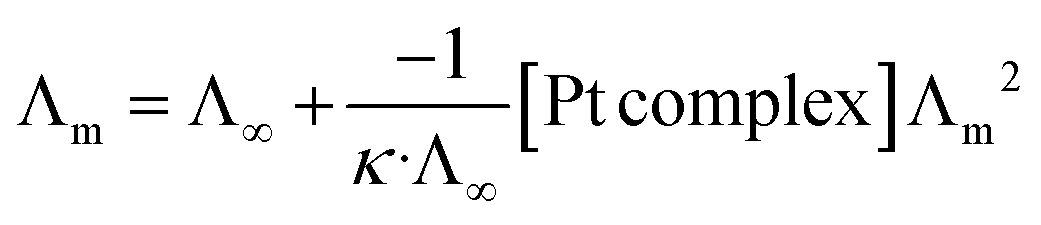DOI:
10.1039/D2RA04992A
(Paper)
RSC Adv., 2022,
12, 27582-27595
Tuning the anticancer properties of Pt(II) complexes via structurally flexible N-(2-picolyl)salicylimine ligands†
Received
9th August 2022
, Accepted 17th September 2022
First published on 27th September 2022
Abstract
Three tridentate Schiff base ligands were synthesized from the reactions between 2-picolylamine and salicylaldehyde derivatives (3-ethoxy (OEt), 4-diethylamino (NEt2) and 4-hydroxy (OH)). Complexes with the general formula Pt(N^N^O)Cl were obtained from reactions between the ligands and K2PtCl4. The ligands and their complexes were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. Further confirmation of the structure of Pt-OEt was achieved by single-crystal X-ray diffraction. The DMSO/chlorido exchange process at Pt-OEt was investigated by monitoring the change in conductivity, revealing very slow dissociation in DMSO. Moreover, solvent/chlorido exchange for Pt-OEt and Pt-NEt2 were investigated by NMR spectroscopy in DMSO and DMSO/D2O; Pt-NEt2 forms an adduct with DMSO while Pt-OEt forms adducts with DMSO and water. The DNA-binding behaviour of the platinum(II) complexes was investigated by two techniques. Pt-NEt2 has the best apparent binding constant. The intercalation mode of interaction with ct-DNA was suggested by molecular docking studies and the increase in the relative viscosity of ct-DNA with increasing concentrations of the platinum(II) complexes. However, the gradual decrease in the relative viscosity over time at constant concentration of platinum(II) complexes indicated a shift from intercalation to a covalent binding mode. Anticancer activities of the ligands and their platinum(II) complexes were examined against two cell lines. The platinum(II) complexes exhibit superior cytotoxicity to that of their ligands. Among the platinum(II) complexes, Pt-OEt possesses the best IC50 against both cell lines, its cytotoxicity being comparable to that observed for cisplatin. Cell cycle arrest in the HepG2 cell line upon treatment with Pt-OEt and Pt-NEt2 was investigated and compared to that of cisplatin; the change in the cell accumulation patterns supports the presumption of an apoptotic cell death pathway. The optimized structures of the B-DNA trimer adducts with the platinum complexes showed hydrogen-bonding interactions between the ligands and nucleobases, affecting the inter-strand hydrogen bonding within the DNA, and highlighting the strong ability of the complexes to induce conformational changes in the DNA, leading to the activation of apoptotic cell death. In summary, the current study demonstrates promising new anticancer platinum(II) complexes with highly flexible tridentate ligands; the functional groups on the ligands are important in tuning their DNA binding/anticancer properties.
Introduction
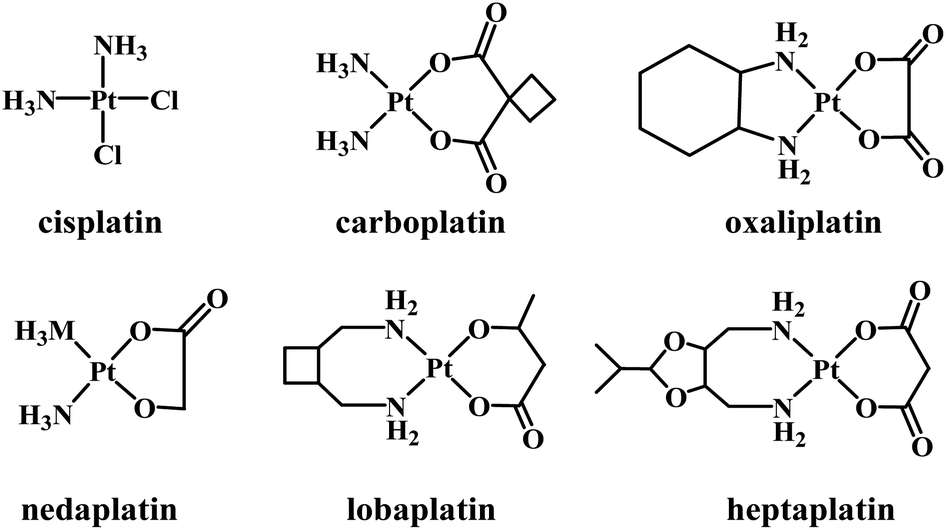
Several platinum(II) complexes have been approved for clinical use as chemotherapeutic agents in treating a large range of cancer types. The first approved Pt-based drug was cisplatin (Fig. 1) which is known to bind to DNA covalently. Binding occurs following the hydrolysis of its labile chlorido ligands, producing cis-[Pt(NH3)2Cl(H2O)]+.1 Inside the cell, aqua/N7-guanine ligand exchange proceeds, producing the cis-[Pt(NH3)2Cl(DNA)]+ adduct.2 The second chlorido ligand is replaced by another guanine nucleobase, forming an intrastrand and interstrand cross-linked DNA.3 The intrastrand cross-linking causes DNA to bend, leading to apoptosis,4 while the interstrand cross-linking unwinds DNA and bends the helix, promoting cytotoxicity.5 The major problems with cisplatin include a lack of selectivity toward cancer cells, acquired resistance, and severe side-effects.6–10 Second- and third-generation platinum(II) complexes have been synthesized that are structurally related to cisplatin. Normally, the ammine and/or chlorido ligands are substituted by either mono- or bidentate ligands, resulting in complexes with altered electronic and steric effects and basicity, but such complexes still form coordinate bonds with DNA.11
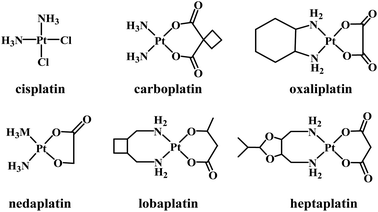 |
| | Fig. 1 Some clinically trialled/approved Pt-based anticancer drugs. | |
Carboplatin (a second-generation complex) and oxaliplatin (third-generation) are clinically approved Pt-based drugs that operate in a similar manner to cisplatin. However, the use of bidentate leaving groups slows the complex activation compared to cisplatin, leading to significantly lower toxicity.12–15 Several other compounds of general formula cis-[PtX2(ammine)2] (where X is an anionic leaving ligand) have been widely studied, with some gaining regional clinical approval including nedaplatin, lobaplatin and heptaplatin.11
In the search for more effective and less toxic platinum(II) complexes, examples that bind to DNA through non-covalent binding modes including electrostatic interactions, hydrogen bonding, and dipole, van der Waals and stacking (intercalation) interactions have been explored. Other ligands can be used to generate platinum complexes that can both intercalate and covalently bind. The most important of the resultant complexes are those that contain labile ligands at the platinum and a tether that ends with an intercalating moiety.16–18 Terpyridine and its derivatives have been employed in synthesizing Pt(II) intercalators that have been reported to be efficient DNA binders and active antitumor agents.19,20 Binding studies of the complex [Pt(tpy)Cl]+ have supported the intercalation of the ligand with DNA, which is followed by covalent bonding with one of the nucleobases (via loss of the labile chlorido ligand).21,22 In contrast, complexes of formula [Pt(tpy)(L)]+ (L = thiols) exhibit DNA-binding via intercalation alone,23 and some examples have shown better cytotoxic effects than carboplatin against human ovarian cancer cell lines.24,25 In contrast to the rigid terpyridine ligand, the more flexible 4-methyl-2-N-(2-pyridylmethylene)aminophenol was employed to synthesize two complexes, Pt(N^N^O)Cl with a coordinated phenolic moiety, and Pt(N^N)Cl2 with non-coordinating phenol. Both complexes interact with DNA via intercalation; Pt(N^N)Cl2 possesses better binding affinity, but Pt(N^N^O)Cl was found to be more cytotoxic, its anticancer efficiency being better than that of cisplatin towards three different cancer cell lines.26
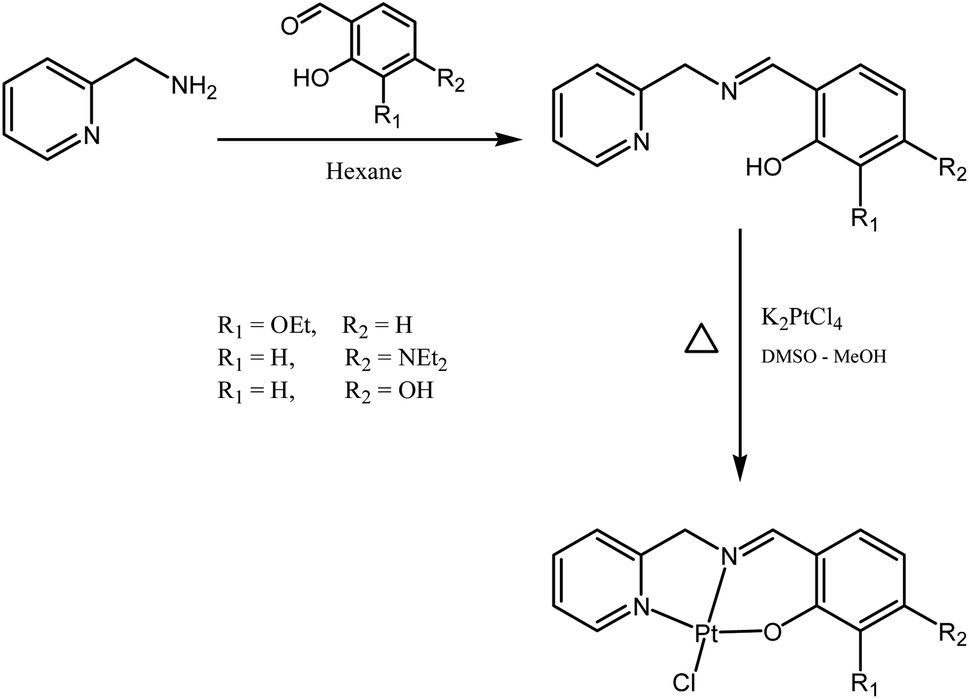
Multi-functional platinum(II) binders designed to simultaneously bind DNA covalently and non-covalently are of significant topical interest. The current work explores tridentate Schiff base ligands accessed from the reactions between 2-picolylamine and salicylaldehyde derivatives. We show that these ligands afford complexes of formula PtCl(N^N^O) that have potential for covalent binding with DNA as well as hydrogen bonding, dipole interactions and/or hydrophobic interactions, and that such interactions may lead to modification of the physiochemical/pharmacological properties.
Materials and methods
Materials
All the reactions were carried out under an inert atmosphere, using standard Schlenk techniques with oven-dried glassware. All the solvents were dried over molecular sieves (4A) for at least 48 hours before use. Propidium iodide (PI) was obtained from Thermo-Fisher Scientific. All other reagents were obtained from Sigma-Aldrich and used without further purification.
