
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal conductivity of polyurethane sheets containing beryllium oxide nanofibers
Md. Shakhawat Hossain ab,
Anamul Hoque Bhuiyanac and
Koji Nakane*a
ab,
Anamul Hoque Bhuiyanac and
Koji Nakane*a
aFrontier Fiber Technology and Science, University of Fukui, Bunkyo 3-9-1, Fukui, 910-8507, Japan. E-mail: nakane@matse.u-fukui.ac.jp
bDepartment of Textile Engineering, Khulna University of Engineering & Technology, Khulna, 9203, Bangladesh. E-mail: shakhawat.shaon@te.kuet.ac.bd
cDepartment of Textile Engineering, Dhaka University of Engineering and Technology, Gazipur, 1700, Bangladesh. E-mail: anamulhb@duet.ac.bd
First published on 21st October 2022
Abstract
Polyvinyl alcohol/beryllium sulfate/polyethyleneimine (PVA/BeSO4/PEI) precursor nanofibers (NFs) was first fabricated to obtain PVA/BeSO4/PEI electrospun NFs by electrospinning technology, finally manufactured beryllium oxide (BeO) NFs followed by various heat treatment methods. The minimum calcination temperature for pure BeO NFs was 1000 °C, and the minimum specific surface area (5.1 m2 g−1) and pore volumes (0.0128 cm3 g−1) were at 1300 °C. 46.18% Be and 53.82% O was measured in BeO NFs by X-ray photoelectron spectroscopy. BeO NFs were then impregnated with polyurethane (PU) aqueous solution to make PU/BeO NFs heat-dissipating sheet. This heat-dissipating sheet showed superior thermal conductivity (14.4 W m−1 K−1) at 41.4 vol% BeO NFs content. The electrical insulating properties of the heat-dissipating sheet were likewise excellent (1.6 × 1012 Ω □−1). In this study, the author attempted to create a thermally conductive but electrically insulating PU/BeO NFs heat-dissipating sheet that could effectively eliminate generated heat from electric equipment.
Introduction
Electronic devices and other high-performance energy-saving home appliances are now significantly smaller, with higher integration and power. Continuous using such devices generates a large amount of heat. The electronic components may create various problems, such as increased failure frequency, fire, and smoke, when the increased temperature exceeds the thermal stability limit of this device. Many researchers tried to develop heat-dissipating materials to improve the thermal conductivity of electronic devices. Some researchers used metal as heat-dissipating materials for electronics devices because they possess excellent thermal conductivity and superior mechanical strength. Although, electrically insulating properties are vital for developing electric devices.1–4Polymeric materials possess excellent electric insulation properties, good moldability, and thermal insulating properties. So, polymeric materials can be used as heat-dissipating sheets if the thermal conductivity properties improve. Adding a certain amount of inorganic filler with low thermal conductive polymer (0.01–0.1 W m−1 K−1), the thermal conductivity of polymeric composite will increase significantly.5,6 The most significant advantages of polymer composites are their simple processing technique, very lightweight, meager cost, and resistance against corrosions.7–13 Some inorganic materials possess thermal conductivity with electrical insulation properties, such as silicon nitride (Si3N4),14,15 beryllium oxide (BeO),16,17 hexagonal boron nitride (h-BN),18–20 aluminum nitride (AlN),21,22 alumina (Al2O3)23 and silicon carbide (SiC).24
One of the most critical properties of BeO ceramic is its ultra-high thermal conductivity (330 W m−1 K−1) with excellent electrical insulation (>1013 Ω cm) properties, among other well-known ceramic materials. However, depending on density, the thermal conductivity of BeO ceramic varies from 230 to 330 W m−1 K−1, which is higher than other metals except for copper, silver, and gold.17 Other than high thermal conductive properties, beryllium ceramics can exhibit a unique combination of other physicochemical properties such as high melting point, high intensity, high insulation nature, high chemical and thermal stability, a considerable specific volumetric resistance, transparency for vacuum, infrared, ultraviolet, visible, radiation resistance, low dielectric losses, low dielectric constant, good technology applicability. BeO ceramics can conduct heat from electronic technology while temperatures range from 300 to 630 K of all ceramic materials. This outstanding property makes BeO ceramic a promising material for using new technology fields, contemporary electronics, vacuum electronics technology, nuclear technology, and microelectronics. There is no alternative to BeO ceramics, especially in thermal applications.25,26
There are few reports on BeO nanoparticle synthesis, and extensive structural characterization of produced BeO nanoparticles is limited and not reported.27 Wang et al. prepared BeO nanoparticles by polyacrylamide gel route. They created xerogel by using acrylamide and N,N′-methylene bis acrylamide monomers and mixed with beryllium sulfate tetrahydrate (BeSO4·4H2O) aqueous solution followed by ammonium persulfate aqueous solution. Finally, they prepared BeO nanoparticles in white powder by heating and calcinating at 700−1000 °C.26
NF research, development, and industrial applications have received much attention recently. Many investigations on heat-dissipating sheets constructed of NFs have been undertaken.28,29 Ohgoshi et al. fabricated magnesia (MgO) NFs by calcinating polyvinyl alcohol/magnesium ethoxide (PVA/Mg(OC2H5)2) precursor NFs at different temperatures. They produced PVA/Mg(OC2H5)2 precursor NFs mat by electrospinning technique using PVA and Mg(OC2H5)2 mixtures.30 Nakane et al. created α-alumina NF mats from a PVA/boehmite spinning mix using electrospinning. They discovered that at 5.7% α-alumina content, the α-alumina NFs sheet had thermal conductivity in parallel (1.29 W m−1 K−1) and perpendicular (0.38 W m−1 K−1) directions.31,32 Ohgoshi et al. generated an α-alumina NFs polyurethane sheet with boehmite particles and aqueous PVA solution using an electrospinning method. The resultant α-alumina NFs polyurethane composite sheet had increased heat conductivity (19.7 W m−1 K−1) in a parallel direction when it included 33% alumina.33,34
Electrospinning is becoming a popular technology for polymer NFs since it is simple to make various inorganic NFs. Electrospinning creates continuous polymer strands with nanoscale diameters by applying an external electric field to a polymer solution.35 Compared to commercial textiles, electrospun NFs have a large specific surface area and a tiny pore size. As a result, NFs can be rationally designed to have novel and significantly improved physical, chemical, and biological properties.36 An electric force is applied between a suspended droplet solution at a capillary tip and a collector in the basic process of electrospinning. A charged jet is released and travels to the grounded target when the intensity of the electric field overcomes the surface tension of the polymer solution, creating fibers in the form of nonwoven mats.37
The authors used granular polyvinyl alcohol (PVA) and BeSO4·4H2O salt and polyethyleneimine (PEI) to produce PVA/BeO/PEI precursor NFs in this research work. After impregnating polyurethane (PU), they make PU/BeO NFs heat-dissipating sheets from PVA/BeO/PEI precursor NFs. PU/BeO NFs heat-dissipating sheet may be very effective as a heat sink because BeO ceramic has high thermal conductivity and excellent electrical resistivity. On the other side, this study also focuses on the electrospinning technique to make BeO NFs, followed by the heat treatment method and make PU/BeO NFs composite sheet. This composite sheet is most effective in dissipating heat from electronic devices. So BeO NFs may be very effective for high-power electronic devices dissipating heat.
Experimental section
Raw materials
BeSO4·4H2O (purity > 98%) was purchased from Kanto chemical co. Inc., Tokyo, Japan. PVA, Degree of polymerization = 1500 and molecular weight = 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was purchased from Wako Pure Chemical Ind. Ltd., Japan. PEI (CH2CH2NH)n (30% in water with a molecular weight of 60000–80000) was purchased from Nacalai tesque, Inc., Kyoto, Japan. Deionized water (H2O) was used as received. PU water emulsion containing 30 wt%, solid (Superflex #300), a kind-hearted gifted by DKS Co. Ltd., Japan. All chemicals were used without any further purification.
000 was purchased from Wako Pure Chemical Ind. Ltd., Japan. PEI (CH2CH2NH)n (30% in water with a molecular weight of 60000–80000) was purchased from Nacalai tesque, Inc., Kyoto, Japan. Deionized water (H2O) was used as received. PU water emulsion containing 30 wt%, solid (Superflex #300), a kind-hearted gifted by DKS Co. Ltd., Japan. All chemicals were used without any further purification.
