
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mono- and dinuclear zirconocene(IV) amide complexes for the catalytic dehydropolymerisation of phenylsilane†
Kevin Lindenau,
Anke Spannenberg,
Fabian Reiß * and
Torsten Beweries*
* and
Torsten Beweries*
Leibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: fabian.reiss@catalysis.de; torsten.beweries@catalysis.de
First published on 15th September 2022
Abstract
The dehydropolymerisation of phenylsilane is investigated using group 4 metallocene amide complexes as catalysts. The dinuclear zirconocene amide complex Cp2Zr(NMe2)(μ-Me3SiC3SiMe3)Zr(NMe2)Cp2 (2) (Cp = η5-cyclopentadienyl) shows high activity in dehydrocoupling reactions, producing polyphenylsilanes with molecular weights ranging from 200 to 3000 g mol−1 and linear-to-cyclic product ratios of up to 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. Likewise, different ratios of oligomers and polymers with different tacticities could be described. Ansa-zirconocene amide complexes possessing the ebthi (ebthi = 1,2-ethylene-1,1′-bis(η5-tetrahydroindenyl)) ligand systems were prepared and evaluated for catalytic dehydropolymerisation in comparison to the dinuclear catalyst system.
:20. Likewise, different ratios of oligomers and polymers with different tacticities could be described. Ansa-zirconocene amide complexes possessing the ebthi (ebthi = 1,2-ethylene-1,1′-bis(η5-tetrahydroindenyl)) ligand systems were prepared and evaluated for catalytic dehydropolymerisation in comparison to the dinuclear catalyst system.
Introduction
The catalytic formation of Si–Si bonds to produce inorganic–organic polymers and hybrid materials has been intensively studied since the 1980s.1–5 The special properties of polysilanes such as σ electron delocalisation have led to the application of these materials for the development of new ceramics, semiconductors and nonlinear optical materials.4 For converting silane-based monomers several different approaches are feasible.6–9 Catalytic dehydrocoupling of hydrosilanes represents an atom-efficient method compared to the Wurtz coupling that produces stoichiometric amounts of salts as waste products and shows a poor functional-group-tolerance.10,11 Early investigations focused on group 4 metallocene-based catalysts, although complexes of later transition metals were also studied and showed comparable catalytic performance in some cases.12–18 The main advantages are generally good yields (>90%), the possibility for ligand variation and post-polymerisation functionalisation.1,19,20 It should however be noted that typically low molecular weight polymers with ill-defined tacticity and poor linear-to-cyclic ratios are obtained.1 However, the utility of this approach for the construction of Si rich polymers containing poly(cyclosilane) units was recently demonstrated by Klausen and co-workers.21,22 For group 4 metallocene catalysis, experimental parameters like concentration effects, catalyst deactivation pathways, optimal metallocene substitution patterns, and the pressure dependence on molecular weight, were studied in great detail in the past, mainly by the groups of Corey, Harrod and Tilley.12–14 The dehydropolymerisation of the primary silane PhSiH3 using the system Cp2ZrCl2/n-BuLi was well-studied in the past, giving >99% polysilane with a mass distribution of Mn/Mw = 3200/1600 g mol−1 and a ratio of linear/cyclic products of 65/35.23–26 These systems require the use of strong base for activation of the precatalyst. Formation of oligomers and polymers was described to occur via σ bond metathesis for group 4 metals.27–29 An alternative approach could be the use of well-defined highly reactive species such as metallocene alkyne complexes, silyl, alkyl, or amide complexes.27,30–33 In the area of dehydropolymerisation of PhSiH3, only a handful of zirconium amide complexes were described as catalysts.34–36 Corey et al. have reported the mononuclear complex Cp2Zr(NMe2)2 (1) as catalyst for the dehydropolymerisation of phenylsilanes (Scheme 1).34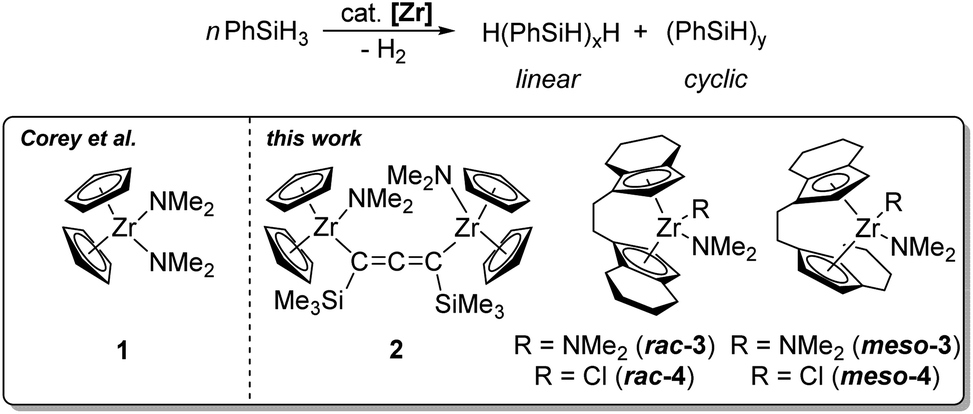 | ||