Methods and instrumentation
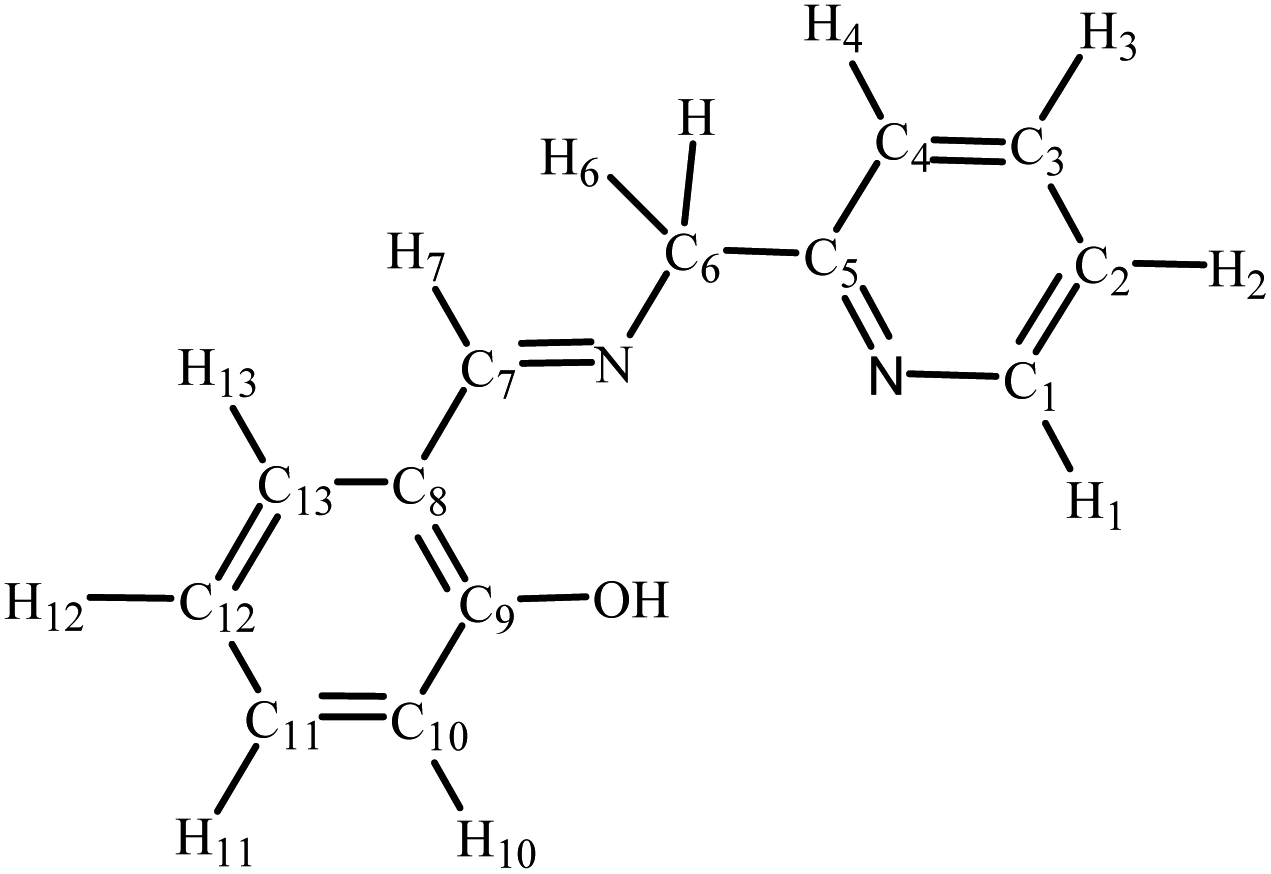
The high-resolution electrospray ionization (ESI) mass spectra were recorded (positive ionization mode) using a Bruker Apex 4.7 FTICR-MS instrument. The elemental analyses were carried out at King Abdulaziz University. The infrared (IR) spectra were collected as KBr disks on a PerkinElmer Spectrum 100 instrument; the reported peaks are in cm−1. 1H NMR (600 MHz), 13C NMR (121 MHz) and 31P NMR (242 MHz) spectra were performed on a Bruker Avance 600 MHz spectrometer equipped with a BBO probe; the spectra were referenced to residual chloroform or DMSO (7.26 or 2.50, 1H), CDCl3 or DMSO-d6 (77.0 or 39.5 13C), or external H3PO4 (0.0 31P).27 Atom labelling in NMR spectra follows the numbering scheme in Chart 1. UV-vis absorption spectra were obtained as chloroform solutions with a Genesys-10s UV-VIS spectrophotometer (Thermo Fisher), using 1 cm path-length quartz cells; the wavelengths of the absorption maxima are given in nanometers.
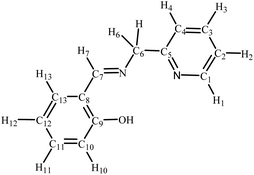 |
| | Chart 1 NMR labelling scheme for compounds in this work. | |
Synthesis and characterization
General synthetic procedure for the Schiff base ligands. Equimolar amounts of 2-picolylamine and salicylaldehyde derivative were stirred in 10 mL of hexane at room temperature. After reaction completion, the precipitate was collected by filtration, washed with hexane, and vacuum-dried.
Ligand 1 (L-OEt). 2-Picolylamine (0.250 g (0.245 mL), 2.31 mmol) and 3-ethoxysalicylaldehyde (0.384 g, 2.31 mmol) reacted to yield L-OEt as a yellow powder (0.539 g, 91%). IR (cm−1): 1576 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1589 ν(CN aromatic), 1631 ν(CN aliph), 3451 ν(OH). 1H-NMR (d6-DMSO) δ: 13.63 (s, 1H, OH), 8.73 (s, 1H, H7), 8.56 (d, JHH = 3 Hz, 1H, H1), 7.83 (t, JHH = 5 Hz, 1H, H3), 7.41 (d, JHH = 5 Hz, 1H, H4), 7.33 (t, JHH = 5 Hz, 1H, H2), 7.08 (d, JHH = 5 Hz, 1H, H13), 7.05 (d, JHH = 5 Hz, 1H, H11), 6.82 (t, JHH = 6 Hz, 1H, H12), 4.92 (s, 1H, H6), 4.03 (q, 2H, JHH = 5 Hz, OCH2), 1.31 (t, JHH = 5 Hz, 3H, CH3). 13C-NMR (d6-DMSO) δ: 167.5 (C7), 157.6 (C5), 151.6 (C1), 149.2 (C9), 147.0 (C10), 136.9 (C3), 122.4 (C4), 123.3 (C13) 121.9 (C2), 118.0 (C11), 117.3 (C8), 116.3 (C12), 63.9 (C6), 63.3 (CH2), 14.7 (CH3). HR ESI MS calcd for [C15H17N2O2]+: 257.1285, found: 257.1278. Anal. calcd for C15H16N2O2: C, 70.29; H, 6.29; N, 10.93%; found: C, 69.94; H, 6.03; N, 10.67%.
C), 1589 ν(CN aromatic), 1631 ν(CN aliph), 3451 ν(OH). 1H-NMR (d6-DMSO) δ: 13.63 (s, 1H, OH), 8.73 (s, 1H, H7), 8.56 (d, JHH = 3 Hz, 1H, H1), 7.83 (t, JHH = 5 Hz, 1H, H3), 7.41 (d, JHH = 5 Hz, 1H, H4), 7.33 (t, JHH = 5 Hz, 1H, H2), 7.08 (d, JHH = 5 Hz, 1H, H13), 7.05 (d, JHH = 5 Hz, 1H, H11), 6.82 (t, JHH = 6 Hz, 1H, H12), 4.92 (s, 1H, H6), 4.03 (q, 2H, JHH = 5 Hz, OCH2), 1.31 (t, JHH = 5 Hz, 3H, CH3). 13C-NMR (d6-DMSO) δ: 167.5 (C7), 157.6 (C5), 151.6 (C1), 149.2 (C9), 147.0 (C10), 136.9 (C3), 122.4 (C4), 123.3 (C13) 121.9 (C2), 118.0 (C11), 117.3 (C8), 116.3 (C12), 63.9 (C6), 63.3 (CH2), 14.7 (CH3). HR ESI MS calcd for [C15H17N2O2]+: 257.1285, found: 257.1278. Anal. calcd for C15H16N2O2: C, 70.29; H, 6.29; N, 10.93%; found: C, 69.94; H, 6.03; N, 10.67%.
Ligand 2 (L-NEt2). 2-Picolylamine (0.250 g (0.245 mL), 2.31 mmol) and 4-diethylaminosalicylaldehyde (0.426 g, 2.31 mmol) reacted to yield L-NEt2 as a yellow powder (0.627 g, 95%). IR (cm−1): 1524 ν(CC), 1580 ν(CN aromatic), 1614 ν(CN aliph), 3447 ν(OH). 1H-NMR (d6-DMSO) δ: 13.60 (s, 1H, OH), 8.54 (d, JHH = 3 Hz, 1H, H1), 8.40 (s, 1H, H7), 7.80 (t, JHH = 7 Hz, 1H, H2), 7.35 (d, JHH = 5 Hz, 1H, H13), 7.30 (dd, JHH = 3 and 6 Hz, 1H, H3), 7.16 (d, JHH = 6 Hz, 1H, H4), 6.21 (dd, JHH = 5 Hz, 1H, H12), 5.69 (s, 1H, H10), 4.78 (s, 2H, H6), 3.33 (q, JHH = 5 Hz, 2H, NCH2), 1.09 (t, JHH = 5 Hz, 3H, CH3). 13C-NMR (CDCl3) δ: 165.5 (C9), 164.4 (C7), 158.4 (C5), 150.9 (C1), 149.1 (C11), 136.8 (C3), 133.1 (C13), 122.3 (C4), 121.7 (C2), 107.8 (C8), 102.9 (C12), 97.1 (C10), 62.4 (C6), 43.7 (CH2), 12.4 (CH3). HR ESI MS calcd for [C17H22N3O]+: 284.1757, found: 284.1749. Anal. calcd for C17H21N3O: C, 72.06; H, 7.47; N, 14.83%; found: C, 71.69; H, 7.30; N, 14.34%.
Ligand 3 (L-OH). 2-Picolylamine (0.250 g (0.245 mL), 2.31 mmol) and 2,4-dihydroxybenzaldehyde (0.319 g, 2.31 mmol) reacted to yield L-OH as a yellow powder (0.487 g, 92%). IR (cm−1): 1481 ν(CC), 1544 ν(CN aromatic), 1623 ν(CN aliph), 2400–3400 ν(OH). 1H-NMR (d6-DMSO) δ: 13.69 (s, 1H, OH), 10.01 (s, 1H, OH), 8.51 (s, 1H, H7), 7.79 (t, JHH = 5 Hz, 1H, H2), 7.37 (d, JHH = 6 Hz, 1H, H1), 7.30 (t, JHH = 5 Hz, 1H, H3), 7.24 (d, JHH = 6 Hz, 1H, H4), 6.31 (dd, JHH = 2 and 6 Hz, 1H, H12), 6.18 (d, JHH = 2 Hz, 1H, H13), 5.75 (s, 1H, H10), 4.82 (s, 2H, H6). 13C-NMR (CDCl3) δ: 166.3 (C9), 164.2 (C7), 161.8 (C1), 158.0 (C5), 149.1 (C11), 136.9 (C3), 133.4 (C13), 122.9 (C4), 122.4 (C2), 111.2 (C8), 106.9 (C12), 102.4 (C10), 62.6 (C6). HR ESI MS calcd for [C13H13N2O2]+: 229.0972, found: 229.0969. Anal. calcd for C13H12N2O2: C, 68.41; H, 5.30; N, 12.27%; found: C, 68.55; H, 5.23; N, 12.08%.
General synthetic procedure for the Pt(II) complexes. Equimolar amounts (0.241 mmol) of the particular ligand (L-OEt, L-NEt2 or L-OH) and NaOAc were dissolved in 15 mL methanol at reflux. A solution of K2PtCl4 (0.241 mmol) in a mixture of 1 mL DMSO and 5 mL methanol was then added in one portion and the resultant mixture was stirred at reflux for 24 h. The mixture was cooled to room temperature and reduced in volume to ca. 5 mL. 15–20 mL diethyl ether was added with stirring. The product was collected by filtration, washed with diethyl ether, and dried.
Complex 1 (Pt-OEt). K2PtCl4 (100 mg, 0.241 mmol) and L-OEt (62 mg, 0.241 mmol) reacted to yield Pt-OEt as a green-yellow powder (96 mg, 82%). IR (cm−1): 1546 ν(CC), 1603 ν(CN aromatic), 1621 ν(CN aliph). 1H-NMR (d6-DMSO) δ: 9.93 (d, JHH = 4 Hz, 1H, H1), 8.73 (s, 1H, H7), 8.13 (t, JHH = 6 Hz, 1H, H2), 7.75 (d, JHH = 6 Hz, 1H, H4), 7.50 (t, JHH = 5 Hz, 1H, H3), 7.21 (d, JHH = 6 Hz, 1H, H13), 7.04 (d, JHH = 6 Hz, 1H, H12), 5.82 (s, 2H, H6), 4.03 (q, JHH = 5 Hz, 2H, OCH2), 1.33 (t, JHH = 5 Hz, 3H, CH3). 13C-NMR (d6-DMSO) δ: 162.4 (C7), 154.5 (C5), 152.4 (C9), 149.9 (C1), 148.8 (C10), 148.4 (C2), 138.4 (C3), 125.4 (C8), 123.9 (C4), 121.3 (C13), 116.5 (C12), 115.2 (C11), 66.0 (CH2), 63.8 (C6), 14.8 (CH3). HR ESI MS calcd for [PtC15H15N2O2Cl]+: 486.0543; found: 486.0544. Anal. calcd for C15H15ClN2O2Pt: C, 37.08; H, 3.11; N, 5.77%; found: C, 36.97; H, 2.93; N, 5.48%.
