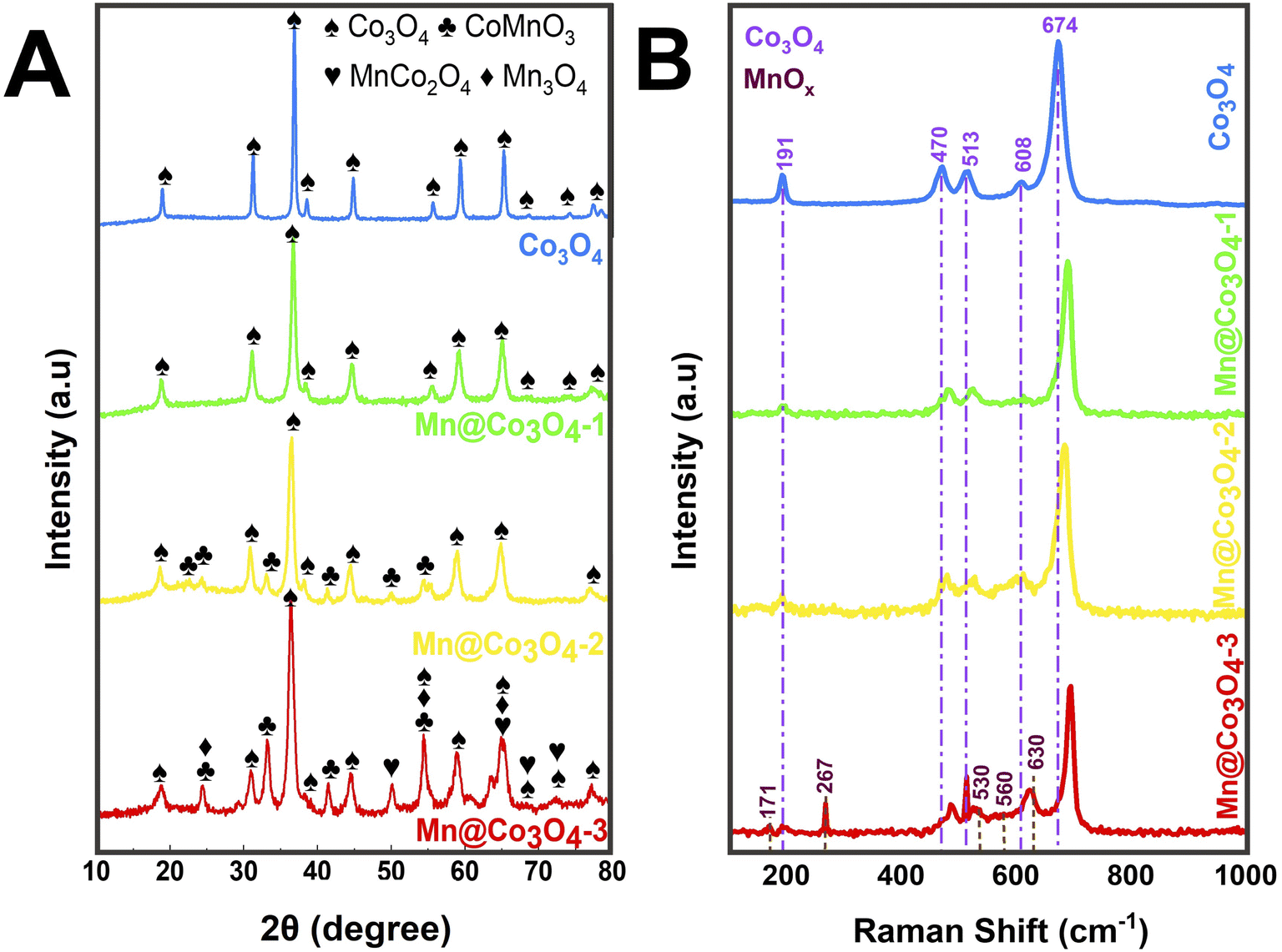 | ||
| Fig. 1 (A) Powder X-ray diffraction patterns, and (B) Raman spectra of the Mn-doped Co3O4. | ||
| Catalyst | Major phasea | Contenta (wt%) | Crystallite sizea (nm) | SBET b (m2 g−1) | Vp b (cm3 g−1) | dp b (nm) |
|---|---|---|---|---|---|---|
| a Determined by XRD Rietveld refinement for the major phase.b Determined by the nitrogen adsorption–desorption isotherms. | ||||||
| Co3O4 | Co3O4 | 100 | 50.0 | 15.2 | 0.159 | 42.08 |
| Mn@Co3O4-1 | Co3O4 | 97.0 | 19.4 | 65.9 | 0.299 | 18.14 |
| Mn@Co3O4-2 | Co3O4 | 82.8 | 16.1 | 53.2 | 0.213 | 16.79 |
| Mn@Co3O4-3 | Co3O4 | 46.8 | 16.0 | 61.3 | 0.304 | 19.85 |
A thorough analysis of the samples containing Co3O4 revealed that there was a shift to lower values of 2θ with the increase of Mn:Co molar ratio, indicating an increase of the lattice cell parameters (Table S1†), similar effect has been described in previous works.30,31 For instance, the lattice parameter a is 8.09 nm in the sample Co3O4 and increased accordingly to 8.11, 8.12, and 8.13 nm in Mn@Co3O4-1, Mn@Co3O4-2, and Mn@Co3O4-3, respectively. This displacement can be attributed to the substitution of Co2+ (72 pm) or Co3+ (62 pm) by Mn2+ (82 pm) or Mn3+ (73 pm) ions that have similar ionic radius, but can cause distortions in the lattice.31 As consequence, the crystallite size of Co3O4 decreased with the increase of Mn:Co, for instance, 50.0 nm for Co3O4 and only 16.0 nm for Mn@Co3O4-3. The enlargement of the Co3O4 peaks corroborates with the replacement of Co by Mn in the spinel lattice. A similar effect was observed for Mn-promoted mesoporous Co3O4,6 and in MnxCo3−xO4 spinel oxides of different Mn:Co molar ratio,20 due to the expansion of the unit cell caused by lattice distortion and Co3O4 crystallization inhibition.
N2 physisorption measurements demonstrate the inversely proportional relationship between the BET surface area and crystallite size of the main phase determined by XRD (Table 1). The pure spinel presented a typically low surface BET area of 15.2 m2 g−1. The SBET increased for the Mn-doped Co3O4, for Mn@Co3O4-1 (97% Co3O4) the SBET was 65.9 m2 g−1 and for Mn@Co3O4-2 (82.8% Co3O4) the SBET was 53.2 m2 g−1, while Mn@Co3O4-3 (46.8% Co3O4) presented a SBET of 61.3 m2 g−1 with a pore size of 19.85 nm and a pore volume of 0.304 cm3 g−1.
Raman spectra of the oxides are shown in Fig. 1B. The spectrum of Co3O4 presents five bands at 191, 470, 513, 608, and 674 cm−1 related to the three active vibrational modes of the spinel structure, being: the F2g mode to the first, third, and fourth peaks, the Eg mode to the second peak and the A1g mode to the fifth peak.18,32–34 It can also be seen that the Raman peaks at 470, 513, and 674 cm−1 increased in intensity with the increase of Co content. Displacements were observed at the peak positions of all five active Co3O4 modes due to the phonon symmetries of the Raman peaks. This phenomenon can be assigned to the optical phonon confinement effect that can be caused by vibrations of the spinel structure, in which Co2+ and Co3+ cations are respectively situated at tetrahedral and octahedral sites in the cubic lattice.18 The Raman spectra of MnOx showed the presence of Mn3O4 peaks at 530 and 630 cm−1 related the vibration of the Mn–O bond, the peaks at 171, 267 and 560 cm−1, which are characteristic of CoMnO3.35–37 Similar trends to the phonon confinement effect observed for the Co3O4 were reported for the Mn3O4,38 and it was also observed in our work. The Raman spectra of Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3 showed the same vibrational modes of the Co3O4 and MnOx.