BeO NFs preparation by electrospinning machine
10 g of PVA were added with 90 g of deionized water and boiled for 4 hours at 90 °C to make a 10% PVA solution. To make the PVA/BeSO4 solution, BeSO4·4H2O salt was added directly to the previously produced 10% PVA solution and kept at room temperature for 7 hours with vigorous stirring. PEI was added with PVA/BeSO4 solution, finally making PVA/BeSO4/PEI composite precursor solution. The electrospinning apparatus was used to make PVA/BeSO4/PEI precursor NFs. The prepared precursor solutions were placed in a 3 mL plastic syringe with a 0.5 mm pinhead inner diameter. The electrospinning settings in this experiment were as follows: 20 kV operating voltage, 0.008 mL min−1 injection rate, and 15 cm needle tip to collector distance. The electrospun PVA/BeSO4/PEI composite NFs were collected on parchment paper. PVA/BeSO4/PEI precursor NFs were calcined at varied heat treatment temperatures (700 °C, 800 °C, 900 °C, 1000 °C, 1100 °C, and 1200 °C) for 5 hours in the air with an electric furnace to eliminate organic components (NHK-170, Nitto Kagaku Co. Ltd., box-type furnace, Japan). Finally, the BeO NFs were then obtained.Formation of PU sheets containing BeO NFs
The BeO NFs were impregnated with a 5% PU solution and cured for 10 hours by vacuuming the solution at room temperature until completely absorbed. Fig. 1 depicted all of the appropriate steps schematically.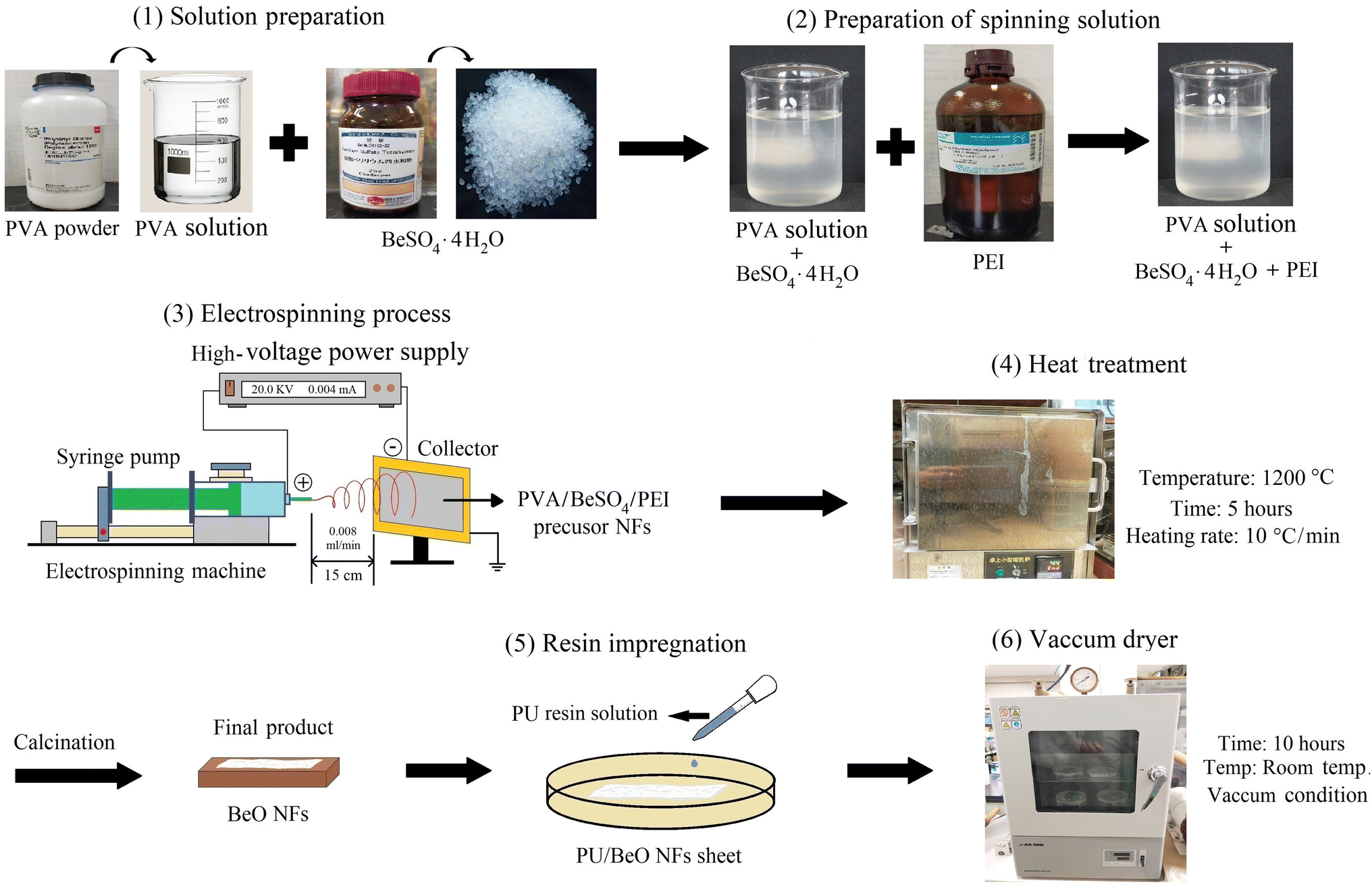 | ||
| Fig. 1 Experimental scheme for the preparation of BeO NF mats. | ||
Characterization
The morphological structure of both precursor and BeO NFs was examined at a voltage of 5 kV using a Keyence scanning electron microscope (VE-9800, Keyence Co. Ltd., Japan). The samples were initially coated with Au/Pd sputtering under vacuum using an ion coater (SC-701; Sanyu Electron Co. Ltd., Japan). Adobe Photoshop CS3 extension software measurements of the precursor and BeO NFs (n = 100) were used to calculate the mean fiber diameter D and standard deviation. An X-ray diffractometer (XRD, Rigaku Mini Flex II, Japan) was used to measure X-ray diffraction using Ni-filtered CuK radiation (30 kV, 15 mA) as an X-ray source to investigate the samples’ crystalline nature. The BeO content (wt%) in the dissipation sheets was assessed by thermogravimetric (TG) analysis carried out in the air atmosphere with a heating speed of 5 °C min−1 from 30 to 600 °C using a differential TG analyzer (Shimadzu DTG-60, Japan). A BELSORP-mini II (MicrotracBEL Corp., Japan) was used to monitor nitrogen adsorption isotherms at −196 °C. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. The Barrett–Joyner–Halenda (BJH) algorithm was used to calculate the pore-size distribution (PSD) curves from the isotherm. A JEOL JPS-9010 (Nippon Electronics Co. Ltd., Japan) equipment was used to perform X-ray photoelectron spectroscopy (XPS) using an Mg-Ka (hv = 1253.6 eV) source at a residual gas pressure of 5 × 10–6 Pa. Hiresta-UP (Mitsubishi Chemical Analytics, Japan) was used to measure the electrical resistivity of the composite sheet by ASTM D257. Thermal diffusivity (m2 s−1) was measured at room temperature in the planar and thickness directions using a thermowave analyzer TA35 (Bethel Co., Ltd., Japan). Thermal diffusivity is measured via temperature wave analysis in this system. We determined four different average and relative standard deviation (RSD) measurements.Results and discussion
