
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bond networks in gas-phase complex anions†
Zhisheng Laia,
Minhui Shena,
Yong Shena,
Yu-Xin Yea,
Fang Zhu a,
Jianqiao Xu*a and
Gangfeng Ouyang*abc
a,
Jianqiao Xu*a and
Gangfeng Ouyang*abc
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry/KLGHEI of Environment and Energy Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: xujq27@mail.sysu.edu.cn; cesoygf@mail.sysu.edu.cn
bCollege of Chemistry, Center of Advanced Analysis and Gene Sequencing, Zhengzhou University, Zhengzhou, 450001, China
cGuangdong Provincial Key Laboratory of Emergency Testing for Dangerous Chemicals, Guangdong Institute of Analysis (China National Analytical Center Guangzhou), Guangdong Academy of Sciences, Guangzhou, 510070, China
First published on 13th October 2022
Abstract
Hydrogen bond networks (HBNs) have piqued the interest of the scientific community due to their crucial roles in nature. However, HBNs that are isolated from complicated backgrounds for unraveling their characteristics are still scarce. Herein, we propose that HBNs exist in complex anions formed between α-cyclodextrin (α-CD) and four benzoic acids (RBAs) in the gas phase. The complex anions are facilely extracted from solutions via the electrospray ionization technique, and subsequently activated through collision for the investigation of their transition dynamics. It is revealed that the generation of deprotonated α-CD and neutral RBAs is the unexpected dominant dissociation pathway for all the four complex anions, and the complex anions formed from more acidic RBAs exhibit higher stabilities. These dissociation results are successfully explained by the cooperative stretching dynamics of the proposed HBNs that are formed involving the intramolecular HBN of α-CD and the intermolecular hydrogen bonds (HBs) between α-CD and RBAs. Furthermore, the rarely observed low barrier HBs (LBHBs) are suggested to be present in the HBNs. It is believed that the present complex anions can serve as a facilely accessible and informative model for studying HBNs in the future.
1 Introduction
Hydrogen bond networks (HBNs) continue to attract wide-spread attention since their discovery more than one hundred years ago.1 They play eminent roles in determining the functions of receptors,2 enzymes,3 and proton channels,4 and are also responsible for the unexpected high mass transfer rates of protons and hydroxide ions in water.5 However, it remains challenging to elucidate the HBNs in natural systems like proteins and bulk water. Although vibrational spectroscopy has long been used to study the structures and dynamics of the HBNs in bulk water and water–air interphases,6 the diffuse nature of the key absorption bands in water requires application of isotope labelling method or multiple-dimension spectroscopy to resolve the characteristic signatures.7,8 As for the characterization of the HBNs in protein pockets, the widely used X-ray diffraction methods could not visualize protons. The subtle characteristics of HBNs are always predicted highly dependent on the high-resolution distances between two heteroatoms.9 Besides, the X-ray diffraction methods could only detect the steady states other than tracing the dynamic transitions of HBNs.10 Although ultraviolet-visible (UV-vis) spectroscopy is applicable in characterizing proton transfer processes of HBNs in the catalytic cavities of enzymes,11 the background interferences may obstruct the analysis.To get deep insights into HBNs, isolated models at molecular levels have been established, as they are small enough for accurate spectroscopic characterization and quantum calculations.12 Water clusters are the most studied HBN models. Remarkable progress has been made in resolving their geometries and cooperative dynamics.12–14 And they have thrown valuable light upon many-body physics.13,14 However, equivalent HBN models that are facilely accessible are still extremely scarce. Some putative models seem difficult to be studied in any experiment frames, as they may not be maintained after being isolated from the surrounding environment.15 On the other hand, the studies focused on single hydrogen bonds (HBs) are much more abundant.16–18 For example, it is pioneered by Jiang and co-authors using scanning tunneling microscope (STM) to reveal the nuclear quantum effects (NQEs) embedded in HBs.19 Nonetheless, unraveling the cooperative characteristics of HBNs is still one of the upper-most interests among the scientific community.
The scientific community has long been conscious that the hydroxyl groups fixed on the primary rim of a single cyclodextrin (CD) molecule are stereochemically possible to form an intramolecular HBN,20 which is inspiring for seeking isolated HBN models alternative to the water clusters. However, the intramolecular HBNs formed on the primary rims of CD molecules have seldom been studied in depth previously. In fact, the flexible primary hydroxyl groups of CDs prefer to form intermolecular HBs with guest molecules and crystal water in condensed phases.21 Thus, it is only expectable that the large intramolecular HBNs on the primary rims come into being by extracting CD molecules from condensed phases.
