
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic water oxidation on CuO–Cu2O modulated cobalt-manganese layered double hydroxide†
Arslan Hameed a,
Faiza Zulfiqara,
Waheed Iqbala,
Hassan Alia,
Syed Shoaib Ahmad Shah*b and
Muhammad Arif Nadeem*ac
a,
Faiza Zulfiqara,
Waheed Iqbala,
Hassan Alia,
Syed Shoaib Ahmad Shah*b and
Muhammad Arif Nadeem*ac
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan. E-mail: manadeem@qau.edu.pk; Tel: +92-51-9064-2062
bDepartment of Chemistry, School of Natural Sciences, National University of Science and Technology, Islamabad, 44000, Pakistan. E-mail: shoaib03ahmad@outlook.com
cPakistan Academy of Sciences, 3-Constitution Avenue Sector G-5/2, Islamabad, Pakistan
First published on 11th October 2022
Abstract
Layered double hydroxides (LDH) are potential electrocatalysts to address the sluggish oxygen evolution reaction (OER) of water splitting. In this work, copper oxide (CuO/Cu2O) nanoparticles are integrated with cobalt-manganese layered double hydroxide (CoMn-LDH) to enhance their performance towards OER. The catalyst is synthesized by growing CoMn-LDH nanosheets in the presence of CuO/Cu2O nanoparticles that were obtained by the calcination of the copper containing metal–organic framework (HKUST-1). The synthesized CoMn-LDH@CuO/Cu2O electrocatalyst shows excellent activity towards OER with an overpotential of 297 mV at a catalytic current density of 10 mA cm−2 and have a Tafel slope value of 89 mV dec−1. Moreover, a slight decrease in the performance parameters is observed until the 15 h of continuous operation. We propose that the conductive strength of CuO/Cu2O and its synergistic effect with the CoMn-LDH are responsible for the improved OER performance of the desired electrocatalyst.
1. Introduction
In pursuit of clean and economical energy technologies, the water splitting reaction is of prime importance. The two half reactions of water splitting are oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). Compared to HER, OER needs more attention due to its slow kinetics.1 Commercial OER electrocatalysts are precious metal containing nanomaterials such as, Pt/C, RuO2, and IrO2.2,3 High cost and less stability under certain conditions are some of the problems associated with these catalysts.2,4 So, development of an efficient electrocatalyst with high activity, long-term stability, and relatively low cost, for renewable energy devices is crucial for practical applications.Transition metal containing layered double hydroxides (LDHs) have recently shown their potential as electrocatalysts for water splitting in alkaline media.5,6 LDHs have a general formula of [MII1-xMIIIx(OH)2]X+[An−x/n−·yH2O]X, where M = metal cations coordinated to hydroxide anions, whilst An− anions are present between the LDH layers.7,8 In recent literature, various LDH composites have shown considerable electrocatalytic performance for the OER process; these include FeNi-LDH,9–11 NiCo-LDH,12,13 CoFe-LDH,14 ZnFe-LDH,15 and NiCoFe-LDH.16 It has been reported that individual LDHs structures have limited electrocatalytic performance due to the lack of exposed active sites and insufficient electrical conductivity.7 To address this problem, integration of LDH with the conductive substrate such as carbon fiber, Vulcan carbon, carbon nanotubes, multi walled carbon nanotubes, carbon quantum dots, and nickel foam has been widely utilized to enhance their electrocatalytic performance.17–22
Previously, we have reported that copper based metal–organic framework, MOF (HKUST-1) can be converted into copper oxides (CuO/Cu2O) under certain reaction conditions.23 Such nanosized copper oxides have unique properties in terms of their particle size, surface area and conductivity. Many researchers also utilized copper oxides as an aid to electrocatalyst to enhance its efficiency. For instance, Niu et al. reported a porous carbon supported Cu/CuO nanoparticles for nitrophenol reduction, synthesized via calcination of a copper MOF.24 The Cu/ZnO catalyst was synthesized by Zheng et al. via calcination of Cu–Zn MOF under reducing environment.25 Cu/ZnO catalyst synthesized by using MOF-derived methodology was more active and stable than Cu/ZnO synthesized by other techniques. Peng et al. compared CuO produced through pyrolysis of HKUST-1 (CuO-p) and CuO obtained via co-precipitation (CuO-c).26 It was concluded that CuO-p was more catalytically active than CuO-c for selective catalytic reduction (SCR) of NO. From these reports, we anticipated that the in situ. Controlled growth of LDHs on the MOF-derived metal-oxides may lead to enhance the OER activity due to the combination of inherited features of both materials.
Herein, we report a novel cobalt-manganese layered double hydroxide (CoMn-LDH), synthesized in the presence of copper-based metal–organic framework, C18H6Cu3O12, (KHUST-1) derived CuO/Cu2O nanoparticles to form CoMn-LDH@CuO/Cu2O. This composite has shown promising OER activity which is superior to the commercial Pt/Ir based catalysts. The production of ultrathin nanosheets of highly OER active CoMn-LDH structure and its interaction with CuO/Cu2O, which form an interconnected electrically conducting network, are the important aspects of this composite.