Complex 2 (Pt-NEt2). K2PtCl4 (100 mg, 0.241 mmol) and L-NEt2 (68 mg, 0.241 mmol) reacted to yield Pt-NEt2 as an orange powder (109 mg, 89%). IR (cm−1): 1513 ν(CC), 1565 ν(CN aromatic), 1600 ν(CN aliph). 1H-NMR (d6-DMSO) δ: 9.27 (d, JHH = 4 Hz, 1H, H1), 8.24 (s, 1H, H7), 8.11 (t, JHH = 5 Hz, 1H, H2), 7.69 (d, JHH = 6 Hz, 1H, H4), 5.46 (s, 1H, H6), 7.30 (d, JHH = 6 Hz, 1H, H13), 7.47 (t, JHH = 5 Hz, 1H, H3), 6.21 (d, JHH = 6 Hz, 1H, H12), 6.02 (s, 1H, H10), 4.98 (t, JHH = 5 Hz, 3H, CH3), 3.34 (q, JHH = 5 Hz, 2H, NCH2). 13C-NMR (d6-DMSO) δ: 162.9 (C7), 162.3 (C5), 152.5 (C9), 152.1 (C1), 143.9 (C13), 148.7 (C11), 137.9 (C3), 123.8 (C2), 120.9 (C4), 111.5 (C10), 103.9 (C12), 98.1 (C8), 65.1 (C6), 43.8 (CH2), 12.5 (CH3). HR ESI MS calcd for [PtC17H20N3OCl]+: 513.1016, found: 513.1017. Anal. calcd for C17H21ClN3OPt: C, 39.81; H, 3.93; N, 8.19%; found: C, 39.71; H, 3.68; N, 7.89%.
Complex 3 (Pt-OH). K2PtCl4 (100 mg, 0.241 mmol) and L-OH (55 mg, 0.241 mmol) reacted to yield Pt-OH as a yellow powder (91 mg, 83%). IR (cm−1): 1490 ν(CC), 1539 ν(CN aromatic), 1621 ν(CN aliph), 3000–3450 ν(OH). 1H-NMR (d6-DMSO) δ: 10.03 (s, 1H, OH), 9.25 (d, JHH = 4 Hz, 1H, H1), 8.44 (s, 1H, H7), 8.11 (t, JHH = 6 Hz, 1H, H2), 7.48 (t, JHH = 5 Hz, 1H, H3), 7.71 (d, JHH = 5 Hz, 1H, H13), 7.41 (d, JHH = 7 Hz, 1H, H4), 6.29 (s, 1H, H6), 6.21 (dd, JHH = 2 and 5 Hz, 1H, H12), 5.50 (s, 1H, H6). 13C-NMR (d6-DMSO) δ: 162.8 (C7), 162.7 (C5), 162.6 (C1), 153.7 (C9), 148.7 (C11), 138.2 (C3), 135.2 (C13), 123.9 (C4), 120.9 (C8), 114.6 (C2), 107.6 (C12), 104.1 (C10), 65.5 (C6). HR ESI MS calcd for [M–Cl + MeCN]+ = [PtC15H14N3O2]+: calcd 463.0734, found: 463.0724. Anal. calcd for C13H11ClN2O2Pt: C, 34.11; H, 2.42; N, 6.12%; found: C, 33.68; H, 2.09; N, 5.61%.
Crystallographic structure determination
Single crystals of Pt-OEt were grown by vapor diffusion of n-pentane into a saturated solution of the compound in dichloromethane containing one drop of DMSO at 3 °C. A suitable single crystal was selected and mounted on a Bruker APEX2 microsource diffractometer. The crystal was kept at 120.00 K during data collection, using Mo Kα (λ = 0.71073) radiation. Using Olex2,28 the structure was solved with the SHELXT29 structure solution program using the intrinsic phasing method and refined with the SHELXL30 refinement package using least squares minimization. Table 1 summarizes the crystal structure refinement details of Pt-OEt·H2O. Crystals of L-OEt were obtained by slow evaporation of a dichloromethane solution of the ligand, but the quality of collected data of L-OEt was low. We have included this study in the ESI† as a proof of the ligand structure and for completeness.
Table 1 Crystal data and structure refinement for Pt-OEt
| Empirical formula |
C15H17ClN2O3Pt |
| Formula weight |
503.84 |
| Crystal system, space group |
Monoclinic, P21/n |
| a, b, c (Å) |
6.6651(3), 22.2825(12), 10.6562(6) |
| α, β, γ (°) |
90, 97.288(2), 90 |
| Volume (Å3) |
1569.82(14) |
| Z |
4 |
| ρcalc (g cm−3) |
2.132 |
| μ (mm−1) |
9.122 |
| F(000) |
960.0 |
| Crystal size (mm3) |
0.313 × 0.097 × 0.086 |
| 2θ range for data collection (°) |
5.312 to 56.674 |
| Index ranges |
−8 ≤ h ≤ 8, −29 ≤ k ≤ 29, −14 ≤ l ≤ 14 |
| Reflections collected |
26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 214 214 |
| Independent reflections |
3851 [Rint = 0.0300, Rsigma = 0.0191] |
| Data/restraints/parameters |
3851/0/204 |
| Goodness-of-fit on F2 |
1.203 |
| Final R indexes [I ≥ 2σ(I)] |
R1 = 0.0171, wR2 = 0.0353 |
| Final R indexes [all data] |
R1 = 0.0187, wR2 = 0.0360 |
| Largest diff. peak/hole (e Å−3) |
0.81/−0.56 |
Solvolysis
The solvent/chlorido ligand exchanges (solvent = DMSO) of the platinum(II) complexes were monitored using conductivity measurements to calculate the dissociation rate of Pt-OEt (as it is a weak electrolyte). Solutions of the complexes of three concentrations, 3.0 × 10−4, 1.5 × 10−4, and 7.5 × 10−5 mM, were prepared in dimethylsulfoxide, and their conductivities were recorded at 25 °C in μS cm−1, employing an OHAUS STARTER3100C with an STCON3 conductivity electrode. Furthermore, 1H NMR was utilized to investigate the DSMO/chlorido and aqua/chlorido exchange processes for Pt-OEt and Pt-NEt2.
DNA-binding studies
Competitive fluorescence quenching of ethidium bromide-DNA adduct. A solution containing 100 μM ct-DNA and 10 μM ethidium bromide (EB) was prepared in an aqueous Tris–HCl/EDTA buffer system (pH = 7.3). Aliquots from the ct-DNA–EB solution were incubated for at least 24 h. The aliquots were treated with different concentrations of each of the platinum complexes in DMSO (the amount of DMSO was ca. 10% V/V), while maintaining the ct-DNA and EB concentrations. After 5 min incubation, the changes in the emission spectra of these solutions were followed between 500 nm and 750 nm upon excitation at 390 nm. The Stern–Volmer quenching constants (Ksv) were calculated using eqn (1), where F0 and F are the emission intensities in the absence and the presence of the samples, respectively. The [Pt complex] was plotted against [F0/F], and the Ksv value obtained from the slope:31,32| | |
F0/F = 1 + Ksv[Pt complex]
| (1) |
| | |
KEB[EB] = Kapp[Pt complex]50%
| (2) |
Apparent binding constant (Kapp) can be obtained from eqn (2), using the concentration of the platinum complexes that quenches 50% of the EB fluorescence33,34 (the binding constant for EB (KEB) = 1 × 105: see ref. 34). As we started with 10 μM ethidium bromide, we can assume that Ksv and Kapp are almost the same.
Determination of binding mode by changes in relative viscosity of DNA. The viscosity studies were carried out employing an Ostwald viscometer. Micro volumes (10 μL) of platinum compounds were added to a solution of ct-DNA in buffer, with the [Pt compounds]/[DNA] ratio constrained to the range 0.033–0.23. The resultant solutions were measured after 10 and 20 min incubation in a 25 °C water bath. The flow times for the solutions were recorded and replicated. The relative viscosities (η/η0)1/3 were plotted against the ratio of platinum compounds to the ct-DNA concentration, where η0 and η represent the specific viscosity of the ct-DNA alone and the ct-DNA–Pt adduct, respectively. The specific viscosities η and η0 were calculated using the formulation [(t − tb)/tb] where t is the observed flow time and tb is the buffer flow time.31,32,35
Computational studies
The docking studies were undertaken to evaluate the interactions between the DNA and platinum complexes with the formula [PtCl(L)] and [Pt(H2O)(L)]+ during the intercalation process. All the docking studies were carried out using Molecular Operating Environment (MOE) 2008.10 software. The docking scores were obtained with the London dG function in the MOE and then the scores were optimized by two other unrelated refinement methods. Grid-min pose and force-field were used to confirm the geometrical structures of the refined poses. Geometries around the platinum atoms, nitrogen atoms and the aromatic planarity were constrained while allowing bond rotations for other groups; the best five binding poses were then tested. To evaluate the binding free energy of the complexes toward DNA, the docking poses of the platinum compounds and the co-crystallized structure of the B-DNA were docked (RSCP PDB code: 1BNA). Assessment of the best binding pose was achieved by selecting poses that involve intercalation interaction with DNA and have the lowest RMSD value.
The covalent binding properties of the platinum complexes and the DNA conformational changes were investigated theoretically employing the Gaussian09 suite,36 and visualized by Gaussview software.37 Density functional theory (DFT) was used with the B3LYP functional,38 the SVP basis set39,40 for the light atoms (N, P, C, O, H), and the SDD basis set for Pt.41 First, the fragment d(CpGpCpGpCpG) was obtained from the crystal structure of the dodecamer B-DNA (RSCP PDB code: 1BNA), and then platinum (with the tridentate ligand) was covalently linked to N7 of the guanine. Partial symmetry constraints were applied in the geometry optimizations on the DNA fragment of the DNA–Pt adduct, and the final geometries were confirmed to be the minimum potential energy structures through frequency calculations. Weak interactions have been included in the energy evaluations using Grimme D3 corrections.42
Assessments of IC50 by MTT assay
Two human cell lines, liver (HepG2) and breast (MCF-7), were provided by the Tissue Culture Unit in the Department of Biochemistry at the Faculty of Science, King Abdulaziz University. Attached human cell lines were cultured for 24 h in a flask containing Dulbecco's Modified Eagle's Medium (DMEM). JK human cell lines were cultured in Roswell Park Memorial Institute Medium (RPMI 1640), which contains 10% fetal bovine serum and 1% antibiotic, at 37 °C and 95% humidity in a sterile 5% CO2 incubator. DMEM and RPMI 1640 were purchased from Life Technologies Gibco. A solution of 0.25% trypsin with EDTA (4 mL) was added to the attached cells after 90% of the confluent cells were collected, and placed in a CO2 incubator for 5 min. With the addition of 5 mL of complete medium, the trypsin reaction was stopped. After centrifugation of the unattached cells containing the medium, the pellets were washed twice with sterile phosphate buffer saline (PBS).43
The number of cells was determined with a hemocytometer and counted in the four major squares after 20 μL of the cell-containing media were stained with 20 μL of 0.4% trypan blue. A 96-well microplate was filled with 0.1 mL of 5000 cells suspended in complete media, and the plate was incubated for 24 h. For each of the platinum complexes, different concentrations (3.5, 7, 14, 28, 56 μM) were added to the media when 70% of the cells in each well were confluent. Each concentration was assessed four times. The plate was incubated for 48 h.
In each well, the media were replaced with 100 μL of 0.5 mg MTT per mL free media for 4 h in the incubator. Each well was filled with 100 μL DMSO and incubated at room temperature for 15 min before being monitored using a microplate reader at 595 nm (Bio-RAD microplate reader, Japan). The half maximal inhibitory concentration (IC50%) of the platinum complexes treated with the cell lines were calculated from the curve of the percentage of cell viability versus different concentration of the platinum complex using GraphPad Prism 9.44
Flow cytometry analysis of the cell cycle of HepG2 treated with platinum complexes
Propidium iodide (PI) is a DNA binder, utilized in DNA staining; it is used in quantifying cellular aggregation in each phase of the cell cycle by flow cytometry.45 Around 1 × 106 HepG2 cells were seeded into a 6-well plate for 24 h. The medium was treated with Pt-OEt and Pt-NEt2 at IC50 concentrations. After 24 h incubation time, treated HepG2 cells were collected by adding 0.5 mL 0.25% trypsin and the activity of trypsin was terminated by adding 0.5 mL complete medium to each well. After that, centrifugation of the suspended cells for 5 min at 1500 rpm was followed by washing the cells twice with PBS. The cells were fixed with 1 mL of 70% ethanol for 4 h at −20 °C, and then 100 μL of suspended cells with cold PBS with RNase A was stained with PI solution (50 mg per mL PI) and incubated in the dark for 1 h. All stained cells were screened using a flow cytometer (Applied Bio-system, USA).