XPS experiments were carried out to characterize the oxidation state of the surface species as well as to examine the atomic surface composition of the catalysts. The survey XP spectra shown in Fig. S2† confirmed that the surface was mainly composed of Co, Mn, and O, as summarized in Table 2. It was observed an increase of Mn content with the increase of Mn:Co ratio, as expected. The surface Mn/Co ratio is higher than the weighted ratio estimated from the Rietveld refinement, indicating predominance of the Mn-containing phases at the surface of the oxide.
| Catalyst | Major phase contenta (wt%) | Surface atomic compositionb (at%) | Binding energyd (eV) | Co3+/Co2+ e | νMn f | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| O | Mn | Co | Mn/Coc | Mn 2p1/2 | Mn 2p3/2 | Co 2p1/2 | Co 2p3/2 | ||||
| a Determined by Rietveld refinement from powder XRD data.b Determined by survey spectra.c The values between brackets correspond to the weighted Mn/Co ratio estimated from the phases determined by XRD (Table S1).d Determined by high-resolution spectra of Co 2p and Mn 2p.e Estimated from the deconvoluted high-resolution Co 2p3/2 spectra.f Estimated by the linear equation proposed by Beyreuther and co-workers.39 — Not detected. | |||||||||||
| Co3O4 | Co3O4 | 70.7 | 0.0 | 29.3 | — | — | — | 779.7 | 797.0 | 1.6 | — |
| 100 | 781.4 | 803.2 | |||||||||
| Mn@Co3O4-1 | Co3O4 | 67.2 | 4.5 | 28.3 | 0.16 | 654.0 | 642.2 | 780.4 | 795.4 | 0.5 | 2.6 |
| 97.0 | (0.010) | 644.4 | 781.5 | 797.0 | |||||||
| Mn@Co3O4-2 | Co3O4 | 91.7 | 2.0 | 6.3 | 0.31 | 653.9 | 642.1 | 781.0 | 796.1 | 0.9 | 2.3 |
| 82.8 | (0.094) | 644.0 | 782.3 | 797.8 | |||||||
| Mn@Co3O4-3 | Co3O4 | 63.2 | 13.4 | 23.4 | 0.57 | 653.6 | 641.7 | 780.1 | 795.3 | 0.8 | 2.9 |
| 46.8 | (0.48) | 643.6 | 781.5 | 797.0 | |||||||
The high-resolution XP spectra of Mn 2p and Co 2p for all catalysts are depicted in Fig. 2 and the corresponding binding energies of Mn 2p3/2, Co 2p3/2, Mn 2p1/2, and Co 2p1/2 are shown in Table 2.
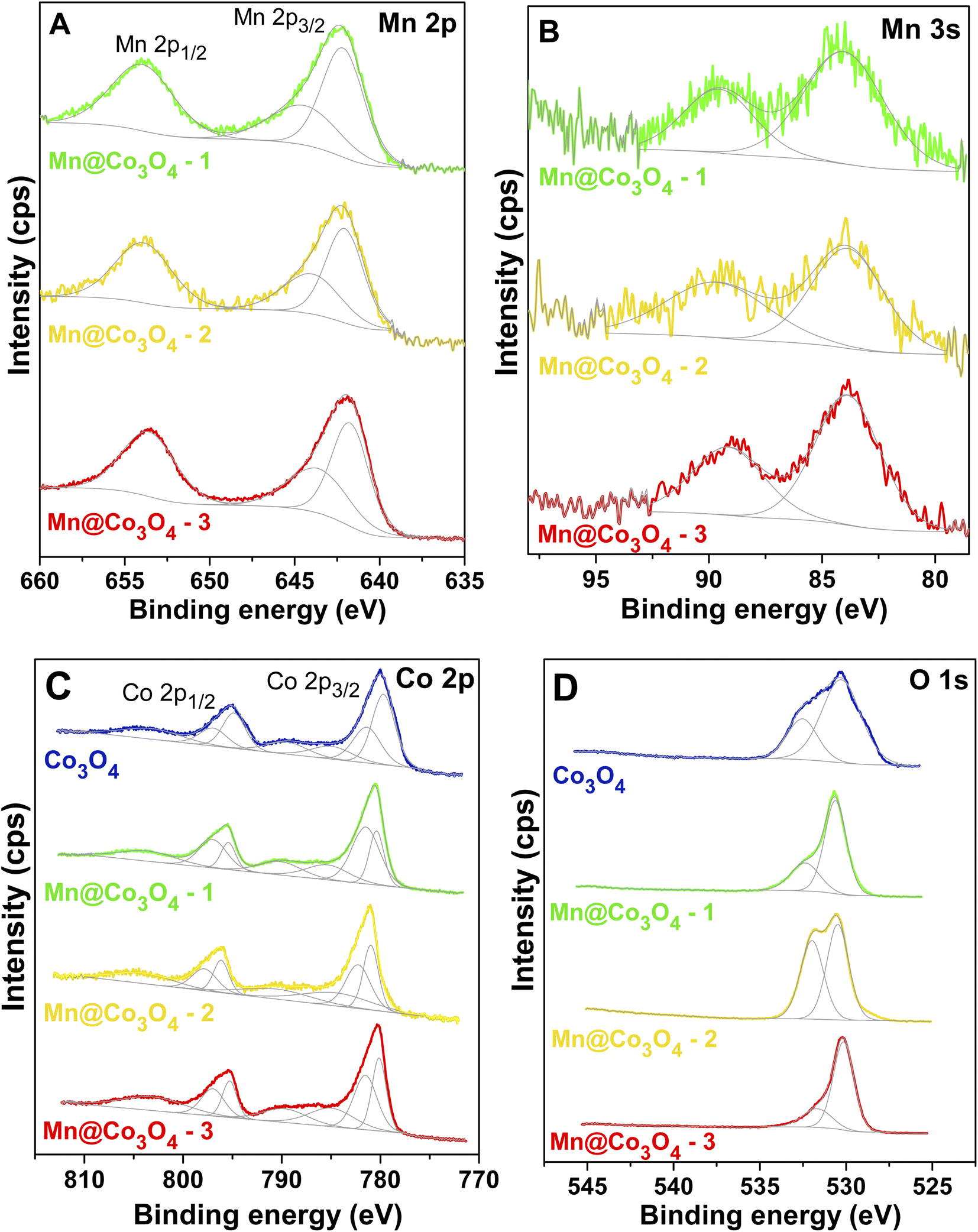 | ||
| Fig. 2 High-resolution XPS spectra of the studied catalysts. (A): Mn 2p, (B): Mn 3s, (C): Co 2p and (D): O 1s. | ||
Mn 2p XP spectra of all catalysts were quite similar (Fig. 2A), presenting a main doublet (Mn 2p3/2 and Mn 2p1/2 lines). The spin–orbit splitting energies between the two Mn 2p peaks, presented in Table S2†, were very close and similar to those shown by similar Mn–Co materials in literature.40–42 The oxidation states of manganese were evaluated from high-resolution XP spectra for Mn 3s (Fig. 2B), because it is described in the literature that this analysis is a more reliable way to assign the Mn oxidation state than by investigating the Mn 2p spectrum.43,44 Beyreuther and co-workers suggested that the value of the Mn 3s exchange splitting (ΔE3s) may be used to estimate the manganese valence (νMn) according to the following linear equation (eqn (7)).39
| νMn = 9.67 − 1.27ΔE3s | (7) |
The values for the manganese valence of all samples are presented in Table 2. It was verified a mixed Mn2+/3+ state, with valence values of +2.9, +2.3, and +2.6 for Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3, respectively. The mixed oxide MnxCo3−xO4 also resulted in a ΔE3s that lies between Mn2+ and Mn3+ cations.45
The Co 2p XP spectrum of Co3O4 sample (Fig. 2C) presents two characteristic peaks with binding energies of Co 2p1/2 and Co 2p3/2 that are separated by approximately 15.0 eV.46,47 The spin–orbit splitting energy (2p1/2–2p3/2) in Co 2p XPS spectra of Co–Mn mixed oxides were very similar to the value verified for Co3O4 oxide, as can be seen in Table S2.† Co3O4 spinel can present weak satellite peaks, while in CoO spectra these peaks are of higher intensity.48 The ratios between shake-up satellite peak areas and Co 2p total area were very close for all catalysts, 0.4 (Mn@Co3O4-3 and Mn@Co3O4-2) and 0.3 (Mn@Co3O4-1 and Co3O4). For CoO a ratio of 0.9 was reported,47,48 thus the small-intensity satellites suggests a more Co3O4-like composition than CoO for the catalysts,49,50 as indicated by XRD data. The binding energy values of these shake-up satellite peaks are summarized in Table S2.†
Co 2p peak fittings were performed considering two doublet pairs with equal full width at half maximum (FWHM) and are ratio of 2:1 for the Co 2p3/2 and Co 2p1/2 components of each pair. Three shake-up satellite peaks were also included for the fitting of Co 2p spectra, setting equal FWHM for the two peaks with lower binding energy values.42,51 According to the literature, the presence of two peaks at the Co 2p3/2 and Co 2p1/2 core-level region is due to Co3O4 being a mixed-valence material, where the component with lower binding energies is assigned to Co(III), while the other with higher is related to Co(II).40,52 The Co3+/Co2+ ratios were estimated from the areas of the curves of Co 2p3/2 peaks, as represented in Table 2. Co3O4 presents a Co3+/Co2+ ratio of 1.6, close to the nominal value, but it decreases around twice with the manganese presence. The trend observed for Co3+/Co2+ ratios did not follow the estimative of average oxidation state of Co in the mixed oxides considering the crystalline phase distribution, 2.7, 2.6, 2.9, and 2.8 eV for Co3O4, Mn@Co3O4-1, Mn@Co3O4-2, Mn@Co3O4-3 (Table S1†), respectively, which may indicate a different cationic distribution in the surface compared to bulk for these materials as well as lower concentration of Co3+ species at the surface.