| Scheme 1 Catalytic dehydropolymerisation of PhSiH3 using different zirconocene amide complexes. | ||
Modification of the cyclopentadienyl scaffold is a common approach in group 4 metallocene chemistry, to synthesise optically active compounds or polymers with well-defined tacticity.37–39 Ansa-zirconocene amide complexes have been reported by Jordan et al., who demonstrated the selective synthesis of [rac/meso-(ebi)Zr(NMe2)2] (ebi = 1,2-ethylene-1,1′-bis(η5-indenyl)) by amine elimination.40,41 Likewise, this route furnishes other zirconocene amide complexes with an indenyl ligand system.42,43 Such complexes are used for a wide range of applications in asymmetric catalysis.44 Jordan et al. described the polymerisation of propene by using [rac-(ebi)Zr(NMe2)2] and [rac-(sbi)Zr(NMe2)2] (sbi = dimethylsilyl-1,1′-bis(η5-indenyl)) with additional MAO activation.45 The group showed that amide complexes are significantly less active compared to the chloride-related systems.45 Kim et al. described the formation of various cationic zirconocene species by using [rac-(ebi)Zr(NMe2)2] as catalyst for polymerisation of propene.46,47
Recently, our group synthesised a set of dinuclear zirconocene complexes, including the amide species Cp2Zr(NMe2)(μ-Me3SiC3SiMe3)Zr(NMe2)Cp2 (2) (Scheme 1) which exhibited high activity in the dehydrocoupling of H3B·NMe2H.48–50 Dinuclear cooperative reactivity of both metal centers was discussed as an important mechanistic feature. Intrigued by the high activity of this complex at room temperature, we aimed at the study of this species for related dehydrocoupling reactions such as the well-known dehydropolymerisation of PhSiH3. In this contribution, we describe the application of 2 as catalyst for the dehydropolymerisation of phenylsilane and discuss its performance in comparison to mononuclear complexes. Furthermore, the synthesis of a series of previously unknown ansa-zirconocene amide complexes is described, as well as the application of these complexes as single-component catalysts.
Results and discussion
Synthesis and characterisation of ansa-zirconocene amides
Based on the previously reported reaction conditions for the preparation of the dinuclear amide complex 2,48 we carried out the reaction of [rac-(ebthi)ZrCl2] (ebthi = 1,2-ethylene-1,1′-bis(η5-tetrahydroindenyl)) with two equivalents of LiNMe2 in a similar fashion (Scheme 2). Formation of one main compound that shows three characteristic resonances in the 1H NMR spectrum in 2:2:12 ratio (δ 6.08, 5.31 and 2.97 ppm, Fig. S4†) was observed. A single crystal X-ray diffraction (SC-XRD) analysis using crystals that were obtained from toluene at −78 °C confirms the assignment as [rac-(ebthi)Zr(NMe2)2] (rac-3) (Fig. 1). 13C NMR analysis shows all typical resonances of the ebthi ligand system and a resonance at δ 48.5 ppm for the NMe2 ligand which is in line with the value of δ 51.6 ppm found for complex 2 (Fig. S5†).
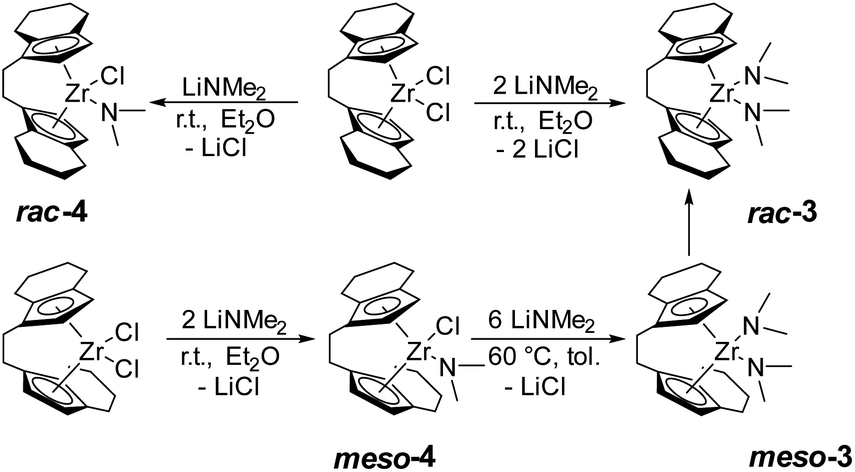 | ||
| Scheme 2 Synthesis of ansa-zirconocene amide complexes. | ||
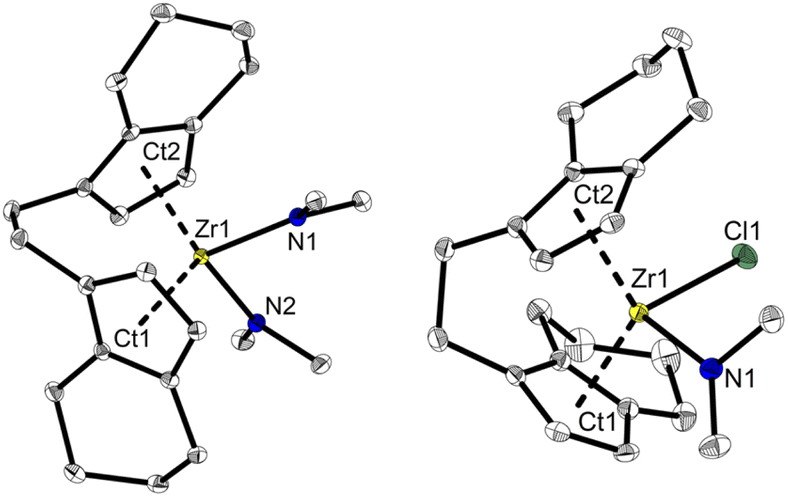 | ||
| Fig. 1 Molecular structures of complexes rac-3 and meso-4. Selected bond lengths and angles 3: Zr1–N1 2.1074(13), Zr1–N2 2.1065(13), Ct1–Zr1 2.284, Ct2–Zr1 2.286 [Å]; N1–Zr1–N2 95.09(5), Ct1–Zr1–Ct2 121.14, Cind1–Cet1–Cet2–Cind2–40.78(19)°. 4 Zr1–Cl1 2.4772(8), Zr1–N1 2.064(3), Ct1–Zr1 2.232, Ct2–Zr1 2.275 [Å]; N1–Zr1–Cl1 96.78(8), Ct1–Zr1–Ct2 123.55, Cind1–Cet1–Cet2–Cind2–46.2(4)°. Thermal ellipsoids correspond to 30% probability. Hydrogen atoms are omitted for clarity.51 | ||