2. Experimental
2.1 Synthesis of CuO/Cu2O
HKUST-1 was synthesized using a modified technique previously described by Wang et al.27 The solution A was made by dissolving 0.84 g of benzene tricarboxylic acid (BTC) in 24 mL ethanol. The solution B was prepared by dissolving 1.75 g of Cu(NO3)·6H2O in 24 mL deionized water, and vigorously stirred. After that, both the solutions (A & B) were mixed and transferred to a Teflon-lined autoclave. The autoclave was kept in an oven at 120 °C for 12 h, and then allowed to cool down to room temperature. Blue colored HKUST-1 crystals were filtered and washed several times with ethanol and deionized water and dried overnight at 80 °C. The synthesized HKUST-1 was then calcined at 900 °C for 3 h in an oxygen environment to produce CuO/Cu2O particles.2.2 Synthesis of CoMn-LDH@CuO/Cu2O
The composite CoMn-LDH@CuO/Cu2O was synthesized using a hydrothermal method. Typically, 100 mg of prepared CuO/Cu2O was completely dispersed in 15 mL of deionized water. The precursors of CoMn-LDH i.e., 218 mg of Co (NO3)2·6H20, 297 mg of MnCl4·4H2O and 180 mg of Urea was added in above solution and stirred for 3 h. After that, the solution was transferred to a Teflon lined autoclave and heated at 130 °C for 24 h. The precipitates produced were centrifuged, washed several times with deionized water and dried at 60 °C in a drying oven.2.3 Electrochemical measurements
All electrochemical experiments were performed at room temperature using a Gamry Interface 5000E electrochemical potentiostat configured with the prepared catalyst as the working electrode, Pt wire as the counter electrode, and an Ag/AgCl electrode as the reference electrode. Prior to performing electrochemical tests, 1 M KOH alkaline electrolyte was purged with N2 gas for 30 min.28,29 Chronoamperometry measurements were used to determine the long-term stability, and linear sweep voltammetry (LSV) experiments were carried out to investigate the current density/overpotential performance parameters at a scan rate of 50 mV s−1. Impedance (EIS) experiments in 1 M KOH were carried out across a frequency range of 0.01 Hz to 100 kHz. To calibrate the reference electrode (Ag/AgCl) and converted to RHE, the following equation was used: E(RHE) = E(Ag/AgCl) + 0.197 V + 0.059 PH.30 Characterization details are in ESI.†3. Results and discussion
To investigate the lattice parameters of the synthesized catalysts, powder X-ray Diffraction (pXRD) patterns were taken. Fig. 1 showed the characteristic diffraction peaks at 11.54°, 23.76°, and 33.5°, which confirmed the layered morphology and could be indexed to the (003), (006) and (009) planes of LDHs (JCPDS # 01-089-0460).31 The diffraction peaks at 2θ angles of 35.6°, 38.9°, 48.8°, 53.7°, 58.6°, 61.9°, 65.6°, 67.8°, correspond to the indices (002), (111), (−202), (020), (202), (−113), (−311) and (220) reflections of CuO lattice planes, respectively (JCPDS # 00-002-1040).32 The diffraction peaks at 36.6°, 42.5° and 73.4° are attributed to the (111), (200) and (311) lattice planes of Cu2O, respectively (JCPDS # 00-001-1142). The intensity of the typical diffraction peaks of Cu2O (111) and CuO (111) planes shows that CuO is the predominant specie produced, with traces of Cu2O.33–35 The pXRD pattern of CoMn-LDH@CuO/Cu2O have all the characteristics peaks of CoMn-LDH and MOF derived CuO/Cu2O which reveals that dispersion of CuO/Cu2O does not change the chemical composition of the catalyst.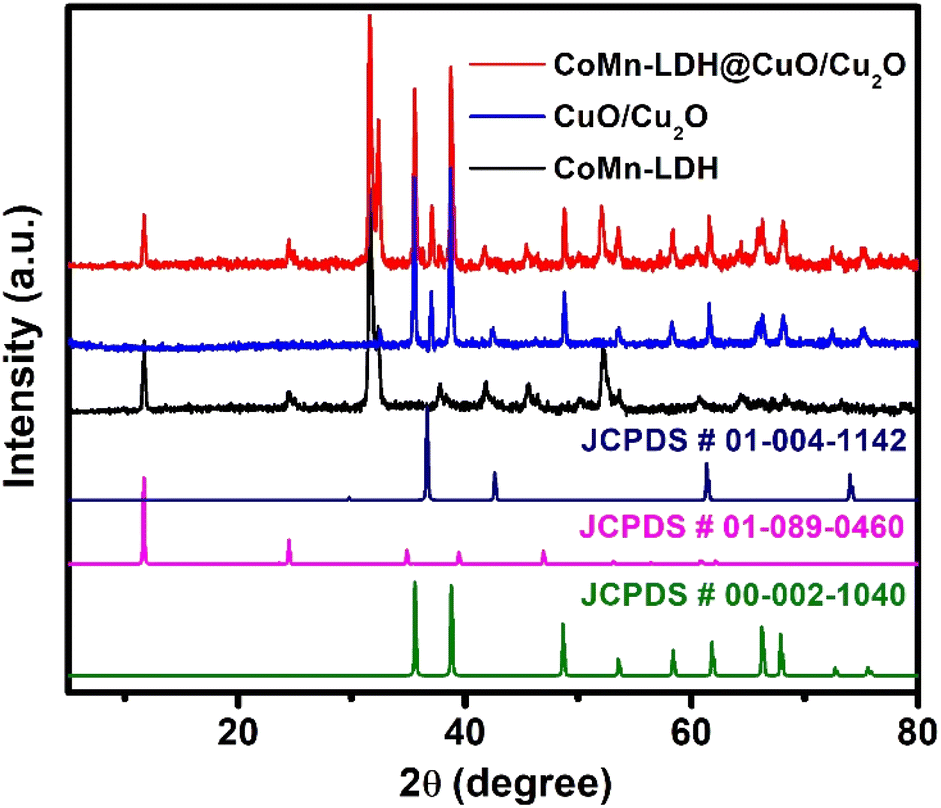 | ||
