
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne pot tandem dehydrogenative cross-coupling of primary and secondary alcohols by ruthenium amido-functionalized 1,2,4-triazole derived N-heterocyclic carbene complexes†
Anuj Kumar‡
a,
Sabyasachi Ta‡a,
Chandrasekhar Nettem‡a,
Joseph M. Tanskib,
Gopalan Rajaraman *a and
Prasenjit Ghosh*a
*a and
Prasenjit Ghosh*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400 076, India. E-mail: pghosh@chem.iitb.ac.in; anujchem007@gmail.com; sabyasachita@iitb.ac.in; rajaraman@chem.iitb.ac.in; Fax: +91 22 2572 3480
bDepartment of Chemistry, Vassar College, 124 Raymond Avenue, Poughkeepsie, NY 12604, USA. E-mail: jotanski@vassar.edu
First published on 13th October 2022
Abstract
One-pot tandem dehydrogenative cross-coupling of primary and secondary alcohols was catalyzed by three ruthenium complexes [1-(R)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]Ru(p-cymene)Cl [R = Et (1b), i-Pr (2b), Bn (3b)], of amido-functionalized 1,2,4-triazole derived N-heterocyclic carbene (NHC) ligands. Density Functional Theory (DFT) calculations were employed for the ruthenium (1b) precatalyst to understand this reaction mechanism completely, and the mechanisms adapted are divided categorically into three steps (i) nucleophilic substitution of chloride ions by alcohols, (ii) dehydrogenation of primary and secondary alcohols, and (iii) olefin and ketone hydrogenation. Our mechanistic study reveals that the formation of a deprotonated Ru–alcoholate (A) or (E) intermediate is favorable compared to the protonated form (A′) or (E′) from (1b) by associative nucleophilic substitution. Though an ionic pathway that proceeds through (A′) or (E′), has less barriers in the dehydrogenation and olefin/ketone hydrogenation steps than that of the neutral pathway, proceeding through (A) or (E), a steep energy barrier was observed in the first nucleophilic substitution step, prohibiting the reaction to proceed via the intermediate (A′) or (E′). Thus, our thorough mechanistic study reveals that the reaction proceeds via deprotonated Ru–alcoholate (A) or (E) species. Furthermore, the 1,4 addition of an α,β-unsaturated carbonyl compound is kinetically and thermodynamically favorable over the 1,2 addition, and the experiments support these observations. As a testimony towards practical application in synthesizing bio-active flavonoid based natural products, five different flavan derivatives (16–20), were synthesized by the dehydrogenative coupling reaction using the neutral ruthenium (1–3)b complexes.
Introduction
Step-efficient multi-sequence reactions, performed in one-pot, are an attractive proposition for synthesizing complex target molecules in organic synthesis, and, when performed catalytically are even more desirable.1–3 One such reaction is the alcohol–alcohol coupling that provides direct access to a large variety of long-chain and branched alcohols of interest to industry and academia alike.4 A related approach includes Guerbet coupling, that is performed both homogeneously5,6 and heterogeneously6–8 for producing bio-based alcohol feedstocks.9 The attraction of the alcohol–alcohol coupling resides on its, (i) ability to achieve oxidation of secondary and primary alcohols to the corresponding carbonyl compounds, (ii) subsequent cross-aldol condensation between them to form the α,β-unsaturated ketone, and (iii) finally the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds to secondary alcohol, all occurring in one-pot and that too, in a tandem fashion.10–12 Needless to say, that the alternate approach of achieving the target product would involve oxidation of the secondary alcohols, alkylation with alkyl halides, and the reduction of the α-alkylated ketones as independent reactions, thus undermining the overall reaction yields.1–3 Additionally, the alcohol–alcohol coupling provides a greener synthetic approach involving the elimination of water as the only byproduct of the reaction.11,13
C and CO bonds to secondary alcohol, all occurring in one-pot and that too, in a tandem fashion.10–12 Needless to say, that the alternate approach of achieving the target product would involve oxidation of the secondary alcohols, alkylation with alkyl halides, and the reduction of the α-alkylated ketones as independent reactions, thus undermining the overall reaction yields.1–3 Additionally, the alcohol–alcohol coupling provides a greener synthetic approach involving the elimination of water as the only byproduct of the reaction.11,13
Transition metals spear-headed the development of the homogeneously catalyzed alcohol–alcohol coupling reactions with a variety of first row transition metals like Mn,14 Fe,4,15 Co,16,17 Cu,18–20 and Ni,21,22 second row transition metals like Ru10,23–26 and Rh,27 and third row transition metal namely Ir,11,25,28,29 been reported. With our recent interest on ruthenium,30,31 we decided to focus on ruthenium for the alcohol–alcohol coupling reactions.32 The ruthenium catalyzed alcohol–alcohol coupling reactions have been developed on the back of different ancillary ligands that primarily range from a variety of [PCP],33 [NNN],10,34 [NPN],35 [NNP]26 and [NNC]26 pincer ligands to tris-chelating tris(pyrazolyl)borate (Tp) ligands23 to bis-chelating NN-bipyridine36 and [NC]-pyrimidine based NHC ligands25 to mono-chelating phosphine ligands.37 Proceeding further along the lines we set out to study the catalytic utility of the less explored triazole derived N-heterocyclic carbenes in the dehydrogenative cross-coupling reaction.
In this regard, the earlier DFT calculations performed for cross-coupling reactions on Ru(II)–NHC complexes suggested participation of ruthenium hydride active species proceeding by a metal–ligand non-cooperative pathway.24,38 However, for the ruthenium complexes of different ligand architectures, the formation of ligand protonated ruthenium hydride active species involving an outer-sphere hydrogen atom transfer in the dehydrogenation step, in accordance with a metal–ligand cooperative mechanism, has found credence based on experimental results.26,39,40 Keeping this in mind, we have explored both the possibilities, i.e. the metal–ligand cooperative and non-cooperative mechanisms, in our calculations.
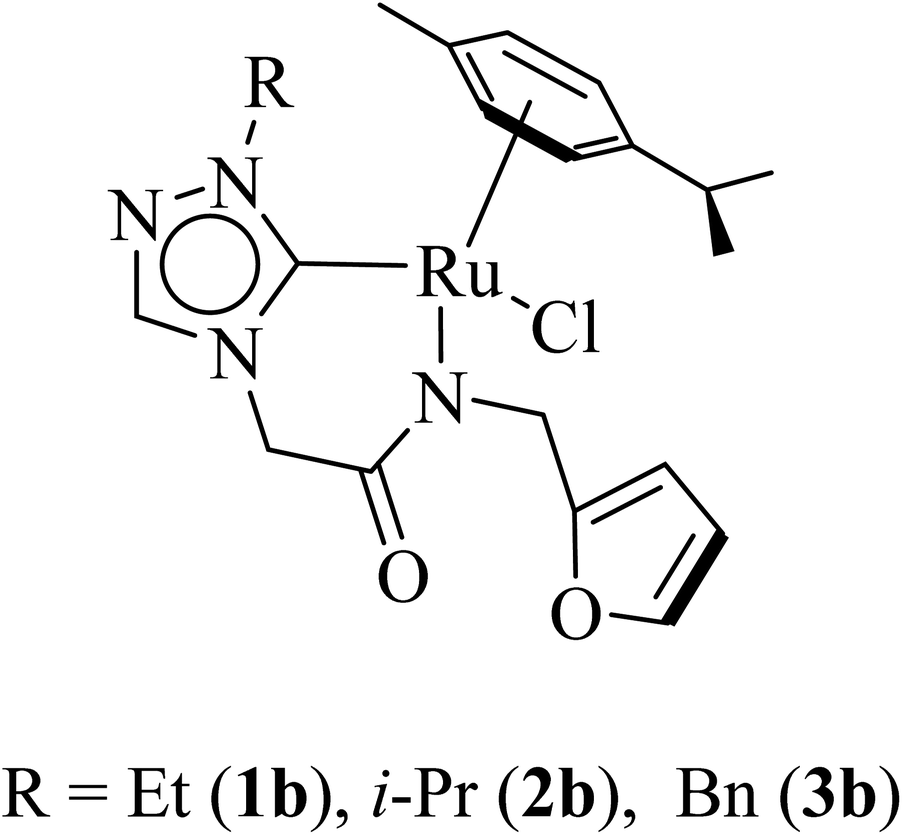 | ||
| Fig. 1 Synthesized neutral amido functionalized 1,2,4-triazole based Ru–NHC (1–3)b complexes. | ||
Combined experimental and theoretical investigations about the development of the homogeneously catalyzed one-pot tandem alcohol–alcohol coupling reactions with less explored triazole derived ruthenium N-heterocyclic carbene complexes are undertaken, and which, we believe, will provide valuable insight in the development of the domain. Here in this manuscript we report a series of neutral ruthenium (1–3)b complexes of amido functionalized 1,2,4-triazole based N-heterocyclic carbenes for dehydrogenative cross-coupling of primary and secondary alcohols (Fig. 1). The study further provides insights on the key mechanistic pathways based on the experimental and computational studies. Additionally, the utility of the synthetic approach of performing multistep sequences in one-pot tandem fashion has been realized by synthesizing a variety of plant based bioactive flavonoids (16–20).
Results and discussions
Three ruthenium complexes [1-(R)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]Ru(p-cymene)Cl [R = Et (1b), i-Pr (2b), Bn (3b)], stabilized over amido-functionalised 1,2,4-triazole derived N-heterocyclic carbene ligands were synthesized for exploring their utility in the tandem alcohol–alcohol coupling reaction between secondary and primary alcohols. These ruthenium (1–3)b complexes were conveniently synthesized from their respective silver complexes, {[1-(R)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− [R = Et (1a),41 i-Pr (2a)41 and Bn (3a)] (Scheme 1). The silver complex (3a) was obtained by treatment of the 1-(Bn)-4-N (furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride (3) salt with Ag2O (Scheme 1) ca. 81% yield, and was confirmed by appearance of diagnostic Ag–Ccarbene resonance at 183.3 ppm in the 13C{1H} NMR. The IR spectrum of 3a showed the amido–CO stretching frequency significantly red shifted at 1667 cm−1, as compared to that the free ligand 3 (1595 cm−1).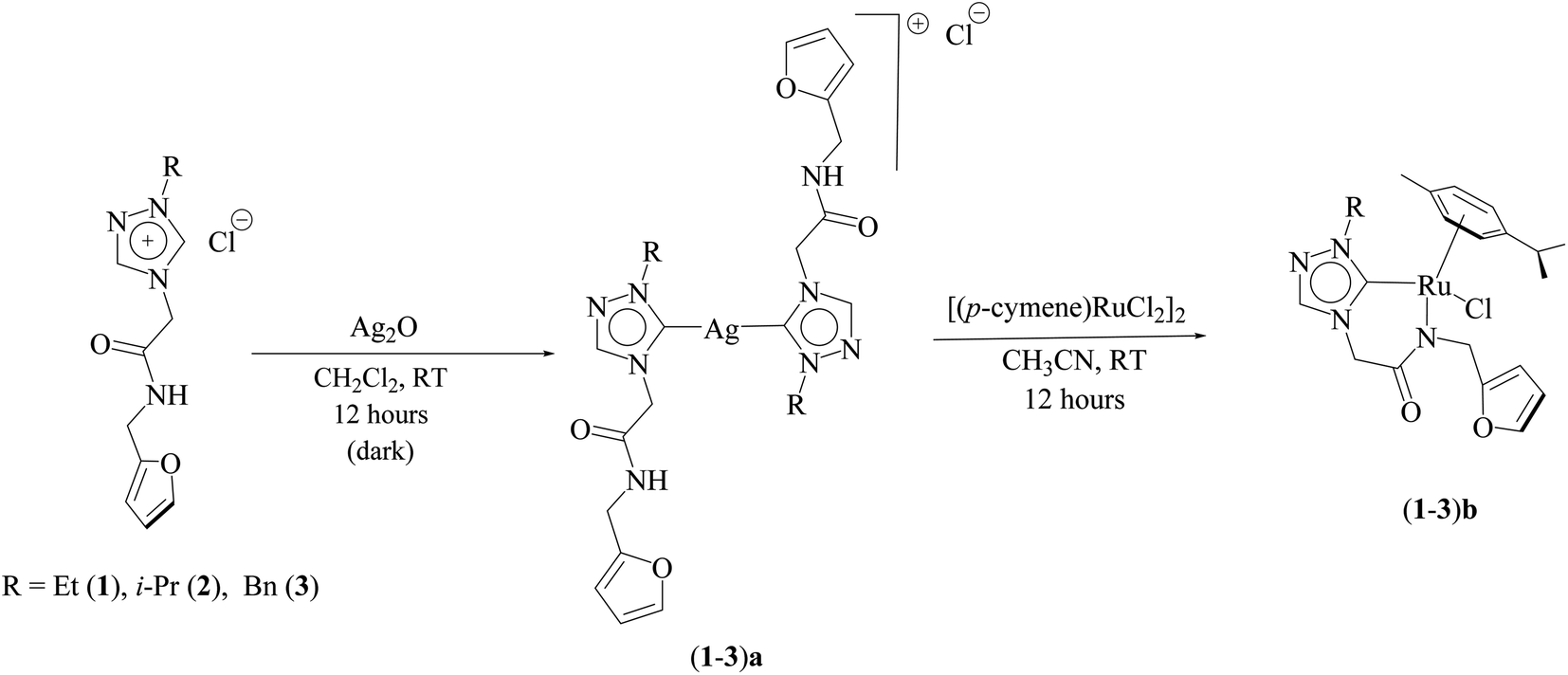 | ||
| Scheme 1 Synthetic route to amido-functionalized Ru–NHC complexes (1–3)b. | ||
The synthesis of the 1,2,4-triazole derived N-heterocyclic carbene complexes of ruthenium (1–3)b were similar to that of the related imidazole based N-heterocyclic carbene analogous reported by us.30,32 The 1H NMR spectrum of the (1–3)b complexes showed the conspicuous absence of the amido (CON![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ) resonance, unlike that of its starting silver (1–3)a complexes and there by suggesting the deprotonation of the (CON) moiety leading to the chelation of the amido sidearm to the ruthenium centre. As expected, the two (C2) moieties of the ruthenium (1–3)b complexes showed two pairs of diastereotopic resonances in the 1H NMR. For example, the (C2) moiety of the amido sidearm appeared as two sets of doublets at ca. 4.98–5.07 ppm and ca. 4.30–4.64 ppm exhibiting a two bond geminal coupling (2JHH) of ca. 15–16 Hz, similar to that seen in the related benzimidazole based analogous namely [{1-(N-R1-2-acetamido)-3-(R2)-benzimidazol-2-ylidine}Ru(p-cymene)Cl]Cl, where {R1 = 2,6-(i-Pr)2C6H3, R2 = i-Pr; (ca. 5.32 and 5.31 ppm and 2JHH ca. 14 Hz);31 R1 = 2,6-(i-Pr)2C6H3, R2 = Et; (ca. 5.41 and 5.40 ppm and 2JHH ca. 14 Hz);31 R1 = 2,4,6-(CH3)3C6H2, R2 = Et; (ca. 5.18 and 5.02 ppm and 2JHH ca. 15 Hz).31 The other (C2) moiety of the furan sidearm in the ruthenium appeared at ca. 4.39–4.63 ppm and ca. 4.30–4.35 ppm with a two bond geminal coupling (2JHH) of ca. 15–16 Hz in the 1H NMR spectra.