Characterization of BeO NFs
In our previous work, we heated PVA/BeSO4/PEI precursor NFs’ from 700 °C to 1200 °C.38 However, here we start heating from 600 °C to 1300 °C. The XRD curves of the residues calcinated from 600 °C to 1300 °C are shown in Fig. 2. Few low-intensity BeO crystal peaks are observed after heating from 600 °C to 900 °C. However, the most pronounced BeO crystal peaks were detected at temperatures of 1000 °C or higher, and their intensity increased as the temperature grew. The main reason is that all organic components are thermally decomposed at high temperatures (1000 °C to 1300 °C). However, BeO residues is very brittle while heated at 1200 °C and above.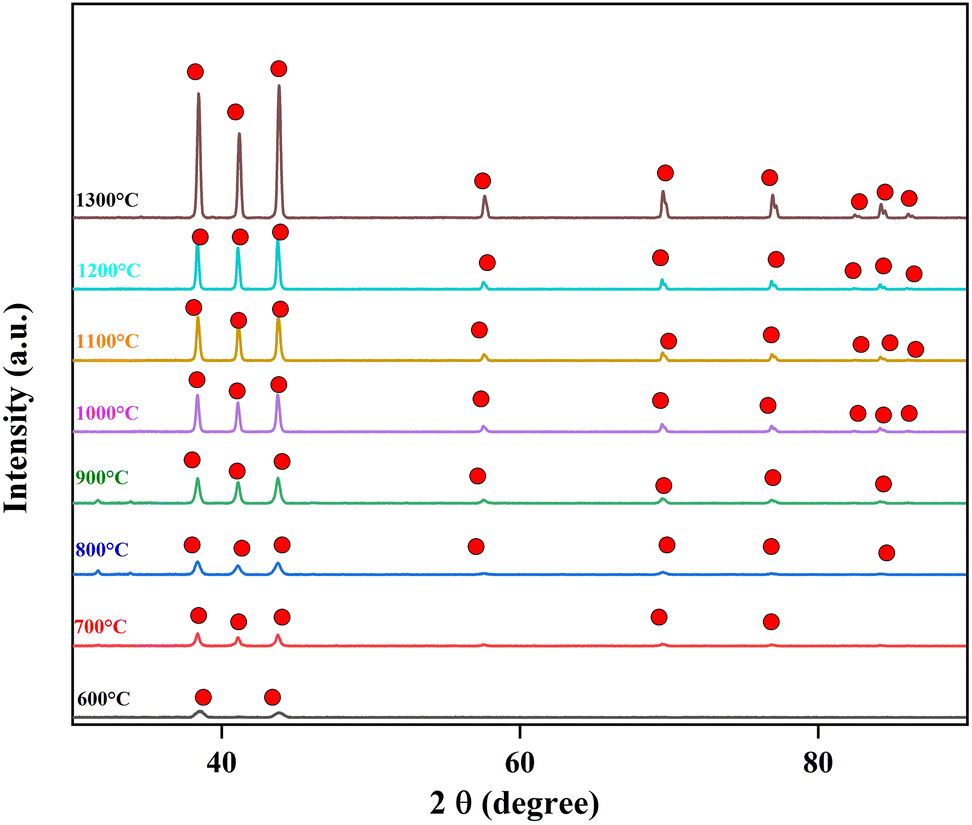 | ||
Fig. 2 XRD curves of residues after calcinating PVA/BeSO4/PEI precursor NFs ( : BeO crystal). : BeO crystal). | ||
The calcination temperatures can influence the surface area and morphologies of fibers, tuning the crystalline structure. PVA/BeSO4/PEI precursor NFs’ specific surface area (36.3 m2 g−1) increases significantly to 145 m2 g−1 for BeO NFs while calcinated at 600 °C. Furthermore, within the mesopore range of 2–50 nm, a mesoporous structure with an average pore diameter of 9.3 nm is seen. However, as the calcination temperature rises to 1000 °C, 1100 °C, 1200 °C, and 1300 °C, the specific surface area reduces dramatically to 23.0 m2 g−1, 17.0 m2 g−1, 11.9 m2 g−1, and 5.1 m2 g−1, respectively. The findings show that removing organics improves specific surface area at an adequate calcination temperature. A high calcination temperature will hinder the increase in specific surface area, resulting in a low specific surface area. Sintering or metal particle obstruction could be the cause. At temperatures of 1000 °C or higher, BeO NFs can be employed as heat-dissipating sheets. However, when heat-treated at 1200 °C and 1300 °C temperatures, samples containing BeO crystal became brittle, as the fiber shape was shattered. Based on nitrogen adsorption investigations, Table 1 summarizes the specific surface area, average pore diameter, and pore volume estimates.
| Precursor and calcinated samples | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|
| PVA/BeSO4/PEI precursor NFs | 36.9 | 0.0638 |
| BeO NFs at 600 °C | 145.0 | 0.3393 |
| BeO NFs at 700 °C | 122.0 | 0.4318 |
| BeO NFs at 800 °C | 77.4 | 0.2891 |
| BeO NFs at 900 °C | 50.8 | 0.1827 |
| BeO NFs at 1000 °C | 23.0 | 0.0643 |
| BeO NFs at 1100 °C | 17.0 | 0.0382 |
| BeO NFs at 1200 °C | 11.9 | 0.0275 |
| BeO residues at 1300 °C | 5.1 | 0.0128 |
The electrospinning method fabricated the PVA/BeSO4/PEI precursor NFs. On the other hand, BeO crystals were formed by calcinating the PVA/BeSO4/PEI precursor NFs at 1000 °C or above temperatures at a dry ratio of PVA/BeO was 90/10 wt%;38 the NFs remained in their shape, and the average fiber diameter was reduced due to heat treatment up to 1200 °C temperatures.39 SEM images and histogram of BeO residue after calcinating PVA/BeSO4/PEI precursor NFs at 600 °C, 1100 °C, and 1300 °C temperatures are shown in Fig. 3. At 600 °C and 1100 °C temperatures, BeO remaining good NFs shape, interestingly, BeO crystal became very compact and cubic in shape at 1300 °C temperatures, as shown in Fig. 3(c).
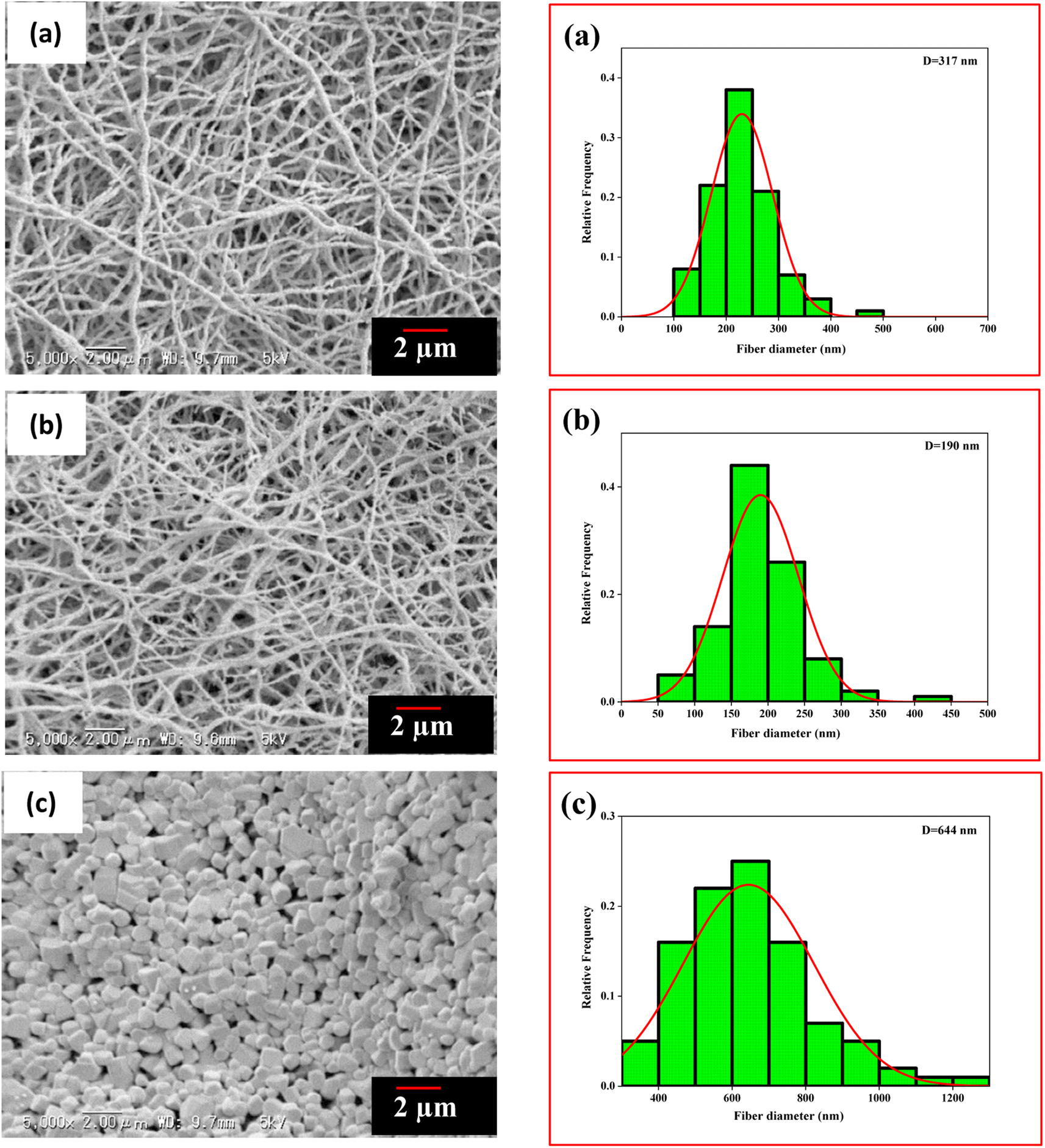 | ||