We next investigated the reaction of the meso isomer [meso-(ebthi)ZrCl2] with two equivalents of LiNMe2 in Et2O at room temperature, aiming at the synthesis of a related bis(amide) complex. To our surprise, only one of the chloride ligands was replaced under these conditions, as evidenced by the 1H NMR analysis, which shows resonances at δ 6.06, 5.38 and 2.82 ppm in a ratio of 2:2:6 (Fig. S12†). 2D NMR analysis revealed that all signals show correlation, confirming the presence of a single new species (Fig. S14 and S15†). Crystals of the highly air and moisture sensitive compound suitable for an SC-XRD analysis were obtained from benzene solution at room temperature and confirm the proposed structure of [meso-(ebthi)Zr(Cl)(NMe2)] (meso-4) (Fig. 1).
We next varied the reaction conditions to investigate the direct synthesis of a meso-(ebthi) bis(amide) complex. By varying the temperature to 60 °C in Et2O formation of bis(amide) compound meso-3 could be observed along with three new resonances at δ 3.01, 2.97 and 2.93 ppm in a ratio of 6:12:6. As the resonance at δ 2.97 ppm resembles that of the rac-(ebthi) complex rac-3 isomerisation of the metallocene scaffold is possible. Addition of further four equivalents of LiNMe2 results in full conversion of meso-4 to furnish the desired bis(amide) complex [meso-(ebthi)Zr(NMe2)2] meso-3 in 80% NMR yield along with the rac-(ebthi) bis(amide) rac-3 (Fig. S7†). To optimise the synthesis conditions of meso-3 and to avoid the formation of rac-3, we next investigated the influence of different solvents. In aprotic non-coordinating solvents at −40 °C, the reaction was rather unselective and only mixtures containing both isomers could be obtained (Fig. S7†). In benzene, only a complex reaction mixture resulted, from which only a dark yellow oil could be isolated (Fig. S8†). Use of pentane for work up did not result in the formation of defined products (Fig. S9†). During work up in toluene with subsequent crystallisation from toluene/CH2Cl2 at −78 °C only the isomeric mixture could be isolated which converts to the racemic complex rac-3 as the main product at room temperature (Fig. S7†). Every route to separate the meso complex meso-3 only leads to the formation of the thermodynamically preferred rac complex rac-3 (ΔRHθ(meso vs. rac) = −18.5, ΔRGθ(meso vs. rac) = −14.8 kJ mol−1)52 probably via ring-slippage of an indenyl unit.53 To investigate the steric hindrance in the meso compared to the rac ligand system a buried volume analysis was performed.54,55 The spheric sterical hindrance map shows that the global sterical demand is identical for meso-3 (buried volume Vbur = 54.9%) as for rac-3 (Vbur = 55.0%, see ESI†). If the analysed space is decomposed into four equally sized quadrants (NE/SE/SW/NW), the main differences between the two ligand systems become apparent, as expected. The distribution of the space requirement values of Vbur.quad. = 47.1/62.9/47.1/62.9 (rac-3) and 45.2/47.4/63.0/64.1 (meso-3) agree with C2 and CS symmetries of the metallocene scaffolds. Theoretically, meso-4 could exist in two isomeric forms that differ in the position of the chloride and amide ligands relative to the metallocene unit (Nin–Clout and Nout–Clin). As expected, the space requirement analysis of the two quadrants facing away from the metallocene pocket clearly shows the larger space requirement of the amide ligand which can be satisfied in the case of formation of the Nout–Clin isomer [Nout: Vbur(NE/SE) = 42.6/44.8%, Clout: Vbur(NE/SE) 38.2/38.4%, see ESI†]. This was confirmed experimentally as only the thermodynamically preferred isomer Nout–Clin (ΔRHθ(Nin–Clout vs. Nout–Clin) = −33.8, ΔRGθ(Nin–Clout vs. Nout–Clin) = −32.9 kJ mol−1) is present in the crystal. In consequence the chloride ligand in meso-4 is sterically protected in the meso-(ebthi) pocket which explains the necessity of higher temperatures and larger amounts of LiNMe2 for formation of complex meso-3. Notably, attempts to prepare a dinuclear zirconocene amide complex from [(meso-(ebthi)ZrCl)2(μ-Me3SiC3SiMe3)]56 also led to formation of the mononuclear complex meso-3 in low yield (Fig. S6†).