) resonance, unlike that of its starting silver (1–3)a complexes and there by suggesting the deprotonation of the (CON) moiety leading to the chelation of the amido sidearm to the ruthenium centre. As expected, the two (C2) moieties of the ruthenium (1–3)b complexes showed two pairs of diastereotopic resonances in the 1H NMR. For example, the (C2) moiety of the amido sidearm appeared as two sets of doublets at ca. 4.98–5.07 ppm and ca. 4.30–4.64 ppm exhibiting a two bond geminal coupling (2JHH) of ca. 15–16 Hz, similar to that seen in the related benzimidazole based analogous namely [{1-(N-R1-2-acetamido)-3-(R2)-benzimidazol-2-ylidine}Ru(p-cymene)Cl]Cl, where {R1 = 2,6-(i-Pr)2C6H3, R2 = i-Pr; (ca. 5.32 and 5.31 ppm and 2JHH ca. 14 Hz);31 R1 = 2,6-(i-Pr)2C6H3, R2 = Et; (ca. 5.41 and 5.40 ppm and 2JHH ca. 14 Hz);31 R1 = 2,4,6-(CH3)3C6H2, R2 = Et; (ca. 5.18 and 5.02 ppm and 2JHH ca. 15 Hz).31 The other (C2) moiety of the furan sidearm in the ruthenium appeared at ca. 4.39–4.63 ppm and ca. 4.30–4.35 ppm with a two bond geminal coupling (2JHH) of ca. 15–16 Hz in the 1H NMR spectra.
The diagnostic Ccarbene–Ru resonance appeared at ca. 180.5 ppm (1b), 179.3 ppm (2b) and 182.3 ppm (3b) similar to that observed in other reported neutral analogous namely [{1-(benzylacetamido)-3-(R)-imidazole-2-ylidene}Ru(p-cymene)Cl] {R = Me [δ 175.2 ppm], i-Pr [δ 175.4 ppm], and CH2Ph [δ 175.9 ppm]}30 and [1-mesityl-3-(2,6-Me2-phenylacetamido)-imidazole-2-ylidene]Ru(p-cymene)Cl [δ 169.7 ppm].32 The amido–CO stretching frequency appeared at 1589 (1b) cm−1, 1584 (2b) cm−1 and 1587 (3b) cm−1 and was found to be significantly red shifted with respect to the corresponding free NHC ligand precursors, 1 (1686 cm−1),42 2 (1689 cm−1)42 and 3 (1669 cm−1).42
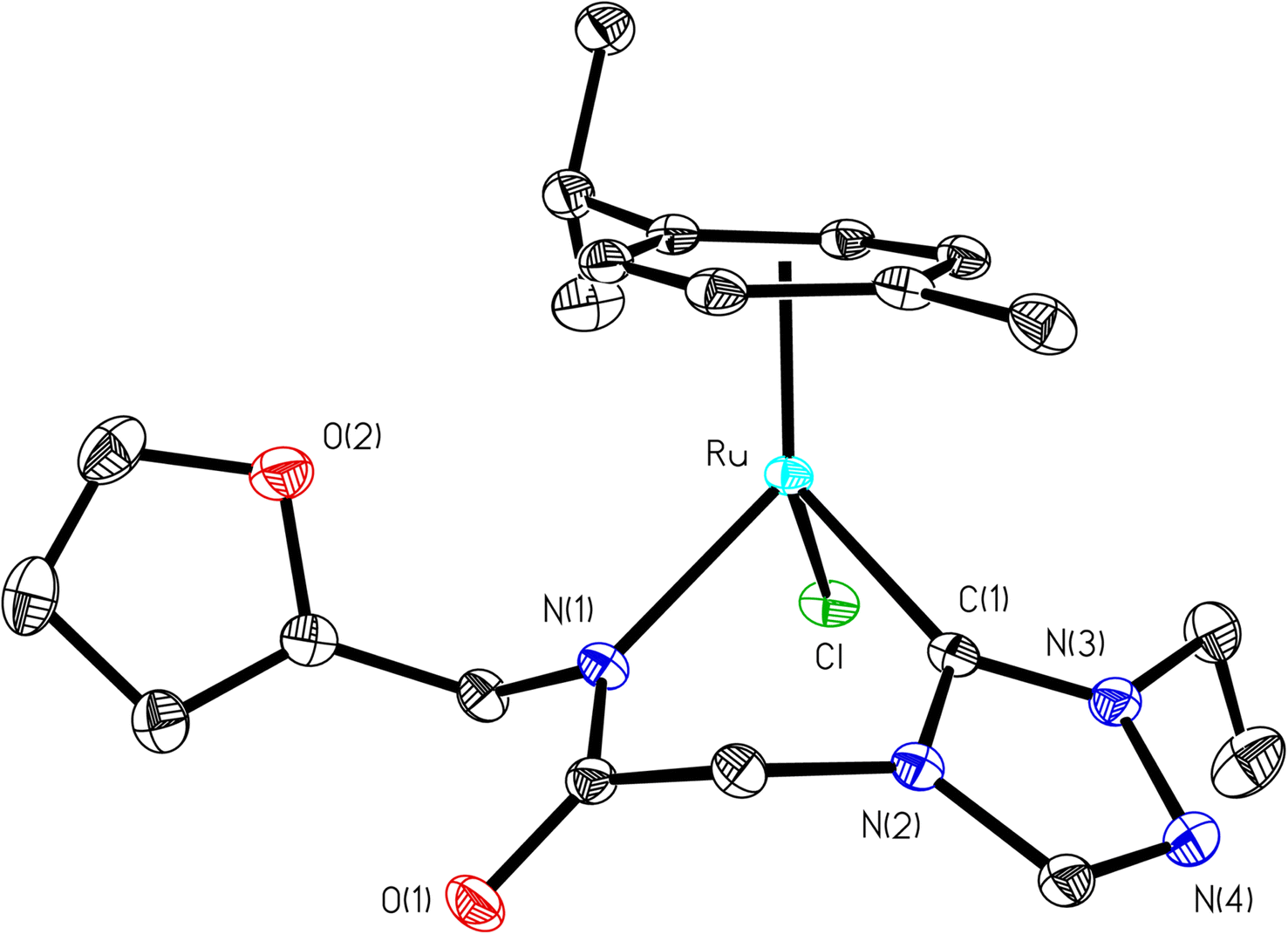 | ||
| Fig. 2 ORTEP diagram of 1b with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): Ru–C1 2.0193(13), Ru–N1 2.1226(11), Ru–Cl 2.4095(3), C1–Ru–N1 82.89(5), N1–Ru–Cl 1 87.16(13). | ||
Significantly enough, the (1–3)b complexes represents the only structurally characterized examples of a triazole based N-heterocyclic carbene ruthenium complex known in the literature (Fig. 2, ESI Fig. S15 and 30†). Hence, a comparison is made with the related imidazole based N-heterocyclic carbene counterparts, and as in all of these complexes the metal centre is bound in a “piano-stool” geometry being attached to the cymene moiety, N-amido, Ccarbene and chloride ligand.
The Ru–Ccarbene bond distance in 1b [2.0193(13) Å], 2b [2.0384(19) Å] and 3b [2.016(3) Å] compared well with that of reported neutral analogous, namely [{1-(benzylacetamido)-3-(R)-imidazole-2-ylidene}Ru(p-cymene)Cl] {R = Me [2.0172(19) Å], i-Pr [2.033(5) Å] and CH2Ph [2.019(3) Å]} (Table S2†),30 and [1-mesityl-3-(2,6-Me2-phenylacetamido)-imidazole-2-ylidene]Ru(p-cymene)Cl [2.087(5) Å] (Table S2†).32 Similar observation is made for the Ru–Cl bond distance in 1b [2.4095(3) Å], 2b [2.4319(5) Å] and 3b [2.4347(7) Å] which, also is in agreement with the related neutral ruthenium complexes [{1-(benzylacetamido)-3-(R)-imidazole-2-ylidene}Ru(p-cymene)Cl] {R = Me [2.4404(7) Å], i-Pr [2.4256(14) Å] and CH2Ph [2.4325(8) Å]},30 and [1-mesityl-3-(2,6-Me2-phenylacetamido)-imidazole-2-ylidene]Ru(p-cymene)Cl [2.4299(14) Å].32 The Ru–N bond distances in (1b) [2.1226(11) Å], (2b) [2.1230(16) Å] and (3b) [2.131(2) Å], were slightly shorter than the sum of individual covalent radii of Ru and N atoms (2.15 Å).43 Also, the Ru–Ccentroid distance in 1b [1.716 Å], 2b [1.724 Å] and 3b [1.714 Å] is comparable with the isostructural neutral ruthenium complexes [{1-(benzylacetamido)-3-(R)-imidazole-2-ylidene}Ru(p-cymene)Cl] {R = Me [1.706 Å], i-Pr [1.712 Å] and CH2Ph [1.719 Å]}30 and [1-mesityl-3-(2,6-Me2-phenylacetamido)-imidazole-2-ylidene]Ru(p-cymene)Cl [1.736 Å].32 For the comparison purpose, the Ru–N bond distances in the related imidazole based ruthenium N-heterocyclic carbene complexes are [{1-(benzylacetamido)-3-(R)-imidazole-2-ylidene}Ru(p-cymene)Cl] {R = Me [2.1074(16) Å], i-Pr [2.125(5) Å] and CH2Ph [2.1074(16) Å]}30 and [1-mesityl-3-(2,6-Me2-phenylacetamido)-imidazole-2-ylidene]Ru(p-cymene)Cl [2.153(4) Å].32
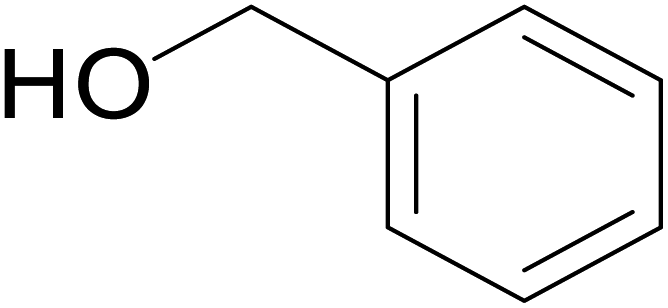
With our recent interest on ruthenium catalyzed one-pot tandem dehydrogenative cross-coupling of primary and secondary alcohols,32 we explored the potential of the 1,2,4-triazole derived N-heterocyclic ruthenium carbene (1–3)b complexes in the reaction. Quite significantly, the ruthenium (1–3)b complexes efficiently carried out the coupling reaction as observed from the good to excellent isolated yields (49–72%) obtained for the secondary alcohol produced in the catalysis. More importantly so, this catalysis provide a greener step efficient pathway for producing secondary alcohols without the generation of any toxic waste as water is the only byproduct. The influence of the N-heterocyclic carbene ligand in the catalysis of the alcohol–alcohol coupling reaction was observed for a representative ruthenium (1b) complex that displayed amplification of the catalysis product [PhCH(OH)CH2CH2Ph] (4), yield by ca. 47% for the substrates, 1-phenylethanol and benzyl alcohol. The yield for this pair of substrates in case of the (1b) complex was ca. 70% while the control experiment performed with [Ru(p-cymene)Cl2]2 gave ca. 23% yield and the blank experiment without any ruthenium initiator showed no product. The homogeneous nature of the catalysis was ascertained from the observation of near equal yields of [PhCH(OH)CH2CH2Ph] (4) obtained in the mercury drop experiment (ca. 61%) and in its absence (ca. 70%) performed for the coupling of the two representative substrates 1-phenylethanol and benzyl alcohol by the (1b) complex (ESI Table S4†).