| Fig. 3 SEM images and histogram of residues after calcinating PVA/BeSO4/PEI precursor NFs at (a) 600 °C, (b) 1100 °C, (c) 1300 °C. | ||
The ATR-FTIR spectra of pure PVA, BeSO4·4H2O salt, PVA/BeSO4/PEI precursor NFs, and BeO NFs were obtained, and the results are shown in Fig. 4. The spectra show strong broadband for pure PVA at 3320 cm−1 and 3210 cm−1 for PVA/BeSO4/PEI precursor NFs. This band was attributed to the O–H stretching vibration of the hydroxyl group of pure PVA and PVA/BeSO4/PEI precursor NF samples because water contains hydrogen bonding. However, no absorbance peak was found for the BeO NFs due to a lack of water-containing hydrogen bonding for BeO NF samples.40 The band corresponding to C–H stretching mode is 2926 cm−1 and 2940 cm−1 for the pure PVA and PVA/BeSO4/PEI precursor NFs.41 The absorption peak at 853 cm−1 for pure PVA has been assigned to C–H rocking (Fig. 4a) and moved to 823 cm−1 for PVA/BeSO4/PEI precursor NFs (Fig. 4c). The sharp band of 1094 cm−1 corresponds to the C–O stretching of acetyl groups present on the PVA backbone that is shifted to 1060 cm−1 and 1080 cm−1 for BeSO4·4H2O salt and PVA/BeSO4/PEI precursor NFs, respectively.40–42 In the complexed system, CH2 bending exhibited at 1426 cm−1 in pure PVA remains the same for PVA/BeSO4/PEI precursor NFs. The acetyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group has an absorbance of 1727 cm−1 in pure PVA, which can be explained by intra/inter molecule hydrogen bonding with the neighboring OH group. For instance, this CO peak is shifted to 1651 cm−1 and 1734 cm−1 for BeSO4·4H2O salt and PVA/BeSO4/PEI precursor NFs, respectively. This demonstrates that the interaction between the salt and PVA is caused by pure PVA’s O–H and C–O groups. Water molecules are likely to be removed at higher calcinating temperatures (1100 °C) could be the reason.40,41 Furthermore, the BeO bending vibration was measured below 1000 cm−1, similar to other inorganic metal oxide compounds.43–48 The low peak is ν (Be–O) vibrations at 725 cm−1.49
O group has an absorbance of 1727 cm−1 in pure PVA, which can be explained by intra/inter molecule hydrogen bonding with the neighboring OH group. For instance, this CO peak is shifted to 1651 cm−1 and 1734 cm−1 for BeSO4·4H2O salt and PVA/BeSO4/PEI precursor NFs, respectively. This demonstrates that the interaction between the salt and PVA is caused by pure PVA’s O–H and C–O groups. Water molecules are likely to be removed at higher calcinating temperatures (1100 °C) could be the reason.40,41 Furthermore, the BeO bending vibration was measured below 1000 cm−1, similar to other inorganic metal oxide compounds.43–48 The low peak is ν (Be–O) vibrations at 725 cm−1.49
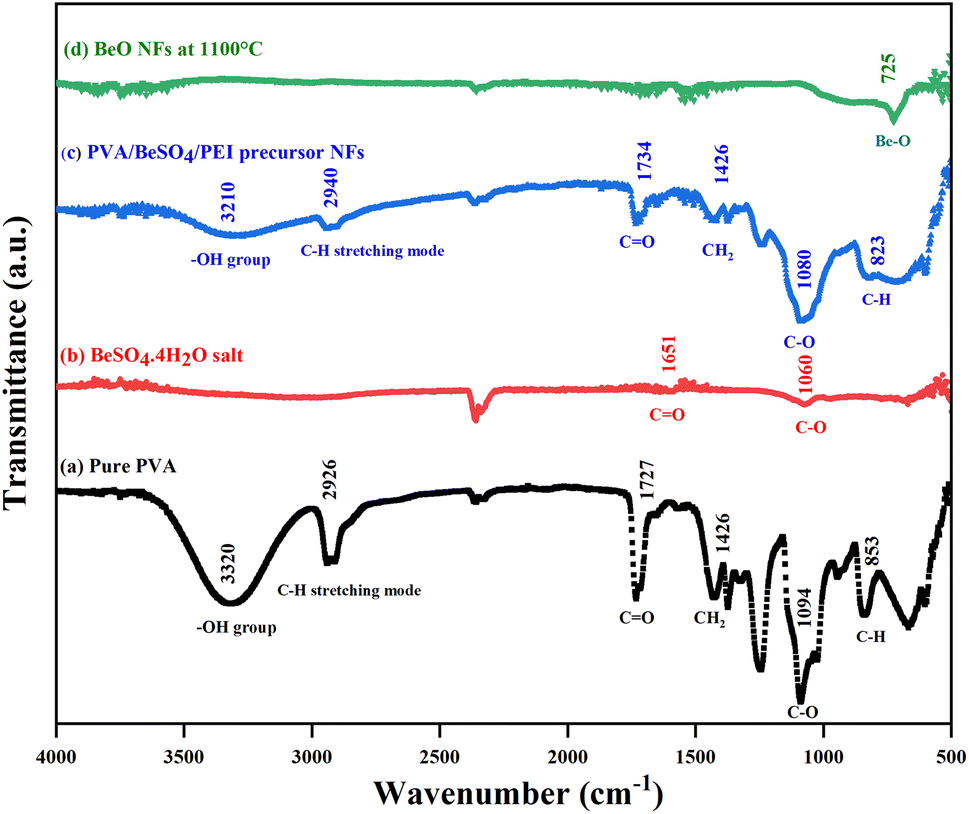 | ||
| Fig. 4 FTIR spectra of (a) pure PVA, (b) BeSO4·4H2O salt, (c) PVA/BeSO4/PEI precursor NFs, and (d) BeO NFs. | ||
BeO NFs were subjected to XPS examination to determine which chemical elements were present on the surface of these compounds. In contrast, BeSO4·4H2O salt, PVA/BeSO4 precursor NFs and PVA/BeSO4/PEI precursor NFs were used as controls. In Fig. 5, BeSO4·4H2O salt, PVA/BeSO4, and PVA/BeSO4/PEI precursor NFs showed peaks of O 1s, C 1s, and S 2p3/2. However, the BeO NFs showed pronounced O 1s peaks where C 1s and S 2p3/2 peaks were absent. Be 1s peaks were present only in the BeO NFs but not in the BeSO4·4H2O salt, PVA/BeSO4 precursor NFs, or PVA/BeSO4/PEI precursor NFs.