Herein, we attach benzoic acids containing different para-substituent groups (RBAs) to α-CD to prepare complex anions in gas phase. The complex anions are activated via collision for the investigation of their transition dynamics. The unexpected dominant collision-induced dissociation (CID) pathways and the stabilities of the complex anions are successfully explained by the adaptive stretching of the intramolecular HBN of α-CD along with the allocation of the bridging protons. What's more, the existence of low barrier HBs (LBHBs) is suggested here. This study provides a new model besides the water clusters for studying HBNs in depth in the future.
2 Methods
2.1 Chemicals and reagents
α-Cyclodextrin (α-CD) was purchased from Macklin Inc. (Shanghai, China). Benzoic acid (denoted as BA, possesses no substitute groups), p-nitrobenzoic acid (pNBA) and p-cynobenzoic acid (pCBA) were purchased from Aladdin Industrial Corporation (Shanghai, China). p-methylbenzoic acid (pMBA) was purchased from Tokyo Chemical Industry Co. Ltd (Tokyo, Japan). HPLC grade methanol was purchased from Thermo Fisher Scientific Ltd (Shanghai, China). Ultrapure water was prepared from a water purification system (Millipore Direct-Q 3UV, Merck, Germany).2.2 Sample preparation
Each benzoic acid together with α-CD was prepared into a sample solution by using the mixed solvent of methanol and water (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The concentrations of the RBA and α-CD were 10 μM in the sample solutions.
:1). The concentrations of the RBA and α-CD were 10 μM in the sample solutions.
2.3 Collision induced dissociation of cluster anions
All the instrumental analysis was run on a triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, MA, USA). An electrospray ionization (ESI) source was used in the triple quadrupole mass spectrometer. Tandem mass spectrometry was performed with nitrogen gas as the collision gas.The sample solutions were injected by a syringe pump at a flow rate of 5 μL min−1. The complex anions were formed in the ESI source under negative mode. For the study of the dissociation manners of the complex anions, the parameters determining the abundances of the [α-CD + RBA–H]−, i.e. the declustering potential and the entrance potential, were first optimized. Then, the target complex anions were selected and dissociated under different collision energies. The abundances of the survived parent ions and the daughter ions were subsequently monitored. The kinetic energies of the complex anions in the mass spectrometer were transformed into collision energies in the center-of-mass frame, as shown in eqn (1),22
| Ecm = Ek[m/(m + M)] | (1) |
2.4 Computation details
The docking conformers shown in Fig. 2 were proposed according to our experiments and reasonable assumption. They were optimized using the B3LYP exchange–correlation functional with Grimme's DFT-D3(BJ) empirical dispersion correction and 6-311G** basis set, abbreviated as B3LYP-D3(BJ)/6-311G**.31,32 Subsequently, the counterpoise corrected energies were calculated at the M062X/def2TZVP level of theory with D3 empirical dispersion correction. The Boys and Bernardi's counterpoise (CP) technique was employed to correct the basis set superposition error (BSSE) problem.33,34 These energies of complex anions were used to elucidate the potential energy diagram of two competitive dissociation pathways of complex anion [α-CD + RBA–H]− (Scheme 1 and Table 2).