We next examined the selective formation of the mixed chloride/amide complex [rac-(ebthi)Zr(Cl)(NMe2)] rac-4 by addition of one equivalent of LiNMe2 to [rac-(ebthi)ZrCl2] in Et2O at room temperature. Selective formation of five new resonances at δ 6.16, 6.09, 5.54, 5.12 and 2.87 ppm in a 1:1:1:1:6 ratio suggests the formation of the desired mono(amide) species (Fig. S10†). 13C NMR analysis shows the characteristic amide resonance at δ 48.4 ppm (Fig. S11†). However, it was difficult to separate complex rac-4 from simultaneously formed bis(amide) rac-3 (9:1 ratio, Fig. S10†).
Catalytic dehydropolymerisation of PhSiH3
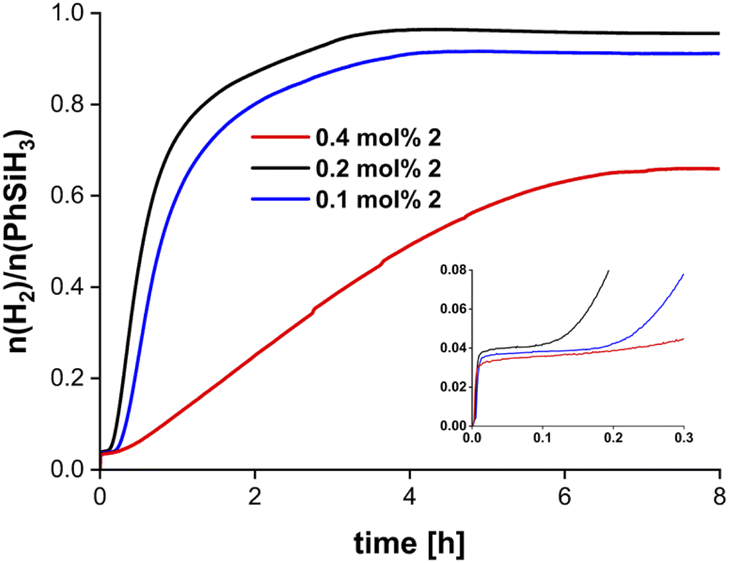 | ||
| Fig. 2 Volumetric curves of dehydropolymerisation of PhSiH3 using complex 2. Conditions: room temperature, no solvent, 1.85 mmol PhSiH3. The inset shows the induction period that was detected in all catalytic runs (units were removed for clarity and are identical to the full plot). | ||
Control experiments showed that an open reaction setup with pressure compensation and continuous stirring of the reaction solution was required to perform the dehydropolymerisation (Fig. S33†). A significantly decreased activity was found when performing the reaction in solution (Fig. S34†).58 To ensure that the catalytic activity of 2 is based on activation and conversion of the silane at the Zr centre we performed NMR scale reactions of PhSiH3 with different amine bases. NMR analysis of PhSiH3 (1.85 mmol) in the presence of tetramethylethylenediamine (0.37 mmol) or LiNMe2 (0.06 mmol) shows no conversion of the substrate (Fig. S43–S48†), indicating that the presence of the metal centre is essential for the dehydropolymerisation of PhSiH3 and the basicity of the NMe2 group is not sufficient to activate the silane. In line with literature data of reactions using group 4 metal complexes, sterically and electronically different secondary aryl silanes or alkyl silanes such as Ph2SiH2, Et2SiH2, or n-BuSiH3 showed no conversion (Fig. S49, S50 and S51†).59–61 When p-tolSiH3 (p-tol = p-CH3-C6H4) was used the reaction time increased drastically and only low conversion could be found at room temperature. The formation of minor amounts of polymer was confirmed by 1H NMR spectroscopy (Fig. S32†).