The spectra of O 1s (Fig. 2D) are quite broad and contain at least two different contributions that can be assigned to oxygen-containing compounds in these catalysts. The binding energy value of each peak is presented in Table S2.† The peaks with lower binding energy correspond to metal–O bonds. Both manganese oxides and cobalt oxides presented peaks assignable to lattice oxygen in this region,42,43,49 and the spectra of CoO, Co3O4 and MnxCo3−xO4 are not distinguishable in terms of binding energy in the O 1s range.48 The peaks in higher binding energy are generally attributed to the presence of non-stoichiometric oxygen on the surface. Some possible species are described in the literature as low-coordinated oxygen ions, weakly adsorbed species, hydroxyl groups, adsorbed oxygen ions on surface oxygen vacancies or contamination like oxygen in retained nitrate species and adsorbed water.40,49,52,53
The XPS analyses results were also employed to evaluate the formation of metal defects in the surface of cobalt-containing materials (i.e. cobalt vacancies and manganese vacancies), using the methodology proposed by Li and coworkers to investigated MnxCo3−xO4 bimetallic spinels.54 The (Mn + Co)/O ratios of the surface were estimated and the results are shown in Table S2.† The stoichiometric (Mn, Co)3O4 was used as a reference. The ratios were higher than 0.75 for Mn@Co3O4-3 and smaller for Mn@Co3O4-2, Mn@Co3O4-1 and Co3O4 suggesting the presence of metal defects in these last catalysts.
The thermal behavior and stability of the samples were verified using TG, detailed discussion is presented in Section S.1.3.†
3.2. Morphological characterization of the mixed oxides
The morphology of the mixed oxide was characterized by FESEM and the obtained images are depicted in Fig. 3 and Section S.1.4.† The FESEM images clearly reflect the different phases formed according to the Co:Mn ratios as observed by XRD and Raman, with the characteristic morphology of nanospheres for Co3O4 and nanorods for MnOx. SEM image of Co3O4 (Fig. 3A and S5†) shows agglomerated nanospheres forming clusters of Co3O4 from 45 to 75 nm. Fig. 3B–D show the FESEM images of the mixed oxides containing both Co3O4 and MnOx, where a change in the morphology of Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3 compared with the pure was observed, with the Co3O4 changing from nanocubes to nanospheres. More images are provided in Fig. S6–S8.†
 | ||
| Fig. 3 SEM images of the oxides. (A): Co3O4, (B): Mn@Co3O4-1, (C): Mn@Co3O4-2, (D): Mn@Co3O4-3. | ||
AFM measurements were also carried out to characterize the surface of the prepared films. With these measurements, it was possible to access the mean representativeness of the surface roughness of all samples (Fig. 4). Kelvin probe force microscopy measurements (KPFM) were also performed as a complementary technique for mapping the areas with different capacitive coupling (∂C/∂Z) of the films related to the dielectric constant of the material. In this work, contrast areas were observed with different capacitive coupling in the mapping (∂C/∂Z) for all analyzed films: Mn@Co3O4-3, Mn@Co3O4-2, Mn@Co3O4-1 and Co3O4, related to spatial variations in the local capacitance gradient. Areas with accumulated and dissipated loads with low electron mobility are shown as darker contrast and areas with high electron mobility are shown as lighter contrast, according to the scale shown on the right side of each image.
 | ||
| Fig. 4 AFM images of ∂C/∂z of the catalysts films. (A): FTO, (B): Mn@Co3O4-3, (C): Mn@Co3O4-2, (D): Mn@Co3O4-1, (E): Co3O4, (F): zoom Mn@Co3O4-2 areas. | ||
The Co3O4 film showed more conductive areas. The mapped surface was quite heterogeneous for Mn@Co3O4-3 (Fig. 4B), with very distinct light and dark areas, and it is not possible to clearly distinguish the characteristic morphology of each oxide (cubic for Co3O4 oxide and needle for MnOx), where it is possible to observe in Fig. 4C the predominant morphological characteristic of square-faced structures associated with cobalt as well as some few areas of rod-shaped ripples associated with manganese. In addition, it was possible to confirm that the film materials are within the magnitude of nanometer as shown by SEM.