Results and discussion
Synthesis and characterization
Schiff bases are typically prepared from condensation reactions between a primary amine and an aldehyde in an organic solvent. The reaction involves a dehydration process from the hemiaminal intermediate, and is acid-catalyzed and reversible. The reaction is pH-sensitive; the formation rate is generally maximized at ca. pH 5, and decreases (or stops completely) at higher and lower pH. In our case, the Schiff base reaction of 2-picolylamine with different salicylaldehyde derivatives in dry polar solvents (ethanol and 2-propanol) was not successful, even at reflux. We also tried to run the reaction in the presence of anhydrous magnesium sulfate or molecular sieves, in both cases without success. This lack of success may be due to the ability of pyridine to stabilize the intermediate by hydrogen bonding, disfavoring dehydration. However, when we changed to nonpolar solvents, we obtained good results: mixing the two starting materials in hexane resulted in precipitation of the desired ligand almost immediately (Fig. 2). The ligands were characterized by 1H NMR, with the disappearance of the aldehyde signals around 10 ppm and the appearance of the imine signals in the range 8.4–8.8 ppm indicating reaction progression. The 1H NMR spectra for the ligands contain three distinctive groups of resonances: O–H in the range 10–13 ppm, aromatic protons in the range 6–9.5 ppm, and aliphatic protons in the range 1–5 ppm. Attaching the ligands to the platinum centres produce complexes with 1H NMR spectra matching the appearance of those of the ligands (but slightly shifted), except with the loss of the phenolic proton signal due to the formation of the O–Pt bonds (note that Pt-OH contains one OH group oriented away from the coordination sphere). The resonances of the CHN and CH2N groups clearly shift following coordination of the nitrogen to the platinum. Mass spectra of all ligands and complexes exhibit [M]+ or [M + H]+ except Pt-OH which shows [M–Cl + MeCN]+ as the major peak in the mass spectrum.
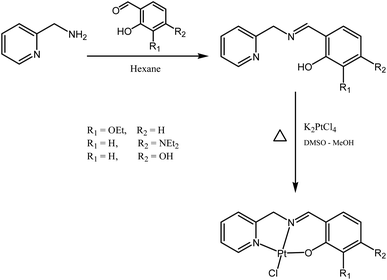 |
| | Fig. 2 Syntheses of the ligands and the Pt(II) complexes. | |
Crystal structures of L-OEt and Pt-OEt
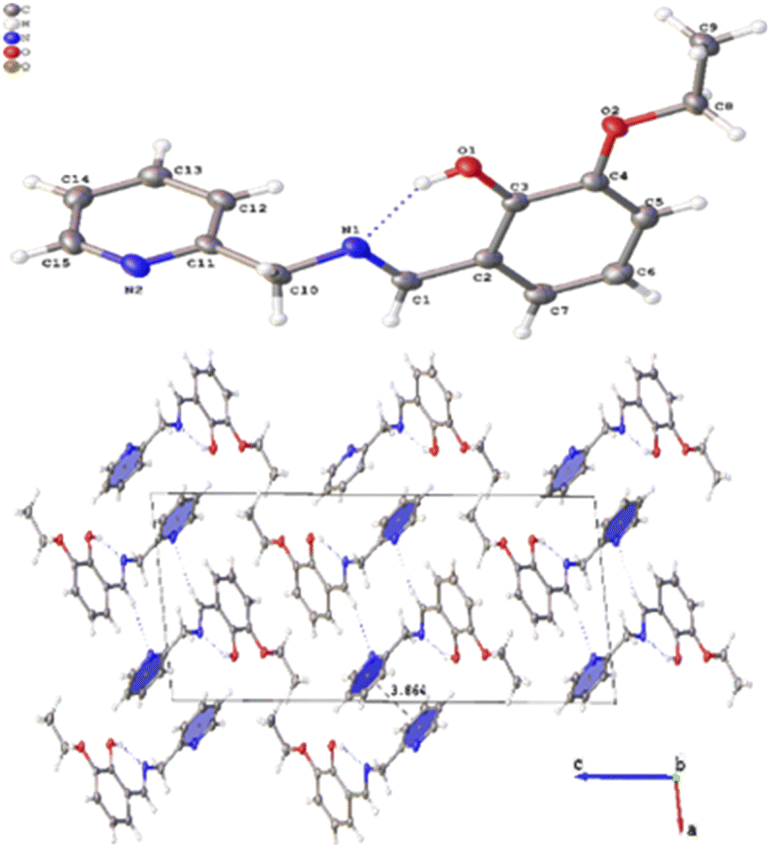
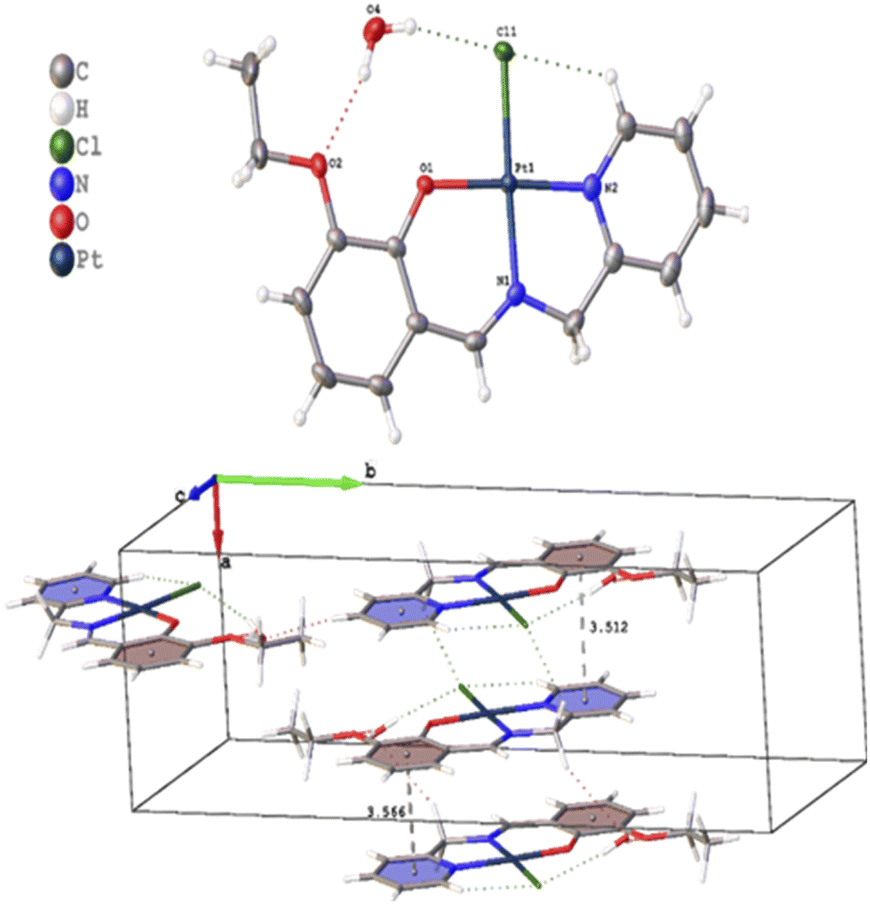
Several crystals of L-OEt ligand were measured using single crystal XRD method. Unfortunately, the data quality was inferior. Nevertheless, we were able to get a reliable structural model of the L-OEt ligand from which some structural information could be deduced (Fig. 3). Intramolecular hydrogen bonding is observed between OH and the nitrogen of the Schiff base. Two different types of intermolecular interactions are responsible for the crystal packing: (a) π–π stacking between two pyridyl rings of adjacent molecules, and (b) hydrogen bonding between NC–H and the pyridyl nitrogen. Other structural data are available in the ESI.† The structure of Pt-OEt is illustrated in Fig. 4 and shows a distorted square planar geometry at the platinum(II) center due to the relatively large N1–Pt–O bond angle (94.69(8)°), resulting in angles of 82.73(9)°, 95.05(7)°, and 87.55(5)° for N1–Pt–N2, N2–Pt–Cl, and Cl–Pt–O, respectively. The bond lengths around the Schiff base nitrogen are 1.286(3) Å (for CN) and 1.471(3) (for N–CH2) (Table 2). The Pt–N bond lengths are 1.954(2) Å (for the Schiff base) and 2.003(2) Å (for the pyridyl), consistent with slightly better electron donation from the Schiff base nitrogen (Table 2). The Pt–Cl bond length is 2.3279(6) Å which is in the range of similar platinum complexes (e.g. the Pt–Cl length is 2.296(2) Å for [Pt(terpy)Cl]Cl·2H2O).46 The crystal packing shows that the aromatic rings are involved in π–π stacking while the water molecules, ethoxy groups and the chlorido ligands are engaged in extensive hydrogen bonding (Fig. 4).
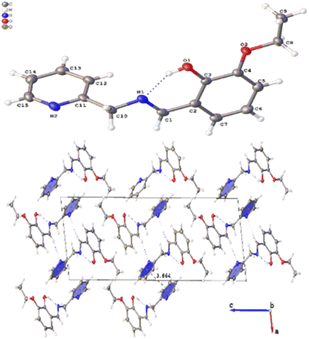 |
| | Fig. 3 Crystal structure of L-OEt showing the intramolecular H-bonding and the crystal packing of the molecule. | |
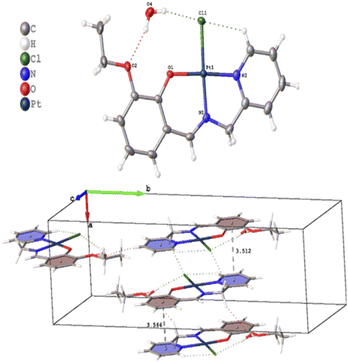 |
| | Fig. 4 Crystal structure and crystal packing of Pt-OEt·H2O. | |
Table 2 Selected bond lengths and angles for Pt-OEt
| Atom |
Atom |
Length/Å |
Atom |
Atom |
Atom |
Angle/° |
| Pt1 |
Cl1 |
2.3279(6) |
O1 |
Pt1 |
Cl1 |
87.55(5) |
| Pt1 |
O1 |
1.9982(17) |
O1 |
Pt1 |
N2 |
177.35(8) |
| Pt1 |
N2 |
2.003(2) |
N2 |
Pt1 |
Cl1 |
95.05(7) |
| Pt1 |
N1 |
1.954(2) |
N1 |
Pt1 |
Cl1 |
177.50(6) |
| C1 |
N1 |
1.286(3) |
N1 |
Pt1 |
O1 |
94.69(8) |
| C10 |
N1 |
1.471(3) |
N1 |
Pt1 |
N2 |
82.73(9) |
| |
|
|
C1 |
N1 |
Pt1 |
124.78(18) |
| |
|
|
C15 |
N2 |
Pt1 |
125.80(18) |
Solvolysis
Chlorido/aqua ligand exchange is believed to be the critical step in activating cisplatin as well as several other platinum(II) and ruthenium(II) arene anticancer agents, the aquation step being followed by covalent DNA-binding.47 The chlorido ligand should not be highly labile, or the anticancer agent is activated in the bloodstream before it reaches its target. On the other hand, it should not be completely inert, or the complex is inactive at the nuclear DNA.48 With this consideration in mind, it is of interest to correlate the rate of the solvent/chlorido process of the new platinum complexes against their anticancer profile. Solvolysis of the neutral platinum complexes affords cationic platinum species and free chloride in solution, and hence the electrical conductivity increases over time. In the present work we have only measured Pt-OEt because its dissociation involves one step; the other two complexes possess OH or NEt2 functional groups, which behave as weak electrolytes themselves, and hence their conductivity cannot be directly linked to the chlorido/solvent exchange process. The variation in Pt-OEt conductivity with time was monitored and found to plateau at 150–180 minutes, which emphasizes the low lability of the system (Fig. 5). Addition of 5% and 20% water to the DMSO caused a slight decrease in the conductivity, which reflects the fact that the chlorido/DMSO exchange is more favorable than the chlorido/aqua exchange. The platinum complex shows low dissociation in DMSO, which can be related to the rate of the chlorido/solvent exchange through the equation for a weak electrolyte derived from Ostwald's law of dilution (eqn (3)):| |
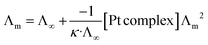 | (3) |
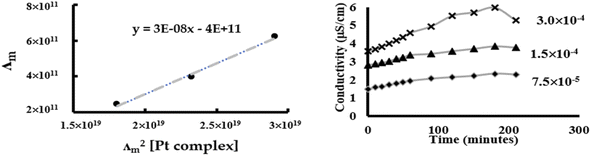 |
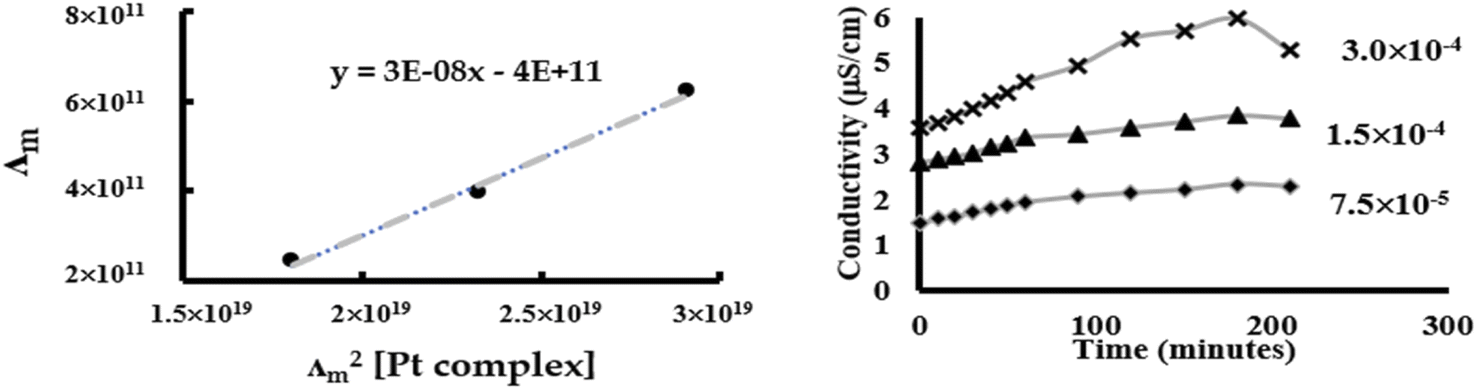
| | Fig. 5 Conductivity measurements of Pt-OEt to quantify chlorido/DMSO ligand exchange. | |
From eqn (3), a linear relationship exists between 1/Λm and Λm [Pt complex], where Λm is the molar conductivity in μS cm−1 (Fig. 5). Ostwald's law of dilution therefore affords the molar conductivity at infinite dilution (Λ∞) from the intersection point of the line with the Y axis, and the dissociation constant (κ) by dividing the square of Λ∞ by the slope of the line (Fig. 5). The value of κ (dissociation rate) obtained for Pt-OEt is 8.33 × 10−5.