In contrast immediate H2 evolution could be observed in the catalytic reaction using complex rac-4 (0.4 mol%) at room temperature with subsequent formation of a highly viscous brownish waxy solid. Volumetric measurement showed a reaction time of three hours with approximately 0.95 eq. of H2 evolved before the stirring bar was fixed in the highly viscous polymer (Fig. S53†). Full conversion of PhSiH3 could be determined by NMR spectroscopy (Fig. S40†). The significant difference in reactivity between isomeric meso-4 and rac-4 could also be attributed to the steric protection of the chloride ligand in meso-4 (vide infra). In consequence, this could result in more facile abstraction of the chloride ligand in rac-4, that could lead to highly reactive zirconocene(III) fragments.62 Such species were discussed as an active species in dehydrocoupling of silanes for the less sterically demanding zirconocenes [Cp2Zr] and [CpCp*Zr].25,63 In summary, both the mono chloride functionality as well as the accessibility of this in rac-4 could contribute to the higher activity of this ansa zirconocene.
Molecular weight of polysilanes
The molecular weights of the obtained polymers were determined by size exclusion chromatography (SEC) with calibration using polystyrene as standard. Polymer samples were isolated as glassy yellow solids, soluble in most organic solvents such as benzene or THF and analysed by SEC and 29Si NMR spectroscopy (vide infra). The molecular weights of polymers isolated from dehydropolymerisation reactions with 2 as catalyst ranged from Mn = 2100–3000 g mol−1, depending on the catalyst concentration (Table 1). SEC profiles of the polymers show minor tailing in the low molecular weight region (Fig. S63–S65†). Formation of low molecular weight cyclic byproducts in combination with linear polysilanes and the subsequent separation was described before for related systems and could serve as an explanation for this observation.12–14,23,29,64 The exact linear-to-cyclic rations (L/C) were determined by SEC and from direct integration of 1H NMR signals corresponding to Si–H fragments in linear (δ 4.3–4.9 ppm) and cyclic (δ 4.9–5.3 ppm) structures (Table 1).23,65,66An increase in catalyst concentrations results in the formation of higher molecular weight polymers, which is in line with previously reported results using the mononuclear complex 1 (Table 1, Fig. S61 and S62†).34,67 Moreover, higher catalyst concentration leads to longer reaction times and a decrease in conversion (Fig. 2, vide supra). Thus, catalyst concentrations of 0.2 mol% were found to be optimal with respect to monomer conversion and molecular weight distribution. Polydispersity indices (PDI) are in the range of 1.22–1.97, suggesting the presence of relatively well-defined polysilanes with narrow mass distribution (Fig. S61–S65†).
We have next analysed the kinetics of polymer growth. Therefore, we quenched the dehydropolymerisation reactions (0.2 mol% 2) at different stages of conversion by addition of THF, which inhibits the reaction. SEC analysis of the reaction mixture after 60 minutes shows a broad low intensity peak corresponding to molecular weights in the range Mn = 150–400 g mol−1 (Table 2, Fig. S67†). After 90 minutes a significant increase in Mn = 1300–1500 g mol−1 was found (Table 2, Fig. S68†). Molecular weights determined at full conversion (t = 3 h) correspond to the values given in Table 2 (Fig. S69†). The presence of low molecular weight oligomers at early stages of the reaction and the significant increase in Mn after 60 min indicates that a chain growth mechanism is not likely for the dehydropolymerisation of PhSiH3 with 2. Instead, a step growth mechanism could operate. The same scenario could play a role for complex 1, which showed Mn = 200–400 g mol−1 when measured after 5 minutes (Table 2).
| [Zr] | c [mol%] | Mn [g mol−1] | Mw [g mol−1] | PDI | Time [min] |
|---|---|---|---|---|---|
| 1 | 0.4 | 200 | 400 | 2.02 | 5 |
| 2685 | 3935 | 1.46 | 20 | ||
| 0.2 | 140–400 | 240–400 | 1.70 | 60 | |
| 2 | 1340–1550 | 2850–3000 | 2.00 | 90 | |
| 2900–3000 | 5600 | 1.95 | 180 |
When performing the dehydropolymerisation with higher catalyst concentrations we could observe that the catalyst was still present in the polymeric mixture (Fig. S22†). We have therefore added a second portion of PhSiH3 (1.85 mmol) after a first catalytic run (0.4 mol% 2) and found that this leads to further conversion to glassy yellow polymer (Fig. S25†). 1H NMR analysis of the isolated polymer using 0.4 mol% 2 showed that the catalyst was still intact after further monomer addition (Fig. S25–S27†). The addition of a second portion of monomer does not lead to an increase in molecular weight (Mn = 2500–2600 g mol−1, Fig. S66†), indicating that although the Zr catalyst is still present, reactive polysilane chain ends are not available for further polymer growth.
SEC analysis of polymer fractions obtained with 0.8 mol% rac-3 at 60 °C shows only the formation of low molecular weight oligomers (Mn = 200–450 g mol−1) (Fig. S70†). Polymer isolated using 0.4 mol% rac-4 as catalyst showed two SEC signals (2100–2300 and 260–400 g mol−1, Fig. S71†). Compared to 2 low molecular weight fragments dominate, suggesting the formation of cyclic/linear oligomers (Fig. S71†).