Additional, insights on the multi-cycle nature of the tandem catalysis came from the time dependence profile of the dehydrogenative cross-coupling between the two representative substrates, 1-phenylethanol and benzyl alcohol, as catalyzed by the ruthenium (1b) complex. The study showed the reaction yield modulating between the CC hydrogenated ketone intermediate, (4′), and the fully CC and CO hydrogenated alcohol product, (4), as a function of time in the catalysis mixture (Fig. 3 and ESI Table S3 and Fig. S109†). The ketone intermediate, PhCOCH2CH2Ph (4′) was observed at an early stage of the reaction at 30 minutes, and after 3 hours the formation of the alcohol product PhCH(OH)CH2CH2Ph (4) dominated. This observation suggested that the alcohol product, PhCH(OH)CH2CH2Ph (4), was formed from the ketone intermediate, PhCOCH2CH2Ph (4′), and on extending the reaction time further to over ca. 120 hours, a systematic modulation occurred between the CC hydrogenated ketone intermediate, PhCOCH2CH2Ph (4′), and the CC and CO hydrogenated alcohol product, PhCH(OH)CH2CH2Ph (4). The ruthenium hydride catalytic species (D), remains active in the catalysis mixture and is behind the repeated interconversions of alcohol to carbonyl compounds using transfer hydrogenation mechanism. Similar phenomenon of interconversions between the alcohol species and the carbonyl species have recently been reported in the literature29 including us.32
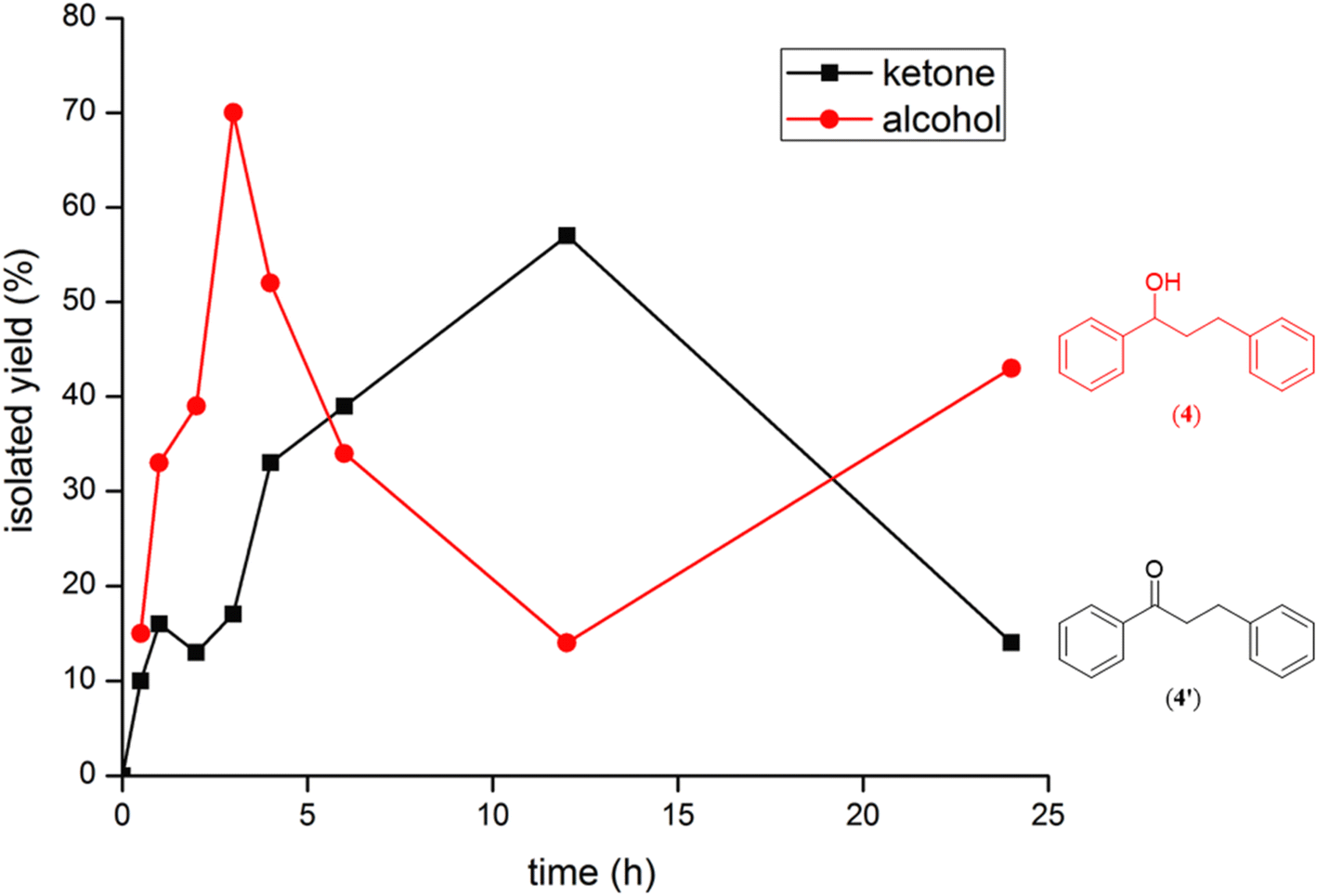 | ||
| Fig. 3 An overlay of the formation of (4) and (4′) as a function of time in the reaction of 1-phenylethanol and benzyl alcohol as catalyzed by the Ru–NHC complex (1b). | ||
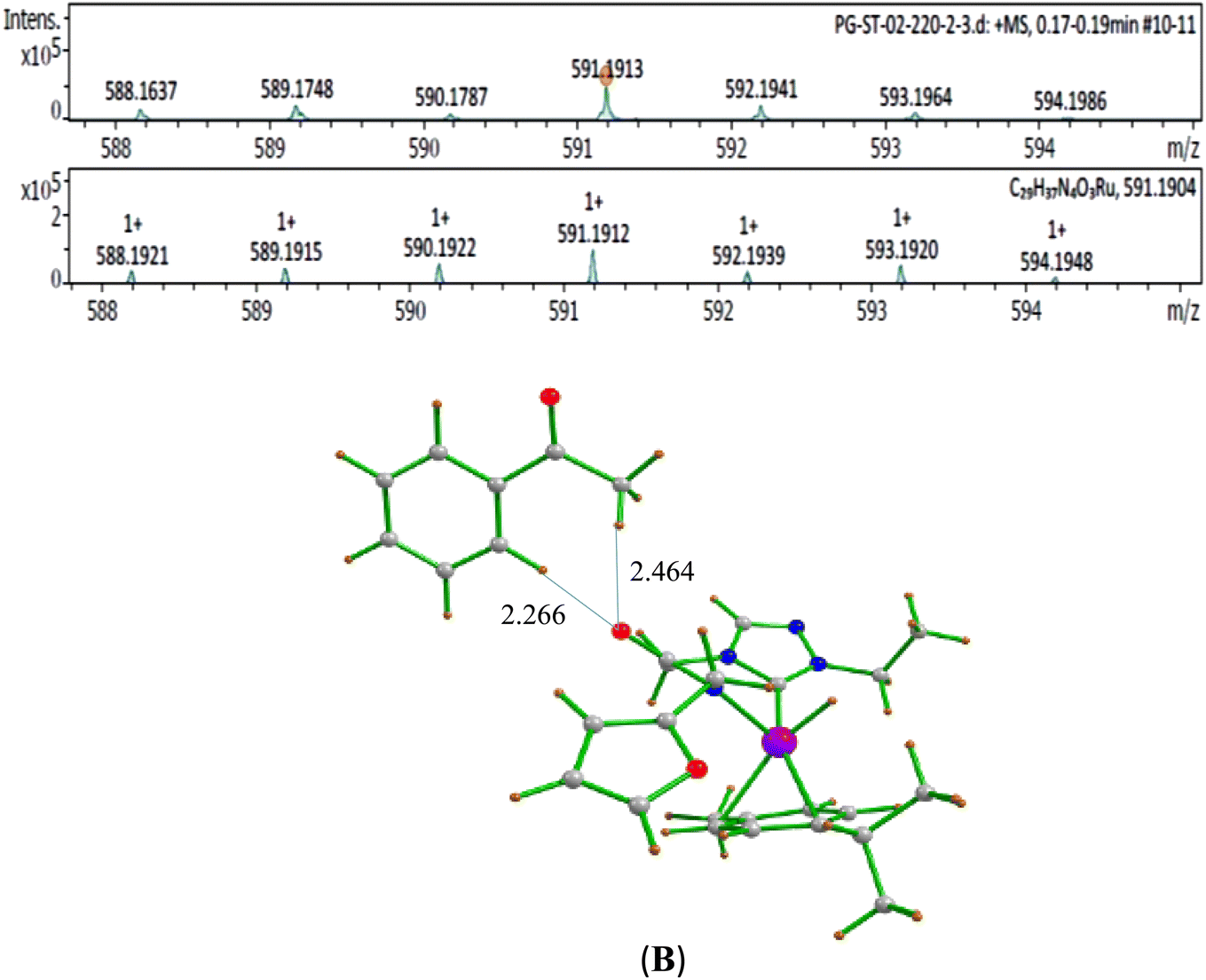 | ||
Fig. 4 HRMS data of the acetophenone bound Ru–H specie (B) detected in the reaction mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio of benzyl alcohol:1-phenylethan-1-ol:NaOiPr 0.1 mmol, 1 mol% of (1b), 2.0 mL of toluene at 110 °C for 5 min [(a) Experimental and (b) simulated pattern of HRMS data]. :1:1 ratio of benzyl alcohol:1-phenylethan-1-ol:NaOiPr 0.1 mmol, 1 mol% of (1b), 2.0 mL of toluene at 110 °C for 5 min [(a) Experimental and (b) simulated pattern of HRMS data]. | ||