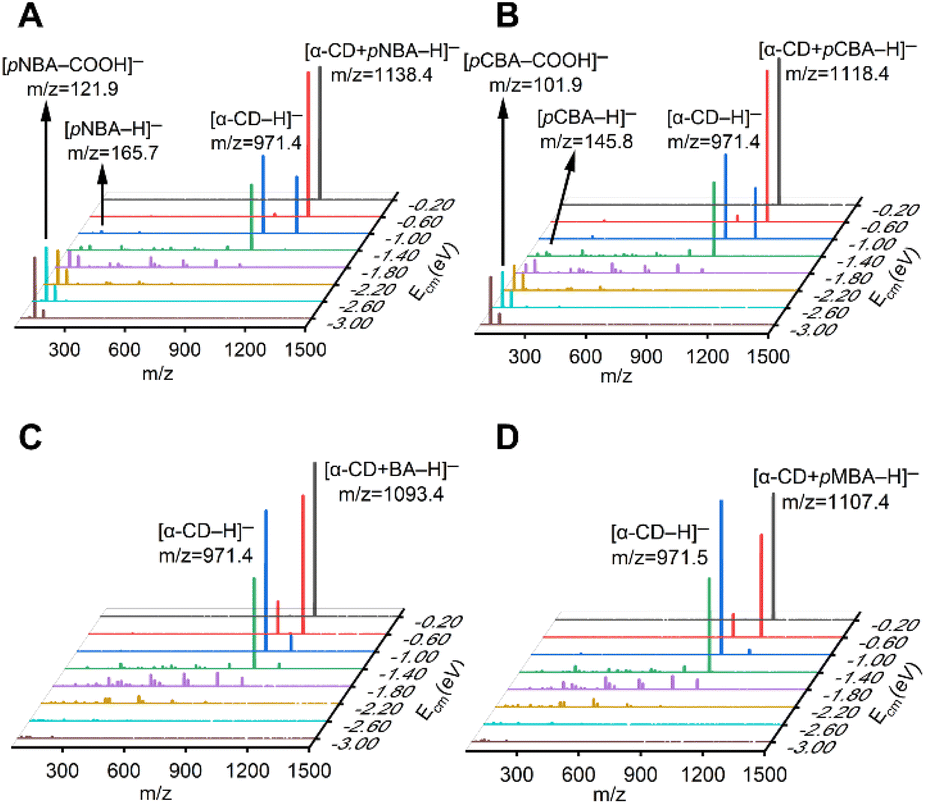 | ||
| Fig. 1 Tandem mass spectra of the (A) [α-CD + pNBA–H]−, (B) [α-CD + pCBA–H]−, (C) [α-CD + BA–H]− and (D) [α-CD + pMBA–H]− complex anions with increasing Ecm. | ||
 | ||
| Fig. 2 Hypothetical conformations of (A and E) [α-CD + pNBA–H]−, (B and F) [α-CD + pCBA–H]−, (C and G) [α-CD + BA–H]− and (D and H) [α-CD + pMBA–H]−. (A to D) Top views and (E to H) side views. | ||
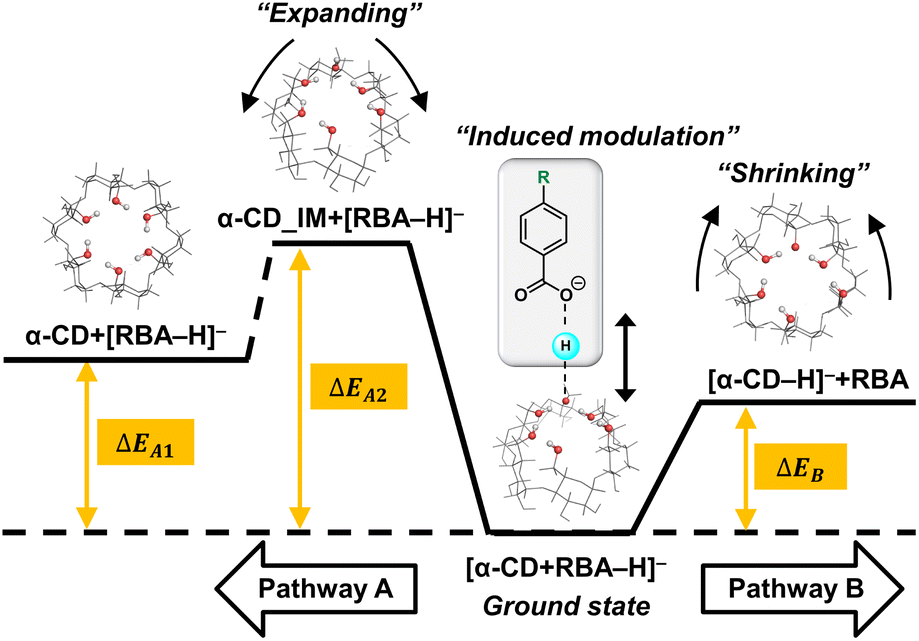 | ||
| Scheme 1 Potential energy diagram of two competitive dissociation pathways of complex anion [α-CD + RBA–H]–. | ||
3 Results and discussion
Four RBAs were selected for preparing the complex anions together with α-CD via the electrospray ionization technique, i.e. p-nitrobenzoic acid (pNBA) and p-cyanobenzoic acid (pCBA) that contained electron-withdrawing groups, p-methylbenzoic acid (pMBA) that contained an electron-donating group, and the benzoic acid that contained no substitute groups (BA). The complex anions were dissociated through collision with nitrogen gas in a tandem mass spectrometer. Fig. 1 shows the superimposed tandem mass spectra of the mass-selected complex anions [α-CD + RBA–H]− under gradient-increasing center-of-mass energy (Ecm). In the low Ecm stage (0.20 eV to 1.40 eV), the intensity of [α-CD−H]− initially grew while that of [α-CD + RBA–H]− declined. With further increasing Ecm, [α-CD−H]− tended to dissociate into fragments. It is notable that [α-CD + RBA–H]− dominantly yielded daughter ion [α-CD−H]− under low Ecm. The rest moieties of the complex anions should be present as neutral molecules, i.e. neutral RBAs (Fig. S1†). Thus, they were not detected in the mass spectrometer under low Ecm. It could be concluded from these dissociation results that α-CD showed apparently higher gas-phase acidity than all the four RBAs. [RBA–H]− could be detected only when the complex anions were activated with higher collision energies. In the high Ecm stage (1.40 eV to 3.00 eV), the anions of the more acidic RBAs, [pNBA–H]− and [pCBA–H]−, were visible in the spectra together with their decarboxylated forms, i.e. [pNBA–COOH]− and [pCBA−COOH]− (Fig. 1). The suspected [pMBA–H]− and [BA–H]− were also detected, if the corresponding mass-to-charge ratios were focused for detecting these daughter anions in the tandem mass spectrometer (Fig. S2†). However, the signal-to-noise ratios of [pMBA–H]− and [BA–H]− were much lower than that of [pNBA–H]− and [pCBA–H]−. Furthermore, it was observed that [α-CD + pNBA–H]− and [α-CD + pCBA–H]− were more stable than [α-CD + BA–H]− and [α-CD + pMBA–H]− (Fig. S2 and Table S1†).Any complex anions formed involving additional solvent molecules were not detected in the mass spectra, which was probably owing to the low stabilities of such complex anions. They were also not presented in previous studies.40–42 Moreover, the fragmentation pathways of deprotonated CDs have not been reported. At the high Ecm stage, α-CD might also be fragmented via an open chain structure, as supposed to be present in the fragmentation processes of protonated CDs.43 However, α-CD was believed to be in its intrinsic cyclic structure in the complex anions before they were activated via collision, since the ether bonds in α-CD are much stronger than the intermolecular HBs.