| Fig. 1 Comparison of powder XRD patterns of CoMn-LDH, CuO/Cu2O and CoMn-LDH@CuO/Cu2O. | ||
The surface elemental composition was confirmed by the XPS analysis. Fig. 2a shows two peaks at 781.6 eV and 798.2 eV correspond to the binding energies (BEs) of Co 2p3/2 and Co 2p1/2, respectively, and two satellite peaks at 786.8 eV and 803.4 eV were also observed. The value of Co 2p3/2 is distinct from Co0 (777.6 0.8 eV) but near to Co2+ (780.9 0.4 eV), suggesting that the valence state of cobalt is Co2+. Mixed oxidation states of Mn have been observed. The binding energy values of 641.4 eV, 642.8 eV and 644.7 eV correspond to Mn2+, Mn3+ and Mn4+, respectively. Mn 2p1/2 has peaks at 647.3 eV (Fig. 2b). Cu 2p3/2 and Cu 2p1/2 binding energies have two main peaks at 933.8 eV and 953.5 eV, respectively, with spin–orbit splitting of around 20.2 eV. The appearance of two satellite peaks at higher binding energies than the Cu 2p3/2 and Cu 2p1/2 peaks and shoulder peak of Co 2p3/2 suggests the presence of Cu2+ and Cu1+ states. Deconvolution of the O 1s peak results in the formation of two sub peaks: ‘O’ in CuO (532.3 eV) and ‘O’ in chemisorbed hydroxyl group (533.5 eV) (Fig. 2d).
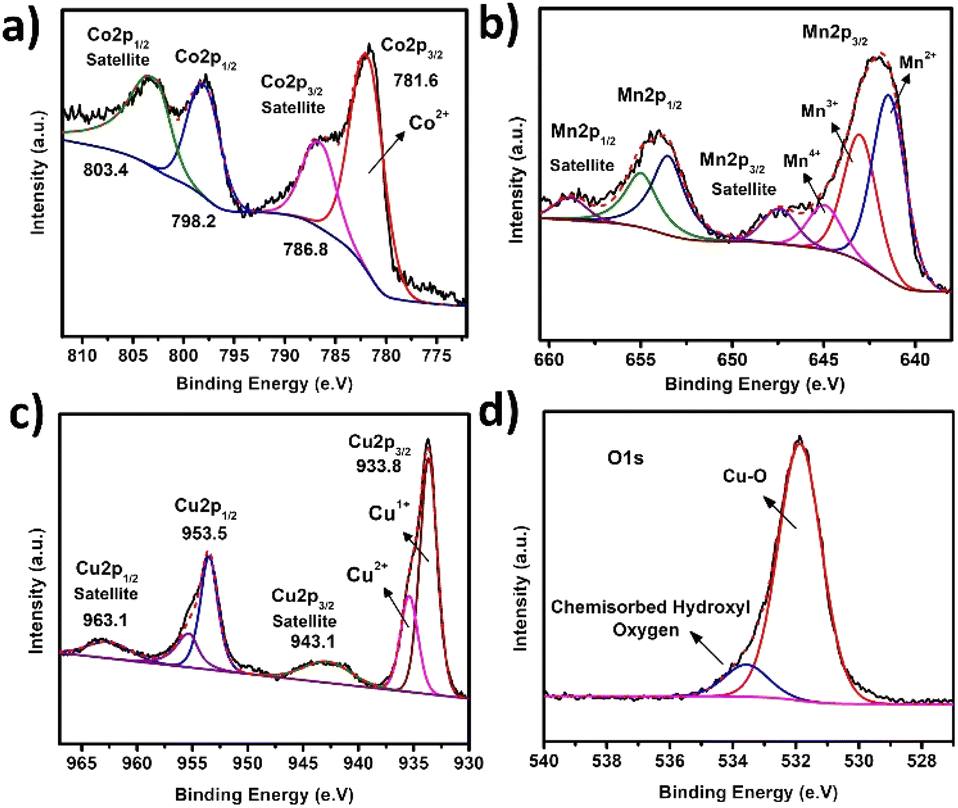 | ||
| Fig. 2 XPS spectra of CoMn-LDH@CuO/Cu2O; (a) Co 2p (b) Mn 2p (c) Cu 2p (d) O 1s. | ||
The size, morphology and structure of the resulting materials were characterized by scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM). The interior structure of as synthesized CoMn-LDH and composite CoMn-LDH@CuO/Cu2O is examined by the high resolution transmission electron microscope (HRTEM). In Fig. 3a and c SEM and TEM results of CoMn-LDH shows hexagonal platelets which further confirms the layered structure of CoMn-LDH.31
 | ||
| Fig. 3 SEM and TEM images of CoMn-LDH (a and c), CoMn-LDH@CuO/Cu2O (b and d), elemental mapping images of cobalt (e), manganese (f), copper (g), oxygen (h). | ||
Fig. 3b and d shows SEM and HRTEM images of composite CoMn-LDH@CuO/Cu2O revealing the full dispersion of Cu-BTC derived CuO/Cu2O across the layers of LDH. This dispersion facilitates mass and charge transfer which results in high ECSA. Lattice fringes can also be seen in Fig. 3d with the d-spacing (d = 0.23 nm) and (d = 0.245 nm) which correspond to (111) plane of CuO and (111) plane of Cu2O respectively in XRD spectra of CuO/Cu2O.31,33,35 The energy dispersive X-ray spectroscopic analysis shown in (Fig. S2†) and elemental mapping shown in Fig. 3e–h indicates the uniform distribution of Co, Mn, Cu and O elements, thus confirming the formation of CoMn-LDH@CuO/Cu2O.