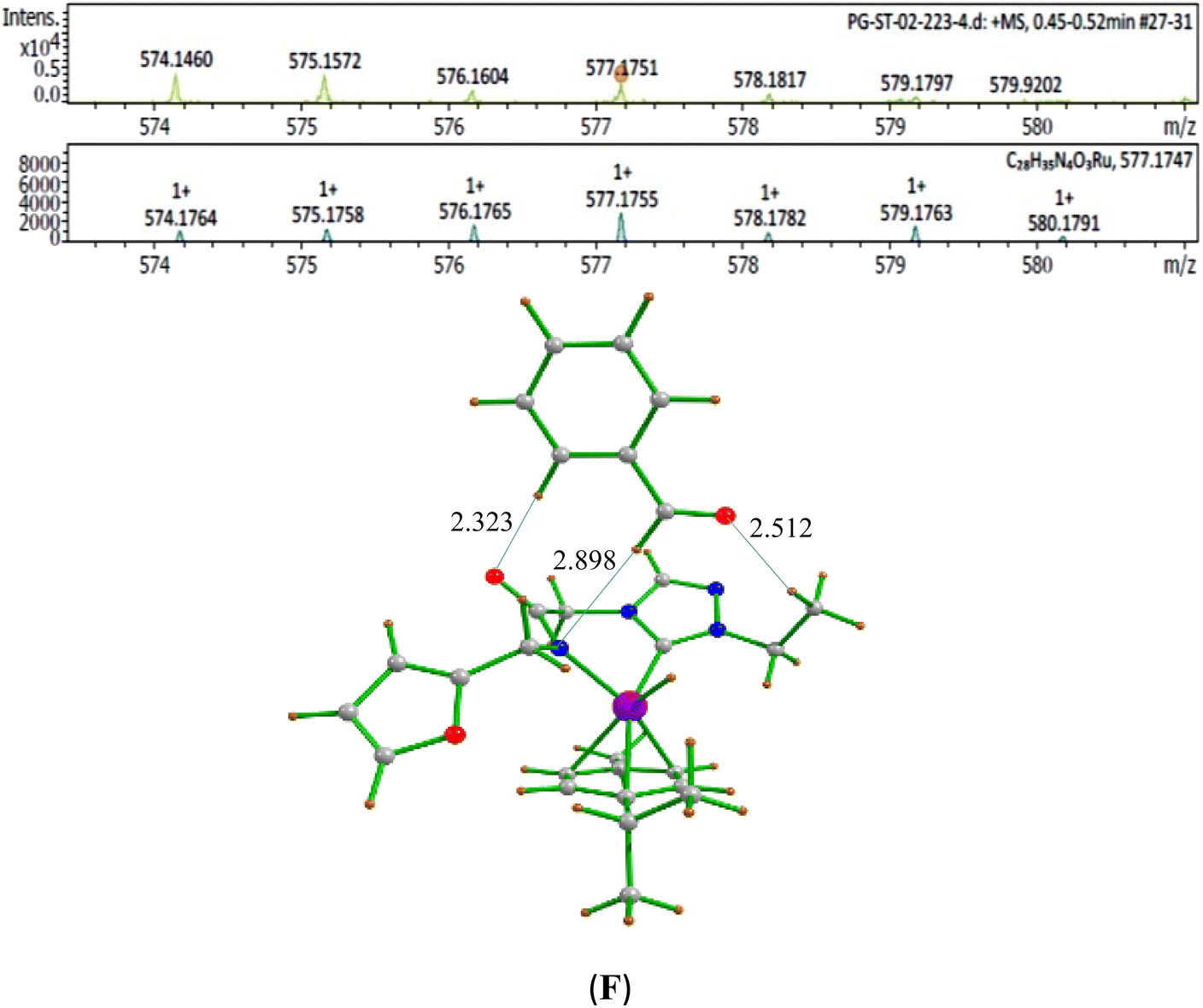 | ||
| Fig. 5 HRMS data of the benzaldehyde bound Ru–H species (F) detected in the reaction mixture of 1:1:1 ratio of benzyl alcohol:1-phenylethanol:NaOiPr 0.1 mmol, 1 mol% of (1b), 2.0 mL of toluene at 110 °C for 20 min [(a) Experimental and (b) simulated pattern of ESI-MS data]. | ||
Significantly enough, the neutral ruthenium (1–3)b complexes successfully carried out the one-pot tandem dehydrogenative cross-coupling of primary and secondary alcohols yielding secondary alcohols at 1 mol% of the ruthenium complex loading in presence of 1 equivalent of NaOiPr as a base at 110 °C in toluene in 3 hours of reaction time. (Table 1) Several combinations of secondary alcohols namely, 1-phenylethanol, 1-(2-methoxyphenyl)ethanol, 1-(benzo[1,3]dioxol-5-yl)ethanol, 1-(naphthalen-2-yl)ethanol and 1-(4-chlorophenyl)ethanol, where successfully alkylated with a group of primary alcohols namely, benzyl alcohol, 4-methylbenzyl alcohol, furan-2-ylmethanol, thiophen-2-ylmethanol, 4-methoxylbenzyl alcohol (o-bromophenyl)methanol, cyclohexylmethanol, and cyclopentaylmethanol by ruthenium (1–3)b complexs in moderate to good yields (ca. 49–72%). A careful scrutiny of the substrate scope study (Table 1) revealed that neutral ruthenium (2b) complex gave a maximum yield of ca. 72% for the reaction of 1-phenylethanol with benzyl alcohol. More interestingly, ruthenium (1–3)b complexes successfully carried out the dehydrogenative cross-coupling of heterocyclic substrates namely, furfuryl alcohol and thiophen-2-ylmethanol, with 1-phenylethanol in moderate to good yield of ca. 49–66%. In this context, the two aliphatic pair of substrates namely cyclohexylmethanol with 1-phenylethanol, and cyclopentylmethanol with 1-phenylethanol, exhibited yields of ca. 61–68% and ca. 62–67% by ruthenium (1–3)b complexes. The reactions of the alcohol substrates bearing nitro, and amine substituents gave no product (entries 13–14 Table 1).
|
||||||
|---|---|---|---|---|---|---|
| S. No. | 2° Alcohol | 1° Alcohol | Product | Ru–NHC (1b) | Ru–NHC (2b) | Ru–NHC (3b) |
| Yieldb | Yieldb | Yieldb | ||||
| a Reaction conditions: 1:1:1 ratio of 1°-alcohol:2°-alcohol:base 1.00 mmol, 1 mol% of catalyst (1b)/(2b)/(3b), 2.0 mL of toluene at 110 °C for 3 hours.b Isolated yields (%). |