For illustrating the apparently high gas-phase acidity of α-CD, the free energy changes of the deprotonation reactions in gas phase were first calculated to evaluate the intrinsic acidities of α-CD and RBAs. The pristine results of the theoretical calculations are presented in the ESI (Table S2).† The theoretical calculations presented that a closed cyclic HBN and an open cyclic HBN were located on the primary rims of the structurally optimized α-CD and [α-CD−H]− (Fig. S3†), which were never observed in condensed phases. The open cyclic HBN in [α-CD−H]− stabilized the deprotonated primary hydroxyl group through intramolecular HBs. As shown in Table 1, although the lower intrinsic acidities of BA and pMBA were observed as expected, the intrinsic acidities of α-CD, pNBA and pCBA were close, which were inconsistent with the observed apparently higher gas-phase acidity of α-CD. In other words, the dominant dissociation pathways of [α-CD + pNBA–H]− and [α-CD + pCBA–H]− could not be well explained based on these thermodynamic values. Thus, it became necessary to analyze the dissociation procedures at molecular level other than solely analyzing the intrinsic acidities. It is notable that the dissociation pathways were well consistent with the intrinsic acidities in the previous study,40 in which more acidic benzoic acids were not considered. Herein, by taking the RBAs possessing electron-withdrawing groups (pNBA and pCBA) into consideration, more information behind the dissociation pathways was indicated.
Subsequently, molecular dynamics (MD) simulations and quantum-mechanical calculations were carried out to figure out the structures of the complex anions (for more details, see the ESI†), which were inevitable for inspecting the dissociation procedures. As shown in Fig. S5,† the low-energy conformers of the complex anions possessed inclusion conformations. The primary hydroxyl groups of neutral α-CD mainly formed intermolecular HBs with [RBA–H]− in the complex anions, and the major part of [RBA–H]− was presented inside the cavity of α-CD. If supposing these conformers really presented prior to collision, there must exist multiple conformation adjustment steps for these complex anions to produce the stable [α-CD−H]−, which should contain the rupture of intermolecular HBs and formation of intramolecular HBs. Multiple intermediates and transient states should be present on the potential energy surfaces, which were too complicated to be fully figured out.