3.1 Oxygen evolution reaction studies
All the electrochemical studies were carried out in 1 M N2-saturated KOH solution, the catalytic performance of the CoMn-LDH@CuO/Cu2O were investigated using a three-electrode system. To provide a fair comparison, the mass loading of the CoMn-LDH@CuO/Cu2O catalyst was set to the same value as that of the CoMn-LDH and CuO/Cu2O samples (1 mg cm−2). The linear sweep voltammetry (LSV) curves of catalysts versus reversible hydrogen electrode (RHE) for OER without IR correction are shown in Fig. 4a. As predicted, in 1 M KOH solution, the bare electrode exhibits negligible OER activity. Among all the three synthesized electrocatalysts, the CoMn-LDH@CuO/Cu2O exhibits the lowest onset potential for OER and the highest current density at the same overpotential, indicating the highest electrochemical activity towards water oxidation. The current density of 10 and 20 mA cm−2 was achieved by the CoMn-LDH@CuO/Cu2O at an overpotential of 297 mV and 320 mV respectively, which is far less than CoMn-LDH (380 mV) and CuO/Cu2O (540 mV) at the same current density. For comparison OER activity of RuO2 was also checked under same circumstances and it required overpotential of 320 mV@10 mA cm−2. Indeed, when compared to previously reported electrocatalysts, CoMn-LDH@CuO/Cu2O shows superior OER activity. As shown in Fig. 4b the Tafel slope was calculated to check the kinetics of the reaction and CoMn-LDH@CuO/Cu2O exhibits low value of Tafel slope of 89 mV dec−1, as compared to 97 mV dec−1 for RuO2, 190 mV dec−1 for CuO/Cu2O and 160 mV dec−1 for CoMn-LDH. We computed turnover frequency (TOF), another essential parameter, to assess the intrinsic characteristic of the electrocatalyst. The TOF value of CoMn-LDH@CuO/Cu2O is (0.012 s−1) at onset overpotential.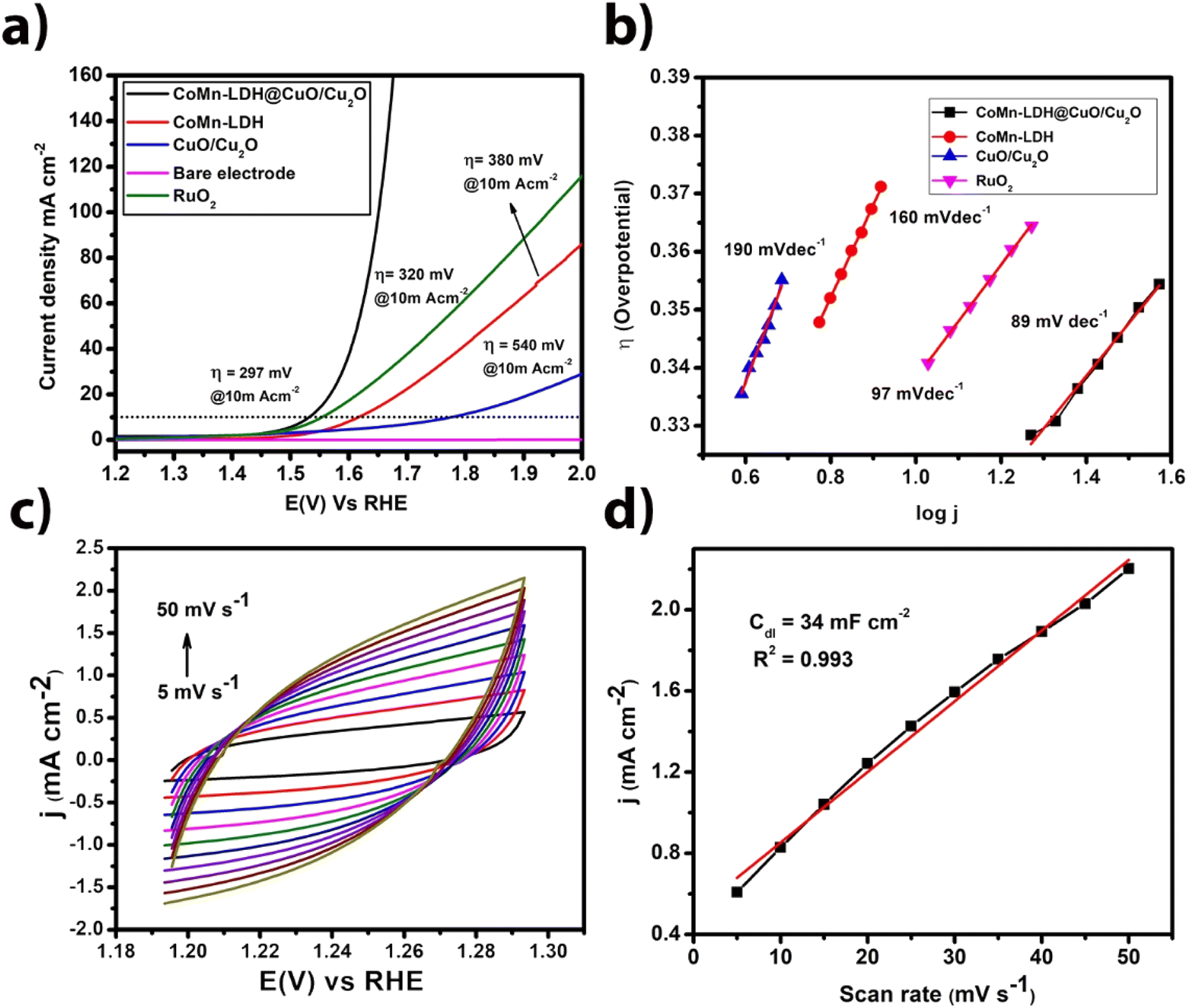 | ||
| Fig. 4 (a) Linear sweep voltammograms of CoMn-LDH, CuO/Cu2O, CoMn-LDH@CuO/Cu2O, RuO2 and B are electrode (b) Tafel slopes of CoMn-LDH, CuO/Cu2O, CoMn-LDH@CuO/Cu2O and RuO2 (c) non-faradaic region cyclic voltammograms at various scan rates (d) anodic current density plot vs. scan rate of CoMn-LDH@CuO/Cu2O fabricated electrode in 1 M KOH solution as an electrolyte. | ||
The electrochemical surface area (ECSA) of an electrocatalyst is a critical parameter because it determines the extent to which active sites for positive/negative ions are exposed to the electrode surface when anodic/cathodic potentials are applied solely in the non-faradaic region. The area of exposed active sites is directly related to double layer capacitance (Cdl) of modified working electrode. To determine the capacitance of the double layer, we used cyclic voltammetry in the non-faradaic region at various scan rates. Cyclic voltammograms shown in Fig. 4c, demonstrates increase in current density with the increase in the scan rate. A single potential value was computed by detecting it in the capacitive potential window that is non-Faradic. In the region of 5 to 50 mV s−1, plotting