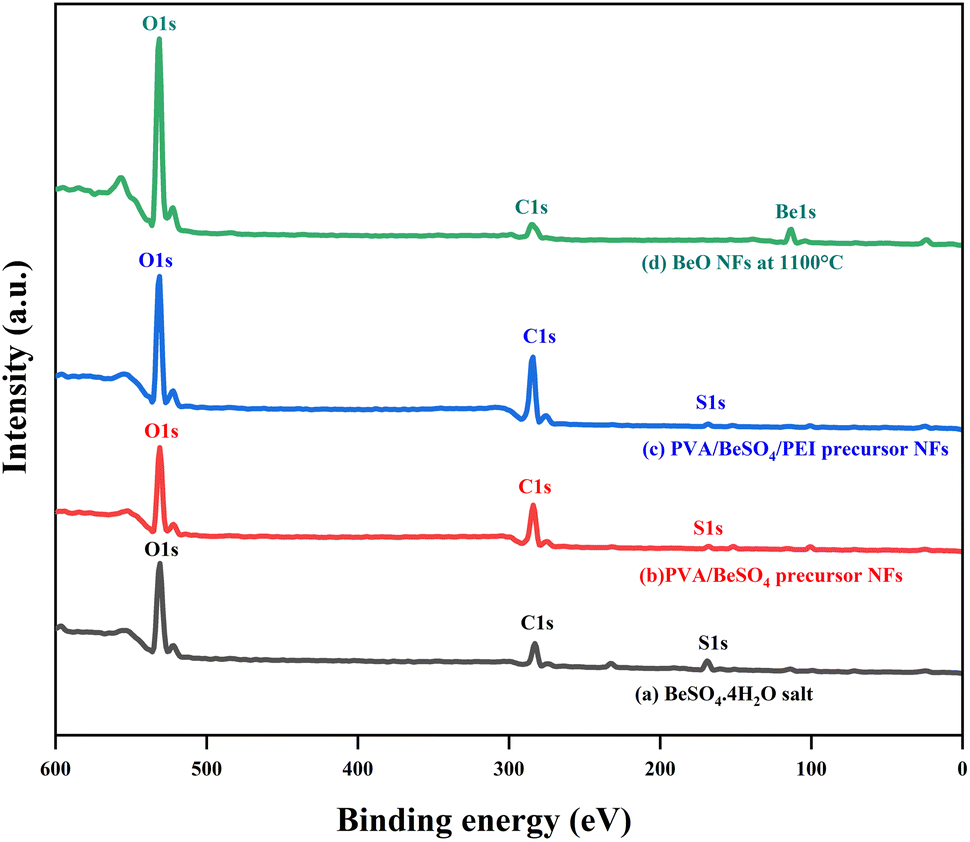 | ||
| Fig. 5 Wide-scan XP spectra of BeSO4·4H2O salt, PVA/BeSO4 precursor NFs, PVA/BeSO4/PEI precursor NFs and BeO NFs. | ||
Fig. 6 shows the BeSO4·4H2O salt, PVA/BeSO4 precursor NFs, PVA/BeSO4/PEI precursor NFs and BeO NFs samples C 1s, O 1s, Be 1s, and S 2p3/2 XP spectra. The PVA/BeSO4/PEI precursor NFs sample spectra showed strong C–C bonding at 284.2 eV (Fig. 6a). Compared with the BeO NF samples, the C 1s peak increased in the PVA/BeSO4/PEI precursor NF samples. The main reason is the significant amount of carbon in precursor samples. The O 1s peaks were detected in all instances; however, the sharpest rise (531.7 eV) for BeO NFs (Fig. 6b). The main reason is that a higher calcinating temperature (1100 °C) reduces organic components. On the other hand, Be 1s peaks were detected at a very high calcinating temperature (1100 °C), only for BeO NFs at 113.7 eV (Fig. 6c). In addition, S 2p3/2 peaks were absent in all samples (PVA/BeSO4 precursor NFs, PVA/BeSO4/PEI precursor NFs and BeO NFs) except BeSO4·4H2O salt (Fig. 6d) at 168.1 eV.
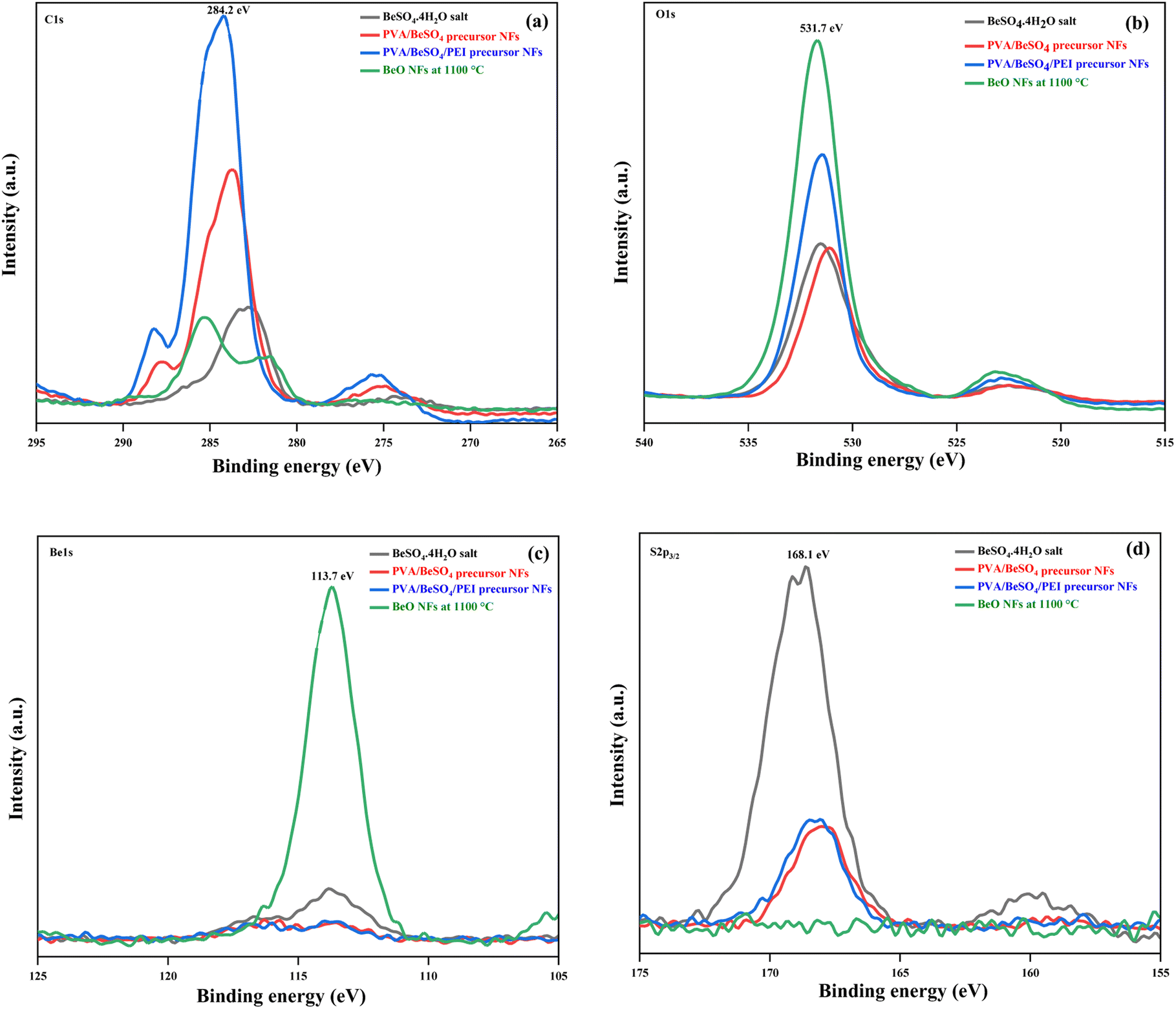 | ||
| Fig. 6 XP spectra of BeSO4·4H2O salt, PVA/BeSO4 precursor NFs, PVA/BeSO4/PEI precursor NFs and BeO NFs (a) C 1s spectra, (b) O 1s spectra, (c) Be 1s spectra, and (d) S 2p3/2 spectra. | ||
The elemental composition contents (Table 2) of C, O, Be, S, and N were analyzed according to the XPS results (Fig. 5) to corroborate the variations in the temporal composition content of each sample. Table 2 shows that no elements other than Be (46.18%) and O (53.82%) could be detected. Be’s weight percentage was significantly increased at BeO NFs compared to BeSO4·4H2O salt and precursor NFs. The possible reason is that the Be content increased dramatically with decreasing C content after calcination, which may lead to BeO bonds.