3.3. Electrochemical characterizations of the films
To verify the electrochemical behavior of the prepared mixed oxides, CV was performed for all FTO films at a scan rate of 50 mV s−1, at different pH values, as presented in Fig. S9.† Characteristic peaks of Con+/Con+1 redox processes were observed. Fig. 5A presents in detail the CV of Co3O4 film in pH 7, as these are very distinct and signature of the voltammetric profile of the pure Co3O4, where two oxidation peaks and two reduction peaks were observed. The redox processes, potentials and attribution for the Co3O4 showed oxidation peaks at 1.25 and 1.49 V vs. RHE and reduction peaks at 1.30 and 1.47 V vs. RHE, probably related to the CoIICoIII ↔ CoIII2 ↔ CoIV2 process of the Co3O4 spinel.55–59 At pH 7 and 13, only the samples Mn@Co3O4-1 and Co3O4 showed redox peaks, which were shifted to higher potentials with increasing pH.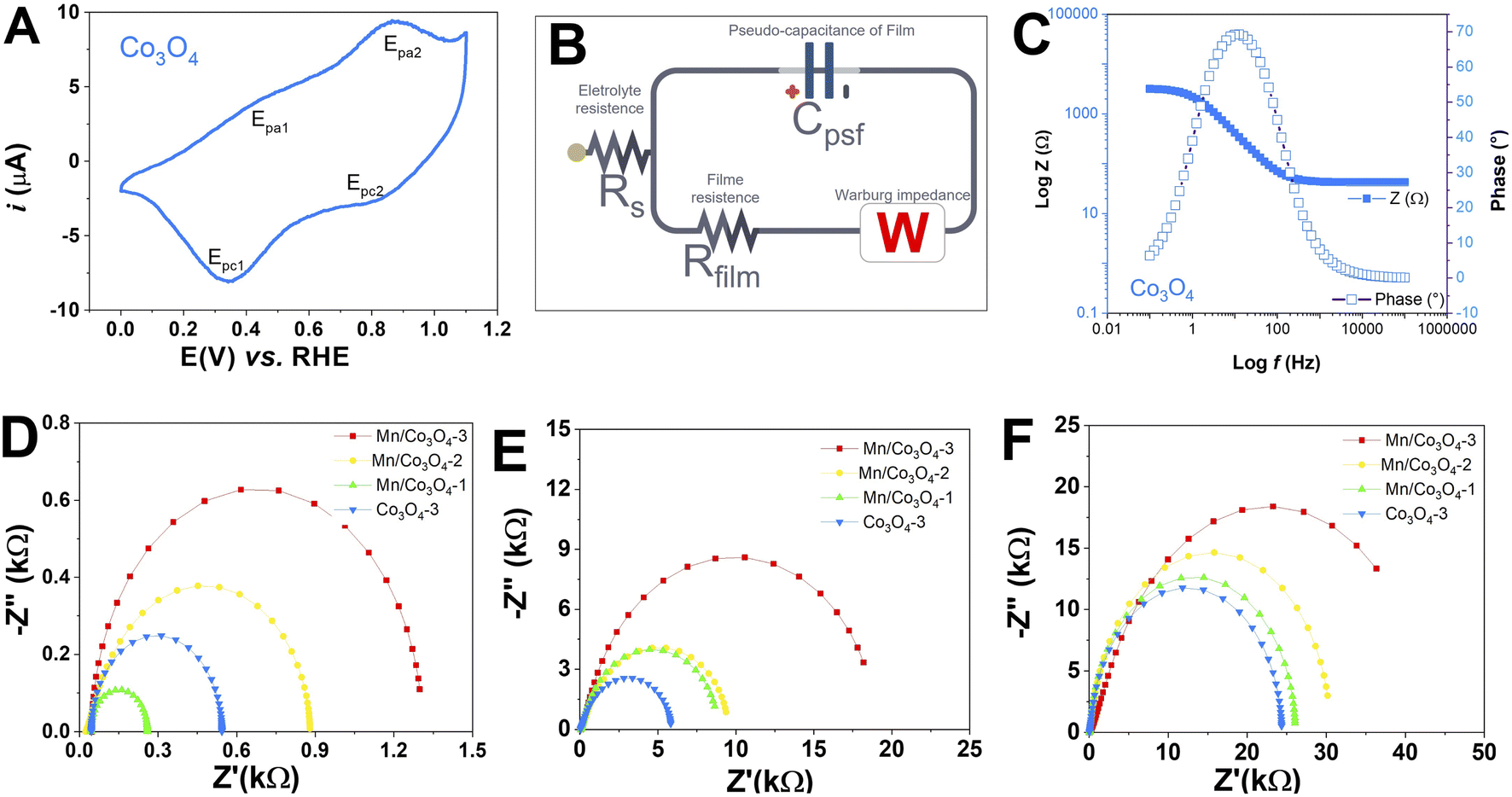 | ||
| Fig. 5 (A): Voltammetric profile of the Co3O4 in 0.1 M Na2SO4 pH 7 with a scan rate at 50 mV s−1; (B): electrical equivalent circuit derived from Nyquist plots; (C): Bode plot for Co3O4 in 0.1 M Na2SO4 pH 7; Nyquist plots derived from EIS measurements of the catalysts films at: 0.1 mol L−1 HNO3 pH 1 (D); 0.1 mol L−1 Na2SO4 pH 7 (E); 0.1 mol L−1 NaOH pH 13 (F), at a scan rate of 5 mV s−1. | ||
In addition, EIS was performed at high applied potential (1.1 V vs. RHE), between 10−2 and 105 Hz before the OER tests, to get some insight into film resistance before the OER. Fig. 5D–F shows the Nyquist plots, Rct (Table 3), Rs and CPE (Table S3†), were obtained from an equivalent circuit model shown in Fig. 5B. First, we can observe that the magnitude of electron transfer resistance differs considerably with pH, higher values of Rct were obtained as the pH increased, which means that the oxides behave as worse conductive materials at alkaline pH. The Rs values follow the same trend. Furthermore, the Rct of Co3O4 is significantly lower than that Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3 for all pHs, indicating that oxides with a higher Co3O4 ratio own the faster charge transfer as described in the literature.18,19 This trend was confirmed by the Bode plot Fig. 5C and emphasized at Fig. S10–S12†. The only exception was Mn@Co3O4-1 that presented the lowest Rct at pH 1, probably due to the presence of MnCo2O4 phase. Unlike what was described in the literature,6 in this work with the increase of the Mn molar ratio, there was also an increase in the diameter of the semicircle in the Nyquist plot for all pHs. The Rct observed by EIS corroborates the AFM measurements, in both techniques Co3O4 was the most conductive.
| Catalyst | η10 (mV) | TOF450 (s−1) | Tafel (mV dec−1) | Rct (kΩ cm2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 1 | pH 7 | pH 14 | pH 1 | pH 7 | pH 14 | pH 1 | pH 7 | pH 14 | pH 1 | pH 7 | pH 14 | |
| Co3O4 | 761 | 490 | 240 | 1.38 × 10−4 | 4.54 × 10−3 | 1.01 × 10−1 | 123 | 79 | 40 | 0.543 | 5.818 | 24.339 |
| Mn@Co3O4-1 | 752 | 673 | 320 | 9.03 × 10−5 | 3.72 × 10−4 | 3.31 × 10−2 | 121 | 109 | 52 | 0.259 | 8.637 | 26.047 |
| Mn@Co3O4-2 | 774 | 650 | 370 | 5.78 × 10−5 | 5.62 × 10−4 | 2.55 × 10−2 | 125 | 105 | 60 | 0.879 | 9.372 | 30.167 |
| Mn@Co3O4-3 | 780 | 562 | 390 | 4.82 × 10−5 | 2.59 × 10−3 | 1.34 × 10−2 | 129 | 90 | 64 | 1.298 | 18.184 | 46.789 |
3.4. OER electrocatalysis