Thus, learnt from the CID results, a similar docking structure with a single proton bridge between two anionic moieties was proposed for all the four complex anions (Fig. 2). The rupture at the two sides of the proton bridge could lead to two dissociation pathways for each complex anion, which produced [α-CD−H]− and neutral RBA, or reversely [RBA–H]− and neutral α-CD. These structures were supposed to be the stable conformers that were closest to the detected dissociation products. Potential energy diagram was depicted for the two competitive dissociation pathways (Scheme 1). The intermediate α-CD_IM, resulted from the approaching of the bridging proton to [α-CD−H]−, was introduced to elaborate the dissociation in Pathway A. At this state, the other moiety [RBA–H]− completely left the complex anion, while the intramolecular HBs of the obtained α-CD_IM remained similar to [α-CD−H]−. Owing to the approaching of the bridging proton, the lengths of the intramolecular HBs of α-CD_IM were elongated in comparison to [α-CD−H]−. In comparison, the approaching of the bridging protons to the other moieties ultimately produce [α-CD−H]− and neutral RBAs, as shown in Pathway B. As the bridging proton left, the lengths of intramolecular HBs of [α-CD−H]− were shortened in comparison to the intramolecular HBs in the complex anions.
The energy diagram of the two competitive pathways is shown in Table 2. The accuracy of the calculated energies was thoroughly validated as shown in Table S3.† Pathway B was preferred over Pathway A due to the much lower energy barriers. By comparing the ΔEB values for different complex anions, the calculated stabilities of the complex anions in gas phase were in excellent agreement with the CID results (Fig. S2 and Table S1†). Moreover, the fact that [pNBA–H]− and [pCBA–H]− were more facilely detected than [pMBA–H]− and [BA–H]− in the CID experiment could also be successfully explained by the smaller differences between the energy barriers of the two pathways (ΔΔE, Table 2). The success in well explaining the experimental results strongly suggested the existence of the proposed conformations shown in Fig. 2. Furthermore, it is notable that the adaptive stretching of the HBN on the primary rim of [α-CD−H]− along with the allocation of the bridging proton was observed, which highlighted the cooperative characteristics of extended HBNs in the complex anions in determining the energy landscapes of the dissociation pathways.
For further illustrating the cooperative dynamics of the HBNs during dissociation with more details, wave function analysis was performed in Multiwfn.44–46 Then, HB interactions, bond paths (BPs) and bond critical points (BCPs) were visualized in the VMD program (Fig. 3A and B and S6†). The different colors in the isosurfaces represent the compositions of the HBs. Meanwhile, the bond energies of the HBs were calculated (Fig. 3D). The lengths and energies of HB1 to HB5 in the complex anions were between that of α-CD_IM and [α-CD−H]− (Fig. 3C and D, Table S5†), which clearly showed that the adaptive stretching of the HBN on the primary rim of [α-CD−H]− was associated with the allocation of the bridging proton. Besides, it also indicated that α-CD_IM was not stable enough and needed to transform into the stable conformer of α-CD via multiple conformation adjustment steps (Pathway A in Scheme 1).
 | ||
| Fig. 3 (A) Detailed view and (B) overview of the visualized HBs in [α-CD−H]−. HBs are labeled counterclockwise. (C) The HB lengths and (D) the HB energies. HB0 and HBs1-5 denote the inter- and intra- HBs, respectively. The HB lengths and the average HB energy of the structurally optimized α-CD were within the pink band and shown by the blue dashed line, respectively. | ||
Furthermore, it is highly remarkable that the intermolecular HB0 of the four complex anions and the intramolecular HB1 of [α-CD−H]− were LBHBs, which were characterized by the O–O distances smaller than 2.5 Å.47 LBHBs were rarely observed before. The present five LBHBs in the anions could be roughly divided into three groups by cursory inspection of the O–O distances and the functional groups involved, which represented a valuable library for revealing the embedded characteristics of LBHBs.