| Sample name | Elemental composition contents (%) | ||||
|---|---|---|---|---|---|
| C 1s | Be 1s | O 1s | S 2p3/2 | N 1s | |
| BeSO4·4H2O salt | 24.75 | 18.83 | 48.97 | 7.45 | — |
| PVA/BeSO4 precursor NFs | 53.28 | 7.94 | 37.11 | 1.67 | — |
| PVA/BeSO4/PEI precursor NFs | 57.07 | 5.14 | 36.42 | 1.20 | 0.17 |
| BeO NFs heated at 1100 °C | — | 46.18 | 53.82 | — | — |
Formation of PU/BeO NFs composite sheet
SEM images of the PU sheet containing the BeO NFs are shown in Fig. 7. PU is impregnated into the calcinated BeO NF mats, making the sheet significantly closer. Here, the PU successfully filled the gap between the NFs, and the NFs were dispersed homogeneously in the PU matrix. NFs were also found on the sheet’s surface, indicating that the fibers were neither fractured nor detached once soaked with PU emulsion. However, there are very few heat transmission channels for BeO in the thickness direction of the sheet. We assumed that the produced sheets would have a superior thermal conductivity in the plane direction than in the thickness direction. | ||
| Fig. 7 SEM images of PU sheet containing aligned BeO NFs (a: 700 °C, b: 900 °C, c: 1100 °C, (left: sheet surface, right: cross-section)). | ||
Thermal conductivity of PU/BeO NF composite sheet
The brittleness of the as-prepared BeO NF mats prevented measuring their thermal conductivity. However, the thermal conductivity of PU/BeO NF composite sheets were measured as shown in Table 3. It was observed that the thermal conductivity was much higher while calcinating at 1100 °C for both plane and thickness directions than at low calcinating temperatures (700 °C and 900 °C). The possible reason is that due to high calcination temperatures, organic content removes, and BeO NFs behave as mesoporous NFs.| Temperature (°C) | BeO content (%) | Thermal conductivity (W m−1 K−1) | |
|---|---|---|---|
| Plane direction | Thickness direction | ||
| 700 | 31.2 | 3.0 ± 0.17a | 1.2 ± 0.75e |
| 40.5 | 2.0 ± 0.20a | 0.6 ± 0.62f | |
| 51.1 | 4.0 ± 0.10a | 0.5 ± 0.26g | |
| 900 | 21.8 | 3.1 ± 0.43b | 2.6 ± 0.78h |
| 27.5 | 3.8 ± 0.44c | 2.3 ± 1.35a | |
| 29.8 | 4.0 ± 0.46a | 2.5 ± 1.32g | |
| 1100 | 16.6 | 1.4 ± 0.36d | 2.6 ± 0.65h |
| 21.5 | 5.0 ± 1.39c | 4.4 ± 1.68i | |
| 22.9 | 4.5 ± 0.62a | 0.8 ± 0.53b | |
| 41.4 | 14.4 ± 0.78a | 1.4 ± 0.56e | |
For all the listed data, the standard deviations of thermal conductivity at the plane direction were less than 1.39, and the thickness direction was less than 1.68, which was presented as percentages of Ls (mean ± SD). To measure the statistical significance, a two-tailed t-test was conducted between the datasets, and p values were derived, where p > 0.05 indicates no significant difference and p ≤ 0.05 indicates a significant difference between the datasets. Here, the statistical analysis showed that, in most cases, the datasets are significantly different (p ≤ 0.05). So, it can be said that changing calcination temperatures significantly affected (p ≤ 0.05) the thermal conductivity of PU/BeO NF sheets. Superscripts indicate the test results of the ANOVA. Different superscripts showing datasets are significantly different, and similar superscripts showing datasets are similar. Here total nine (9) superscripts has been measured (a = 0.00, b = 0.12, c = 0.19, d = 0.16, e = 0.11, f = 0.31, g = 0.26, h = 0.01, and i = 0.02).
It also observed that thermal conductivity was much higher in the planner direction than in the thickness direction. The possible reason is that the thermal conductivity in the thickness direction is almost similar to conventional spherical filler particles. The contact area of traditional spherical fillers is limited, and an efficient heat conduction path is difficult to implement. On the other side, the thermal conductivity parallel to the aligned BeO NFs rose in proportion to the NF content. This improvement was due to the BeO NFs’ elongated shape, which provides excellent heat pathways for the BeO NFs in the resin. The whole mechanism of low and high thermal conductivity is shown in Fig. 8.
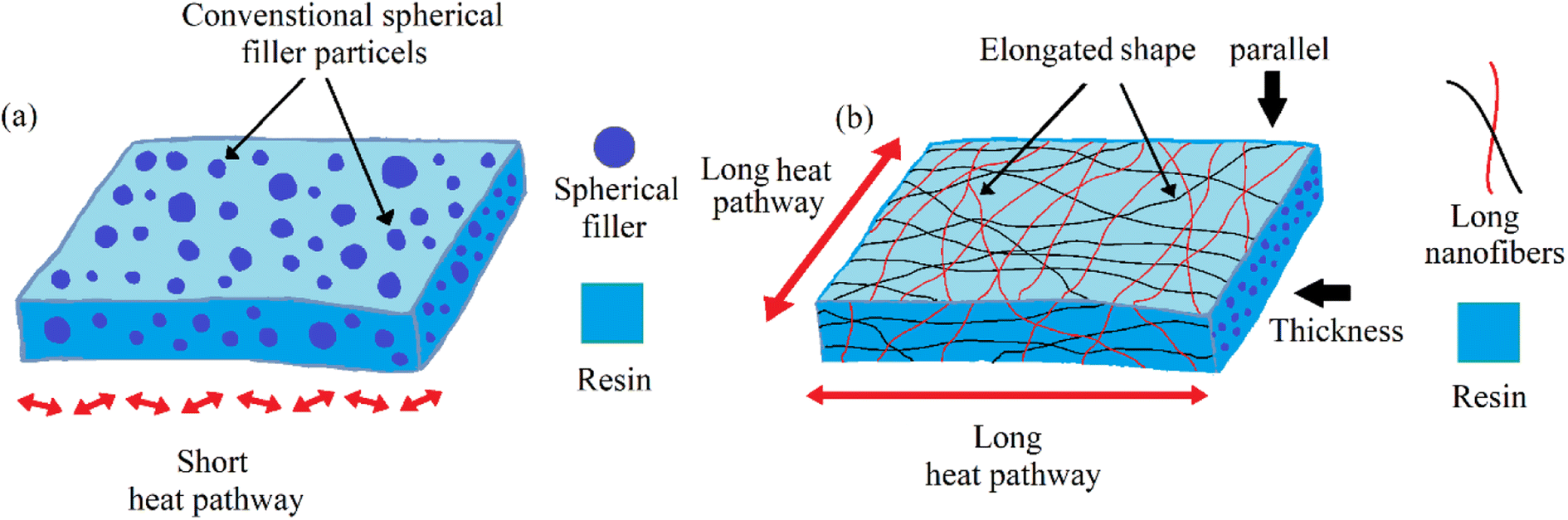 | ||
| Fig. 8 Mechanism of (a) low thermal conductivity by conventional spherical fillers and (b) improved thermal conductivity by the elongated shape of BeO NFs. | ||
The thermal conductivity of pure is PU, 0.022 W m−1 K−1.31 Adding BeSO4·4H2O salt with pure PU increases the thermal conductivity of the PU/BeO NF sheets. The lowest thermal conductivity (1.4 W m−1 K−1) of the PU/BeO NFs sheet was at 16.6 vol% BeO content, which was increased at 21.5, 22.9, 23.4 vol% BeO content. At 41.4 vol% BeO content, the highest thermal conductivity (14.4 W m−1 K−1) was found in the plane direction. However, the relationship between BeO% and thermal conductivity is not always regular. The thickness of the highest thermal conductive (14.4 W m−1 K−1) BeO NF sheet was 81 μm at 41.4 vol% and content. Fig. 9 shows the thermal conductivity of the PU/BeO NF sheet at 1100 °C temperatures in both plane and thickness directions.
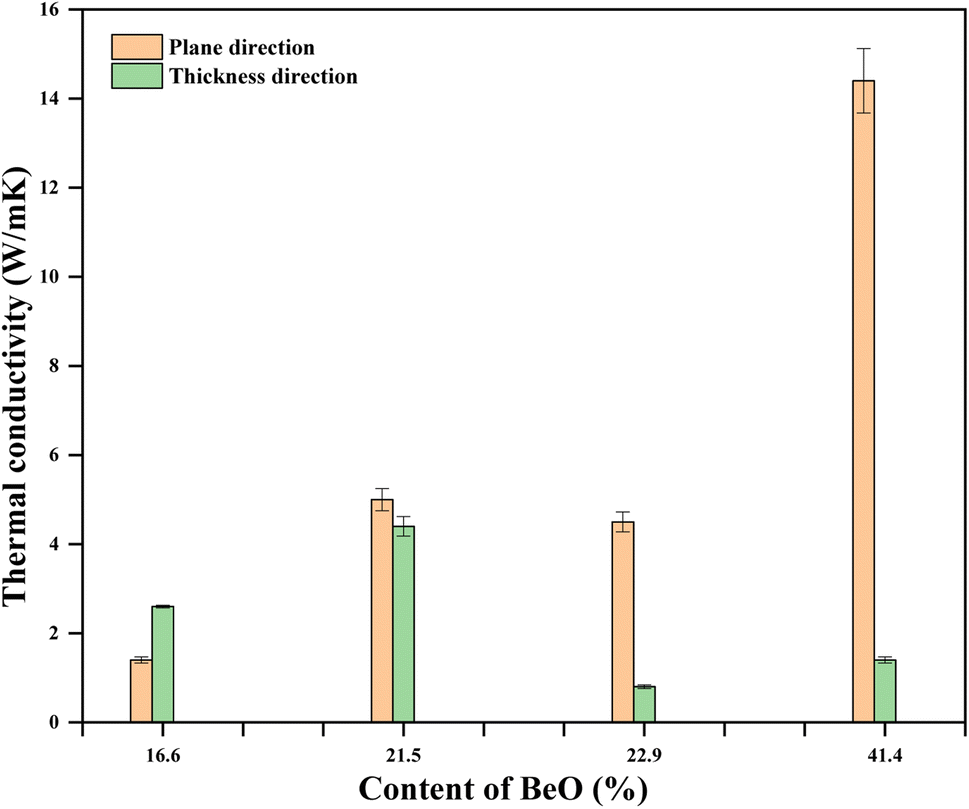 | ||
| Fig. 9 Effect of BeO content of PU/BeO NF sheets on the thermal conductivity at 1100 °C. | ||
The authors also measured the thermal diffusivity of the PU/BeO NF sheets to predict cooling processes and to know how quickly a material reacts to a change in temperature. Fig. 10 shows the thermal diffusivity of PU/BeO NF sheets at different temperatures. Here, the thermal diffusivity was lower (7.7 × 10−7 to 16.5 × 10−7 m2 s−1) at 700 °C temperatures compared to 900 °C temperatures (12.9 × 10−7 to 16.7 × 10−7 m2 s−1). However, the thermal diffusivity was much higher (10.8 × 10−7 to 56.7 × 10−7 m2 s−1) at higher 1100 °C temperatures than at low temperatures (700 °C and 900 °C).