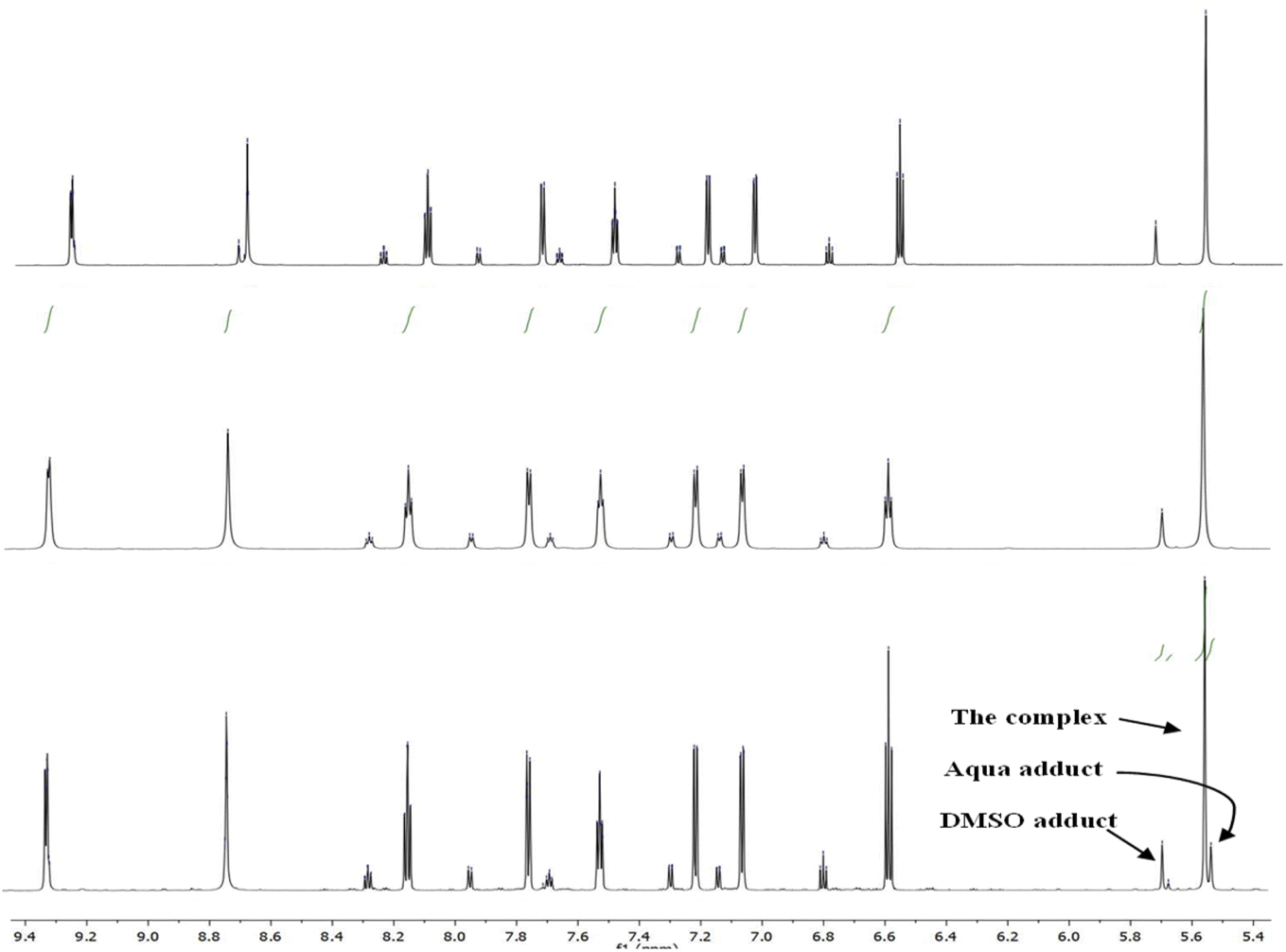
1H NMR data for Pt-OEt and Pt-NEt2 were collected in DMSO after 15 and 30 min and 24 h, showing a gradual increase in the formation of the Pt-DMSO adduct After 24 h, the ratios of the adduct to the complex were (25:75) for Pt-OEt and (35:65) for Pt-NEt2. However, the solvolysis process was affected dramatically when the solvent system was changed to 5% D2O in DMSO. For Pt-NEt2, the observed ratio for the Pt–DMSO adduct against the complex was (20:80); there was no sign of the Pt-aqua adduct in the spectrum. In contrast, the spectrum of Pt-OEt showed the presence of three peaks for the CH2 of the picolyl at 5.53, 5.55 and 5.67 ppm (Fig. 6); their integrations provided the distribution of the species in the solution as 20% (aqua adduct), 65% (the complex), and 15% (DMSO adduct). We conclude that the functional group on the salicyl ring is important in determining the solvent/chlorido exchange outcomes.
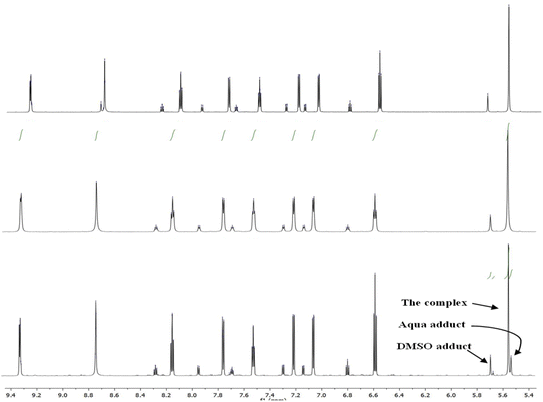 |
| | Fig. 6 1H NMR spectra of Pt-OEt in: DMSO after 24 h (top), DMSO/D2O after 30 min (middle) and DMSO/D2O after 24 h (bottom). | |
DNA-binding studies
While cisplatin covalently binds to DNA, binding studies of [Pt(tpy)Cl]+ have suggested intercalation of the 2,2′:6′,2′′-terpyridine with DNA, followed by covalent bonding between the metal and one of the nucleobases.21–23 In light of the structural similarities of our complexes to [Pt(tpy)Cl]+, we explored their binding with ct-DNA by studying changes in (a) fluorescence behavior of EB–DNA upon addition of platinum(II) complexes, and (b) relative viscosity of ct-DNA upon addition of platinum(II) complexes.
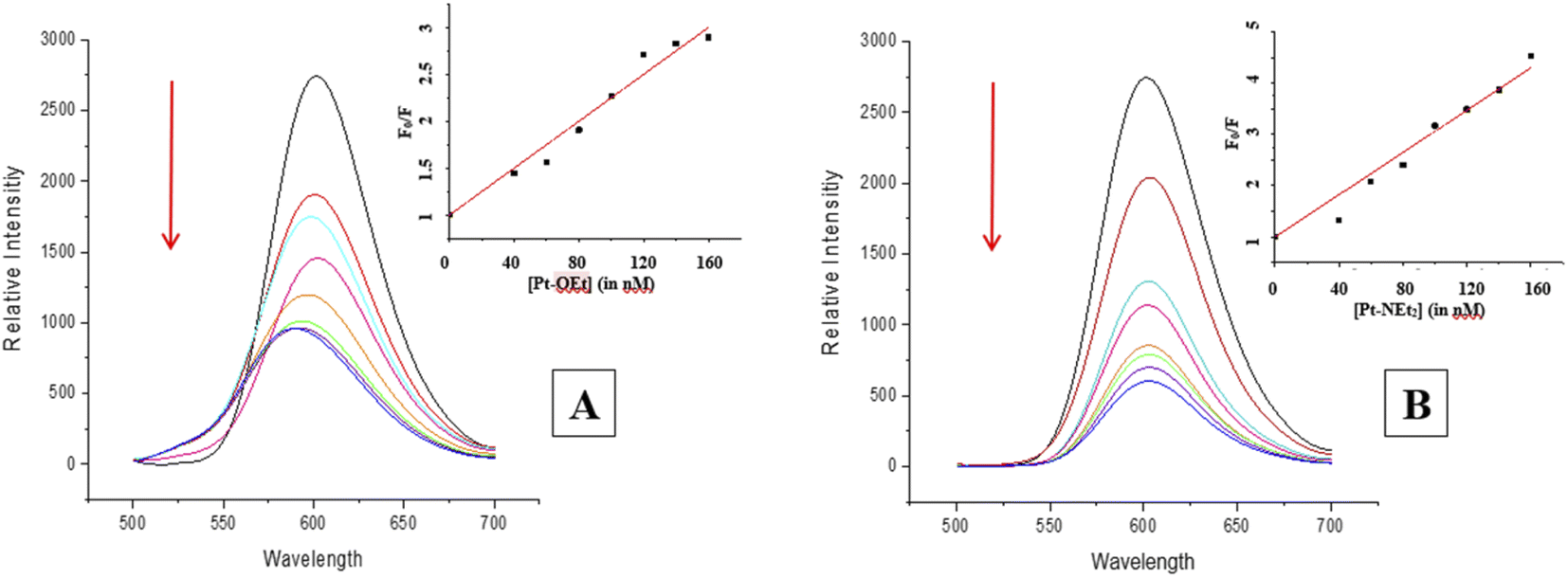
Many intercalators such as ethidium bromide experience a strong enhancement in their fluorescence due to the change in environment (from polar aqueous to hydrophobic between the nucleobases). When EB is intercalated into ct-DNA, a strong emission band at 590 nm is observed.49 The platinum(II) complexes function as quenchers when added, with the EB emission band decreasing due to the destabilization of the EB–DNA adduct as a result of the binding between the platinum(II) complexes and ct-DNA (Fig. 7). All of the complexes in the present study were able to quench the EB–DNA adduct. Stern–Volmer quenching constants (Ksv) were calculated from equation (1) (considering that Ksv ≈ Kapp), the values being listed in Table 3.
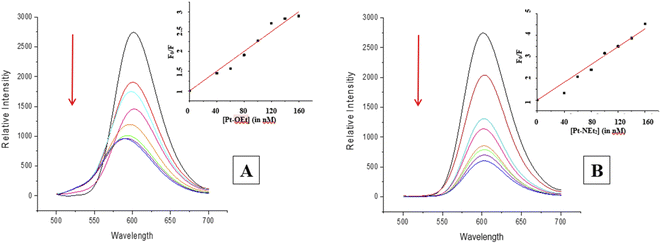 |
| | Fig. 7 Changes in the emission spectrum of the EB–DNA adduct upon increasing the concentration of (A) Pt-OEt and (B) Pt-NEt2. | |
Table 3 Binding constants (Kapp) and binding scores for the platinum complexes
| Compound |
Apparent binding constant (Kapp) |
Binding score |
| For [Pt]–Cl |
For [Pt]–OH2 |
| Pt-OEt |
(1.25 ± 0.40) × 106 |
−5.04 |
−5.25 |
| Pt-NEt2 |
(2.06 ± 0.08) × 106 |
−5.97 |
−5.99 |
| Pt-OH |
(1.54 ± 0.09) × 106 |
−5.18 |
−5.48 |
The values are higher than the apparent binding constant for EB; Pt-NEt2 has the highest apparent binding constant while Pt-OEt has the lowest. The quenching process with Pt-OEt ceased with a distinct blue shift of the emission maximum following addition of 120 nM, in contrast to Pt-OH and Pt-NEt2. We suggest that steric hindrance caused by the ethoxy group results in conformational changes near the EB and hence changes its emission maximum.