||||||
| 1 | 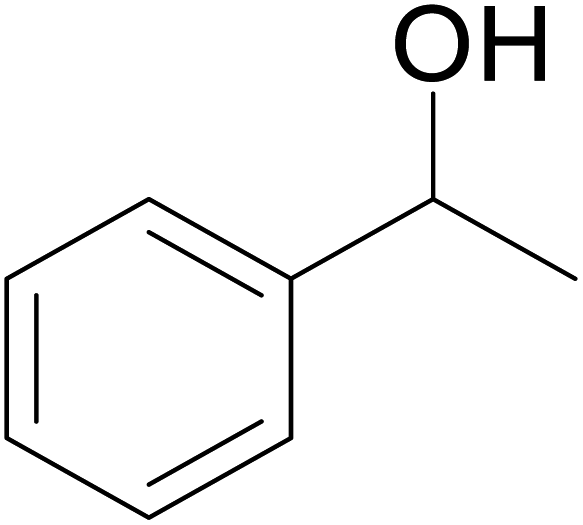 |
 |
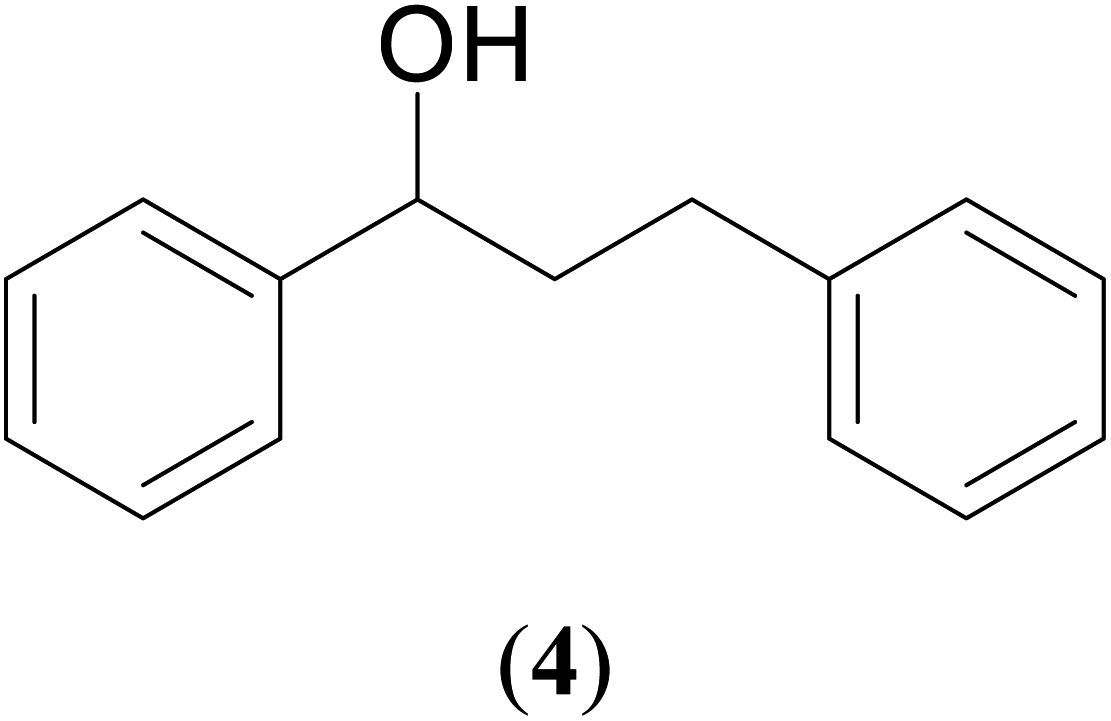 |
70 | 72 | 68 |
| 2 | 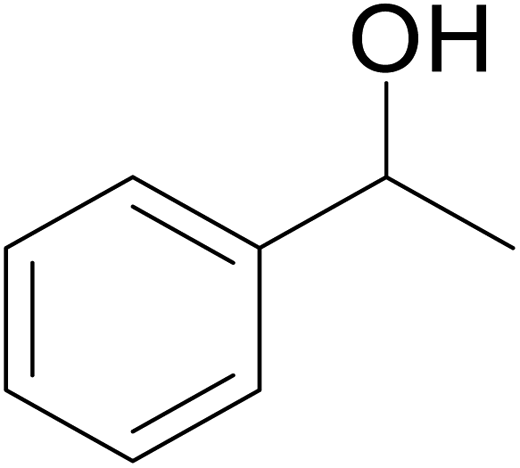 |
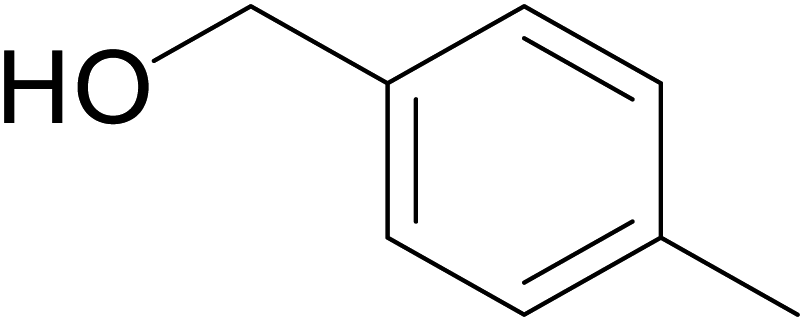 |
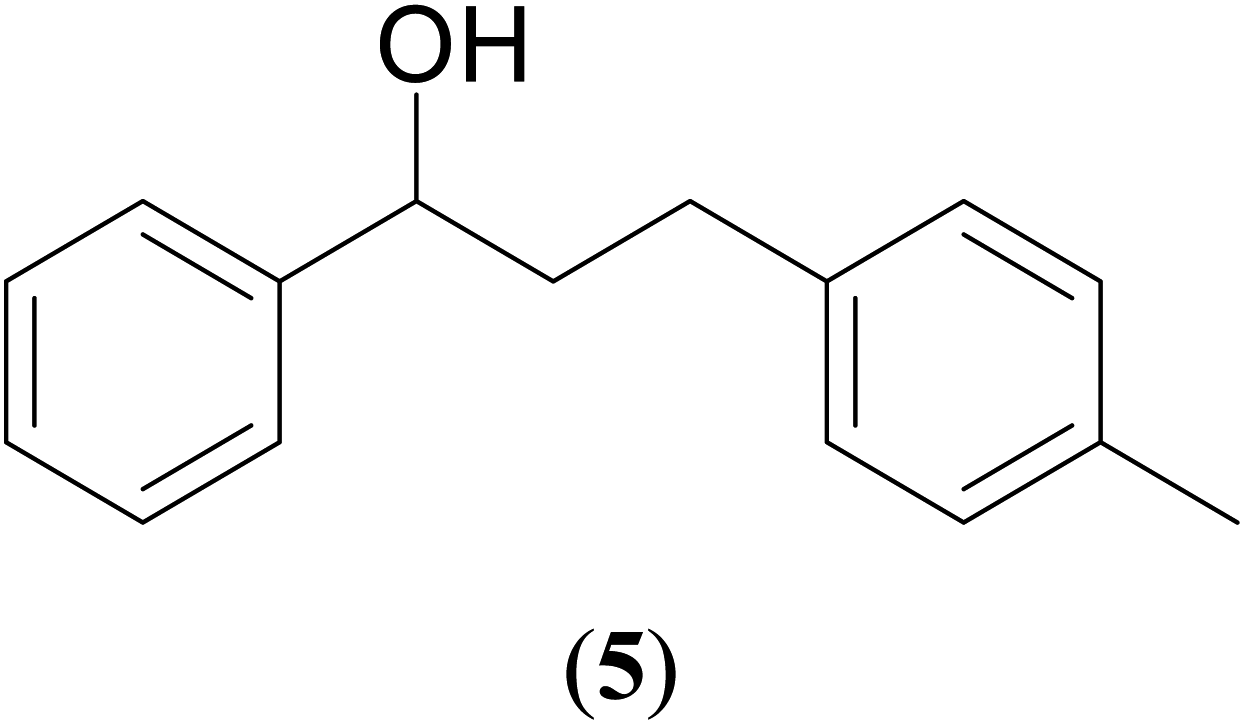 |
53 | 61 | 63 |
| 3 | 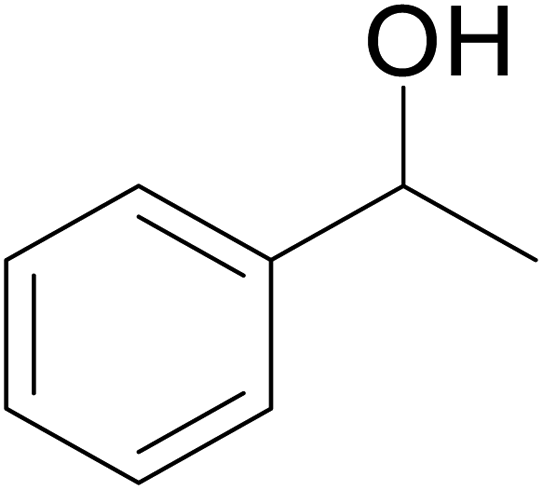 |
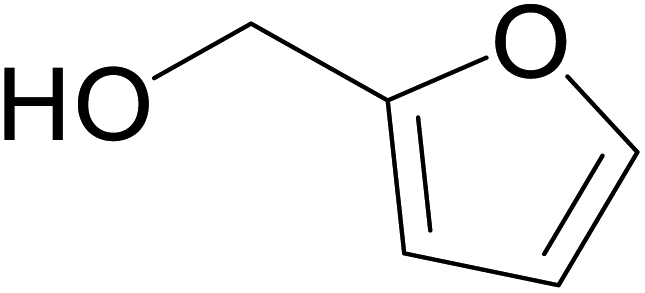 |
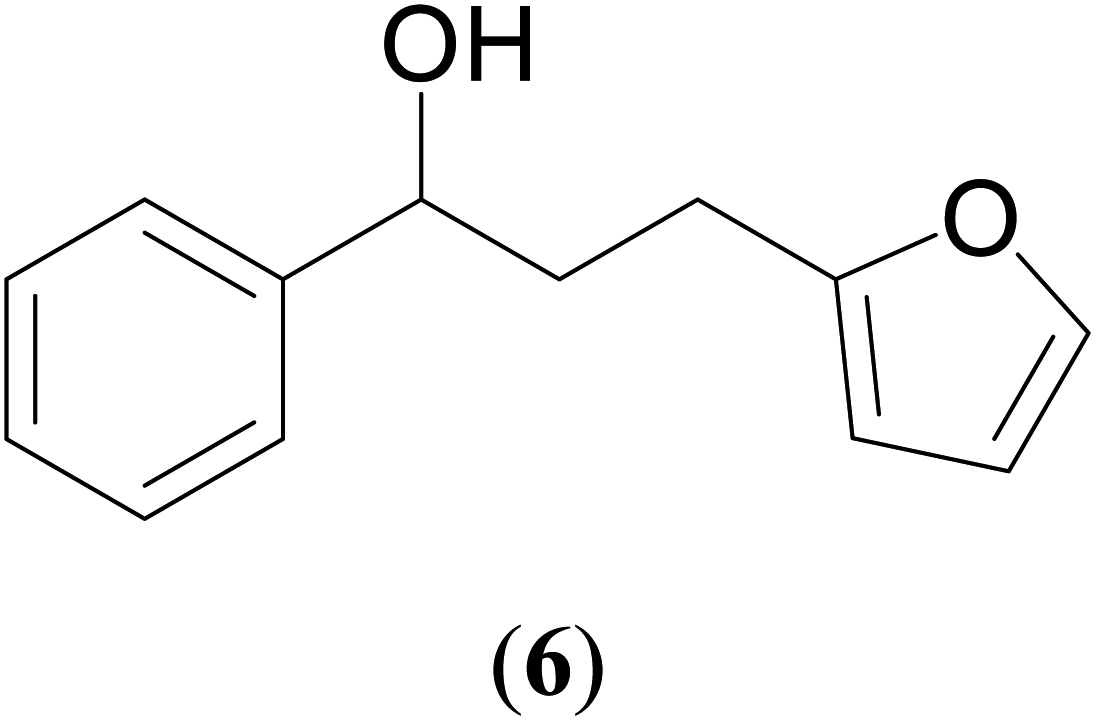 |
49 | 62 | 66 |
| 4 | 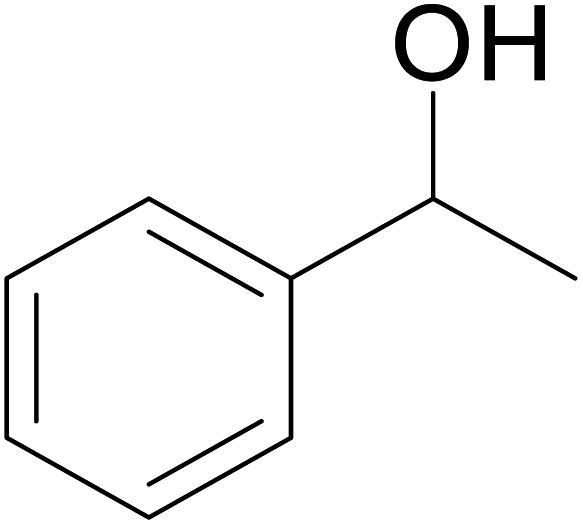 |
 |
 |
64 | 63 | 61 |
| 5 |  |
 |
 |
66 | 59 | 63 |
| 6 |  |
 |
 |
64 | 64 | 62 |
| 7 | 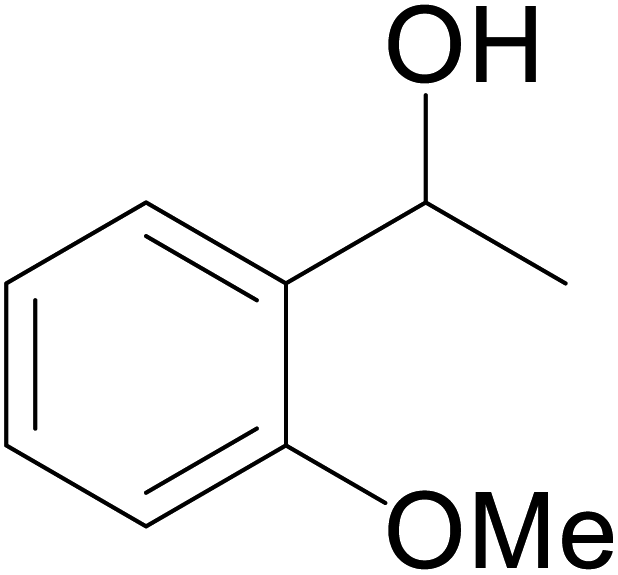 |
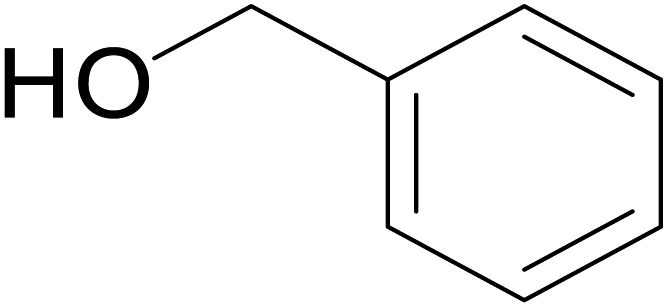 |
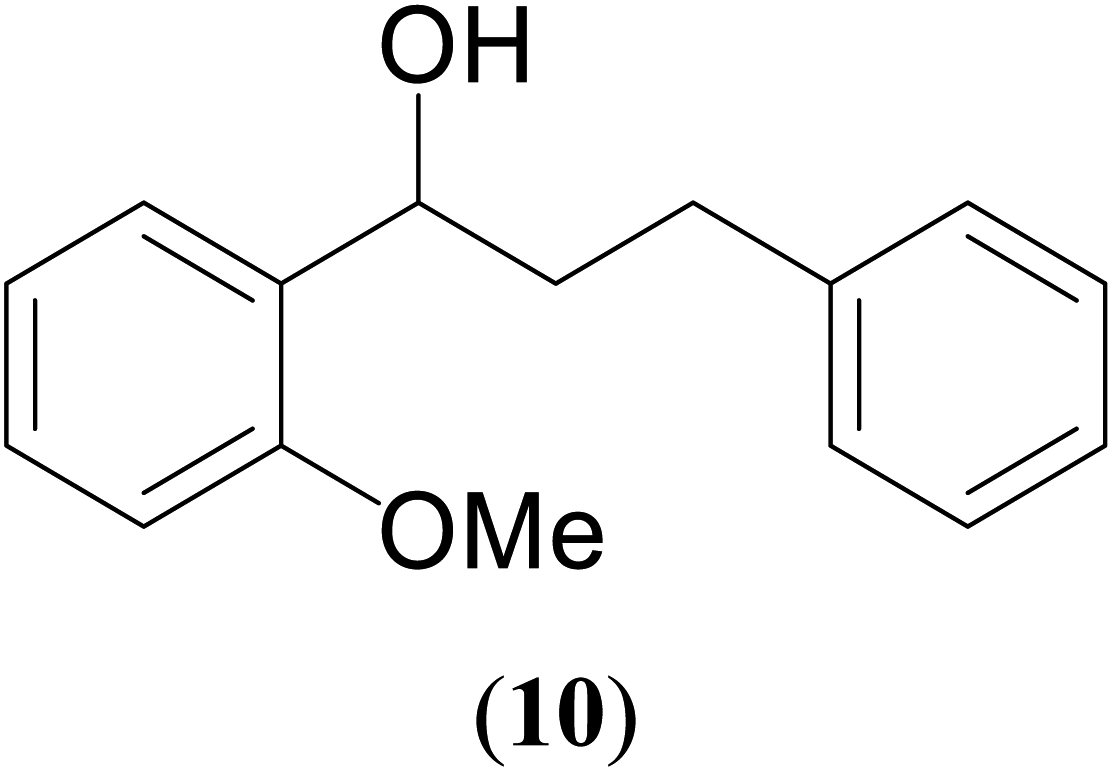 |
60 | 70 | 67 |
| 8 | 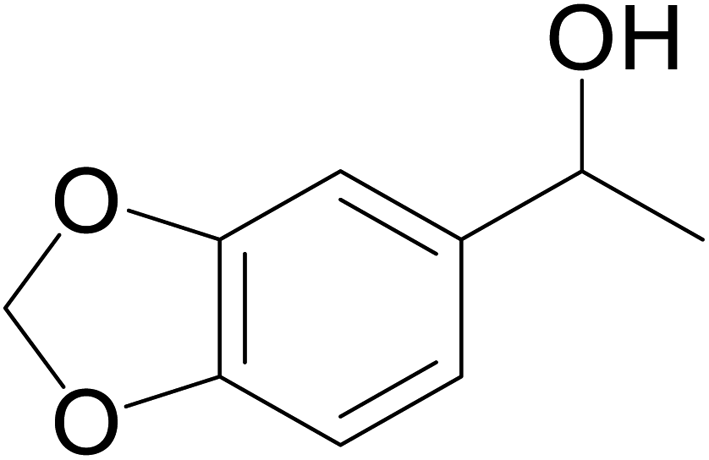 |
 |
 |
67 | 66 | 63 |
| 9 |  |
 |
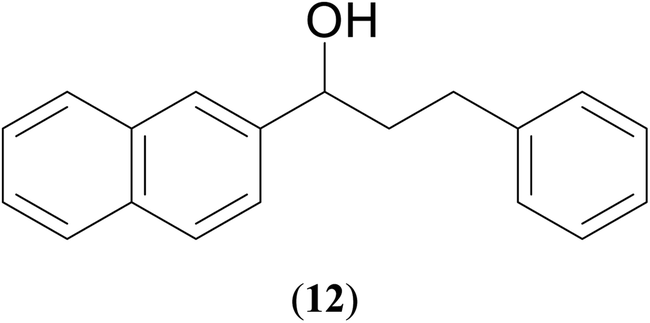 |
71 | 65 | 69 |
| 10 | 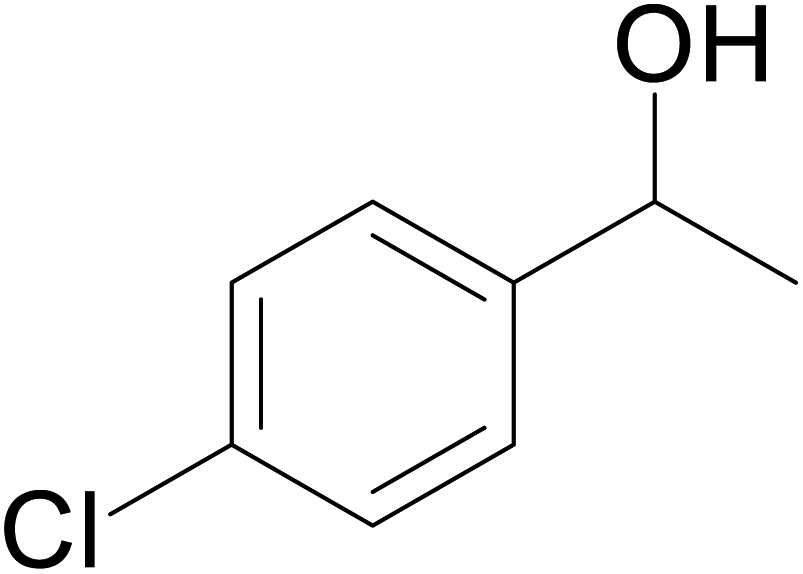 |
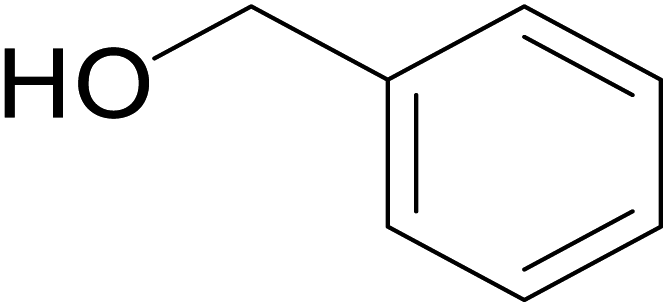 |
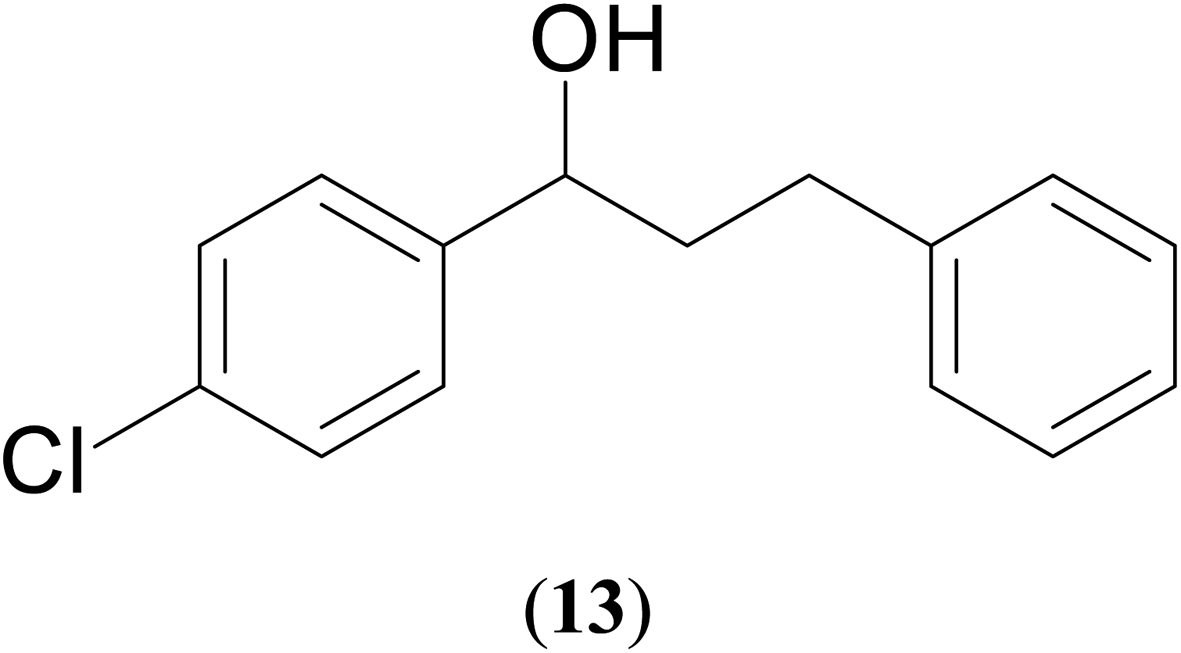 |
57 | 62 | 51 |
| 11 |  |
 |
 |
68 | 61 | 65 |
| 12 |  |
 |
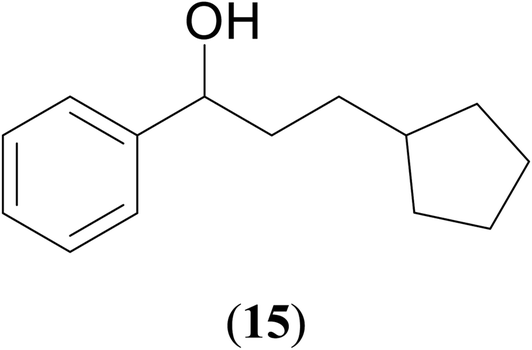 |
67 | 62 | 65 |
| 13 | 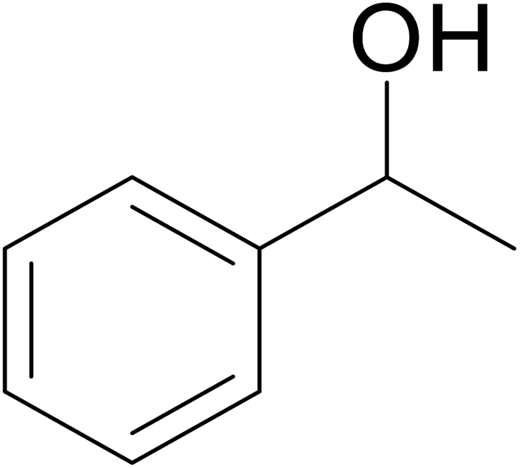 |
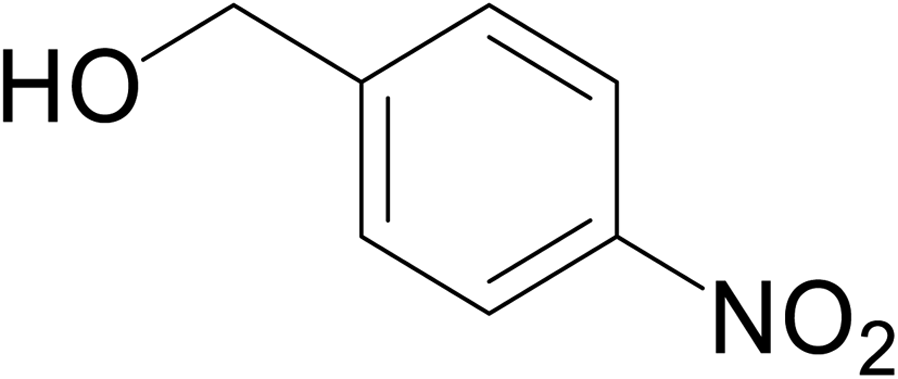 |
No product | — | — | — |
| 14 | 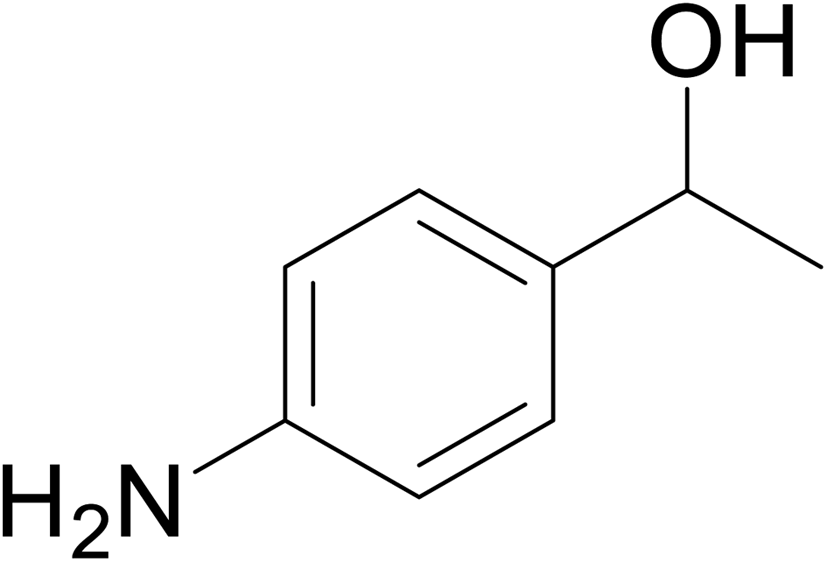 |
 |
No product | — | — | — |

The relevance of the catalytic activities of the ruthenium (1–3)b complexes in the dehydrogenative cross-coupling of primary and secondary alcohols can be gauged from the fact that till date there exist only six structurally characterized examples of well-defined ruthenium complexes reported in the literature. (Table 2)24,32,44 Out of these six structurally characterized ruthenium complexes, two have been reported earlier from our group.32 Furthermore in situ NMR yields have been reported in case of three complexes (entries 1, 2 and 3 of Table 2),44 while isolated yields have been reported for the remaining entries including the ruthenium (1–3)b complexes. Significantly enough, the imidazole based neutral and cationic N-heterocyclic carbene ruthenium complexes32 and the 1,2,4-triazole based neutral N-heterocyclic carbene ruthenium (1–3)b complexes exhibited shorter reaction time of 3 hours as compared to that of ca. 8–40 hours reported for other ruthenium catalyst (entries 1, 2, 3 and 4 of Table 2).24,44