The electrocatalytic OER activity of the Mn-doped Co3O4 oxides was evaluated by linear sweep voltammetry in different pH values, with the oxide films prepared by drop casting over FTO glass substrate (data shown in Section S2†) and Pt RDE (Fig. 6A–C). The overpotential defined at j = 10.0 mA cm−2 (η10) was measured according to the geometric area of the working electrodes,60 and the TOF values were calculated at η = 450 mV, the results are summarized in Table 3.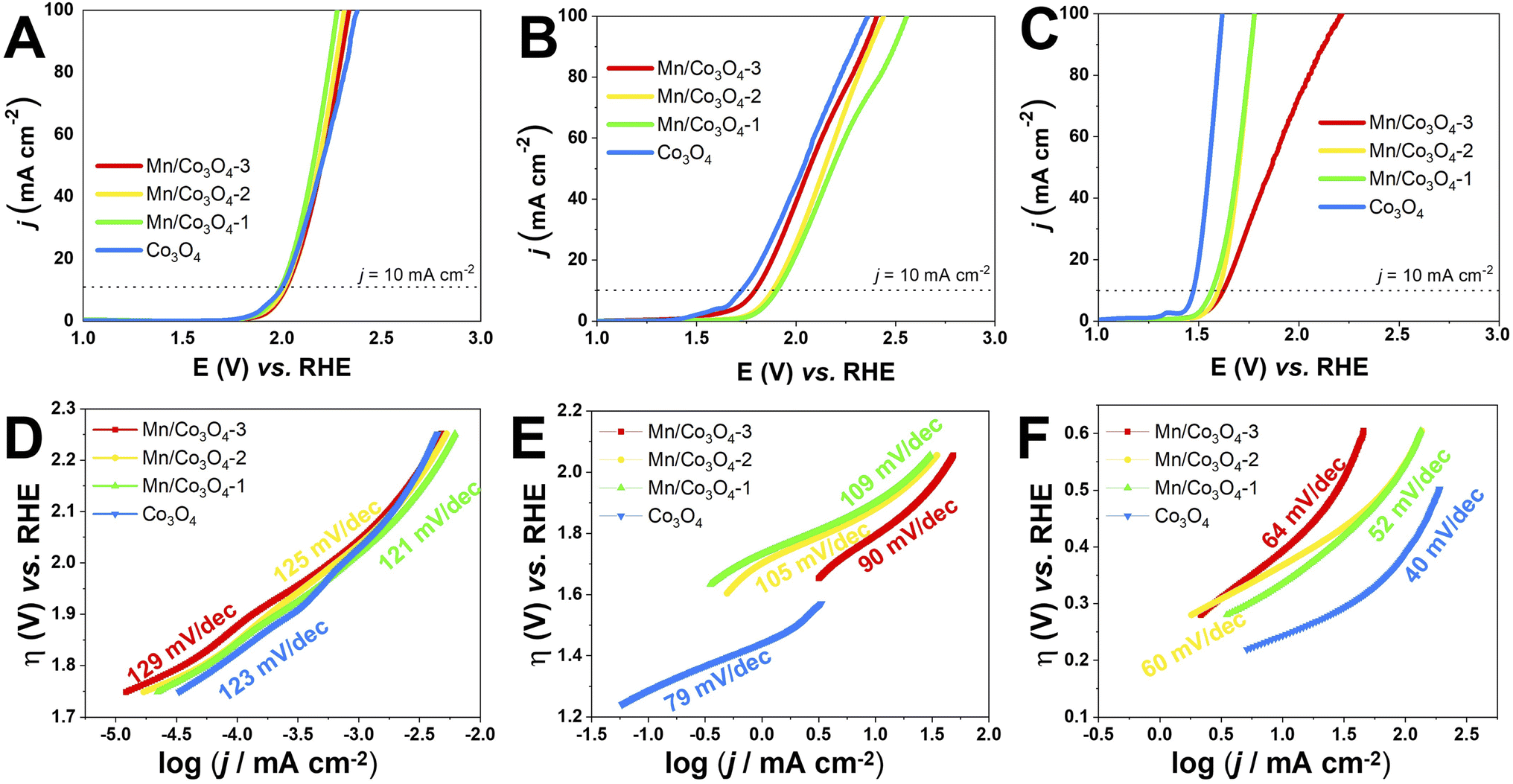 | ||
| Fig. 6 Polarization LSV curves for oxygen evolution reactions of the catalysts films RDE electrode. (A): pH 1, (B): pH 7, (C): pH 14; and respective Tafel plots: (D): pH 1, (E): pH 7, (F): pH 14, at a scan rate of 0.005 V s−1. | ||
The effect of pH in the electrocatalysis was investigated, low η10 and high TOF values were achieved at pH 14, followed by pH 7, and pH 1. Nevertheless, considerably high OER activity was still obtained at neutral or acidic pH. The development of highly efficient OER electrocatalysts under neutral and acid conditions is very important for safe and low-cost water splitting electrolyzer.4,29 In respect to the Co and Mn content, the catalysts with higher weight percentage of Co3O4 phase showed the highest activities in all pHs. Fig. 7A evidences the effect of pH and Co3O4 content in the η10.
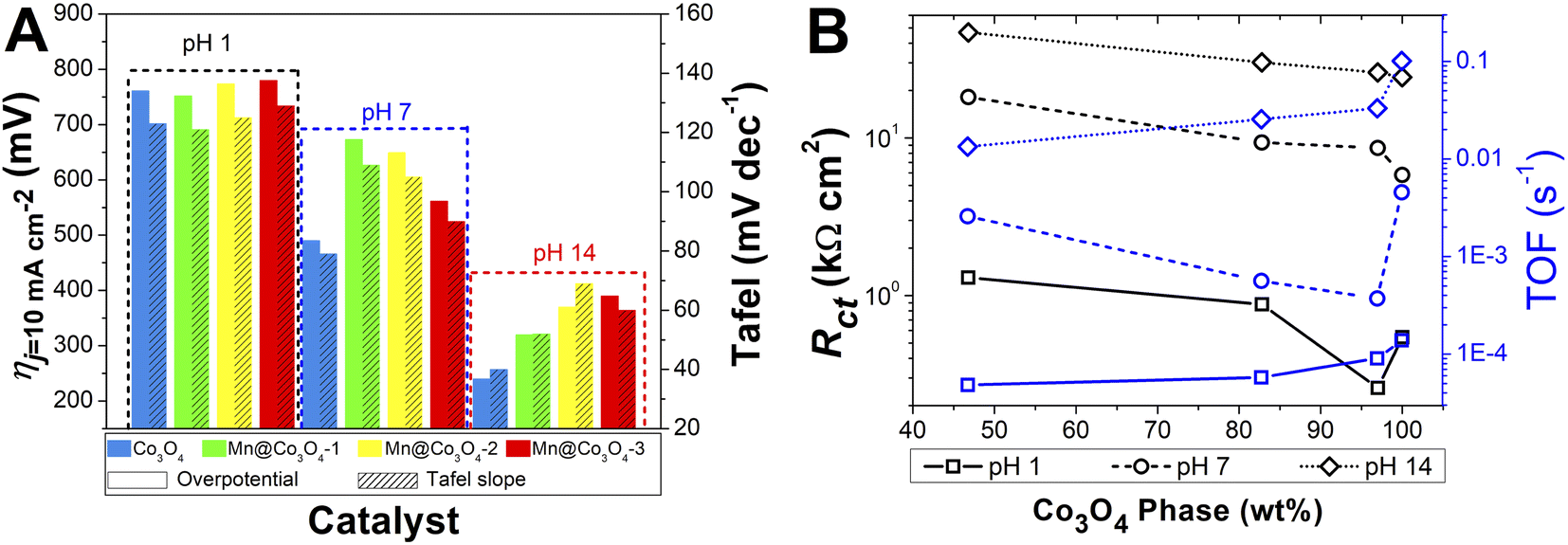 | ||
| Fig. 7 (A): Histograms of the required overpotentials at 10 mA cm−2 and Tafel slopes for all pHs. (B): Correlation of Rct and TOF with the percentage of Co3O4 phase. | ||
Pure Co3O4 presented very low η10 of 761, 490 and 240 mV at pH 1, 7 and 14, respectively. The same tendency is observed for the TOF values at η = 450 mV, 1.38 × 10−4, 4.54 × 10−3, and 1.01 × 10−1 s−1 at pH 1, 7, and 14, respectively. The TOF values difference between the pHs was remarkable, being a 10-folds increment from 1 to 7, and 20-folds from 7 to 14. These overpotentials are amongst the smallest reported for these types of materials. For instance, Co-borate ultrathin nanosheet/graphene hybrid displayed ηonset = 235 mV at neutral conditions, although at low overpotential η10 of 290 mV at pH 14.4 Mesoporous Co3O4 presented η10 of 396 mV,6 and Co3O4 presented η10 = 368 mV,20 both at pH 14. More examples of reported materials for OER are shown in Table S9.†
Remarkable OER electroactivity was also achieved for the mixed oxides Mn@Co3O4-1, Mn@Co3O4-2 and Mn@Co3O4-3. At pH 14, the respective η10 was 320, 370 and 390 mV, with TOF values of 3.31 × 10−2, 2.55 × 10−2 and 1.34 × 10−2 s−1, 10-folds lower than pure Co3O4. Mn@Co3O4-1 reached a very low η10 of 752 mV at pH 1. The results are comparable to reported Mn–Co oxides at pH 14, mesoporous 20% Mn-mesoCo3O4 presented ηonset of 320 mV at pH 14,6 while the spinel MnCo2O4 presented η10 of 365 mV at pH 14,19 and Mn2CoO4 and Mn0·5Co2·5O4 presented η10 = 399 mV and 345 mV, respectively.20
Co3O4 is the material with lower SBET, so TOF/SBET was calculated (Table S5†) and pure Co3O4 continues to be the most active oxide. Accordingly, when only the Co3O4 phase was taken into account for the TOF and TOF/SBET calculation, even higher values were found (Tables S6 and S7†), but still the pure Co3O4 was the most active showing that the higher efficiency of the pure spinel is not due exclusively to the higher surface area neither to the percentage of the phase.