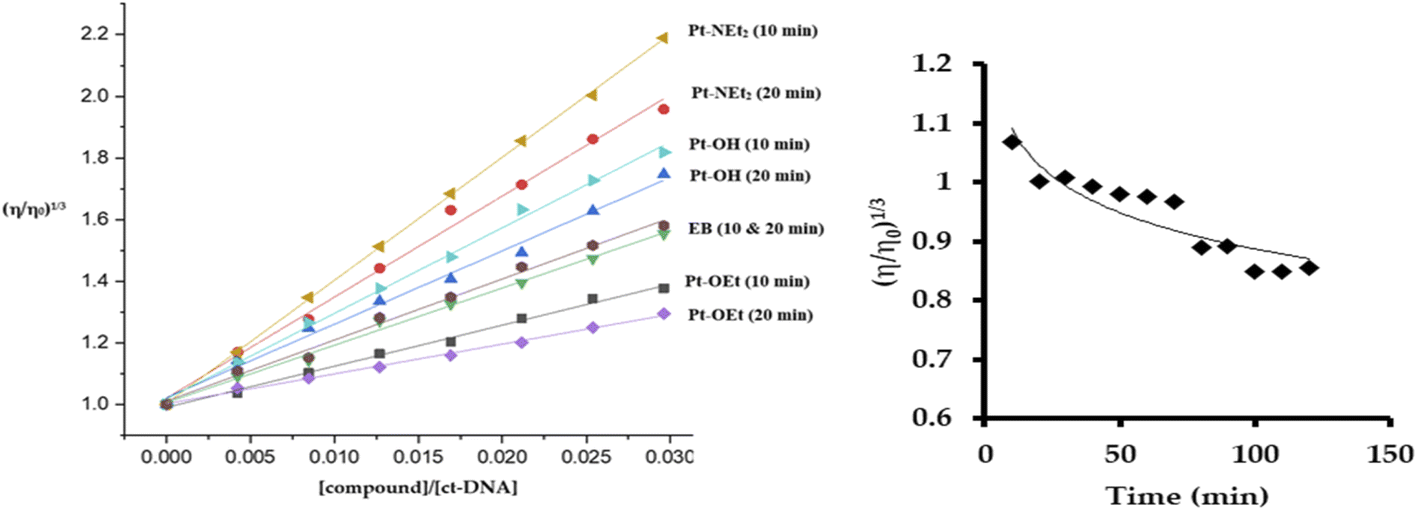
The quenching of emission of the EB–DNA adduct is an indication of the strength of the binding and is sometimes used as an indication of intercalation, but it is not conclusive in that regard. The changes induced in the viscosity of ct-DNA by a molecule can also suggest the type of interaction. Electrostatic- and groove-binders do not affect the relative viscosity of the ct-DNA solution.50,51 In contrast, intercalators produce increases in the relative viscosity due to the elongation of the DNA double strands,51 while covalently bound molecules decrease the relative viscosity by unwinding the DNA double helix.52 Upon treating the ct-DNA solution with increasing concentrations of the platinum(II) complexes, as well as EB as a positive control, strong increases in the relative viscosity of DNA were observed after 10 min of mixing, which supports intercalation as the mode of interaction (Fig. 8). Pt-NEt2 caused the strongest increase in the relative viscosity of ct-DNA while Pt-OEt induced the least impact on the relative viscosity. This trend is consistent with trends observed in the apparent binding constants. Changing the mixing time to 20 min before measuring the changes in the relative viscosity revealed shorter flow times of the solutions compared to 10 min mixing (except in the case of EB). This indicates that, after intercalation, the platinum(II) compounds start to form covalent interactions which counteracts the elongation process of the DNA. Pt-OEt has been chosen as an illustrative example (Fig. 8). The changes in relative viscosity over time shed light on the time frame for covalent interaction; from the measurements, it seems that the compound needs around 90 min to reach the equilibrium of the covalent binding reaction.
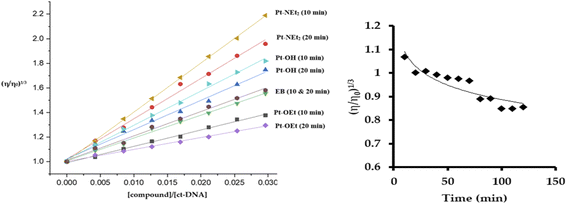 |
| | Fig. 8 Changes in relative viscosity of ct-DNA upon treatment with platinum(II) complexes (left). Change in relative viscosity of ct-DNA upon addition of 10 μM Pt-OEt over 120 min (right). | |
Computational studies
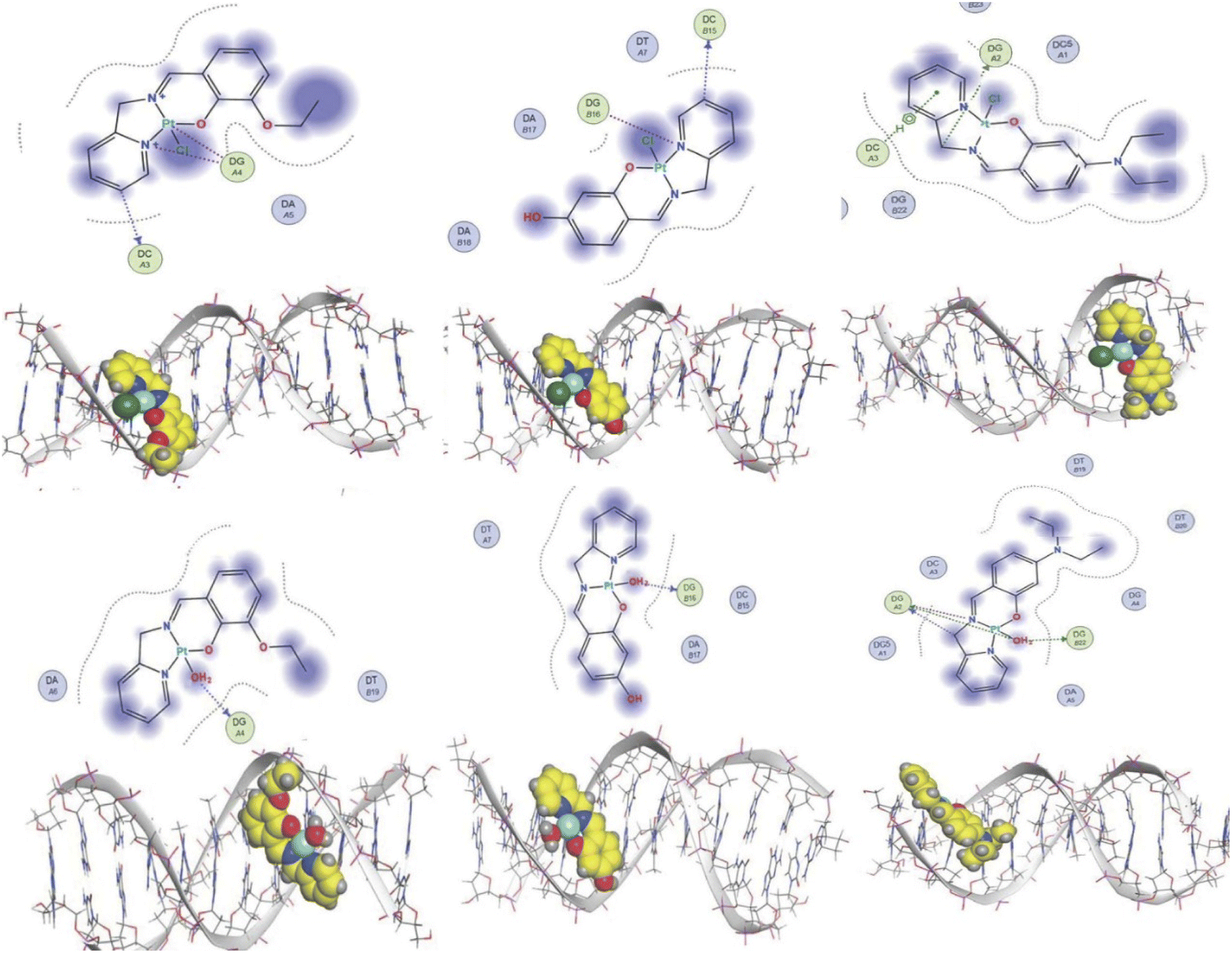
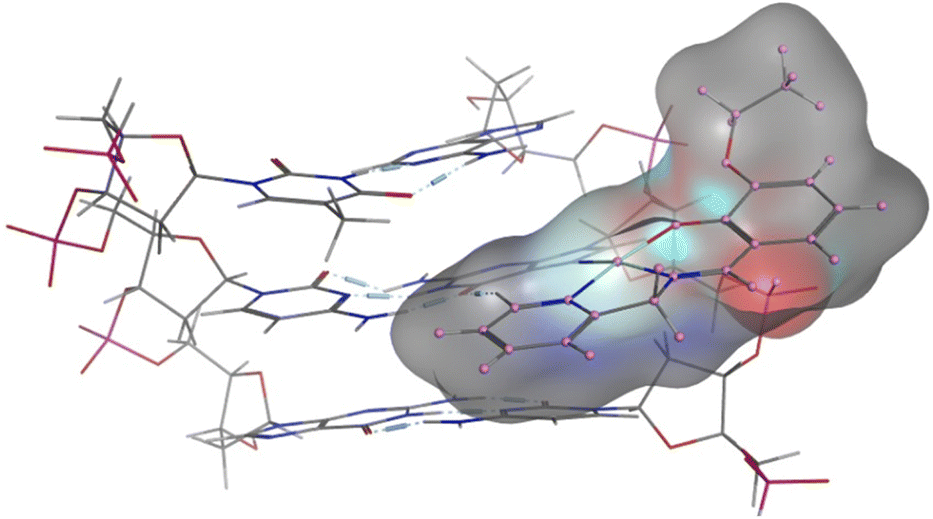
The experimental data suggest that the platinum(II) complex bindings occur over two stages (a fast intercalation followed by a relatively slow covalent bonding). Computational studies were undertaken to study each stage individually. Molecular docking is a strong tool in suggesting the key noncovalent binding modes of drugs with biomolecules. Docking can be explored to confirm experimental results, predict binding sites on the targeted biomolecule, and extract structure–property correlations which are crucial for planning further optimization of new drugs.53 To evaluate the role of the differently functionalized ligands on the platinum–DNA intercalation, the [PtCl(L)] and [Pt(OH2)(L)]+ forms were docked with B-DNA; all complexes were able to insert partially between the nucleobases. Functional groups (3-ethoxy, 4-hydroxy, and 4-diethylamino) clearly alter the electronic and steric properties of the complexes, which causes pronounced differences in their interactions (Fig. 9). The chlorido-containing complexes interact with different nucleobases via H-donor and electrostatic interactions, while the aqua-platinum adducts exhibit H-donor and electrostatic interactions with the nucleobases via the oxygen of the aqua ligand. The aqua adducts have much better binding scores than the chlorido-platinum adducts (Table 3), highlighting the importance of the solvolysis process in improving the targeting of the DNA.