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| S. No. | Catalyst | Base | Time (h) | Solvent | Catalyst loading | Temperature | Yield (%) | TON | Reference |
| a With respect to each metal of the tetra nuclear complex. | |||||||||
| 1 | 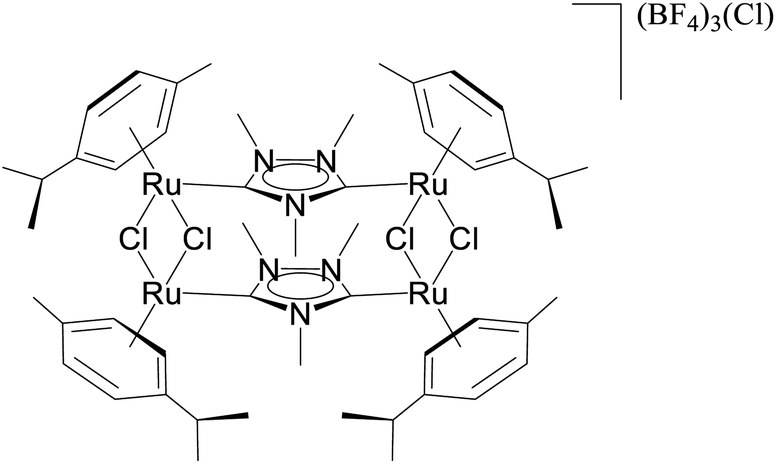 |
KOH (2.5 mmol) | 40 | Toluene (0.3 mL) | 1 mol% | 110 °C | 100 (by 1H NMR) | 95a | 44 |
| 2 | 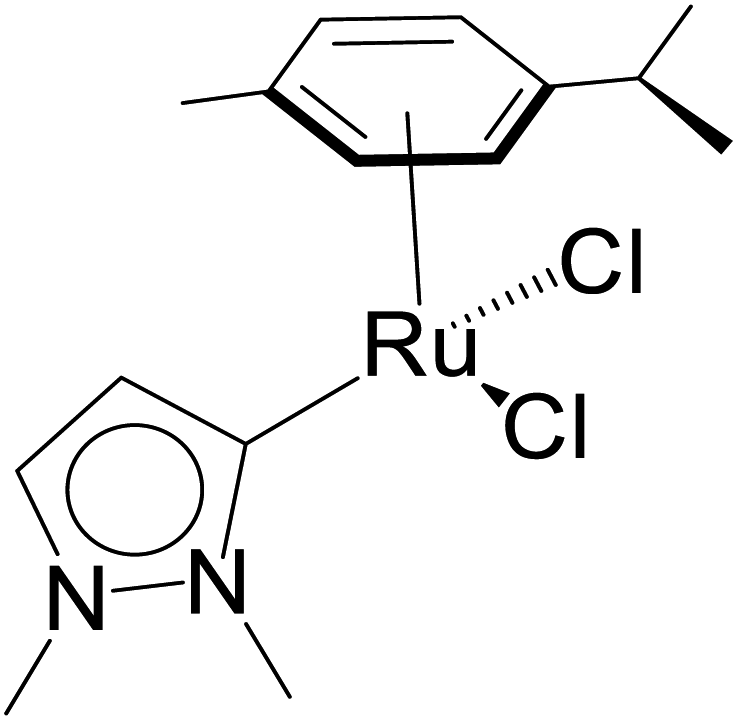 |
KOH (1 mmol) | 8 | Toluene (0.3 mL) | 1 mol% | 110 °C | 92 (by 1H NMR) | 92 | 45 |
| 3 | 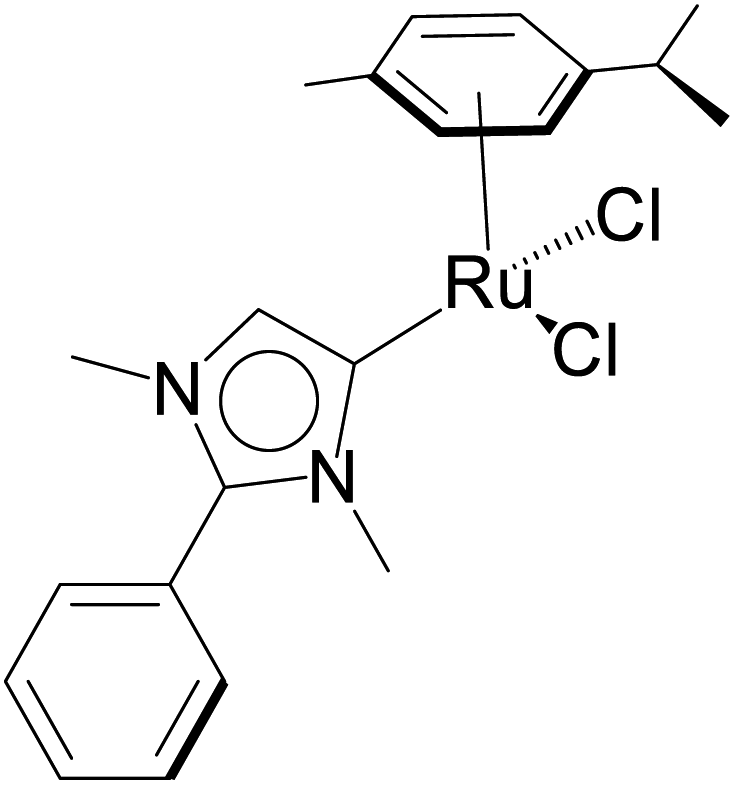 |
KOH (1.0 mmol) | 8 | Toluene (0.3 mL) | 1 mol% | 110 °C | 88 (by 1H NMR) | 88 | 45 |
| 4 |  |
NaOiPr (0.8 mmol) | 12 | Solvent free | 0.001 mol% | 125 °C | 97 (isolated) | 97000 |
24 |
| 5 |  |
NaOiPr (1.00 mmol) | 3 | Toluene (2.0 mL) | 1 mol% | 110 °C | 72 (isolated) | 72 | 32 |
| 6 |  |
NaOiPr (1.00 mmol) | 3 | Toluene (2.0 mL) | 1 mol% | 110 °C | 68 (isolated) | 68 | 32 |
| 7 | 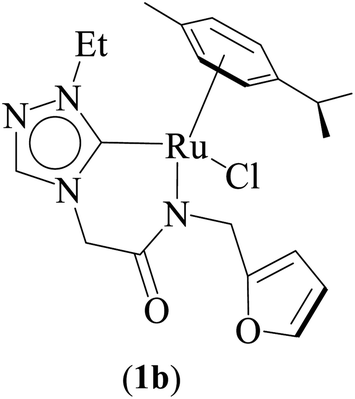 |
NaOiPr (1.00 mmol) | 3 | Toluene (2.0 mL) | 1 mol% | 110 °C | 70 (isolated) | 70 | Present work |
| 8 | 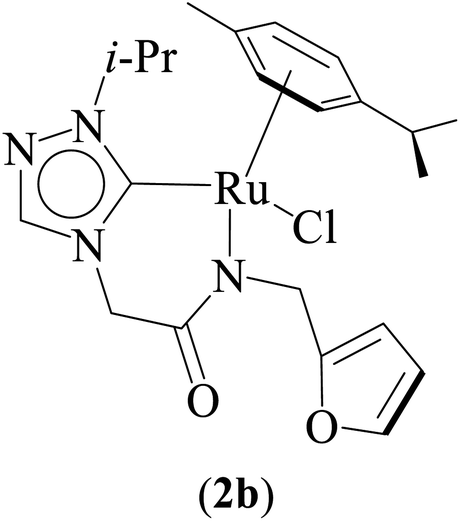 |
NaOiPr (1.00 mmol) | 3 | Toluene (2.0 mL) | 1 mol% | 110 °C | 72 (isolated) | 72 | Present work |
| 9 | 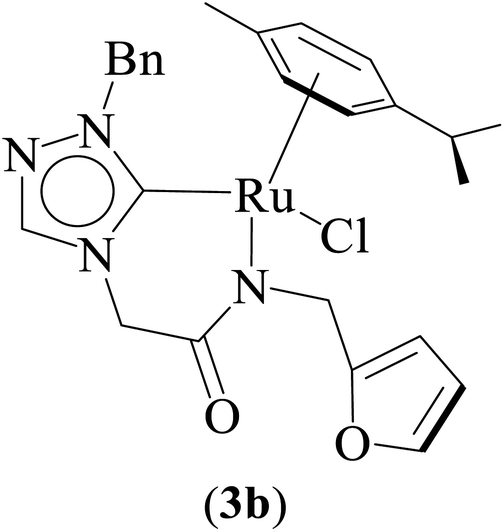 |
NaOiPr (1.00 mmol) | 3 | Toluene (2.0 mL) | 1 mol% | 110 °C | 68 (isolated) | 68 | Present work |

Furthermore, a careful comparison of the catalytic activities of the 1,2,4-triazole-based (1–3)b complexes with that of the earlier reported imidazole-based neutral and cationic ruthenium–NHC complexes32 for the same pair of substrates under analogous catalysis conditions revealed that the imidazole-based neutral and cationic ruthenium–NHC complexes32 exhibited superior yields for the two heterocycle bearing substrates namely furfuryl alcohol and thiophen-2-ylmethanol. Specifically, for the pair of furfuryl alcohol and 1-phenylethanol substrates, the imidazole-based ruthenium complexes32 exhibited comparatively higher yields of ca. 68–70% than the triazole-based (1–3)b complexes (ca. 