anodic current versus scan rates yielded a straight line whose slope is equal to Cdl. The Cdl values were calculated for CoMn-LDH@CuO/Cu2O (34 mF cm−2), CoMn-LDH (14.4 mF cm−2) and CuO/Cu2O (9.2 mF cm−2) (Fig. S6†). It was observed that CoMn-LDH@CuO/Cu2O had the highest value of double layer capacitance which indicates high electrochemical active surface area (ECSA). The double layer capacitance values of some reported materials also support our Cdl studies i.e. NiCo·P/C nanocubes (145 mF cm−2), NiCo LDH nanocubes (9.16 mF cm−2) and NiCo·P nanoboxes (28.94 mF cm−2).36 The ECSA has also been calculated from double layer capacitance. We noticed that CoMn-LDH@CuO/Cu2O exhibits highest electrochemical active surface area (425 cm2) compared to CoMn-LDH (180 cm2) and CuO/Cu2O (115 cm2) materials. The high value of Cdl and ECSA indicates that CoMn-LDH@CuO/Cu2O facilitate mass and charge transfer.
Conventionally, the electrochemical water oxidation process primarily entails three intermediate stages: (1) the adsorption of water molecule onto the electrode surface, (2) the separation of water into molecular oxygen, and (3) the evolution of oxygen. The CoMn-LDH nanosheets have a distinct layered structure and the sheets are usually stacked on over the other. Since LDH has a wide interlayer gap, the hydroxide units and molecules of water can diffuse across the layers and move randomly, enabling close contact among the catalyst and active species. The addition of CuO/Cu2O in the catalyst not only help in enhancing the conductivity and thus charge transfer but also to expose the active sites via preventing LDH sheets from coagulation. Therefore, the electrochemical active surface area increases, and reaction occur at more accessible sites. In addition, as the interlayer spacing will make it easier for O2 to transit and to evolve from the electrode surface, which is essential for the third phase of the OER process.
As predicted by the Pourbaix diagrams of Co37 and Mn,38 the high-valent cations (Co3+/4+, or Mn3+/4+) are highly crucial for OER.39 X-ray absorption spectroscopy (XAS),40 in situ Raman spectroscopy,41 and density functional theory (DFT) calculations42 have demonstrated that Co hydroxide electrocatalysts can be oxidized in situ to the catalytically active CoOOH in alkaline conditions, which has been regarded as the preferred metal species for OER. The role of manganese cation on the other hand is to exchange oxygen atoms with the electrolyte at relevant potentials.43 This characteristic of Mn may facilitate OER. In alkaline media, the water oxidation reaction often comprises proton-coupled electron transfer (PCET) activities, namely the PCET transformation of Co3+/OH to Co4+/O, prior to the formation of molecular oxygen.44 So, we can say that under given conditions, cobalt center is the actual active catalytic site, however, manganese center and copper oxide species are proposed to boost the OER ability of cobalt center via different mechanistic roles, as stated above.