Microstructure of isolated polysilanes
The microstructures of isolated polymers were analysed by 29Si DEPT45 and DEPT135 NMR spectroscopy. Polymers that were isolated from reactions using 0.1–0.4 mol% 2 show a complex pattern of resonances in the 29Si DEPT spectra (Fig. S16–S24†). Broad signals in the range of δ −55 to −65 ppm correspond to the typical, mainly atactic structure of the polymer mixture (Fig. 3).29,68 29Si DEPT135 analysis shows that signals from δ −60 to −65 ppm correspond to tertiary Sin(Ph-SiH)Sin units (Fig. 3).23,29,65 The DEPT135 experiment also confirmed the phase-shifted resonances at δ −56 to −58 ppm as –SiH2 end groups (Fig. 3).29 Polymer samples synthesised with 0.2 mol% of 2 showed the most well-defined resonance patterns, which can be divided into a high-intensity region from δ −60 to −63 ppm and a lower-intensity region at δ −63 to −65 ppm (Fig. S21†),62,68 the former corresponding to β silyl centres.6 Polymer fractions obtained from reactions using 0.1 and 0.4 mol% 2 show a significantly higher portion of linear/cyclic oligomers in the range δ −50 to −55 ppm (Fig. S17, S18, S23 and S24†).62 In the case of 0.1 mol% 2, this could be the explanation for the lower molecular weight (Table 1, Fig. S17and S18†). | ||
| Fig. 3 Stacked 29Si DEPT135 NMR spectra of poly(phenylsilane) obtained from catalytic dehydropolymerisation of PhSiH3 using 0.2 mol% 2 (top), 0.4 mol% rac-4 (bottom). | ||
Investigations of polymer microstructure have been done before using deconvolution processes.6,29,68 Tertiary Sin(Ph-SiH)Sin units can be categorised into stereospecific triads, namely linear isotatic (mm), linear atactic (mr) and linear syndiotactic polysilane (rr) (Fig. 3).29 Although reasonably good separation of the 29Si DEPT resonances was observed for samples prepared with 0.2 mol% 2 (Fig. 3), the low resolution and the overlap of resonances made it impossible to analyse the tacticity of the polymer on the basis of these NMR spectra (Fig. S16–S24†).
29Si DEPT NMR analysis of polymers obtained from reactions using ansa-zirconocene amide complex rac-3 suggests the presence of an oligomeric mixture with a complex resonance pattern (Fig. S36–S38†). Formation of the products of PhSiH3 dimerisation (δ −61.7 ppm, SiH2), trimerisation (δ −59.1, SiH and −68.5 ppm, SiH2), and tetramerisation (two isomers, two sets of resonances at δ −59.0, SiH and −65.0 to −65.5 ppm, SiH2) could be detected (Fig. S36–S38†).25 The formation of higher oligomers can also be assumed, however the separation of the 29Si DEPT resonances is not sufficient for a detailed assignment (Fig. S37 and S38†). These results are consistent with the previously reported formation of two diastereomers of the tetramer, showing that the coupling of the Si–Si bonds proceeds without stereoselectivity.25 29Si DEPT spectra of samples produced with 0.4 mol% rac-4 show the formation of isotatic and atactic polymer fractions with sharp resonances in the range of δ −61.0 ppm (Fig. 3, S40, S41†). Compared to materials prepared with 2, the distribution of oligomers and polymers was better resolved in the range upfield of δ −61.0 ppm (Fig. 3, S41†). This result is consistent with SEC data indicating a larger amount of oligomers isolated using rac-4 (Fig. S71,† vide infra). The C2 chirality of the ebthi ligand system has a significant effect on the tacticity and the formation of low molecular weight oligomers, as previously shown in the literature.25
Conclusion
The dinuclear zirconocene complex 2 shows selectivity for the dehydropolymerisation of phenylsilane that is comparable to that of the parent mononuclear analog Cp2Zr(NMe2)2, producing a polymer that possesses isotactic, atactic and syndiotactic structures. The molecular weights of the isolated polyphenylsilane products are comparable to those prepared by previously studied related catalyst systems, which again shows the previously discussed limitations of this approach of silane dehydropolymerisation. SEC analysis of the polymers at varying conversion suggests step growth as a likely scenario for polymer formation. In contrast to previously reported highly active mononuclear complex Cp2Zr(NMe2)2, no decomposition of the dinuclear catalyst 2 was observed, and the catalyst retains its activity at high conversion of substrate. As a consequence, we could show that selective polymer formation with a well-defined mass distribution can be observed after addition of further monomer. Ansa-zirconocene amide complexes prepared in this study were tested as catalysts and the activity of these species was found to be strongly dependent on the symmetry of the metallocene fragment and the presence of amide/chloride ligands. Only the C2 symmetric mixed amide/chloride complex rac-4 showed activity that was comparable to that of dinuclear 2. Future studies will aim at evaluation of the potential of the herein reported and further dinuclear group 4 metallocene complexes for dehydrocoupling reactions.Author contributions
K. L., F. R. and T. B. conceived and conceptualised the project. K. L. performed the experiments and analysed the data. A. S. performed the SC-XRD and F. R. the DFT study. F. R. and T. B. supervised the project. T. B. acquired funding. K. L., F. R. and T. B. prepared and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank our technical and analytical staff, in particular Hanan AlHamwi and PD Dr Wolfgang Baumann for assistance. We thank Sihan Li (LIKAT) for providing [(meso-(ebthi)ZrCl)2(μ-Me3SiC3SiMe3)] as well as Lanxess Organometallics GmbH (Bergkamen, Germany) for generously donating ansa-metallocene starting materials. Financial support by the Deutsche Forschungsgemeinschaft (project code 419924354) is gratefully acknowledged.Notes and references
- J. Koe, M. Fujiki, Organosilicon Compounds, ed. V. Y. Lee, Academic Press, 2017, pp. 219–300 Search PubMed.