49–66%). Similarly, for the other heterocyclic substrate, thiophen-2-ylmethanol, the imidazole based ruthenium complexes32 showed higher yields of ca. 63–74% than the triazole-based ones, (1–3)b, (ca. 61–64%). Lastly, even for an electron rich 4-methoxy-benzyl alcohol substrate, the imidazole-based ruthenium complexes32 exhibited the higher yields of ca. 86–89% than (1–3)b (ca. 59–66%). In short, the imidazole-based ruthenium complexes32 are marginally superior to the triazole-based (1–3)b complexes for certain heterocyclic and electron rich substrates. However, owing to very similar steric and electronic requirements of these imidazole-based ruthenium complexes32 and the triazole-based (1–3)b complexes, their catalytic activities are comparable and the exact influences of chelating NHC and the amide substituents cannot be inferred with certainty.
A mechanism of the dehydrogenative cross-coupling of primary and secondary alcohols by a representative 1,2,4-triazole derived amido-functionalised neutral ruthenium (1b) complex is proposed along the lines of the one reported earlier for the related imidazole based neutral and catatonic ruthenium complexes (Scheme 2).32
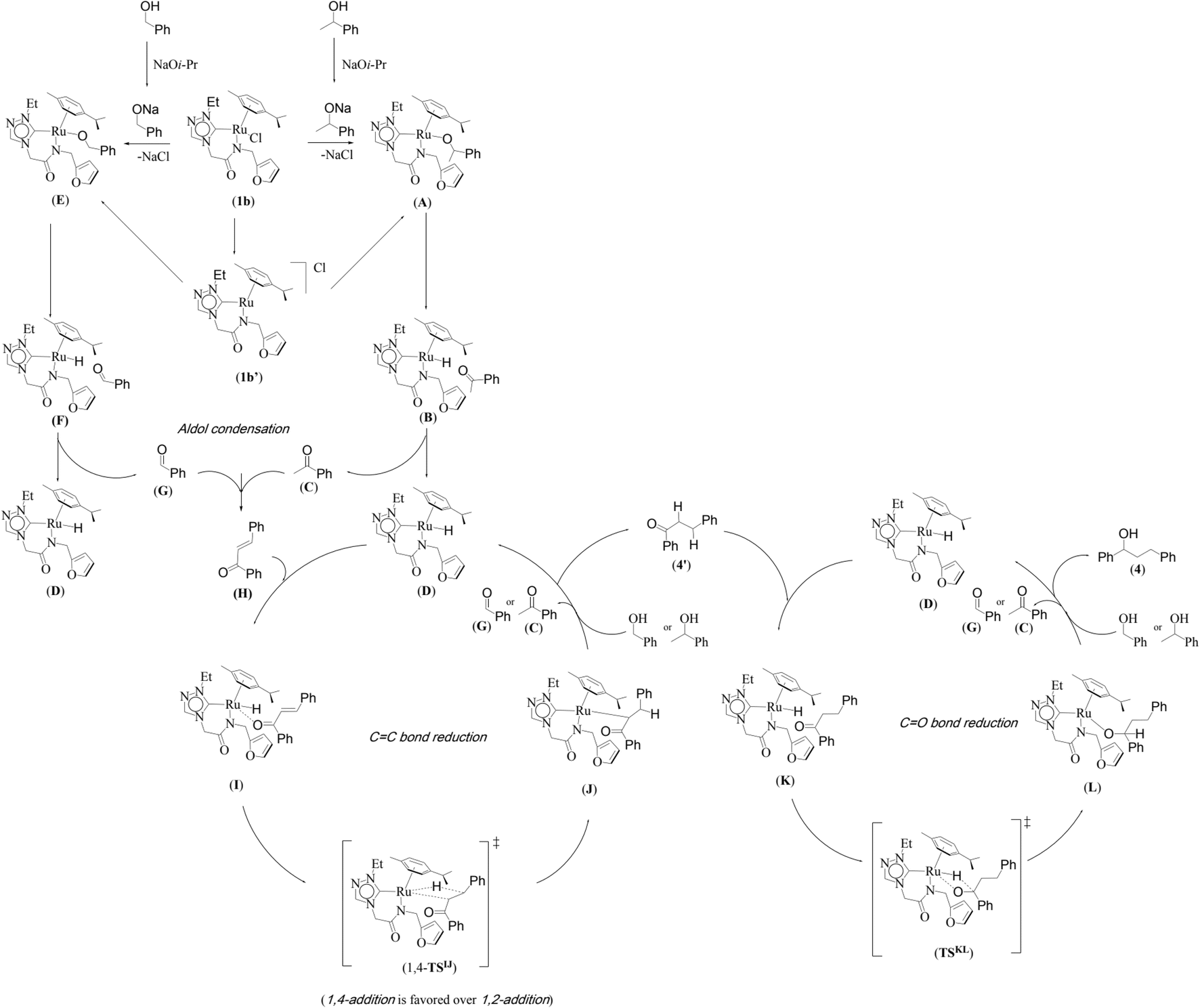 | ||
| Scheme 2 A proposed neutral mechanistic pathway for the Ru–NHC (1b) catalyzed one pot tandem β-alkylation reaction for representative substrates namely 1-phenylethanol and benzyl alcohol is shown. | ||
To begin with, we have optimized the ruthenium (1b) precatalyst and the computed geometrical parameters agree with the X-ray structure, thus offering confidence in the chosen methodology. The computed Ru–C(carbene) bond distances of 2.013 Å, Ru–N(amido) bond distance of 2.101 Å, and Ru–Cl bond distance of 2.472 Å, along with the d-based molecular orbital diagram of (1b) are shown in Fig. 6. The Ru t2g orbitals are found to split due to differences in the nature of ligand donors, with 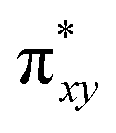 being the lowest, followed by
being the lowest, followed by 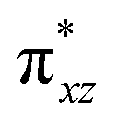 and
and 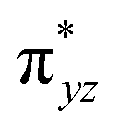 . A strong σ donation from the NHC ligand destabilises the eg orbitals leading to a large HOMO–LUMO gap (4.56 eV).