Determining the faradaic efficiency of a catalyst is a beneficial parameter for elucidating its oxygen generation ability (FE). To obtain the FE, the actual and predicted yields of evolved oxygen are compared by using chronoamperometry. To check the faradaic efficiency of the catalyst, controlled potential electrolysis technique (CPE) was performed utilizing comparable electrochemical reaction conditions at a static potential of 1.45 V versus RHE for a period of 1 h.45 To establish a baseline, DO meter probe was immersed in an electrolyte of airtight anodic chamber and the concentration of dissolved oxygen was measured for an hour. The expected yield of O2 was estimated using Faraday's law and the charge stored during the electrochemical reaction. The faradaic efficiency was computed through the actual and theoretical yield for CoMn-LDH@CuO/Cu2O (92.4%), CoMn-LDH (75.6%), and CuO/Cu2O (62.3%) (Fig. S1 and Table S1†). The current due to the redox reaction of metals is also involved along with water electrolysis.
Electrochemical impendence spectroscopy was used to evaluate the conductivities of CoMn-LDH, CuO/Cu2O, and CoMn-LDH@CuO/Cu2O composite between 0.1 Hz and 100 kHz. The charge transfer resistance (Rct) is related with the semicircle diameter in the Nyquist plot's high frequency region, and a small Rct value indicates a quick reaction rate. In Fig. 5a it can be clearly shown that CoMn-LDH@CuO/Cu2O has small radii of its semi-circle and low resistance to charge transfer (Rct) as compared to individual CoMn-LDH and CuO/Cu2O, thus revealing its fast OER kinetics and low charge transfer resistance. The low charge transfer resistance, which facilitates efficient charge transport, is owing in part to the dispersion of CuO/Cu2O and the synergistic impact of the presence of several transition metals, as well as other intrinsic factors. For commercial and industrial water electrolyzer, the stability of catalyst is a critical criterion. Since catalyst's catalytic activity and selectivity are the key parameters, its effectiveness is critical unless it exhibits adequate long-term stability. For this purpose, a 15 h controlled-potential electrolysis (CPE) study was conducted under constant experimental conditions utilizing chronoamperometry at 1.42 V against RHE to determine the stability and durability of the catalyst CoMn-LDH@CuO/Cu2O. A steady current of 10 mA was maintained by the catalyst until the last minute (Fig. 5b). Linear sweep voltammetry has been performed to check the durability of the catalyst. From the inset of Fig. 5b it is cleared that the onset potential and maximum current density values of the LSV of modified electrode (CoMn-LDH@CuO/Cu2O) that has been coated (pristine and post-catalytic reactions) showed no significant change, indicating that structure of CoMn-LDH@CuO/Cu2O remained the same throughout the catalytic phenomena. XPS analysis of the post catalytic sample has also been performed. There is no significant change observed which indicates that catalyst's structure and physical properties are retained after CPE test of 15 h (Fig. S3–S5†). Table 1 shows the comparative analysis of the as prepared anode with previously reported composites of LDHs. The most probable proposed mechanism for oxygen evolution reaction in alkaline media is shown in Fig. S6.† In the first step metal active site bind with OH- ion from water to form M-OH. Further oxidation leads to the formation of M–O specie followed by M–OOH which is thermodynamically unfavorable intermediate because of the weak interaction between metal and –OOH. In the last step metal oxygen bond breaks leading to the formation of O2. The comparative study of various reported catalysts with the CoMn-LDH@CuO/Cu2O is shown in Table 1.
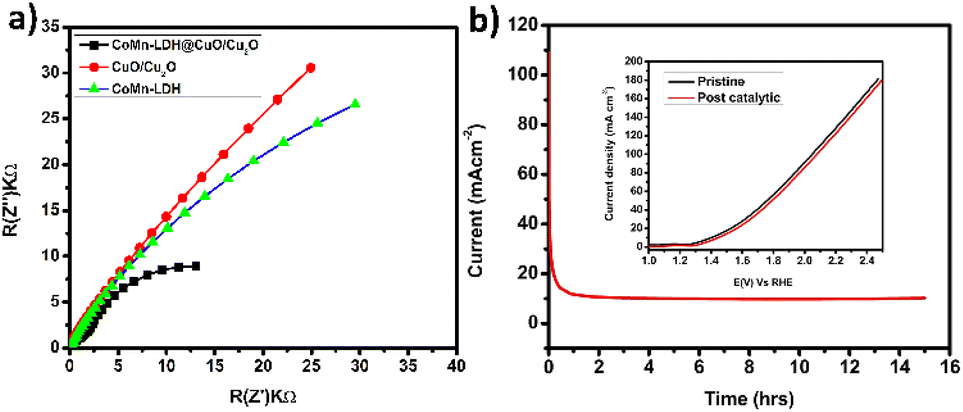 | ||
| Fig. 5 (a) EIS spectra of CoMn-LDH, CuO/Cu2O, CoMn-LDH@CuO/Cu2O between the range of 0.1 Hz and 100 kHz. (b) Controlled potential electrolysis of CoMn-LDH@CuO/Cu2O for 15 h (inset) LSV curve of pristine and post catalytic CoMn-LDH@CuO/Cu2O. | ||
| Catalyst | Method | Overpotential at 10 mA cm−2 (mV) | Ref. |
|---|---|---|---|
| Fabrication of CoFe-LDH over graphitic nitrides | Co-precipitation | 280 | 46 |