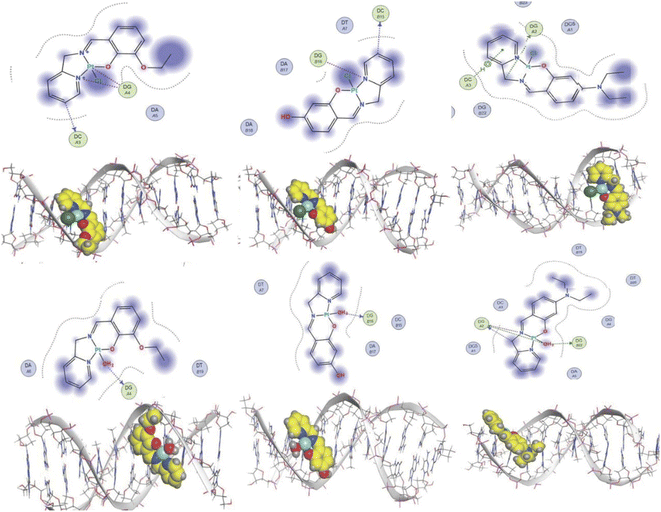 |
| | Fig. 9 2D and 3D structures of [Pt(L)Cl] and [Pt(L)(OH2)]+ docked into B-DNA. | |
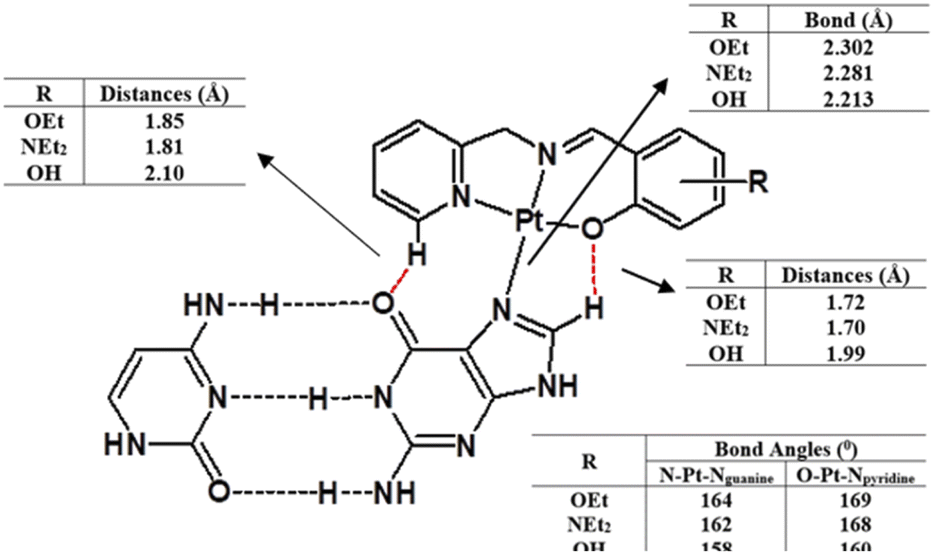
DFT calculations were also undertaken to evaluate the role of the different ligands on the covalent bonding between the platinum(II) and N7 of the guanine, and the resultant changes in the DNA structure. For this purpose, we isolated a trimer from the crystal structure of the B-DNA dodecamer to reduce the computational expense. Platinum complexes were then coordinated to the middle guanine unit. The data suggest that Pt-OH has the shortest Pt–Nguanine bond while Pt-OEt has the longest (Fig. 10). This can be seen as a reflection of the difference in steric hindrance caused by the functional groups. However, hydrogen bonding interactions were observed between the complexes and nucleobases of the DNA, and these are strongly influenced by the functional groups. Electron releasing groups (diethylamino and ethoxy) strengthen the hydrogen bonding interactions, in contrast to the hydroxy group (Fig. 10). The combination of good electron releasing capability with reasonable steric hindrance is the key feature in accessing good DNA binding properties. In Fig. 11, the optimized structure of the B-DNA trimer adduct with Pt-OEt is illustrated. The ability of the ligand in the complex to establish hydrogen interactions affects the inter-strand hydrogen bonding between the guanine and cytosine pair. As a result, the guanine-cytosine pair loses coplanarity, which highlights the strong ability of the complexes to bind with DNA and to induce conformational changes in DNA.
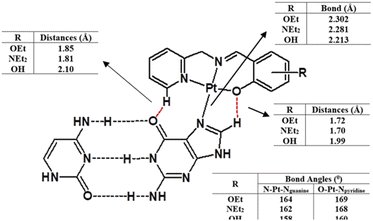 |
| | Fig. 10 Structural differences between the three platinum complexes bound to guanine within the B-DNA trimer, as obtained by DFT calculations. | |
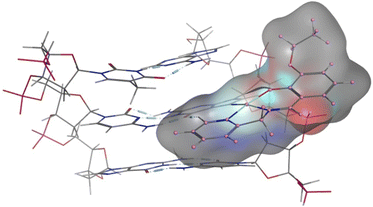 |
| | Fig. 11 Structural differences between the three platinum complexes bound to guanine within the B-DNA trimer, as obtained by DFT calculations. | |
Preliminary in vitro antiproliferative activity
Two cancer cell lines were chosen to examine the anticancer activities of the synthesized compounds. The HepG2 cell line corresponds to human liver carcinoma cells derived from a patient with a hepatocellular carcinoma (HCC), which is the second most common cause of cancer-related deaths worldwide.54 HepG2 cells are non-tumorigenic with high proliferation and are stimulated with human growth hormone. Sorafenib and regorafenib are approved chemotherapeutic agents for HCC, functioning as multikinase inhibitor drugs.55 The metastatic adenocarcinoma breast cancer cell line (MCF7) was chosen because breast cancer is the most common cancer among females worldwide; this cell line has a proliferative response to estrogens.56 Cisplatin or carboplatin are used as treatment for secondary breast cancer, in combination with other chemotherapeutic agents.57 All of the ligands from the present study exhibit weak cytotoxicity towards the two cancer cell lines; the best IC50 values were observed for L-OEt (Table 4). Upon complexation with platinum, strong improvements in IC50 values are seen, which reflects the importance of the metal in the cytotoxic effects (Table 4). The best candidate of the three complexes is Pt-OEt, which exhibits IC50 values 40–70% lower than those of Pt-NEt2 and ca. 80–90% lower than the values observed for Pt-OH; moreover, Pt-OEt has comparable anticancer activities to those observed for cisplatin. Solvolysis is believed to be the activation step to facilitate platinum binding with DNA;2,47 Pt-OEt is capable of exchanging the chlorido ligand for DMSO and aqua ligands. While Pt-OEt has the best anticancer activities of the synthesized complexes, its structural differences make it hard to conclude if the position or the nature of the group is the more dominant factor in its improved cytotoxicity. Pt-NEt2 and Pt-OH have the diethylamino and hydroxy functional groups at the same position; the better cytotoxicity of Pt-NEt2 compared to Pt-OH could be due to one or more of the following factors: (1) the basic nature of the amino group (the hydroxy group of the phenol is weakly acidic), (2) the more lipophilic nature of the amine, induced by the ethyl groups, and/or (3) the lower capability of the nitrogen toward hydrogen bonding (compared to the hydroxy).
Table 4 Anticancer activities of the ligands and their platinum(II) complexes against two cancer cell lines
| Compound |
IC50 (μM) |
| HepG II |
MCF-7 |
| L-OEt |
66.60 ± 9.03 |
79.60 ± 7.54 |
| Pt-OEt |
3.39 ± 0.34 |
6.45 ± 0.35 |
| L-NEt2 |
61.27 ± 5.00 |
102.00 ± 8.32 |
| Pt-NEt2 |
12.53 ± 1.19 |
10.65 ± 1.01 |
| L-OH |
112.30 ± 14.34 |
162.00 ± 6.94 |
| Pt-OH |
23.20 ± 5.25 |
36.00 ± 4.28 |
| Cisplatin |
5.94 ± 1.09 |
5.53 ± 0.61 |
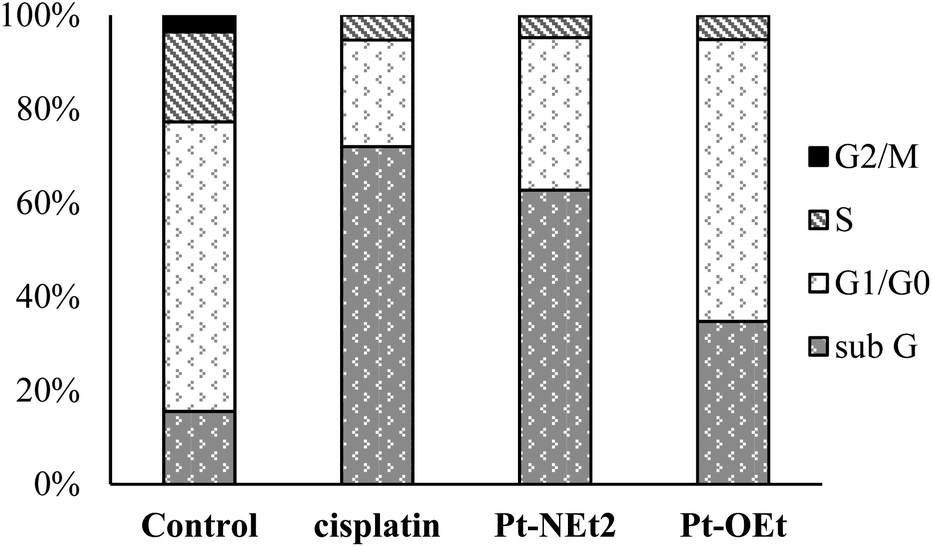
Cell cycle phase analysis of HepG2 treated with Pt-OEt and Pt-NEt2
To explore the Pt-OEt and Pt-NEt2 interference with cell cycle progression, flow cytometry analysis of cell cycle phase distribution was examined in HepG2 cells using PI staining. The impacts of the two complexes and cisplatin were analysed at concentrations around the IC50 values for 24 h. Compared with control cells, the percentages of HepG2 cells at different cell cycle phases were modified, but in different ways, upon treatment with cisplatin, Pt-OEt or Pt-NEt2 (Fig. 12). After 24 h treatment, cisplatin caused an increase in the cell population in the sub-G phase (from 15.5% to 71.9%) with concomitant decreases in the cell population in G1/G0, S and G2/M phases, suggesting the activation of an apoptotic cell death pathway. The cell cycle assay of HepG2 treated with Pt-NEt2 resembles that of HepG2 treated with cisplatin, but with different populations in the sub-G (62.7%) and G1/G0 (32.5%) phases. However, treating HepG2 cells with Pt-OEt produced a moderate increase in the cell population in the sub-G phase, and with almost no change in the cell population in G1/G0 compared to the untreated cells. Moreover, decreases in the cell population were observed in the S and G2/M phases. The data suggest that the complexes activate cell death by an apoptotic pathway. Cisplatin produces inter- and intra-strand crosslinked DNA adducts; the cytotoxicity is facilitated by propagation of DNA damage, involving ATR, p53, p73, and mitogen-activated protein kinases. The DNA damage results in apoptotic cell death.58 However, our complexes only form mono-adducts with DNA, resulting in conformational changes in the DNA. The functional groups (3-ethoxy, 4-diethylamino and 4-hydroxy) cause different levels of conformational changes and activate cell death in ways that differ from that of cisplatin.
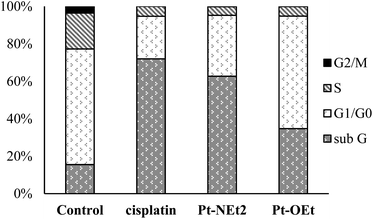 |
| | Fig. 12 Cell cycle assay of HepG2 upon treatment with different compounds. | |
Conclusion
In this work, three tridentate Schiff base ligands were synthesized from the reactions of 2-picolylamine and salicylaldehyde derivatives. The ligands reacted with K2PtCl4 in DMSO/MeOH mixtures to produce complexes of general formula Pt(N^N^O)Cl. The DMSO/chlorido exchange process of Pt-OEt was investigated by monitoring the change in conductivity; the process is very slow, with the increase in conductivity plateauing after ca. 150 min. The platinum complex shows slow dissociation in DMSO, with a dissociation constant (κ) of 8.33 × 10−5. Solvolysis of Pt-OEt and Pt-NEt2 were investigated by NMR spectroscopy in DMSO, showing the formation of Pt-DMSO adducts with ratios (25:75) for Pt-OEt and (35:65) for Pt-NEt2. The addition of D2O to a DMSO solution of Pt-NEt2 decreased the formation of the Pt–DMSO adduct slightly. In contrast, Pt-OEt was able to form an aqua adduct and a DMSO adduct. The binding of our complexes with ct-DNA was investigated from their fluorescence-quenching of the EB–DNA adduct and from the changes in relative viscosity of ct-DNA upon addition of the complexes. All complexes quenched EB–DNA effectively, Pt-NEt2 exhibiting the largest apparent binding constant. Pt-OEt behaves slightly differently from the other two complexes in causing a blue shift in the emission maximum of the EB–DNA adduct, which suggests conformational changes in the ct-DNA. The intercalation mode of interaction with DNA was suggested by molecular docking studies, and this is supported by the increase in the relative viscosity of ct-DNA with increasing concentrations of the platinum(II) complexes. However, gradual decreases in the relative viscosities in the presence of platinum(II) complexes over time were observed, indicating that the binding mode proceeds to involve covalent binding. Anticancer activities of the ligands and their platinum(II) complexes were examined against human liver carcinoma cells (HepG2) and the metastatic adenocarcinoma breast cancer cell line (MCF7). All the ligands exhibit weak cytotoxicity towards the two cancer cell lines; the best IC50 values are observed for L-OEt. The platinum(II) complexes possess much better cytotoxicity than their ligands. Among the platinum(II) complexes, Pt-OEt shows the best IC50 against both cell lines, its cytotoxicity being comparable to that observed for cisplatin. The presence of more than one structural variable prevents the extraction of structure–property relationships at this stage. Cell cycle arrest in the HepG2 cell line upon treatment with Pt-OEt and Pt-NEt2 was investigated, the results being compared to that of cisplatin. After 24 h treatment, cisplatin caused an increase in the cell population in the sub-G phase with concomitant decreases in the cell population in the G1/G0, S, and G2/M phases. The cell cycle assay of HepG2 treated with Pt-NEt2 resembles that of HepG2 treated with cisplatin, but with different populations in the sub-G (62.7%) and G1/G0 (32.5%) phases. However, treating the HepG2 cells with Pt-OEt caused a moderate increase in the cell population in the sub-G0/G1 phase with decreases in the cell populations of the S and G2/M phases. The data suggest that the complexes activate cell death by an apoptotic pathway. The major difference between cisplatin and the two new complexes is the ability of cisplatin to produce inter- and intra-strand crosslinked DNA adducts, while our complexes only form mono-adducts with DNA. Binding of the complexes with the DNA results in conformational changes in the DNA, leading to apoptotic cell death. The functional groups (3-ethoxy, 4-diethylamino and 4-hydroxy) cause different levels of conformational changes and activate cell death in ways that differ from that of cisplatin. The optimized structures of the B-DNA trimer adduct with the different platinum complexes confirms the ability of the ligand in the complex to establish hydrogen interactions, affecting the inter-strand hydrogen bonding between the guanine and cytosine pair. As a result, the guanine-cytosine pair loses coplanarity, which highlights the strong ability of the complexes to bind with DNA and to induce conformational changes. In conclusion, the current study illustrates the importance of the functional groups on the ligands in tuning their DNA binding/anticancer properties; this new class of platinum(II) complexes with highly flexible tridentate ligands are promising tunable anticancer candidates.