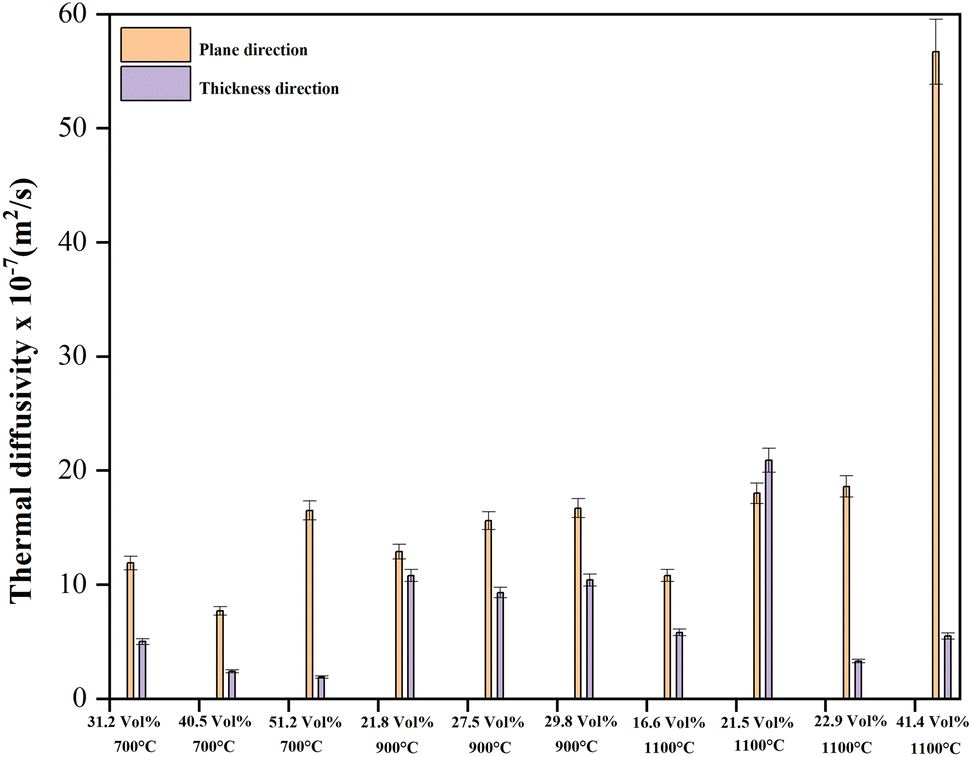 | ||
| Fig. 10 Thermal diffusivity of PU/BeO NF sheets at 700 °C, 900 °C, and 1100 °C. | ||
Our previous work could produce a thermal conductivity (7.9–14.4 W m−1 K−1) PU/Al2O3 NFs heat- dissipating sheet.39 At this time, we can also create PU/BeO NFs heat-dissipating sheet with high thermal conductivity (1.4–14.4 W m−1 K−1). However, we may be able to improve the thermal conductivity of BeO NFs by overcoming two difficulties. The first difficulty is that we couldn't produce completely nonporous BeO NFs.39 Another is that we couldn't create high crystallinity of BeO NFs because, after 1200 °C temperatures, BeO crystal is very brittleness. We may circumvent those two challenges by altering the precursor-making process. In addition, the authors strongly believe that mechanical properties are significant from the application point of view. At this time, the authors couldn’t create high crystallinity of BeO NF because, after 1200 °C temperatures, BeO crystal is very brittle. In the future, the authors will try to modify the precursors-making process to produce high crystallinity of BeO NF at low temperatures.
Electrical insulation properties of BeO/PU sheets (ASTM D257)
Table 4 shows the electrical insulating properties of BeO heat-dissipating sheets at various BeO contents. At 21.4% BeO contents, the heat-dissipating sheets had the maximum resistance (2.6 × 1012 Ω □−1). However, with 23.4% BeO content, BeO heat-dissipating sheets have the minimum electrical resistivity (1.5 × 1012 Ω □−1). The electrical resistivity measurements are all within the normal range. Both PU and BeO have a high electrical resistance could be the reason. As a result, mixed PU and BeO had a higher resistance.50–52 So, it concluded that the BeO heat-dissipating sheet’s electrical insulating qualities were outstanding.| BeO content (%) | Thickness (μm) | Electrical insulation (Ω □−1) | ||
|---|---|---|---|---|
| 700 °C | 900 °C | 1100 °C | ||
| 31.2 | 132 | 1.6 × 1012 | — | — |
| 40.5 | 101 | 2.4 × 1012 | — | — |
| 80.4 | 61 | 1.9 × 1012 | — | — |
| 21.8 | 100 | — | 2.6 × 1012 | — |
| 27.5 | 33 | — | 2.1 × 1012 | — |
| 39.0 | 68 | — | 1.6 × 1012 | — |
| 16.6 | 552 | — | — | 1.8 × 1012 |
| 21.5 | 107 | — | — | 2.1 × 1012 |
| 22.9 | 555 | — | — | 1.8 × 1012 |
| 23.4 | 155 | — | — | 1.5 × 1012 |
| 41.4 | 81 | — | — | 1.6 × 1012 |
Conclusions
In summary, PVA/BeSO4/PEI precursor NFs were successfully formed by electrospinning technique mixing BeSO4·4H2O salt and PEI in PVA aqueous solution, then calcination at different temperatures to produce BeO NF mats. Furthermore, the crystallinity increased dramatically as the calcination temperatures increased from 600 °C to 1300 °C. The calcination temperatures also influenced the specific surface area and average pore area due to the removal of organics PVA. This is beneficial to improving the low specific surface area of precursor PVA/BeSO4/PEI NFs of 36.3 m2 g−1 to 145 m2 g−1 for BeO NFs calcined at 600 °C. On the contrary, a very high calcination temperature of 1300 °C will decrease specific surface area (5.1 m2 g−1) due to sintering by metal particles. This study also investigated the thermal conductivity of PU/BeO NFs composite sheets by adding PU with BeO NF mats. The heat-dissipating sheets showed improved thermal conductivity (1.4–14.4 W m−1 K−1) at 16.6–80.4 vol% BeO NFs content. The possible reason is that the elongated shape of BeO NFs serves as an effective filler, creating linear heat pathways in the resin. In addition, high-performance heat-dissipating sheets showed excellent electrical insulating properties (1.6 × 1012 Ω □−1). This processing technique is very effective in synthesizing BeO NFs for the first time. Electrically insulated thermal conductive PU/BeO NF composite sheets would effectively dissipate heat from electronic gadgets.Author contributions
Md. Shakhawat Hossain: methodology, writing – original draft, investigation, data curation, visualization. Anamul Hoque Bhuiyan: experimental implementation and apparatus use. Koji Nakane (corresponding author): conceptualization, resources, design the research, formal analysis, editing, supervision.Funding
The authors state that they have no known competing financial interests or personal ties that could have influenced the research presented in this study.Conflicts of interest
There are no conflicts of interest declared by the authors.Acknowledgements
Md. Shakhawat Hossain wishes to thank the Japanese government for providing him with the MONBUKAGAKUSHO scholarship, which has allowed him to complete his Ph.D. The authors would like to thank Dr. Akiyoshi Ohgoshi for his kind and technical assistance throughout the project.References
- H. Chen, V. V. Ginzburg, J. Yang, Y. Yang, W. Liu, Y. Huang, L. Du and B. Chen, Prog. Polym. Sci., 2016, 59, 41–85 CrossRef CAS.