| NiFeMn-LDH | Hydrothermal | 289 | 47 |
| Co @ NiFe-LDH | Hydrothermal | 267 | 48 |
| Co intake Fe Ni-LDH | Hydrothermal | 220 | 49 |
| Al @ NiFe-LDH | Solvothermal | 300 | 50 |
| NiMn-LDH@MWCNT | Refluxing | 350 | 51 |
| Ni Fe-LDH on cabon quantum | Solvothermal | 235 | 21 |
| CoFe-LDH @ Cu foam | Electrodeposition | 240 | 52 |
| CoFe-LDH | Hydrothermal | 266 | 53 |
| Enriching vacancy CoFe-LDH | Acid Etching | 302 | 54 |
| CoFe-LDH@g-C3N4 | Hydrothermal | 322 | 55 |
| CoMn-LDH@MWCNT | Refluxing | 300 | 51 |
| CoMn-LDH@CuO/Cu2O | Hydrothermal | 297 | This work |
4 Conclusions
In conclusion, CoMn-LDH@CuO/Cu2O was successfully fabricated by a simple and facile process. CuO/Cu2O nanoparticles produced by the calcination of HKUST-1 were added during the growth of CoMn-LDH nanosheets and their integration is proved using PXRD, SEM, TEM, EDX and XPS techniques. The desired electrocatalyst exhibits High performance OER activity. To drive a current density of 10 mA cm−2, the optimized anode only requires an overpotential of 297 mV with the Tafel slope value (89 mV dec−1) which is remarkably lower than the individual CoMn-LDH and CuO/Cu2O. Additionally, our catalyst displays high stability for OER with negligible current loss during a 15 h period. Our experimental results indicates that the interaction of CoMn-LDH and CuO/Cu2O increased the electrochemical active surface area and decreased the charge transfer resistance, both of which contribute to the excellent OER performance of the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Pakistan Academy of Sciences (PAS) and Higher Education Commission (HEC) of Pakistan (No. 8400/Federal/NRPU/R&D/HEC/2017).Notes and references
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- T. Audichon, T. W. Napporn, C. Canaff, C. u. Morais, C. m. Comminges and K. B. Kokoh, J. Phys. Chem. C, 2016, 120(5), 2562–2573 CrossRef CAS.
- T. Najam, S. S. A. Shah, W. Ding, Z. Ling, L. Li and Z. Wei, Electrochim. Acta, 2019, 327, 134939 CrossRef CAS.
- S. Ibraheem, X. Li, S. S. A Shah, T. Najam, G. Yasin, R. Iqbal, S. Hussain, W. Ding and F. Shahzad, ACS Appl. Mater. Interfaces, 2021, 13(9), 10972–10978 CrossRef CAS PubMed.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9(3), 1314–1352 RSC.
- A. Hameed, M. Batool, Z. Liu, M. A. Nadeem and R. Jin, ACS Energy Lett., 2022, 7, 3311–3328 CrossRef.
- X. Han, Z. Lin, X. He, L. Cui and D. Lu, Int. J. Hydrogen Energy, 2020, 45(51), 26989–26999 CrossRef CAS.
- C. Ziegler, S. Werner, M. Bugnet, M. Wörsching, V. Duppel, G. A. Botton, C. Scheu and B. V. Lotsch, Chem. Mater., 2013, 25(24), 4892–4900 CrossRef CAS.
- C. Tang, H. S. Wang, H. F. Wang, Q. Zhang, G. L. Tian, J. Q. Nie and F. Wei, Adv. Mater., 2015, 27(30), 4516–4522 CrossRef CAS PubMed.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan and G. Qian, Adv. Mater., 2017, 29(17), 1700017 CrossRef PubMed.
- X. Han, N. Suo, C. Chen, Z. Dou, X. He and L. Cui, Int. J. Hydrogen Energy, 2019, 44(57), 29876–29888 CrossRef CAS.
- Q. Zhou, Y. Chen, G. Zhao, Y. Lin, Z. Yu and X. Xu, ACS Catal., 2018, 8(6), 5382–5390 CrossRef CAS.
- J. Yang, C. Yu, C. Hu, M. Wang, S. Li, H. Huang, K. Bustillo, X. Han, C. Zhao and W. Guo, Adv. Funct. Mater., 2018, 28(44), 1803272 CrossRef.
- C. You, Y. Ji, Z. Liu, X. Xiong and X. Sun, ACS Sustainable Chem. Eng., 2018, 6(2), 1527–1531 CrossRef CAS.
- S. Zhang, Z. Liu, D. Chen, Z. Guo and M. Ruan, Chem. Eng. J., 2020, 395, 125101 CrossRef CAS.
- L. Foruzin and Z. Rezvani, Ultrason. Sonochem., 2020, 64, 104919 CrossRef PubMed.
- H. Zhou, F. Wu, L. Fang, J. Hu, H. Luo, T. Guan, B. Hu and M. Zhou, Int. J. Hydrogen Energy, 2020, 45(23), 13080–13089 CrossRef CAS.
- R. A. Senthil, J. Pan, X. Yang and Y. Sun, Int. J. Hydrogen Energy, 2018, 43(48), 21824–21834 CrossRef CAS.
- X. Wen, Int. J. Hydrogen Energy, 2020, 45(29), 14660–14668 CrossRef CAS.
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., 2014, 126(29), 7714–7718 CrossRef.
- D. Tang, J. Liu, X. Wu, R. Liu, X. Han, Y. Han, H. Huang, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2014, 6(10), 7918–7925 CrossRef CAS PubMed.
- L. Huang, L. Ai, M. Wang, J. Jiang and S. Wang, Int. J. Hydrogen Energy, 2019, 44(2), 965–976 CrossRef CAS.