Author contributions
Conceptualization, BAB; data curation, KSA; formal analysis, KSA, EMMA, BD, A-HME, MAA and MJ; funding acquisition, BAB; investigation, KSA and BAB; methodology, KSA, AJ, MAH and A-HME; project administration, BAB; resources, BAB; software, AJ and MAH; supervision, BAB; visualization, KSA and BAB; writing–original draft, BAB and MGH; writing–review & editing, MGH.
Conflicts of interest
The authors declare no conflicts of interest in this publication.
Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, Saudi Arabia, under grant no. (KEP-PhD: 36-130-1443). The authors, therefore, acknowledge with thanks DSR technical and financial support. Computation for the work described in this paper was supported by King Abdulaziz University's High-Performance Computing Center (Aziz Supercomputer).
References
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- D. P. Bancroft, C. A. Lepre and S. J. Lippard, Platinum-195 NMR kinetic and mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to DNA, J. Am. Chem. Soc., 1990, 112, 6860–6871 CrossRef CAS.
- A. M. Fichtinger-Schepman, J. L. van der Veer, J. H. den Hartog, P. H. Lohman and J. Reedijk, Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation, Biochemistry, 1985, 24, 707–713 CrossRef CAS PubMed.
- U.-M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins, Nature, 1999, 399, 708–712 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Direct cellular responses to platinum-induced DNA damage, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- P. J. Sadler and Z. Guo, Metal complexes in medicine: design and mechanism of action, Pure Appl. Chem., 1998, 70, 863–871 CrossRef CAS.
- M. D. Hall, M. Okabe, D.-W. Shen, X.-J. Liang and M. M. Gottesman, The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 495–535 CrossRef CAS PubMed.
- H. Timmer-Bosscha, N. H. Mulder and E. G. de Vries, Modulation of cis-diamminedichloroplatinum(II) resistance: a review, Br. J. Cancer, 1992, 66, 227–238 CrossRef CAS PubMed.
- D.-W. Shen, L. M. Pouliot, M. D. Hall and M. M. Gottesman, Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes, Pharmacol. Rev., 2012, 64, 706–721 CrossRef CAS PubMed.
- M. M. Gottesman, Mechanisms of cancer drug resistance, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Platinum coordination compounds with potent anticancer activity, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- M. M. Regan, E. K. O'Donnell, W. K. Kelly, S. Halabi, W. Berry, S. Urakami, N. Kikuno and W. K. Oh, Efficacy of carboplatin-taxane combinations in the management of castration-resistant prostate cancer: a pooled analysis of seven prospective clinical trials, Ann. Oncol., 2010, 21, 312–318 CrossRef CAS PubMed.
- S. P. Fricker, Metal based drugs: from serendipity to design, Dalton Trans., 2007, 43, 4903–4917 RSC.
- J. T. Hartmann and H.-P. Lipp, Toxicity of platinum compounds, Expert Opin. Pharmacother., 2005, 4, 889–901 CrossRef PubMed.
- F. Lévi, G. Metzger, C. Massari and G. Milano, Oxaliplatin: pharmacokinetics and chronopharmacological aspects, Clin. Pharmacokinet., 2000, 38, 1–21 CrossRef PubMed.
- Ž. D. Bugarčić, F. W. Heinemann and R. van Eldik, Substitution reactions of [Pt(terpy)X]2+ with some biologically relevant ligands. Synthesis and crystal structure of [Pt(terpy)(cyst-S)](ClO4)2·0.5H2O and [Pt(terpy)(guo-N7)](ClO4)2·0.5guo·1.5H2O, Dalton Trans., 2004, 2, 279–286 RSC.
- D. Petrović, B. Stojimirović, B. Petrović, Z. M. Bugarčić and Ž. D. Bugarčić, Studies of interactions between platinum(II) complexes and some biologically relevant molecules, Bioorg. Med. Chem., 2007, 15, 4203–4211 CrossRef PubMed.
- C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Single-stranded nucleic acid-induced helical self-assembly of alkynylplatinum(II) terpyridyl complexes, Proc. Natl. Acad. Sci., 2006, 103, 19652–19657 CrossRef CAS PubMed.
- S. D. Cummings, Platinum complexes of terpyridine: Interaction and reactivity with biomolecules, Coord. Chem. Rev., 2009, 253, 1495–1516 CrossRef CAS.
- P. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Metal complex interactions with DNA, Dalton Trans., 2015, 44, 3505–3526 RSC.
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Baueri, Metallointercalation Reagents. 2-Hydroxyethanethiolato(2,2′,2′′-terpyridine)-platinum(I1) Monocation Binds Strongly to DNA By Intercalation, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839–3843 CrossRef CAS PubMed.
- M. Howe-Grant, K. C. Wu, W. R. Bauer and S. J. Lippard, Binding of Platinum and Palladium Metallointercalation Reagents and Antitumor Drugs to Closed and Open DNAs, Biochemistry, 1976, 15, 4339–4346 CrossRef CAS PubMed.
- P. J. Bond, R. Langridge, K. W. Jennette and S. J. Lippard, X-ray fiber diffraction evidence for neighbor exclusion binding of a platinum metallointercalation reagent to DNA, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4825–4829 CrossRef CAS PubMed.
- K. Becker, C. Herold-Mende, J. J. Park, G. Lowe and R. H. Schirmer, Human thioredoxin reductase is efficiently inhibited by (2,2':6',2'-terpyridine)platinum(II) complexes. Possible implications for a novel antitumor strategy, J. Med. Chem., 2001, 44, 2784–2792 CrossRef CAS PubMed.
- G. Lowe, A. S. Droz, T. Vilaivan, G. W. Weaver, J. J. Park, J. M. Pratt, L. Tweedale and L. R. Kelland, Cytotoxicity of 2,2':6',2'-terpyridineplatinum(II) complexes against human ovarian carcinoma, J. Med. Chem., 1999, 42, 3167–3174 CrossRef CAS PubMed.
- J. M. Saborit, A. Caubet, R. F. Brissos, L. Korrodi-Gregorio, R. Perez-Tomas, M. Martinez and P. Gamez, pH-Driven preparation of two related platinum(II) complexes exhibiting distinct cytotoxic properties, Dalton Trans., 2017, 46, 11214–11222 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. S. Devi, B. Thulasiram, S. Satyanarayana and P. Nagababu, Analytical techniques used to detect DNA binding modes of ruthenium(II) complexes with extended phenanthroline ring, J. Fluoresc., 2017, 27, 2119–2130 CrossRef CAS PubMed.
- W. Villarreal, L. Colina-Vegas, G. Visbal, O. Corona, R. S. Corrêa, J. Ellena, M. R. Cominetti, A. A. Batista and M. Navarro, Copper(I)–phosphine polypyridyl complexes: synthesis, characterization, DNA/HSA binding study, and antiproliferative activity, Inorg. Chem., 2017, 56, 3781–3793 CrossRef CAS PubMed.
- A. Erxleben, Investigation of non-covalent interactions of metal complexes with DNA in cell-free systems, Chimia, 2017, 71, 102–111 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, GC base sequence recognition by oligoimidazolecarboxamide and C-terminus-modified analogs of distamycin deduced from circular dichroism, proton nuclear magnetic resonance, and methidiumpropylethylenediaminetetraacetate-iron(II) footprinting studies, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- S. Alsaedi, B. A. Babgi, M. H. Abdellattif, M. N. Arshad, A.-H. M. Emwas, M. Jaremko, M. G. Humphrey, A. M. Asiri and M. A. Hussien, DNA-Binding and cytotoxicity of copper(I) complexes containing functionalized dipyridylphenazine ligands, Pharmaceutics, 2021, 13, 764–777 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- R. Dennington, R. T. Keith and J. Millam, Gauss View, Version 5, vol Shawnee Mission, Semichem, Inc., 2009 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schaefer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian-basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian-basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the rare earth elements, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- E. M. Ali, A. A. Elashkar, H. Y. El-Kassas and E. I. Salim, Methotrexate loaded on magnetite iron nanoparticles coated with chitosan: Biosynthesis, characterization, and impact on human breast cancer MCF-7 cell line, Int. J. Biol. Macromol., 2018, 120, 1170–1180 CrossRef CAS PubMed.
- J. A. Plumb, Cell sensitivity assays: the MTT assay, Cancer cell culture, Springer, 2004, pp. 165–169 Search PubMed.
- J. Fried, A. G. Perez and B. D. Clarkson, Flow cytofluorometric analysis of cell cycle distributions using propidium iodide. Properties of the method and mathematical analysis of the data, J. Cell Biol., 1976, 71, 172–181 CrossRef CAS PubMed.
- A. Şengül, Crystal and molecular structure of the yellow form of Chloro(2,2':6',2''-terpyridine)platinum (II)chloride dihydrate, [Pt(terpy)Cl]Cl·2H2O, Turk. J. Chem., 2004, 28, 667–672 Search PubMed.
- J. A. Platts, M. Ravera, E. Gabano, M. Sardi, S. Bianco and D. Osella, Solvolysis of a series of cisplatin-like complexes – Comparison between DNA-biosensor and conductivity data, Eur. J. Inorg. Chem., 2012, 2012, 5625–5631 CrossRef CAS.
- T. Lazarevic, A. Rilak and Z. D. Bugarcic, Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef CAS PubMed.
- F. J. Meyer-Almes and D. Porschke, Mechanism of intercalation into the DNA double helix by ethidium, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS.
- R. Kalantari and Z. Asadi, DNA/BSA binding of a new oxovanadium (IV) complex of glycylglycine derivative Schiff base ligand, J. Mol. Struct., 2020, 1219, 128664 CrossRef CAS.
- Z. Seferoğlu, M. M. A. Mahmoud and H. Ihmels, Studies of the binding interactions of dicationic styrylimidazo[1,2-a]pyridinium dyes with duplex and quadruplex DNA, Dyes Pigm., 2016, 125, 241–248 CrossRef.
- N. Busto, M. Martínez-Alonso, J. M. Leal, A. M. Rodríguez, F. Domínguez, M. I. Acuña, G. Espino and B. García, Monomer–dimer divergent behavior toward DNA in a half-sandwich ruthenium(II) aqua complex. Antiproliferative biphasic activity, Organometallics, 2015, 34, 319–327 CrossRef CAS.
- G. M. Morris and M. Lim-Wilby, Molecular docking, Methods Mol. Biol., 2008, 443, 365–382 CrossRef CAS PubMed.
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012, Int. J. Cancer, 2015, 136, 359–386 CrossRef.
- M. Ikeda, C. Morizane, M. Ueno, T. Okusaka, H. Ishii and J. Furuse, Chemotherapy for hepatocellular carcinoma: current status and future perspectives, Jpn. J. Clin. Oncol., 2018, 48, 103–114 CrossRef PubMed.
- A. S. Levenson and V. C. Jordan, MCF-7: the first hormone-responsive breast cancer cell line, Cancer Res., 1997, 57, 3071–3078 CAS.
- P. Prabhakaran, F. Hassiotou, P. Blancafort and L. Filgueira, Cisplatin induces differentiation of breast cancer cells, Front. Oncol., 2013, 3, 134–143 Search PubMed.
- S. Tanida, T. Mizoshita, K. Ozeki, H. Tsukamoto, T. Kamiya, H. Kataoka, D. Sakamuro and T. Joh, Mechanisms of cisplatin-induced apoptosis and of cisplatin sensitivity: potential of BIN1 to act as a potent predictor of cisplatin sensitivity in gastric cancer treatment, Int. J. Surg. Oncol., 2012, 2012, 862879 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ehab M. M. Alibc,
Bambar Davaasurend,
Abdesslem Jedidia,
Abdul-Hamid M. Emwasd,
Maymounah A. Alrayyania,
Mariusz Jaremko
*a,
Ehab M. M. Alibc,
Bambar Davaasurend,
Abdesslem Jedidia,
Abdul-Hamid M. Emwasd,
Maymounah A. Alrayyania,
Mariusz Jaremko