The possible existence of the aforementioned inclusion conformers of the complex anions was reinspected. The inclusion conformers were obtained from MD simulations, which were termed as MD-conformers below. The conformers in Scheme 1 well explained the CID results, thus, they were termed as CID-conformers. The single point energies of the two conformers were compared (Table S6†). It was observed that the MD-conformers were more stable than the corresponding CID-conformers. It was thermodynamically possible that the relatively stable MD-conformers were the precursors of the CID-conformers. Then, the collision energies needed to convert the MD-conformers into the CID-conformers and subsequently dissociate the CID-conformers (Fig. S7†). As the CID-conformers become the intermediates in this circumstance, each complex anion would still be dominantly dissociated into [α-CD−H]− and neutral RBA. However, the relative stabilities of the complex anions were difficult to be compared, since it was difficult to figure out the energy barriers between the MD-conformers and the CID-conformers. The transition of the MD-conformers into the CID-conformers should also contain many currently unknown HB rupture and rebuilding steps, which might involve the conformation adaption of α-CD that was similar to the description in ref. 20. On the other hand, it was also possible that the formation of the complex anions was mainly governed by the docking kinetics.18 That is, α-CD formed intramolecular HBs during the ESI processes, and simultaneously formed the CID-conformers together with the benzoic acids. Then, the CID-conformers were the dominant species, while the MD-conformers were negligible before the collision activation. Advanced hyphenated techniques integrating ion mobility spectrometry, ultraviolet spectroscopy and infrared spectroscopy48 are expected to resolve the conformers of the complex anions in the future.
4 Conclusions
In conclusion, by illustrating the proton allocation during the dissociation of the [α-CD + RBA–H]− complex anions, it was proposed that the [α-CD + RBA–H]− complex anions contained HBNs. The cooperative characteristics of the extended HBNs well explained the dissociation pathways and stabilities of the complex anions. Compared with the water clusters, the geometries of the present HBNs are easier for illumination, as the primary hydroxyl groups are fixed on the scaffold of α-CD, while the positions of the water molecules in the water clusters are variable. The phenomena previously observed in the water clusters are awaited for exploration in the present HBNs in the future. Moreover, five LBHBs came into being in the HBNs. As LBHBs were rarely observed before, the present LBHBs represented a valuable library for revealing the embedded characteristics of LBHBs. Besides a valuable model for studying HBNs proposed herein, this study also indicates that polyhydroxy compounds own the potential to donate protons in gaseous environments or aprotic solvents, which may be inspiring in atmospheric chemistry and synthetic chemistry. It is notable that vibrational spectra are possible to be recorded in gas phase to provide structural information at molecular levels, which might be useable to reveal the structures of the complex anions. The in-depth investigation is now ongoing in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (22122612, 22036003 and 21806188), NSF of Guangdong Province (2018A030313324), Guangzhou Science and technology planning project (201803030018), the NSF of Guangdong Provincial Key R&D Programme (2020B1111350002), and the Fundamental Re-search Funds for the Central Universities (2021qntd24). We also thank Dr Xue-Peng Zhang from Shaanxi Normal University for his kind assistance in theoretical calculations.References
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- S. X. Liu, S. Li, G. M. Shen, N. Sukumar, A. M. Krezel and W. K. Li, Science, 2021, 371, eabc5667 CrossRef CAS PubMed.
- V. V. Welborn and T. Head-Gordon, J. Am. Chem. Soc., 2019, 141, 12487–12492 CrossRef CAS PubMed.
- R. Fu, Y. Miao, H. Qin and T. A. Cross, J. Am. Chem. Soc., 2020, 142, 2115–2119 CrossRef CAS PubMed.
- M. Chen, L. Zheng, B. Santra, H.-Y. Ko, R. A. DiStasio Jr, M. L. Klein, R. Car and X. Wu, Nat. Chem., 2018, 10, 413–419 CrossRef CAS PubMed.
- F. Perakis, L. De Marco, A. Shalit, F. Tang, Z. R. Kann, T. D. Kühne, R. Torre, M. Bonn and Y. Nagata, Chem. Rev., 2016, 116, 7590–7607 CrossRef CAS PubMed.
- C. T. Wolke, J. A. Fournier, L. C. Dzugan, M. R. Fagiani, T. T. Odbadrakh, H. Knorke, K. D. Jordan, A. B. McCoy, K. R. Asmis and M. A. Johnson, Science, 2016, 354, 1131–1135 CrossRef CAS PubMed.
- M. Thamer, L. De Marco, K. Ramasesha, A. Mandal and A. Tokmakoff, Science, 2015, 350, 78–82 CrossRef PubMed.