- J. R. Koe, Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007, pp. 549–649 Search PubMed.
- H. Jamshidi and A. Rahimi, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 2565–2576 CrossRef CAS.
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- J. Koe, Polym. Int., 2009, 58, 255–260 CrossRef CAS.
- R. G. Jones, R. E. Benfield, P. J. Evans, S. J. Holder and J. A. M. Locke, J. Organomet. Chem., 1996, 521, 171–176 CrossRef CAS.
- K. Matyjaszewski, Y. L. Chen and H. K. Kim, J. Inorg. Organomet. Polym., 1988, 360, 78–88 CAS.
- K. Sakamoto, K. Obata, H. Hirata, M. Nakajima and H. Sakurai, J. Am. Chem. Soc., 1989, 111, 7641–7643 CrossRef CAS.
- M. Ishifune, C. Sana, M. Ando and Y. Tsuyama, Polym. Int., 2011, 60, 1208–1214 CrossRef CAS.
- T. J. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed.
- J. F. Harrod, Coord. Chem. Rev., 2000, 206–207, 493–531 CrossRef CAS.
- J. Y. Corey, Adv. Organomet. Chem., 2004, 51, 1–52 CrossRef CAS.
- F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1998, 42, 363–405 CrossRef CAS.
- T. D. Tilley, Acc. Chem. Res., 1993, 26, 22–29 CrossRef CAS.
- M. Minato, T. Matsumoto, M. Ichikawa and T. Ito, Chem. Commun., 2003, 2968–2969 RSC.
- F.-G. Fontaine and D. Zargarian, Organometallics, 2002, 21, 401–408 CrossRef CAS.
- E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Organometallics, 2010, 29, 6527–6533 CrossRef CAS.
- N. T. Mucha and R. Waterman, Organometallics, 2015, 34, 3865–3872 CrossRef CAS.
- J. P. Banovetz, Y. L. Hsiao and R. M. Waymouth, J. Am. Chem. Soc., 1993, 115, 2540–2541 CrossRef CAS.
- Y.-L. Hsiao and R. M. Waymouth, J. Am. Chem. Soc., 1994, 116, 9779–9780 CrossRef CAS.
- F. Fang, Q. Jiang and R. S. Klausen, J. Am. Chem. Soc., 2022, 144, 7834–7842 CrossRef CAS.
- C. P. Folster and R. S. Klausen, Polymer Chemistry, 2018, 9, 1938–1941 RSC.
- Y. Obora and M. Tanaka, J. Organomet. Chem., 2000, 595, 1–11 CrossRef CAS.
- J. Y. Corey and X. H. Zhu, Organometallics, 1992, 11, 672–683 CrossRef CAS.
- V. K. Dioumaev, K. Rahimian, F. Gauvin and J. F. Harrod, Organometallics, 1999, 18, 2249–2255 CrossRef CAS.
- V. K. Dioumaev and J. F. Harrod, Organometallics, 1994, 13, 1548–1550 CrossRef CAS.
- H. G. Woo and T. D. Tilley, J. Am. Chem. Soc., 1989, 111, 3757–3758 CrossRef CAS.
- H. G. Woo, J. F. Walzer and T. D. Tilley, J. Am. Chem. Soc., 1992, 114, 7047–7055 CrossRef CAS.
- B. J. Grimmond and J. Y. Corey, Organometallics, 1999, 18, 2223–2229 CrossRef CAS.
- F. Lunzer, C. Marschner, B. Winkler, N. Peulecke, W. Baumann and U. Rosenthal, Monatsh. Chem., 1999, 130, 215–219 CAS.
- H. Hisako, O. Sachiko and K. Mitsuo, Chem. Lett., 2000, 29, 188–189 CrossRef.
- J. P. Banovetz, H. Suzuki and R. M. Waymouth, Organometallics, 1993, 12, 4700–4703 CrossRef CAS.
- J. F. Harrod and S. S. Yun, Organometallics, 1987, 6, 1381–1387 CrossRef CAS.
- Q. Wang and J. Y. Corey, Can. J. Chem., 2000, 78, 1434–1440 CrossRef CAS.
- X. Liu, Z. Wu, Z. Peng, Y.-D. Wu and Z. Xue, J. Am. Chem. Soc., 1999, 121, 5350–5351 CrossRef CAS.
- K. A. Erickson, M. P. Cibuzar, N. T. Mucha and R. Waterman, Dalton Trans., 2018, 47, 2138–2142 RSC.