As discussed in the EIS and AFM results, lower charge transfer resistance was associated with the increase of the Co3O4 percentage phase in the mixed oxides. The OER electrocatalytic activity has been associated to the higher conductive property of the materials in the literature. Fig. 7B shows that the Rct decrease caused by the higher Co3O4 percentage lead to higher TOF values for all pHs. Accordingly, it was possible to observe that the oxides with higher values of Rct also presented higher η10 in all pHs. Furthermore, from Fig. 7B it is clear that although higher values of Rct are found in higher pH, as the ηj = 10 is lower with increasing pH, the TOF is also lower at higher pH, but inside each electrolyte pH the effect of the Rct is evidenced.
The kinetic parameters that describe the electron transfer reactions at the electrode interface were studied to evaluate the performance of the catalysts by Tafel slope according to Fig. 6D–F for RDE, and the data are summarized in Table 3. Assuming that the OER Tafel slope is the rate at which the current increases versus the overpotential and its value depends mainly on the coefficient of anodic transfer, it can be expressed by eqn (8):
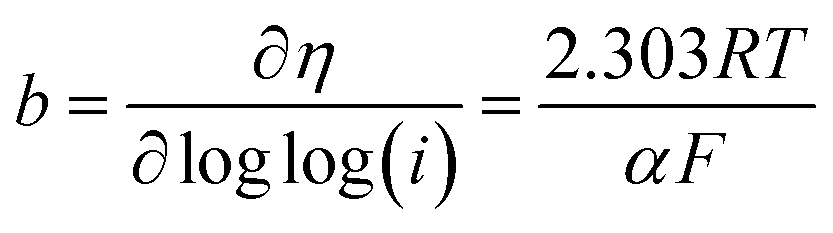 | (8) |
Lower Tafel slope indicates improvement in the OER kinetics because of the enhanced electron transfer capacity and strongly coupled effects.4,61 Low Tafel slopes were obtained in all pHs for the oxides, following the order: pH 14 < pH 7 < pH 1. Fig. 7A shows the pH effect in the Tafel slope, low η10 also lead to low Tafel. The most common OER mechanisms proposed in the literature is a four-electron transfer that takes place at the surface of a metal oxide MOx electrocatalyst. The general mechanisms in acidic and alkaline conditions are shown in Scheme 1.57,62 The Tafel slope is important to know the rate-determining step (RDS), for instance, it is broadly accepted that the OER first step is a one-electron transfer, and if this is the RDS it will correspond to theoretical Tafel slope of 120 mV dec−1. This is the case for the studied Co3O4-based materials at pH 1 (step I), which present Tafel slopes of 121–129 mV dec−1. At pH 14 the Tafel slope is 40–69 mV dec−1. Theoretical Tafel slope of 60 mV dec−1 is characteristic of a chemical step after a one-electron transfer step, indicating that the second (step VI) is the RDS. Similar mechanism was proposed for Co3O4 and Pd-doped Co3O4.57,63
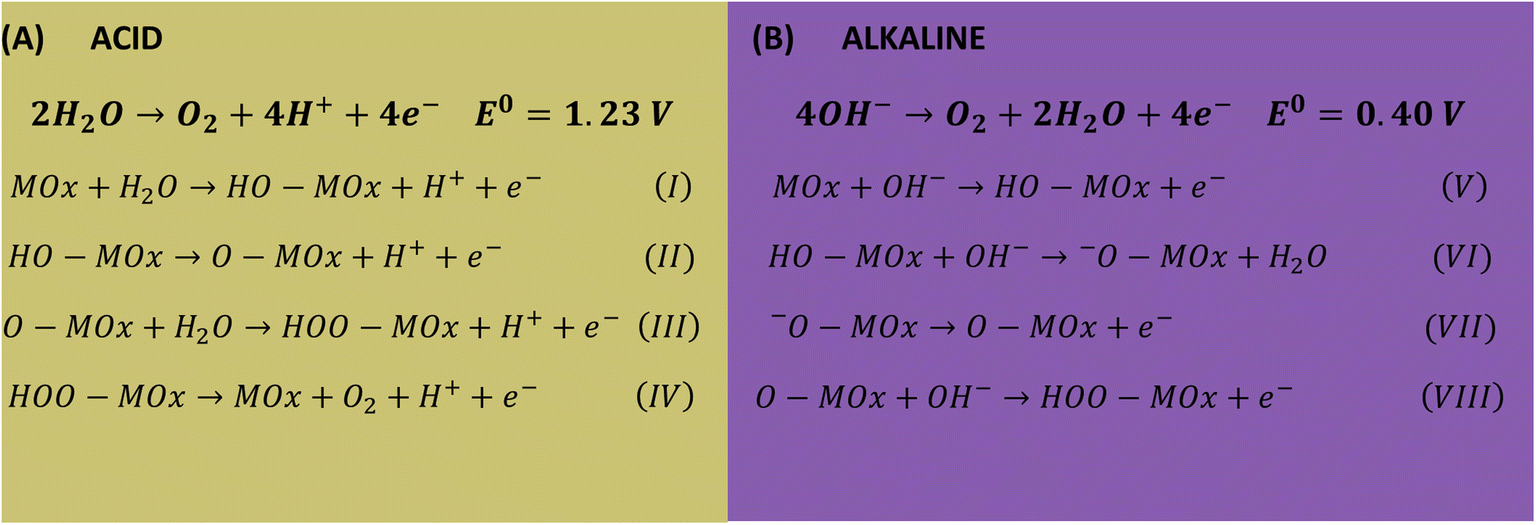 | ||
| Scheme 1 Proposed mechanism for OER in acid (A) and alkaline (B) conditions. | ||
Comparing the presented mixed oxides with other similar catalysts reported in the literature,4–6,13,64 it was observed that their Tafel curves had similar slopes, indicating that the prepared catalysts have favourable kinetics making them efficient and promising for OER reactions. Co-borate ultrathin nanosheet/graphene hybrid presented Tafel of 53 mV dec−1 at pH 14.4 CoOx presented Tafel around 60 mV dec−1 at alkaline pH.5 Meso-Co and 20%Mn–Co presented Tafel at pH 14 of 74.3 and 114 mV dec−1, respectively.6
Stability tests were performed through long-term electrolysis for the most active catalyst Co3O4, the potential versus time curve is shown in Fig. 8. The catalyst over the FTO electrode remained active up to 2 hours, while using the RDE electrode higher stability up to 15 hours were obtained in all pHs. The steady-state density current at pH 1 even increased with time. Unlike what is described in the literature for CoOx, at pH 13 Co3O4 presented high stability. The poor long-term stability has been attributed to partial oxidation and deactivation of the oxide-based electrocatalysts that occur spontaneously under OER alkaline conditions,19 but has shown long-term stability in acidic electrolytes.21,65
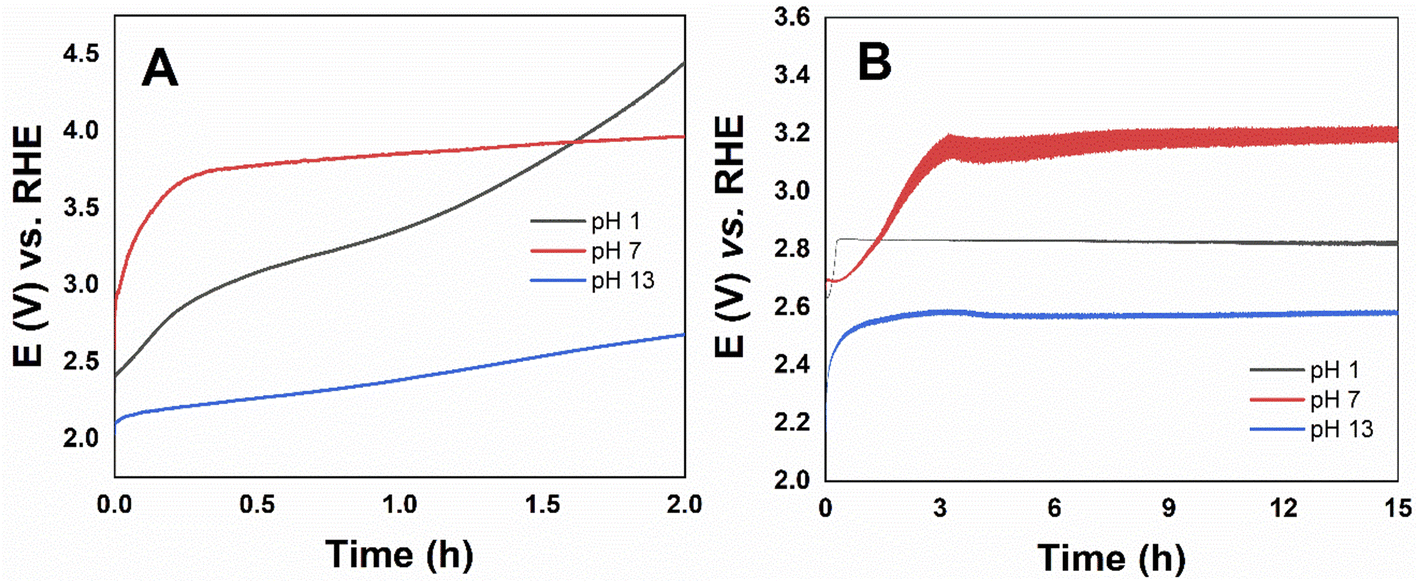 | ||
| Fig. 8 Long-term stability electrolysis in Co3O4 on (A) FTO (j = 10 mA cm−2) and (B) RDE (j = 141 mA cm−2) under a constant current of 10 mA in 0.1 mol L−1 HNO3 pH 1, 0.1 mol L−1 Na2SO4 pH 7, and 0.1 mol L−1 NaOH pH 13. | ||
In order to verify any change in the electrocatalysts after the OER, we carried out FESEM and Raman spectroscopy of the pure Co3O4 and Mn@Co3O4-3 FTO films after the reaction, for all pHs. Fig. S15–S20† shows the FESEM and no differences in the materials morphology was observed. Raman spectra shown in Fig. S21† show the same peaks of the pristine oxides, indicating no considerable changes in their structure or formation of other phases. This results corroborates with the high stability of the materials under the harsh electrocatalytic conditions.