- S. Dai, L. M. Funk, F. R. von Pappenheim, V. Sautner, M. Paulikat, B. Schroder, J. Uranga, R. A. Mata and K. Tittmann, Nature, 2019, 573, 609–613 CrossRef CAS PubMed.
- T. Fransson, Y. Harada, N. Kosugi, N. A. Besley, B. Winter, J. J. Rehr, L. G. M. Pettersson and A. Nilsson, Chem. Rev., 2016, 116, 7551–7569 CrossRef CAS PubMed.
- L. Wang, S. D. Fried, S. G. Boxer and T. E. Markland, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18454–18459 CrossRef CAS PubMed.
- J. A. Fournier, C. J. Johnson, C. T. Wolke, G. H. Weddle, A. B. Wolk and M. A. Johnson, Science, 2014, 344, 1009–1012 CrossRef CAS PubMed.
- J. O. Richardson, C. Perez, S. Lobsiger, A. A. Reid, B. Temelso, G. C. Shields, Z. Kisiel, D. J. Wales, B. H. Pate and S. C. Althorpe, Science, 2016, 351, 1310–1313 CrossRef CAS PubMed.
- W. T. S. Cole, J. D. Farrell, D. J. Wales and R. J. Saykally, Science, 2016, 352, 1194–1197 CrossRef CAS PubMed.
- A. Nemkevich, M. A. Spackman and B. Corry, J. Am. Chem. Soc., 2011, 133, 18880–18888 CrossRef CAS PubMed.
- G. L. Hou, X. B. Wang and M. Valiev, J. Am. Chem. Soc., 2017, 139, 11321–11324 CrossRef CAS PubMed.
- J. B. Peng, J. Guo, P. Hapala, D. Y. Cao, R. Z. Ma, B. W. Cheng, L. M. Xu, M. Ondracek, P. Jelinek, E. G. Wang and Y. Jiang, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- A. Guerrero, T. Baer, A. Chana, J. González and J. Z. Dávalos, J. Am. Chem. Soc., 2013, 135, 9681–9690 CrossRef CAS PubMed.
- J. Guo, J.-T. Lü, Y. Feng, J. Chen, J. Peng, Z. Lin, X. Meng, Z. Wang, X.-Z. Li, E.-G. Wang and Y. Jiang, Science, 2016, 352, 321–325 CrossRef CAS PubMed.
- W. Snor, E. Liedl, P. Weiss-Greiler, A. Karpfen, H. Viernstein and P. Wolschann, Chem. Phys. Lett., 2007, 441, 159–162 CrossRef CAS.
- J. S. Flier, E. Maratos-flier, J. A. Pallotta and D. Mcisaac, Nature, 1979, 279, 343–344 CrossRef PubMed.
- Y. Chen, Z. Zuo, X. Dai, P. Xiao, X. Fang, X. Wang, W. Wang and C.-F. Ding, Talanta, 2018, 186, 1–7 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada, M. Ehara, et al.Gaussian 09 Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- M. Rejzek, C. E. Stevenson, A. M. Southard, D. Stanley, K. Denyer, A. M. Smith, M. J. Naldrett, D. M. Lawson and R. A. Field, Mol. BioSyst., 2011, 7, 718–730 RSC.
- R. Dennington; T. A. Keith and J. M. MillamGaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed.
- T. Lu and Q. X. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- T. Lu and Q. X. Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 27–28 CrossRef.
- Z. Li, E. P. A. Couzijn and X. Zhang, J. Phys. Chem. B, 2012, 116, 943–950 CrossRef CAS PubMed.
- P. Su, A. J. Smith, J. Warneke and J. Laskin, J. Am. Soc. Mass Spectrom., 2019, 30, 1934–1945 CrossRef CAS PubMed.
- Y. Fan, Y. Zhang, Q. Jia, J. Cao and W. Wu, Mass Spectrom. Lett., 2015, 6, 13–16 CrossRef.
- P. S. Bruni and S. Schurch, Carbohydr. Res., 2021, 504, 108316 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and Q. X. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed.
- W. W. Cleland and M. M. Kreevoy, Science, 1994, 264, 1887–1890 CrossRef CAS PubMed.
- L. Voronina, A. Masson, M. Kamrath, F. Schubert, D. Clemmer, C. Baldauf and T. Rizzo, J. Am. Chem. Soc., 2016, 138, 9224–9233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05029c |
| This journal is © The Royal Society of Chemistry 2022 |