- W. Asim, A. S. Waheeb, M. A. Awad, A. M. Kadhum, A. Ali, S. H. Mallah, M. A. Iqbal and M. M. Kadhim, J. Mol. Struct., 2022, 1250, 131925 CrossRef CAS.
- M. A. Bau, S. Wiesler, S. L. Younas and J. Streuff, Chem.–Eur. J., 2019, 25, 10531–10545 CrossRef CAS PubMed.
- A. Razavi, Polyolefins: 50 years after Ziegler and Natta II: Polyolefins by Metallocenes and Other Single-Site Catalysts, ed. W. Kaminsky, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 43–116 Search PubMed.
- G. M. Diamond, R. F. Jordan and J. L. Petersen, Organometallics, 1996, 15, 4030–4037 CrossRef CAS.
- G. M. Diamond, R. F. Jordan and J. L. Petersen, J. Am. Chem. Soc., 1996, 118, 8024–8033 CrossRef CAS.
- J. N. Christopher, G. M. Diamond, R. F. Jordan and J. L. Petersen, Organometallics, 1996, 15, 4038–4044 CrossRef CAS.
- E. Polo, R. M. Bellabarba, G. Prini, O. Traverso and M. L. H. Green, J. Organomet. Chem., 1999, 577, 211–218 CrossRef CAS.
- O. Olabisi, M. Atiqullah and W. Kaminsky, J. Macromol. Sci., Polym. Rev., 1997, 37, 519–554 CrossRef.
- I. Kim and R. F. Jordan, Macromolecules, 1996, 29, 489–491 CrossRef CAS.
- I. Kim, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35, 293–304 CrossRef.
- I. Kim and G. Nam Hwang, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35, 1987–2008 CrossRef.
- K. Lindenau, N. Jannsen, M. Rippke, H. Al Hamwi, C. Selle, H.-J. Drexler, A. Spannenberg, M. Sawall, K. Neymeyr, D. Heller, F. Reiß and T. Beweries, Catal. Sci. Technol., 2021, 11, 4034–4050 RSC.
- M. Trose, M. Reiß, F. Reiß, F. Anke, A. Spannenberg, S. Boye, A. Lederer, P. Arndt and T. Beweries, Dalton Trans., 2018, 47, 12858–12862 RSC.
- F. Reiß, M. Reiß, A. Spannenberg, H. Jiao, W. Baumann, P. Arndt, U. Rosenthal and T. Beweries, Chem.–Eur. J., 2018, 24, 5667–5674 CrossRef PubMed.
- For clarity, we have shown only one isomer of the asymmetric unit. The asymmetric unit contains three isomers with disorders in the ebthi ligand system (Fig. S54†)..
- We optimized the structures, considering empirical dispersion correction, on a double zeta basis set, followed by triple zeta basis set single point calculations. Geometries were confirmed to be local minima on the potential energy surface by harmonic vibration frequency calculations on the same level of theory (B3LYP/GD3BJ/def2svpp//def2tzvp)..
- J. M. OConnor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef CAS.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- X. Shi, S. Li, M. Reiß, A. Spannenberg, T. Holtrichter-Rößmann, F. Reiß and T. Beweries, Chem. Sci., 2021, 12, 16074–16084 RSC.
- T. Beweries, J. Thomas, M. Klahn, A. Schulz, D. Heller and U. Rosenthal, ChemCatChem, 2011, 3, 1865–1868 CrossRef CAS.
- M. Horacek, J. Merna, R. Gyepes, J. Sykora, J. Kubista and J. Pinkas, Collect. Czech. Chem. Commun., 2011, 76, 75–94 CrossRef CAS.
- C. T. Aitken, J. F. Harrod and E. Samuel, J. Am. Chem. Soc., 1986, 108, 4059–4066 CrossRef CAS.
- C. Aitken, J. F. Harrod and E. Samuel, J. Organomet. Chem., 1985, 279, C11–C13 CrossRef CAS.
- W. H. Campbell, T. K. Hilty and L. Yurga, Organometallics, 1989, 8, 2615–2618 CrossRef CAS.
- B. J. Grimmond and J. Y. Corey, Inorg. Chim. Acta, 2002, 330, 89–94 CrossRef CAS.
- V. K. Dioumaev and J. F. Harrod, Organometallics, 1997, 16, 2798–2807 CrossRef CAS.
- J. P. Banovetz, K. M. Stein and R. M. Waymouth, Organometallics, 1991, 10, 3430–3432 CrossRef CAS.
- N. Choi, S.-y. Onozawa, T. Sakakura and M. Tanaka, Organometallics, 1997, 16, 2765–2767 CrossRef CAS.
- P. T. K. Lee, M. K. Skjel and L. Rosenberg, Organometallics, 2013, 32, 1575–1578 CrossRef CAS.
- Details on NMR spectroscopy of polymers using complex 1 are given in the supporting information (Fig. S28–S31†)..
- B. J. Grimmond, N. P. Rath and J. Y. Corey, Organometallics, 2000, 19, 2975–2984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic and spectroscopic details, SEC analysis, and catalytic data. CCDC 2181386 and 2181387. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra04955d |
| This journal is © The Royal Society of Chemistry 2022 |