- I. Majeed, M. A. Nadeem, A. Badshah, F. K. Kanodarwala, H. Ali, M. A. Khan, J. A. Stride and M. A. Nadeem, Catal. Sci. Technol., 2017, 7(3), 677–686 RSC.
- H. Niu, S. Liu, Y. Cai, F. Wu and X. Zhao, Microporous Mesoporous Mater., 2016, 219, 48–53 CrossRef CAS.
- L. Zheng, X. Li, W. Du, D. Shi, W. Ning, X. Lu and Z. Hou, Appl. Catal., B, 2017, 203, 146–153 CrossRef CAS.
- B. Peng, C. Feng, S. Liu and R. Zhang, Catal. Today, 2018, 314, 122–128 CrossRef CAS.
- Q. M. Wang, D. Shen, M. Bülow, M. L. Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55(2), 217–230 CrossRef CAS.
- W. Iqbal, M. Batool, A. Hameed, S. Abbas and M. A. Nadeem, Int. J. Hydrogen Energy, 2021, 46, 25050–25059 CrossRef CAS.
- A. Hameed, M. Batool, W. Iqbal, S. Abbas, M. Imran, I. A. Khan and M. A. Nadeem, Front. Chem., 2021, 9, 379 Search PubMed.
- S. Anantharaj, S. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11(4), 744–771 RSC.
- J. S. Valente, E. Lima, J. A. Toledo-Antonio, M. A. Cortes-Jacome, L. Lartundo-Rojas, R. Montiel and J. Prince, J. Phys. Chem. C, 2010, 114(5), 2089–2099 CrossRef CAS.
- D. Tuncel and A. Ökte, Catal. Today, 2019, 328, 149–156 CrossRef CAS.
- R. Zhang, L. Hu, S. Bao, R. Li, L. Gao, R. Li and Q. Chen, J. Mater. Chem. A, 2016, 4(21), 8412–8420 RSC.
- L. Hu, Y. Huang, F. Zhang and Q. Chen, Nanoscale, 2013, 5(10), 4186–4190 RSC.
- Y. Yang, H. Dong, Y. Wang, C. He, Y. Wang and X. Zhang, J. Solid State Chem., 2018, 258, 582–587 CrossRef CAS.
- P. He, Y. Fang, X. Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56(40), 12202–12205 CrossRef CAS PubMed.
- L. G. Bloor, P. I. Molina, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2014, 136(8), 3304–3311 CrossRef CAS PubMed.
- Z. Zeng, M. K. Chan, Z. J. Zhao, J. Kubal, D. Fan and J. Greeley, J. Phys. Chem. C, 2015, 119(32), 18177–18187 CrossRef CAS.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134(6), 2930–2933 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132(39), 13692–13701 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133(14), 5587–5593 CrossRef CAS PubMed.
- J. Chen and A. Selloni, J. Phys. Chem. C, 2013, 117(39), 20002–20006 CrossRef CAS.
- H. Y. Su, Y. Gorlin, I. C. Man, F. Calle-Vallejo, J. K. Nørskov, T. F. Jaramillo and J. Rossmeisl, Phys. Chem. Chem. Phys., 2012, 14(40), 14010–14022 RSC.
- G. Mattioli, P. Giannozzi, A. Amore Bonapasta and L. Guidoni, J. Am. Chem. Soc., 2013, 135(41), 15353–15363 CrossRef CAS PubMed.
- M. Agote-Arán, I. Lezcano-González, A. G. Greenaway, S. Hayama, S. Díaz-Moreno, A. B. Kroner and A. M. Beale, Appl. Catal., A, 2019, 570, 283–291 CrossRef.
- M. K. Kundu, R. Mishra, T. Bhowmik, S. Kanrar and S. Barman, Int. J. Hydrogen Energy, 2020, 45(11), 6036–6046 CrossRef CAS.
- Z. Lu, L. Qian, Y. Tian, Y. Li, X. Sun and X. Duan, Chem. Commun., 2016, 52(5), 908–911 RSC.
- X. Long, S. Xiao, Z. Wang, X. Zheng and S. Yang, Chem. Commun., 2015, 51(6), 1120–1123 RSC.
- G. Rajeshkhanna, T. I. Singh, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10(49), 42453–42468 CrossRef CAS PubMed.
- H. Liu, Y. Wang, X. Lu, Y. Hu, G. Zhu, R. Chen, L. Ma, H. Zhu, Z. Tie and J. Liu, Nano Energy, 2017, 35, 350–357 CrossRef CAS.
- G. Jia, Y. Hu, Q. Qian, Y. Yao, S. Zhang, Z. Li and Z. Zou, ACS Appl. Mater. Interfaces, 2016, 8(23), 14527–14534 CrossRef CAS PubMed.
- L. Yu, H. Zhou, J. Sun, F. Qin, D. Luo, L. Xie, F. Yu, J. Bao, Y. Li and Y. Yu, Nano Energy, 2017, 41, 327–336 CrossRef CAS.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56(21), 5867–5871 CrossRef CAS PubMed.
- P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2017, 53(86), 11778–11781 RSC.
- M. Arif, G. Yasin, M. Shakeel, M. A. Mushtaq, W. Ye, X. Fang, S. Ji and D. Yan, Mater. Chem. Front., 2019, 3(3), 520–531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05036f |
| This journal is © The Royal Society of Chemistry 2022 |