- F. Yuan, W. Jiao, F. Yang, W. Liu, Z. Xu and R. Wang, RSC Adv., 2017, 7, 43380–43389 RSC.
- X. Pu, H.-B. Zhang, X. Li, C. Gui and Z.-Z. Yu, RSC Adv., 2014, 4, 15297–15303 RSC.
- F. Nazeer, Z. Ma, Y. Xie, L. Gao, A. Malik, M. A. Khan, F. Wang and H. Li, RSC Adv., 2019, 9, 17967–17974 RSC.
- H. Shen, J. Guo, H. Wang, N. Zhao and J. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 5701–5708 CrossRef CAS PubMed.
- Y. Xu, D. D. L. Chung and C. Mroz, Composites, Part A, 2001, 32, 1749–1757 CrossRef.
- C. Huang, X. Qian and R. Yang, Mater. Sci. Eng. R Rep., 2018, 132, 1–22 CrossRef.
- A. R. J. Hussain, A. A. Alahyari, S. A. Eastman, C. Thibaud-Erkey, S. Johnston and M. J. Sobkowicz, Appl. Therm. Eng., 2017, 113, 1118–1127 CrossRef CAS.
- X. Chen, Y. Su, D. Aydin, D. Reay, R. Law and S. Riffat, Energy Build., 2016, 125, 99–108 CrossRef.
- W. Zhou, Thermochim. Acta, 2011, 512, 183–188 CrossRef CAS.
- X. Lu and G. Xu, J. Appl. Polym. Sci., 1997, 65, 2733–2738 CrossRef CAS.
- S. Zhang, X. Y. Cao, Y. M. Ma, Y. C. Ke, J. K. Zhang and F. S. Wang, Exp. Polym. Lett., 2011, 5, 581–590 CrossRef CAS.
- G.-W. Lee, M. Park, J. Kim, J. I. Lee and H. G. Yoon, Composites, Part A, 2006, 37, 727–734 CrossRef.
- W. Cui, Y. Zhu, X. Yuan, K. Chen and F. Kang, J. Alloys Compd., 2012, 540, 165–169 CrossRef CAS.
- T. Kusunose, T. Yagi, S. H. Firoz and T. Sekino, J. Mater. Chem. A, 2013, 1, 3440–3445 RSC.
- G. P. Akishin, S. K. Turnaev, V. Ya. Vaispapir, M. A. Gorbunova, Yu. N. Makurin, V. S. Kiiko and A. L. Ivanovskii, Refract. Ind. Ceram., 2009, 50, 465–468 CrossRef CAS.
- H. Liu and S. Wu, RSC Adv., 2022, 12, 12647–12654 RSC.
- M. Harada, N. Hamaura, M. Ochi and Y. Agari, Composites, Part B, 2013, 55, 306–313 CrossRef CAS.
- K. Sato, H. Horibe, T. Shirai, Y. Hotta, H. Nakano, H. Nagai, K. Mitsuishi and K. Watari, J. Mater. Chem., 2010, 20, 2749–2752 RSC.
- W. Zhou, S. Qi, Q. An, H. Zhao and N. Liu, Mater. Res. Bull., 2007, 42, 1863–1873 CrossRef CAS.
- S. Kume, I. Yamada, K. Watari, I. Harada and K. Mitsuishi, J. Am. Ceram. Soc., 2009, 92, S153–S156 CrossRef CAS.
- Y. Zhou, Y. Yao, C.-Y. Chen, K. Moon, H. Wang and C. Wong, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- R. F. Hill and P. H. Supancic, J. Am. Ceram. Soc., 2004, 87, 1831–1835 CrossRef CAS.
- T. Zhou, X. Wang, G. U. Mingyuan and X. Liu, Polymer, 2008, 49, 4666–4672 CrossRef CAS.
- G. P. Akishin, S. K. Turnaev, V. Y. Vaispapir, M. A. Gorbunova, Y. N. Makurin, V. S. Kiiko and A. L. Ivanovskii, Refract. Ind. Ceram., 2009, 50, 465–468 CrossRef CAS.
- X. Wang, R. Wang, C. Peng, T. Li and L. I. U. Bing, Prog. Nat. Sci.: Mater. Int., 2010, 20, 81–86 CrossRef.
- S. Rezabeyk and M. Manoochehri, RSC Adv., 2020, 10, 36897–36905 RSC.
- C. J. Buchko, L. C. Chen, Y. Shen and D. C. Martin, Polymer, 1999, 40, 7397–7407 CrossRef CAS.
- J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- A. Ohgoshi, K. Takahashi and K. Nakane, J. Mater. Sci.: Mater. Electron., 2019, 30, 20566–20573 CrossRef CAS.
- K. Nakane, S. Ichikawa, S. Gao, M. Seto, S. Irie, S. Yonezawa and N. Ogata, Sen’i Gakkaishi, 2015, 71, 1–5 CrossRef.
- K. Nakane, M. Seto, S. Irie, T. Ogihara and N. Ogata, J. Appl. Polym. Sci., 2011, 121, 1774–1779 CrossRef CAS.
- A. Ohgoshi, S. Gao, K. Takahashi and K. Nakane, J. Text. Eng., 2019, 65, 67–72 CrossRef.
- C. Lu, S. W. Chiang, H. Du, J. Li, L. Gan, X. Zhang, X. Chu, Y. Yao, B. Li and F. Kang, Polymer, 2017, 115, 52–59 CrossRef CAS.
- J. M. Deitzel, J. Kleinmeyer, D. E. A. Harris and N. B. Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- J. Y. Lu, C. Norman, K. A. Abboud and A. Ison, Inorg. Chem. Commun., 2001, 4, 459–461 CrossRef CAS.
- K. H. Lee, H. Y. Kim, Y. J. Ryu, K. W. Kim and S. W. Choi, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1256–1262 CrossRef CAS.
- X. Zhang, Y. Zheng, X. Feng, X. Han, Z. Bai and Z. Zhang, RSC Adv., 2015, 5, 86102–86112 RSC.
- M. S. Hossain and K. Nakane, Results Mater., 2022, 13, 100241 CrossRef CAS.
- Y. Hou, C. Chen, K. Liu, Y. Tu, L. Zhang and Y. Li, RSC Adv., 2015, 5, 24023–24030 RSC.
- A. R. Polu and R. Kumar, Chin. J. Polym. Sci., 2013, 31, 641–648 CrossRef CAS.
- C. Liebenow, Electrochim. Acta, 1998, 43, 1253–1256 CrossRef CAS.
- A. B. Shatan, K. Venclíková, B. A. Zasońska, V. Patsula, O. Pop-Georgievski, E. Petrovský and D. Horák, Pharm. Res., 2019, 36, 1–12 CrossRef CAS PubMed.
- D. Perra, N. Drenchev, K. Chakarova, M. G. Cutrufello and K. Hadjiivanov, RSC Adv., 2014, 4, 56183–56187 RSC.
- Y. Liang, J. Ouyang, H. Wang, W. Wang, P. Chui and K. Sun, Appl. Surface Sci., 2012, 258, 3689–3694 CrossRef CAS.
- F. Mohandes and M. Salavati-Niasari, RSC Adv., 2014, 4, 25993–26001 RSC.
- H. Lulit, S. Natnael, M. DuRe, T. Thriveni, C. Ramakrishna and A. JiWhan, Sustainability, 2019, 11, 3196–3205 CrossRef.
- C. Sarkar and S. K. Dolui, RSC Adv., 2015, 5, 60763–60769 RSC.
- N. Jain, N. Marwaha, R. Verma, B. K. Gupta and A. K. Srivastava, RSC Adv., 2016, 6, 4960–4968 RSC.
- Y. Mobarak, M. Bassyouni and M. Almutawa, Adv. Mater. Sci. Eng., 2013, 2013, 1–6 CrossRef.
- A. Ş. Demirkiran, R. Artir and E. Avci, Ceram. Int., 2010, 36, 917–921 CrossRef CAS.
- J.-C. Zhao, F.-P. Du, X.-P. Zhou, W. Cui, X.-M. Wang, H. Zhu, X.-L. Xie and Y.-W. Mai, Composites, Part B, 2011, 42, 2111–2116 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |