
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcise two-step chemical synthesis of molnupiravir†
Vinícius R. D. Pereiraa,
Marco A. M. Bezerrab,
Mauro R. B. P. Gomezb,
Guilherme M. Martins c,
Adilson D. da Silvaa,
Kleber T. de Oliveirac,
Rodrigo O. M. A. de Souza*b and
Giovanni W. Amarante*a
c,
Adilson D. da Silvaa,
Kleber T. de Oliveirac,
Rodrigo O. M. A. de Souza*b and
Giovanni W. Amarante*a
aDepartment of Chemistry, Federal University of Juiz de Fora, Campus Martelos, Juiz de Fora, Minas Gerais 36036-900, Brazil. E-mail: giovanni.amarante@ufjf.edu.br
bChemistry Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Rio de Janeiro 22041-909, Brazil
cDepartment of Chemistry, Federal University of São Carlos, Campus São Carlos, São Carlos, São Paulo 13565-905, Brazil
First published on 21st October 2022
Abstract
A concise synthesis of molnupiravir in a one-pot two-step approach starting from uridine is described. Formally, herein, two sets of one-pot two-reaction steps introducing simplicity for purifications and using chemically available reagents are presented. In this context, molnupiravir was obtained in up to 68% overall yield and multigram-scale. In addition, HPLC analysis showed the molnupiravir purity above 99%.
1. Introduction
The ongoing COVID-19 pandemic caused by the SARS-CoV-2 virus has impacted people's health and the economy worldwide, presently with more than 500 million confirmed cases and 6 million deaths.1 Therefore, in addition to vaccines, there is an urgent need for drugs that can be utilized to reduce the high burden of COVID-19 infections and combat potential new virus variants.2 Molnupiravir (MK-4482, EIDD-2801) is an orally available antiviral prodrug that was recently approved for emergency antiviral use against COVID-19 in the United States, Japan, and the European Union.3 It is proposed that this drug impairs SARSCoV-2 replication by increasing the frequency of viral RNA mutations. This happens when the virus RNA-dependent RNA-polymerase (RdRp) uses the tautomers of the active form of molnupiravir, β-D-N4-hydroxycytidine (NHC) triphosphate, instead of cytidine triphosphate or uridine triphosphate, leading to mutated RNA products.4 Due to the importance of this drug, there is a wide interest in developing an efficient and cost-effective synthesis route for its production to ensure a sufficient and readily available global supply chain.5In this context, there are many chemical routes already described to molnupiravir (2) from uridine (1) (Scheme 1). In 2019, the patented seminal work developed at Emory University involves a 5-step route to molnupiravir (2) with 17% overall yield after purifications by chromatography.6 Subsequently, Dallinger and Kappe have developed an approach starting from uridine (1) involving triazolation, one-pot acetonide protection/esterification and a telescoped hydroxyamination/continuous flow acetonide deprotection to produce molnupiravir (2) in 61% overall yield.7 Despite the triazole high-cost reagent, it is important to highlight the innovation on the use of a continuous flow set-up for the last deprotection step. In 2021, Das, Yadav and co-workers reported an outstanding improvement, preparing molnupiravir (2) in 62% overall yield.8 However, to this end, a one-pot thionation step was included in the process. A gram-scale experiment was then carried out by taking one equivalent of Lawesson's reagent to produce a thionated intermediate which smoothy gave molnupiravir (2) after purification by chromatography followed by hydroxyamination/deprotection processes. Likewise, in 2021, Fier, Xu and co-workers revisited the original five-step synthesis from Emory. They were able to improve 1.6-fold the overall yield, optimizing each step and this protocol has been adopted to produce tons per year of molnupiravir (2).9 For extra details, see also Section 3 on ESI.†
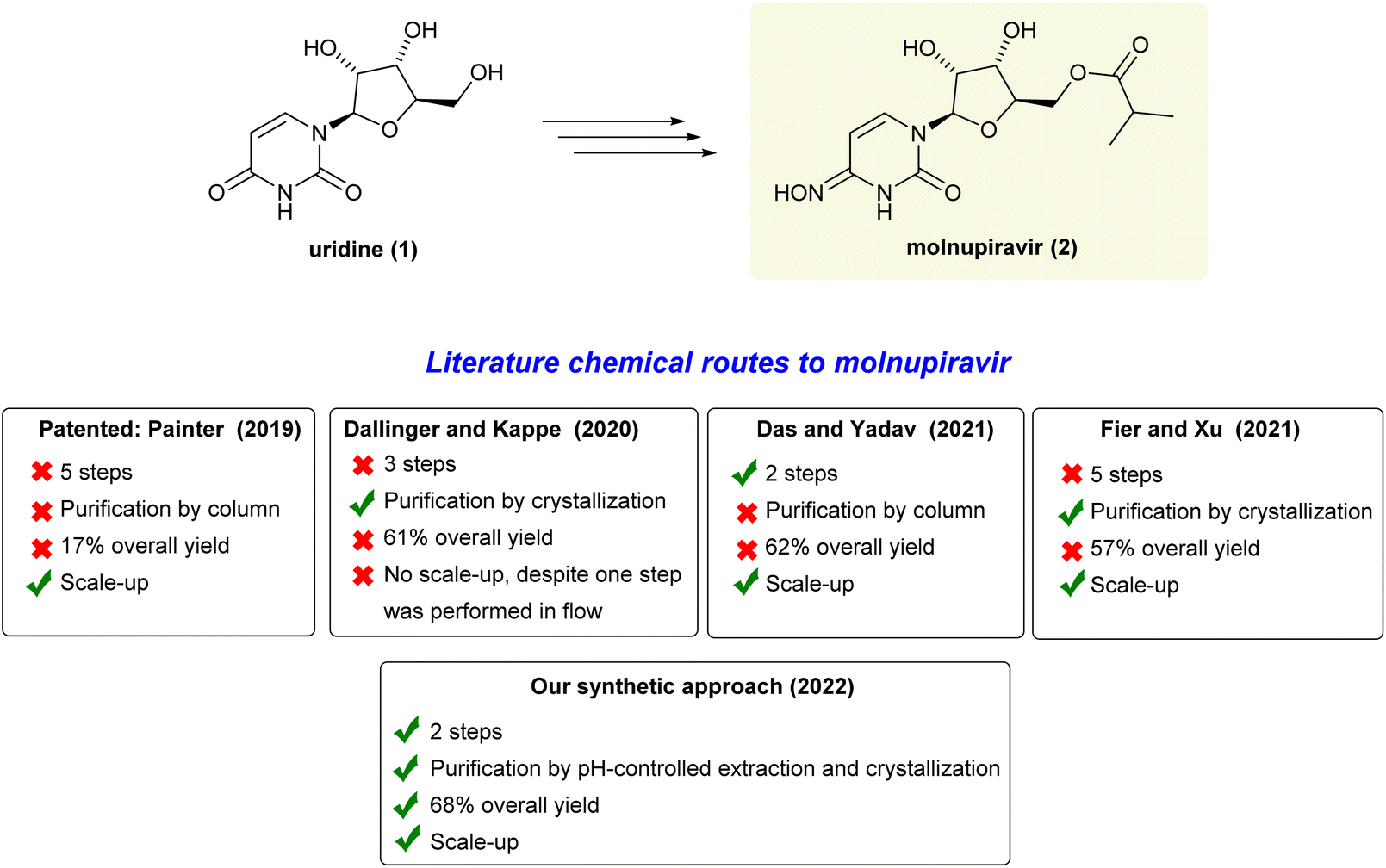 | ||
| Scheme 1 Chemical routes to molnupiravir (2). | ||
Given the importance of all those mentioned chemical syntheses and some inherent limitations of the reported approaches to molnupiravir (2), it is evident that more efficient and inexpensive routes from uridine (1) still need to be developed. As a result, herein, we report a chemical route involving two sequential one-pot steps, using available commodity reagents, affording molnupiravir (2) in a batch mode with 68% overall yield through simple pH-controlled extractions and crystallization as purification procedures.
2. Results and discussion
2.1 One-pot cetalization/esterification process
Despite all the above-mentioned advent in the chemical synthesis of molnupiravir (2), in some cases, serious drawbacks appear in those reported procedures. For instance, toxicity and high-cost chemicals and, most importantly, tedious isolation/purification processes are the most undesired walk to chemically synthesize molnupiravir (2). Thus, we initiated by looking in-depth into each already available batch procedure. Concerning the well-established acid catalyzed cetalization of 1,2-diols, there are some variants in the case of uridine (1) hydroxyl-groups protection. For example, depending on the use of either 2,2-dimethoxypropane or acetone, different equivalents of sulfuric acid is adopted. In special, in our case, different amounts of sulfuric acid gave almost the same isolated yield, with an average of 50%.To our delight, the inclusion of molecular sieves 3 Å allowed us to keep the H2SO4 to 5 mol% and drop the reaction time to 1 h at room temperature (versus 18 h (ref. 6)). Under this optimized reaction condition, the intermediate (3) was isolated in 97% yield after only pH-controlled (pH = around 7–8) during liquid/liquid extraction (Scheme 2).
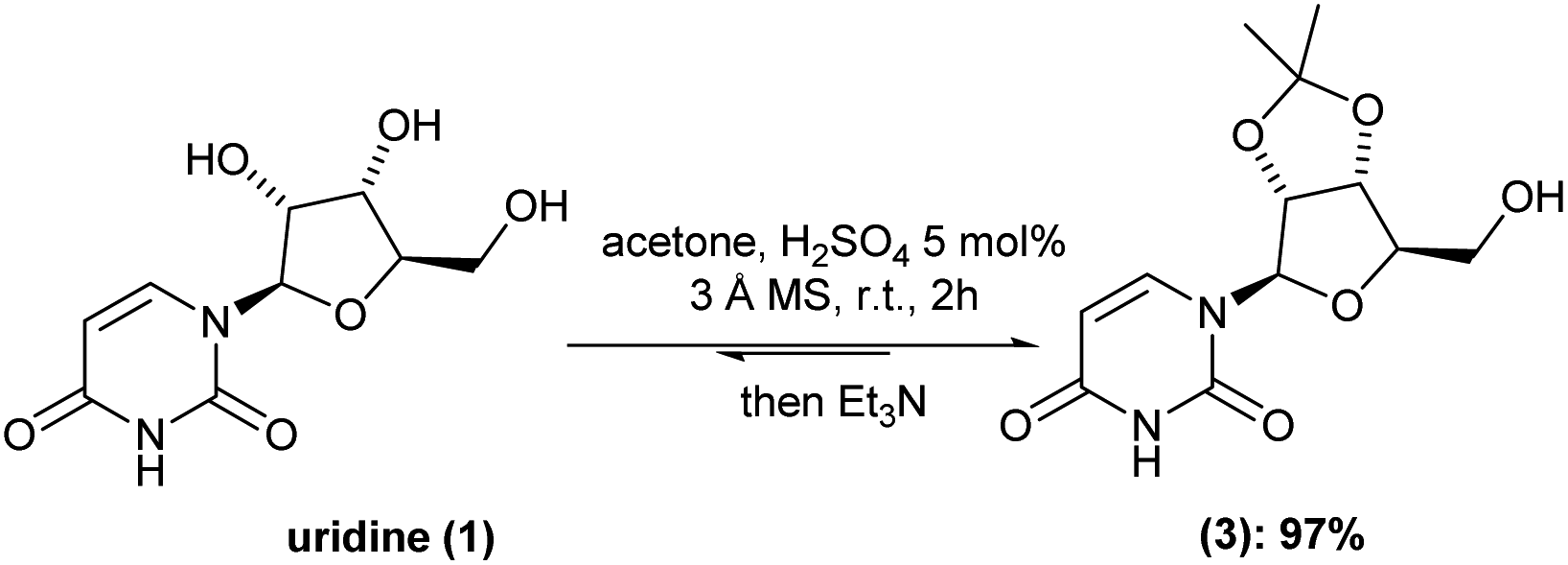 | ||
| Scheme 2 Batch cetalization process of uridine (1). | ||
Having intermediate (3) in hand, we carried out the esterification of the primary hydroxyl group. Here, based on previous reports,6,7 we envisioned a one-pot procedure cetalization/esterification process should be really benefit. Thus, after completion cetalization reaction, the same reaction vessel was charged with triethylamine, 4-DMAP and isobutyric anhydride in this respective sequence. After 1 h reaction, the volatiles were removed under reduced pressure, the crude reaction diluted in ethyl acetate and washed with saturated solution of NaHCO3 (pH around 7–8), water and then brine. This isolation procedure gave the corresponding intermediate (4) in 95% yield with no need of further purification (Scheme 3). It is worth nothing that, the inclusion of molecular sieves 3 Å was essential to successfully reach such high yield.
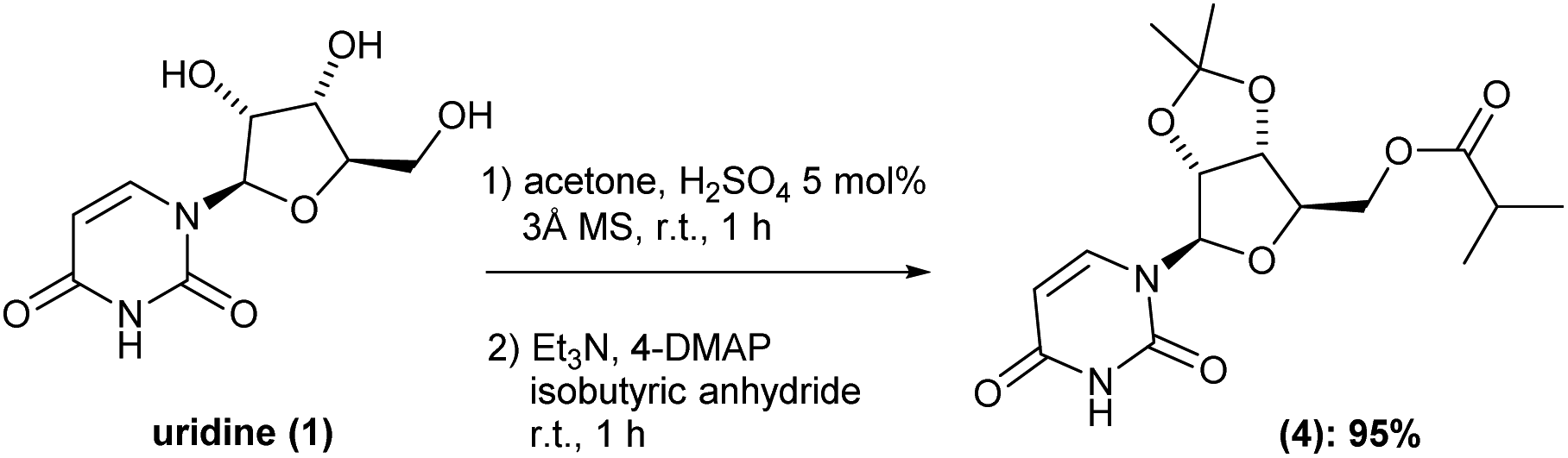 | ||
| Scheme 3 One-pot cetalization and esterification processes of the uridine (1). | ||
Before moving forward with the next step, we decided to scale-up the process to verify the robustness of the developed methodology on upper scales. Starting from uridine (1) at 5 and 10 g-scale, we were able to successfully obtain the intermediate (4) in 6.6 g-scale (92%) and 13.7 g-scale (94% yield), respectively, thus proving the scalability and reproducibility of our one-pot two-reactions protocol. Additionally, during the scale-up it was possible to further optimize the method, reducing the volume of acetone by half and the equivalence of triethylamine from 12 to only 2.
2.2 One-pot oxyamination/deprotection process
The synthesis sequence requires now the oxyamination reaction. Despite the previously reported low yield (29%), 1,2,4-triazol strategy was adopted in the first place.6 This strategy was recently optimized, and the heterocycle intermediate has been achieved in high levels of isolated yield, while 10 equiv. of 1,2,4 triazol is required, increasing the cost and impacting the sustainability of this strategy.7 Recent developments in this step include different carbonyl activation processes, such as the use of trimethylsylil/imidazole complex.10,11 On the other hand, aryl sulfonyl activation mode would be an alternative path to this oxyamination reaction.12 In all these cases, lower yields due to either lower product conversion (messy reaction) or product purification have been in charge to decrease the overall yield of the molnupiravir (2). It is important to highlight that, an outstanding contribution has been recently reported by Das and Yadav, which consists of previous thiolation reaction, followed by an oxyamination/deprotection process.8 Despite having one additional operation, this reaction was carried out in a one-pot three-reactions procedure.In order to start this key step, some aryl sulfonyl activations were considered. However, in our hands, only low yields or traces of the desired oxyamination product was obtained. In addition, in these cases, purification processes were also challenged, even considering column chromatography, which certainly compromised the last step.
The promising solution came out the idea to set up a one-pot procedure. In this scenario, the reaction vessel was charged with HMDS, imidazole, KHSO4 and (NH2OH)2·H2SO4 at room temperature. After 2 h oxyamination reaction, 20 volumes of formic acid 85% was added. Thus, the one-pot deprotection was completed after only 30 min, and Na2CO3 solution was added to the vessel controlling the pH to ca. 7–8, and extracted with ethyl acetate. The volatiles were removed under reduced pressure and the solid was then recrystallized in a mixture of AcOEt 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN v/v. This method gave the molnupiravir (2) in 72% yield (Scheme 4). The recrystallized material was subjected to HPLC analysis to verify impurities and related substances leading to a purity assessment of >99% (Fig. 1).
:1 MeCN v/v. This method gave the molnupiravir (2) in 72% yield (Scheme 4). The recrystallized material was subjected to HPLC analysis to verify impurities and related substances leading to a purity assessment of >99% (Fig. 1).
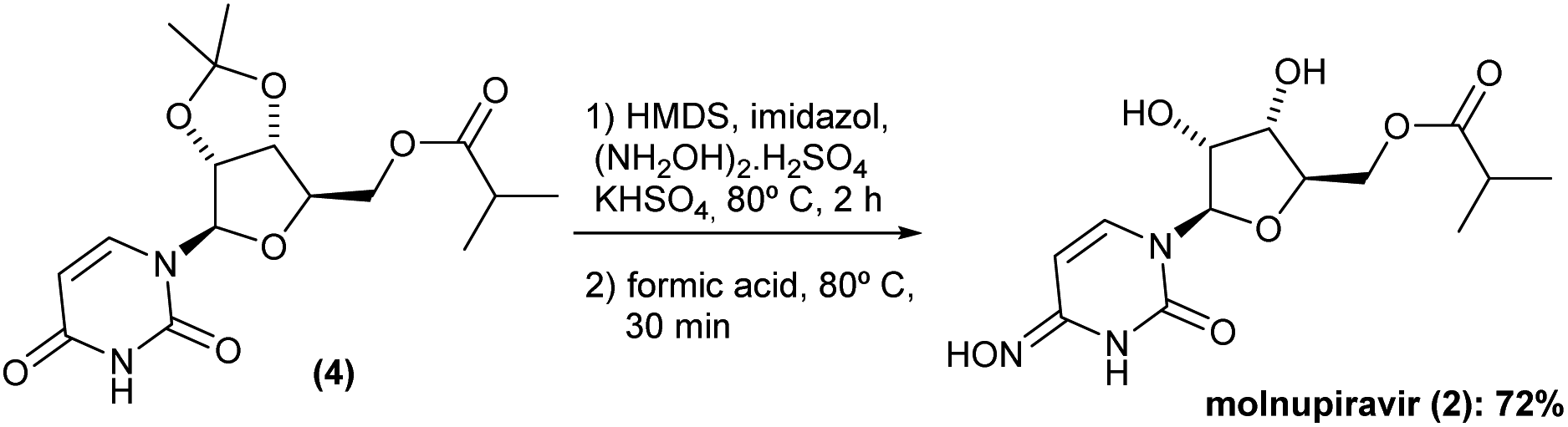 | ||
| Scheme 4 One-pot oxyamination/deprotection reaction to molnupiravir (2). | ||
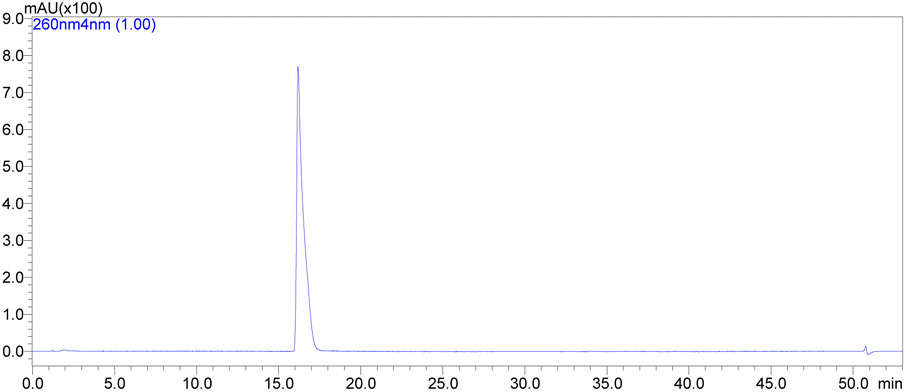 | ||
| Fig. 1 Molnupiravir (2) HPLC analysis conditions: mobile phase A: 40 mM ammonium dihydrogen phosphate aqueous solution; mobile phase B: methanol; column: Agilent Eclipse XDB-C18, 4.6 × 150 mm, 5 μm; injection volume: 10 μL, detector: DAD (260 nm). | ||
It is important to mention that, carrying out these two steps separately, the combined yield was only 43%. Furthermore, the reaction time could be considerably dropped. Only 2.5 h was required for running these two reactions in a one-pot procedure. Finally, again, no purification by chromatography was required, and a simple recrystallization gave the molnupiravir (2) in high purity.
After completing the synthesis of molnupiravir (2) with the highlighted innovative protocol we also scale-up the second part of our approach. Thus, we tested the reaction profile in a multigram-scale experiment starting from both 6.6 and 13.7 g of (4) (previously obtained from 5 and 10 g of uridine (1)). As a result, molnupiravir (2) was obtained in 4.3 g (68% overall yield) and 8.1 g (64% overall yield), respectively, showing the reproducibility of the sequence.
3. Conclusions
An improved two-steps one-pot synthetic approach for the molnupiravir (2) from uridine (1) was developed. This API could be obtained in 68% overall yield, employing commodity reagents and simple pH-controlled extraction and additional recrystallization. Furthermore, scaled-up experiments were demonstrated showing the robustness of our protocol with reproducibility even at multigram-scales.4. Experimental section
4.1 General information
All reactions were performed on the benchtop under ambient atmosphere. Reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, Merck, Neon, Acros Organics, TCI, Reatec, Alphatec, CRQ, Neon, or Vetec) and used without further purification. Molecular sieves 3 Å 8–12 mesh (Aldrich Chemical Company) were used to remove water molecules formed during the reaction. TLC silica gel 60 Å plates (MACHEREY-NAGEL) were used to accompany the progress of the reactions (mobile phase: DCM 9:1 MeOH v/v), and UV light 254 nm was used to reveal them. NMR spectra were recorded on a Bruker Avance III spectrometer (500 MHz for 1H and 125 MHz for 13C). Chemical shifts were reported in parts per million (δ) relative to the residual solvent resonance (CDCl3 = 7.26 ppm for 1H and 77.0 ppm for 13C, CD3OD = 3.31 ppm for 1H and 49.0 ppm for 13C), and coupling constants (J) in hertz (Hz). The signal multiplicity was denoted by the abbreviations: s (simplet), d (doublet), dd (doublet of doublets), m (multiplet), and sept (septet). Melting points were recorded on a Microquímica MQAPF-302 melting point equipment. LC-HRMS analysis acquired on Dionex LC UltiMate 3000 coupled HRMS Q-Exactive Plus (Thermo Scientific, Frenton, CA, USA).
4.2 Analytical conditions
Analytical protocol was developed based on draft proposal for inclusion of molnupiravir (2) in the International Pharmacopoeia (QAS/21.907), in the following conditions: mobile phase A: 40 mM ammonium dihydrogen phosphate aqueous solution; mobile phase B: methanol; column: Agilent Eclipse XDB-C18, 4.6 × 150 mm, 5 μm; injection volume: 10 μL, detector: DAD (260 nm).4.3 Synthesis procedures
:1 MeCN v/v) and the solid formed was filtered under reduced pressure. Finally, molnupiravir (2) was obtained as a white solid (66.9 mg, 72% yield). Melting point: 156–157 °C (Lit.: 158–160 °C).8 1H NMR (500 MHz, CD3OD) δ 6.91 (d, J = 8.3 Hz, 1H), 5.82 (d, J = 4.8 Hz, 1H), 5.62 (d, J = 8.2 Hz, 1H), 4.29 (d, J = 3.3 Hz, 2H), 4.16–4.06 (m, 3H), 2.62 (sept, J = 6.9 Hz, 1H), 1.18 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CD3OD) δ 178.3, 151.5, 146.2, 131.8, 99.5, 90.4, 82.5, 74.3, 71.5, 64.9, 35.2, 19.33, 19.3. The NMR data matched those previously reported in the literature.7,8,13 ESI-HRMS calc. for C13H19N3O7 [M + H]+ 330.1296 found 330.1290. ESI-HRMS calc. for C13H19N3O7 [M − H]− 328.1150 found 328.1149.:1 MeCN v/v) and the solid formed was filtered under reduced pressure. Finally, molnupiravir (2) was obtained as a white solid (4.26 g, 69% yield). Melting point: 156–157 °C (Lit.: 158–160 °C).8 1H NMR (500 MHz, CD3OD) δ 6.91 (d, J = 8.3 Hz, 1H), 5.82 (d, J = 4.8 Hz, 1H), 5.62 (d, J = 8.2 Hz, 1H), 4.29 (d, J = 3.3 Hz, 2H), 4.16–4.06 (m, 3H), 2.62 (sept, J = 6.9 Hz, 1H), 1.18 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CD3OD) δ 178.3, 151.5, 146.2, 131.8, 99.5, 90.4, 82.52, 74.3, 71.5, 64.9, 35.2, 19.33, 19.3. The NMR data matched those previously reported in the literature.7,8,13:1 MeCN v/v and the solid formed was filtered under reduced pressure. Finally, molnupiravir (2) was obtained as a white solid (8.1 g, 64% yield). Melting point: 156–157 °C (Lit.: 158–160 °C).8 1H NMR (500 MHz, CD3OD) δ 6.91 (d, J = 8.3 Hz, 1H), 5.82 (d, J = 4.8 Hz, 1H), 5.62 (d, J = 8.2 Hz, 1H), 4.29 (d, J = 3.3 Hz, 2H), 4.16–4.06 (m, 3H), 2.62 (sept, J = 6.9 Hz, 1H), 1.18 (d, J = 6.9 Hz, 6H). 13C NMR (125 MHz, CD3OD) δ 178.3, 151.5, 146.2, 131.8, 99.5, 90.4, 82.52, 74.3, 71.5, 64.9, 35.2, 19.33, 19.3. The NMR data matched those previously reported in the literature.7,8,13Author contributions
V. R. D. Pereira has mostly performed the experiments of the chemical approach and part of the scale-up. M. A. M. Bezerra and M. R. B. P. Gomez developed the scale-up and tested some optimized conditions. G. M. Martins, critically reviewed the manuscript and reproduced some batch experiments. G. W. Amarante, R. O. M. A. de Souza, K. T. de Oliveira and A. D. da Silva, conceived the approach, critically discussed during all the development and wrote the manuscript final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Conselho Nacional de Pesquisa – CNPq and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Financial Code 001. G. W. A., K. T. O. and R. O. M. A. S thank the CNPq for the research fellowship (No. 308200/2021-7, 303890/2019-3 and 404973/2018-3, respectively), FAPEMIG and UFJF for their support. K. T. O. and G. M. M. thank the São Paulo Research Foundation FAPESP (grant numbers 2019/27176-8, 2020/06874-6 and fellowship 2022/00074-3).Notes and references
- WHO, WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int/, accessed May 31th, 2022 Search PubMed.
- W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut and S. J. Peacock, Nat. Rev. Microbiol., 2021, 19, 409–424 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2022, 82, 455–460 CrossRef CAS PubMed.
- F. Kabinger, C. Stiller, J. Schmitzová, C. Dienemann, G. Kokic, H. S. Hillen, C. Höbartner and P. Cramer, Nat. Struct. Mol. Biol., 2021, 28, 740–746 CrossRef CAS.
- T. C. Braga, J. A. Santos, P. P. Castro and G. W. Amarante, Quim. Nova, 2022, 45, 53–73 CAS.
- G. R. Painter, G. R. Bluemling, M. G. Natchus and D. Guthrie, WO pat., WO2019113462, 2019.
- A. Steiner, D. Znidar, S. B. Ötvös, D. R. Snead, D. Dallinger and C. O. Kappe, Eur. J. Org. Chem., 2020, 6736–6739 CrossRef CAS PubMed.
- R. Dey, S. Nayak, P. Das and S. Yadav, ACS Omega, 2021, 6, 28366–28372 CrossRef CAS PubMed.
- P. S. Fier, Y. Xu, M. Poirier, G. Brito, M. Zheng, R. Bade, E. Sirota, K. Stone, L. Tan, G. R. Humphrey, D. Chang, J. Bothe, Y. Zhang, F. Bernardoni, S. Castro, M. A. Zompa, J. Taylor, K. M. Sirk, A. Diaz-Santana, I. Diribe, K. M. Emerson, B. Krishnamurthi, R. Zhao, M. Ward, C. Xiao, H. Ouyand, J. Zhan and W. J. Morris, Org. Process Res. Dev., 2021, 25, 2806–2815 CrossRef CAS PubMed.
- T. Benkovics, J. McIntosh, S. Silverman, J. Kong, P. Maligres, T. Itoh, H. Yang, M. Huffman, D. Verma and W. Pan, ChemRxiv, 2020 DOI:10.26434/chemrxiv.13472373.v1 , https://chemrxiv.org/engage/chemrxiv/article-details/60c753480f50db4947397b3f.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS PubMed.
- J. Lu, N.-S. Li, S. C. Koo and J. A. J. S. Piccirilli, Synthesis, 2010, 2708–2712 CAS.
- D. J. Paymode, N. Vasudevan, S. Ahmad, A. L. Kadam, F. S. P. Cardoso, J. M. Burns, D. W. Cook, R. W. Stringham and D. R. Snead, Org. Process Res. Dev., 2021, 25, 1822–1830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05064a |
| This journal is © The Royal Society of Chemistry 2022 |