4. Conclusions
Mn-doped Co3O4 oxides were prepared by hydrothermal synthesis in presence of urea, in different Mn:Co molar ratios. The oxides were thoroughly characterized and the spinel Co3O4 was the major phase in most of the catalysts, which provided important electrochemical properties, such as low electron transfer resistance. The oxides performance in OER was systematically investigated in acidic, neutral and alkaline conditions. Overall, the oxides with higher Co content were more active, with overpotential at j = 10 mA cm−2 of 791, 490 and 240 mV, at pH 1, 7 and 14, respectively, for the pure Co3O4. The mixed oxides are also promising OER electrocatalysts in all pH values, the material with 97 wt% of Co3O4 presented overpotential of 752 mV at j = 10 mA cm−2 at pH 1, while the material with 82.8 wt% of Co3O4 showed overpotential of 673 mV at pH 7, and 320 mV at pH 14. These overpotentials are extremely low in comparison with the electrocatalysts reported in the literature for OER. The Tafel slopes indicated different mechanisms at alkaline and acidic pH, one-electron transfer step and a chemical reaction after a one-electron transfer step as the rate-limiting step at pH 1 and 14, respectively. Moreover, outstanding stability was achieved for Co3O4 in all pHs, up to 15 h at j = 141 mA cm−2. Analysis of the oxides films after OER did not shown any considerable change in the morphology or structure. In summary, this study shows that Mn-doped Co3O4 nano-oxides are very efficient electrocatalysts, with high activity for OER at all pH ranges, especially at acidic conditions that is important to broaden the practical applicability in artificial photosynthesis. Although the Co3O4 was the most active phase, the presence of MnOx was important to improve the activity at acidic pH.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (BR) – CNPq [PQ-2/2018: 313831/2018-1], Fundação Carlos Chagas de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ [JCNE 2018: E-26/203.023/2018; Apoio às Universidades Estaduais: E-26/010.101141/2018; Projetos Temáticos: SEI-260003/001198/2020; PD10 2020: E-26/290.125/2020 post-doctoral Scholarship], and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) [Finance Code 001]. The authors would like to acknowledge NANOFAB/UERJ for the SEM measurements, LCM/CAIPE-UFF for the Raman analyses, G2E-UFF for the RDE electrode, LACES-UFRJ for the thermogravimetric analysis, Multiuser Laboratory of Pontal at the Federal University of Uberlândia for the ASAP analyses (FINEP/2013 INFR13 01.13.0371.00), and LCS/LNNano-CNPEM for the use of AFM in its open facilities.References
- L. Tan, A. Zhang, Z. Liu, P. Wei, P. Yang, H. Guo, H. Fang, J. Han, Y. Zhu and Z. Ren, RSC Adv., 2021, 11, 11779–11785 RSC.
- M. F. Othman, A. Adam, G. Najafi and R. Mamat, Renewable Sustainable Energy Rev., 2017, 80, 694–709 CrossRef.
- W. Liu, M. Kamiko, I. Yamada and S. Yagi, RSC Adv., 2022, 12, 8731–8736 RSC.
- P. Chen, K. Xu, T. Zhou, Y. Tong, J. Wu, H. Cheng, X. Lu, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- W. Song, Z. Ren, S. Y. Chen, Y. Meng, S. Biswas, P. Nandi, H. A. Elsen, P. X. Gao and S. L. Suib, ACS Appl. Mater. Interfaces, 2016, 8, 20802–20813 CrossRef CAS PubMed.
- X. Zhong, C. Shu, X. Su, W. Wang and J. Gong, Mater. Today Commun., 2022, 31, 103708 CrossRef CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- Y.-C. Huang, W. Chen, Z. Xiao, Z. Hu, Y.-R. Lu, J.-L. Chen, C.-L. Chen, H.-J. Lin, C.-T. Chen, K. Thanigai Arul, S. Wang, C.-L. Dong and W.-C. Chou, J. Phys. Chem. Lett., 2022, 13, 8386–8396 CrossRef CAS.
- Y. Zhang, X. Wang, F. Luo, Y. Tan, L. Zeng, B. Fang and A. Liu, Appl. Catal., B, 2019, 256, 117852 CrossRef CAS.
- S. Woo, J. Lee, H. Lee, N. Kwon and B. Lim, ACS Appl. Energy Mater., DOI: DOI:10.1021/acsaem.2c01523.
- H. Simchi, K. A. Cooley, J. Ohms, L. Huang, P. Kurz and S. E. Mohney, Inorg. Chem., 2018, 57, 785–792 CrossRef CAS PubMed.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Energy Mater., 2020, 3, 5439–5447 CrossRef CAS.
- L. Li, Z. Hu, L. Tao, J. Xu and J. C. Yu, ACS Appl. Energy Mater., 2020, 3, 3071–3081 CrossRef CAS.
- W. Jiang, J. Wang, X. Wang, J. Liao, J. Wei, R. Xu and L. Yang, RSC Adv., 2022, 12, 10634–10645 RSC.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- C. Bae, T. A. Ho, H. Kim, S. Lee, S. Lim, M. Kim, H. Yoo, J. M. Montero-Moreno, J. H. Park and H. Shin, Sci. Adv., 2017, 3, e1602215 CrossRef PubMed.
- K. R. Park, J. E. Jeon, G. Ali, Y.-H. Ko, J. Lee, H. Han and S. Mhin, Catalysts, 2019, 9, 564 CrossRef CAS.
- K. R. Park, J. E. Jeon, K. Kim, N. Oh, Y. H. Ko, J. Lee, S. H. Lee, J. H. Ryu, H. S. Han and S. Mhin, Appl. Surf. Sci., 2020, 510, 145390 CrossRef CAS.
- K. Lankauf, K. Cysewska, J. Karczewski, A. Mielewczyk-Gryń, K. Górnicka, G. Cempura, M. Chen, P. Jasiński and S. Molin, Int. J. Hydrogen Energy, 2020, 45, 14867–14879 CrossRef CAS.
- C. R. M. F. Fink, J. Eckhardt, P. Khadke and T. Gerdes, ChemElectroChem, 2020, 7, 4822–4836 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- C. Zhang, J. Chen, Y. Zeng, X. Rui, J. Zhu, W. Zhang, C. Xu, T. M. Lim, H. H. Hng and Q. Yan, Nanoscale, 2012, 4, 3718–3724 RSC.
- H. Chen, J. Wang, F. Liao, X. Han, Y. Zhang, C. Xu and L. Gao, Ceram. Int., 2019, 45, 11876–11882 CrossRef CAS.
- S. Li, Y. Wang, J. Sun, Y. Zhang, C. Xu and H. Chen, J. Alloys Compd., 2020, 821, 153507 CrossRef CAS.
- E. Wu, X. Feng, Y. Zheng, D. Lin, Y. Luo, Y. You, B. Huang, Q. Qian and Q. Chen, ACS Sustainable Chem. Eng., 2020, 8, 5787–5798 CrossRef CAS.
- E. Vila, R. M. Rojas, J. L. Martín De Vidales and O. García-Martínez, Chem. Mater., 1996, 8, 1078–1083 CrossRef CAS.
- R. S. Kalubarme, S. M. Jadhav, B. B. Kale, S. W. Gosavi, C. Terashima and A. Fujishima, Nanotechnology, 2018, 29, 285705 CrossRef PubMed.
- I. Lorite, J. J. Romero and J. F. Fernández, J. Raman Spectrosc., 2012, 43, 1443–1448 CrossRef CAS.
- R. Edla, N. Patel, M. Orlandi, N. Bazzanella, V. Bello, C. Maurizio, G. Mattei, P. Mazzoldi and A. Miotello, Appl. Catal., B, 2015, 166–167, 475–484 CrossRef CAS.
- A. B. Vennela, D. Mangalaraj, N. Muthukumarasamy, S. Agilan and K. v. Hemalatha, Int. J. Electrochem. Sci., 2019, 14, 3535–3552 CrossRef CAS.
- J. E. Post, D. A. McKeown and P. J. Heaney, Am. Mineral., 2020, 105, 1175–1190 CrossRef.
- J. E. Post, D. A. McKeown and P. J. Heaney, Am. Mineral., 2021, 106, 351–366 CrossRef.
- L. Yang, S. Cheng, X. Ji, Y. Jiang, J. Zhou and M. Liu, J. Mater. Chem. A, 2015, 3, 7338–7344 RSC.
- J. Zuo, C. Xu, Y. Liu and Y. Qian, Nanostruct. Mater., 1998, 10, 1331–1335 CrossRef CAS.
- E. Beyreuther, S. Grafström, L. M. Eng, C. Thiele and K. Dörr, Phys. Rev. B, 2006, 73, 155425 CrossRef.
- A. N. Naveen and S. Selladurai, Phys. B., 2015, 457, 251–262 CrossRef CAS.
- W. Tang, W. Li, D. Li, G. Liu, X. Wu and Y. Chen, Catal. Lett., 2014, 144, 1900–1910 CrossRef CAS.
- J. L. Gautier, E. Rios, M. Gracia, J. F. Marco and J. R. Gancedo, Thin Solid Films, 1997, 311, 51–57 CrossRef CAS.
- R. A. Davoglio, G. Cabello, J. F. Marco and S. R. Biaggio, Electrochim. Acta, 2018, 261, 428–435 CrossRef CAS.
- V. R. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. A. Ovechkina, E. Z. Kurmaev, N. I. Lobachevskaya, Y. M. Mukovskii, J. Mitchell and D. L. Ederer, Phys. Rev. B, 2002, 65, 113102 CrossRef.
- O. A. Bulavchenko, T. N. Afonasenko, A. v. Ivanchikova, V. Y. Murzin, A. M. Kremneva, A. A. Saraev, V. v. Kaichev and S. v. Tsybulya, Inorg. Chem., 2021, 60, 16518–16528 CrossRef CAS PubMed.
- F. Babar, U. Mehmood, H. Asghar, M. H. Mehdi, A. U. H. Khan, H. Khalid, N. ul Huda and Z. Fatima, Renewable Sustainable Energy Rev., 2020, 129, 109919 CrossRef CAS.
- K. Guse and H. Papp, Fresenius. J. Anal. Chem., 1993, 346, 84–91 CrossRef CAS.
- S. C. Petitto and M. A. Langell, J. Vac. Sci. Technol., 2004, 22, 1690–1696 CrossRef CAS.
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413–429 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. Restovic, E. Ríos, S. Barbato, J. Ortiz and J. L. Gautier, J. Electroanal. Chem., 2002, 522, 141–151 CrossRef CAS.
- M. Konsolakis, M. Sgourakis and S. A. C. Carabineiro, Appl. Surf. Sci., 2015, 341, 48–54 CrossRef CAS.
- A. C. Co, J. Liu, I. Serebrennikova, C. M. Abel and V. I. Birss, J. Mater. Sci., 2005, 40, 4039–4052 CrossRef CAS.
- K. Li, R. Zhang, R. Gao, G. Q. Shen, L. Pan, Y. Yao, K. Yu, X. Zhang and J. J. Zou, Appl. Catal., B, 2019, 244, 536–545 CrossRef CAS.
- X. H. Xia, J. P. Tu, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Liu, C. Chang, D. Zhang and Y. Wu, Prog. Nat. Sci.: Mater., 2013, 23, 593–597 CrossRef.
- G. Godillot, L. Guerlou-Demourgues, P. L. Taberna, P. Simon and C. Delmas, Electrochem. Solid-State Lett., 2011, 14, 1–6 CrossRef.
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, Adv. Mater., 2014, 26, 5702–5707 CrossRef CAS PubMed.
- C. N. Brodsky, D. K. Bediako, C. Shi, T. P. Keane, C. Costentin, S. J. L. Billinge and D. G. Nocera, ACS Appl. Energy Mater., 2019, 2, 3–12 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B. J. Ni, Nano Energy, 2020, 78, 105270 CrossRef CAS.
- M. Y. Solangi, U. Aftab, A. Tahira, M. I. Abro, R. Mazarro, V. Morandi, A. Nafady, S. S. Medany, A. Infantes-Molina and Z. H. Ibupoto, Int. J. Hydrogen Energy, 2022, 47, 3834–3845 CrossRef CAS.
- R. M. Ramsundar, J. Debgupta, V. K. Pillai and P. A. Joy, Electrocatalysis, 2015, 6, 331–340 CrossRef CAS.
- M. Etzi Coller Pascuzzi, M. van Velzen, J. P. Hofmann and E. J. M. Hensen, ChemCatChem, 2021, 13, 459–467 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI includes detailed material characterization, including XRD Rietveld refinement, XPS, TG curve and discussion, CV curves, EIS fitting results (Nyquist diagram and Bode plots), OER electrocatalysis LSV curves and Tafel plots, stability tests, comparison of OER catalytic activity with other recently reported transition metal-based catalysts, and FESEM images and Raman spectra of the oxides after OER. See https://doi.org/10.1039/d2ra04570b |
| This journal is © The Royal Society of Chemistry 2022 |