. A strong σ donation from the NHC ligand destabilises the eg orbitals leading to a large HOMO–LUMO gap (4.56 eV).
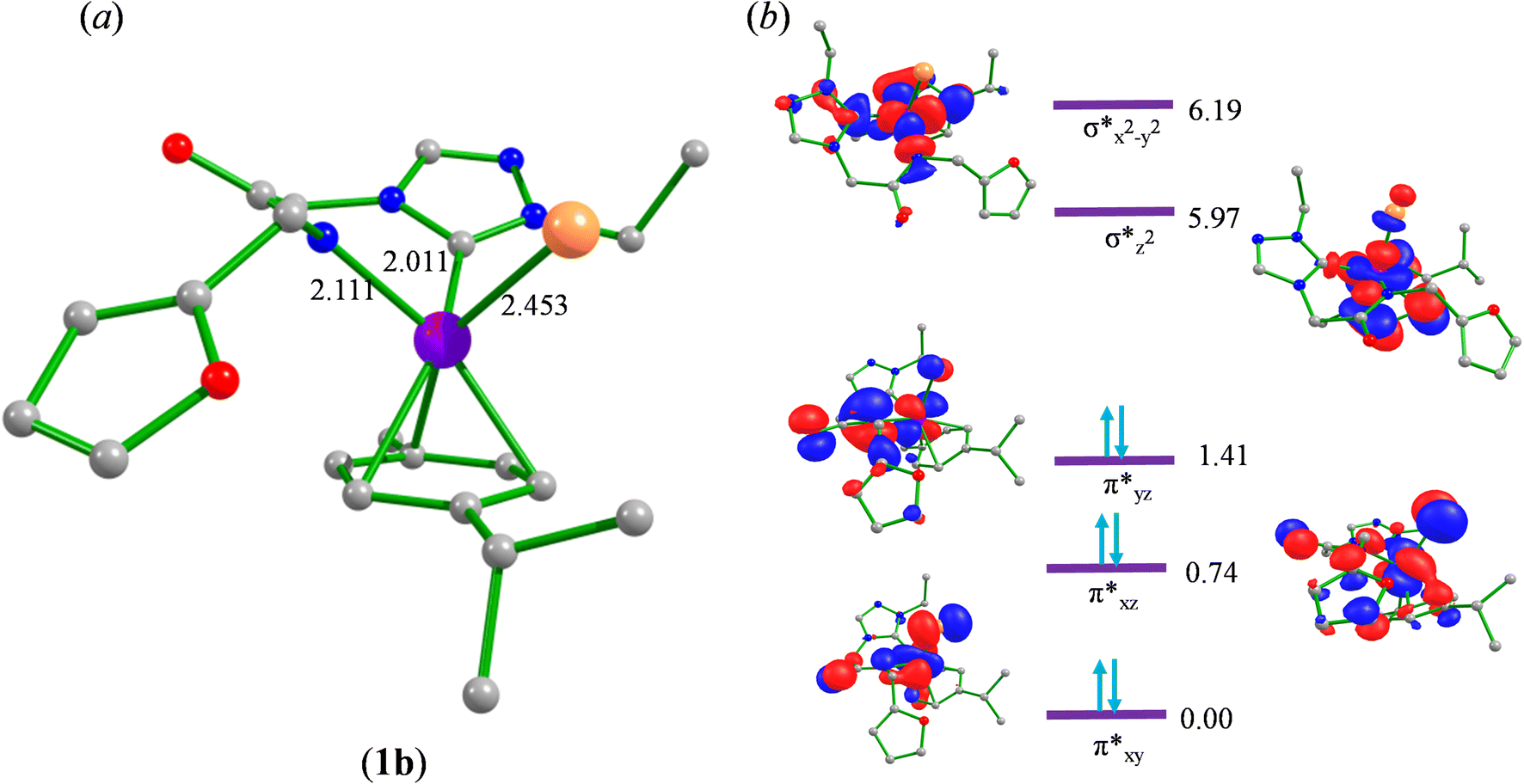 | ||
| Fig. 6 Computed (a) optimized geometry and (b) electronic structure of (1b). | ||
We investigated both the (i) metal–ligand non-cooperative mechanism, proceeding by a neutral pathway (Scheme 2) and the (ii) metal–ligand cooperative mechanism, proceeding by an ionic pathway (ESI Scheme S2†). For each of these two mechanisms, the catalytic cycle involves three steps, namely, (i) the nucleophilic substitution of chloride ion by secondary/primary alcohols, (ii) dehydrogenation of primary/secondary alcohols by Ru–alcoholate intermediate, (iii) hydrogenation of olefin and the hydrogenation of ketone by ruthenium hydride species.
(i) Nucleophilic substitution of chloride ion
The computed potential energy surfaces for both the mechanisms are given in Fig. 7 and ESI Fig. S142.† In particular, the nucleophilic substitution step can occur either via associative or dissociative pathways via either (E) or (A) intermediates in the neutral metal–ligand non-cooperative pathway (Scheme 2 and Fig. 7) or via the (E′) or (A′) intermediates in the ionic metal–ligand cooperative pathway (ESI Scheme S2 and Fig. S142†). In the dissociative pathway, the cleavage of Ru–Cl bond leads to a intermediate (1b′) that is extremely endothermic by 212 kJ mol−1, This is essentially due to the strong Ru–Cl bond and its π-donor capability found to enhance the donation from metal centers to other ligands, as witnessed by its presence in all three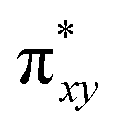 ,
, 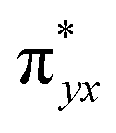 and
and 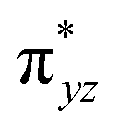 orbitals (Fig. 6). Moreover, the addition of the alcohols to the intermediate 1b′ is found to be endothermic for both primary (+27.1 kJ mol−1) and secondary (+38.2 kJ mol−1) alcohols. For deprotonated alcohols, on the other hand, the addition is exothermic (−60.3 and −46.0 kJ mol−1 for primary and secondary alcohols, respectively). However, a steep energy penalty to generate the (1b′) intermediate suggests that this pathway is very unlikely. In the associative mechanism, the formation of (E) and (A) form (1b) is exothermic by 60.3 kJ mol−1 and 46.0 kJ mol−1 respectively (Fig. 7), whereas in the case of (E′) and (A′), the formation is steeply endothermic by 239.1 kJ mol−1 and 250.2 kJ mol−1 respectively for primary and secondary alcohols, respectively (ESI Fig. S142†). Thus, our calculations suggest that the mechanism proceeds via an associative mechanism with the deprotonated alcohol, whose π-donor capability more than compensate for the loss Ru–Cl moiety.
orbitals (Fig. 6). Moreover, the addition of the alcohols to the intermediate 1b′ is found to be endothermic for both primary (+27.1 kJ mol−1) and secondary (+38.2 kJ mol−1) alcohols. For deprotonated alcohols, on the other hand, the addition is exothermic (−60.3 and −46.0 kJ mol−1 for primary and secondary alcohols, respectively). However, a steep energy penalty to generate the (1b′) intermediate suggests that this pathway is very unlikely. In the associative mechanism, the formation of (E) and (A) form (1b) is exothermic by 60.3 kJ mol−1 and 46.0 kJ mol−1 respectively (Fig. 7), whereas in the case of (E′) and (A′), the formation is steeply endothermic by 239.1 kJ mol−1 and 250.2 kJ mol−1 respectively for primary and secondary alcohols, respectively (ESI Fig. S142†). Thus, our calculations suggest that the mechanism proceeds via an associative mechanism with the deprotonated alcohol, whose π-donor capability more than compensate for the loss Ru–Cl moiety.
(ii) Dehydrogenation of primary/secondary alcohols
The second step in the catalytic cycle is the dehydrogenation of alcohols, as mentioned earlier. The attempts for finding the inner sphere β-hydrogen elimination transition state were not successful. Therefore, we have explored outer-sphere β-hydrogen elimination for both (E) or (A) and (E′) or (A′) leading to the formation of active Ru–H species (D′) and (D) along with their respective ketone/alcohol (Fig. 7 and ESI Fig. S140, S142, S144, and S145†). For the conversion of (E) to (F), the transition state barrier is estimated to be 73.7 kJ mol−1 (for primary alcohol). Our attempts to find the transition state for secondary alcohol were not successful. The Ru–O bond distance is 2.070 Å for (E) and 2.081 Å for (A). The Ru–N bond distance is 2.108 Å for (E) and 2.117 Å for (A). At the transition state TSEF, the Ru–N distance elongates to 2.123 Å and the bond distances of Ru–H and C–H are 1.448 Å and 1.756 Å, respectively. The formation of (F) and (B) from (1b) is exothermic by 83.0 kJ mol−1 and 100.2 kJ mol−1 respectively. Non-covalent interactions are present between aldehyde and ketone and the ligands of (B) and (F) respectively (Fig. 4 and 5). Similar non-covalent interactions are seen between aldehyde and ketone and the ligands of (B′) and (F′) respectively (ESI Fig. S148†). The species (B) and (F) have also been characterized by mass spectrometric analysis of the catalysis mixture that showed the [M + H]+ peak at and 591.1913 [calcd 591.1912] (Fig. 4) and m/z 577.1751 [calcd 577.1755] (Fig. 5) respectively. From species (B) and (F), elimination of aldehyde or ketone leads to the formation of active species (D). The formation of (D) from (1b) is exothermic by 214 kJ mol−1 (Fig. 7).Whereas for the other metal–ligand cooperative mechanism, the conversion from species (E′) and (A′) to (F′) and (B′), a stepwise mechanism is detected with an initial barrier of 39.4 kJ mol−1 and 48.1 kJ mol−1 for the primary and secondary alcohol respectively. (ESI Fig. S142–S145†). At the transition state TSE′E′′, the Ru–O distance shortens to 2.139 Å compared to 2.219 Å at the intermediate (E′), while the Ru–N distance concomitantly elongates to 2.223 Å from 2.112 Å in (E′). Similar behaviour is also detected for the TSA′A′′ species. The newly forming N–H bond distance at the transition state is found to be 1.218 Å while the cleaving O…H distance is found to be 1.308 Å, suggesting at the transition state TSE′E′′, neither the N–H bond is fully formed nor the O–H bond is fully broken. The formations of the intermediates are estimated to be endergonic by 37.0 kJ mol−1 for (E′′) and 28.0 kJ mol−1 for (A′′). In the intermediate (E′′), the Ru–N bond of 2.298 Å is significantly longer than that of 2.112 Å in (E′), while the Ru–O distance of 2.062 Å is much shorter compared to species (E′) (2.219 Å). Further, there is a strong H-bonding between the N–H, and O (alc) was detected at the intermediate E′′(A′′). A shorter and stronger electrostatic interaction of oxygen upon deprotonation with Ru, weakens the Ru–N bond. In the next step, outer-sphere β-hydrogen elimination takes place via the transition state TSA′′B′ or TSE′′F′. For this TSE′′F′ or TSA′′B′ transition state, the barrier height is estimated to be 57.2 kJ mol−1 or 49.0 kJ mol−1. At this transition state TSE′′F′, the Ru–N distance was found to be shortened to 2.226 Å as compared to 2.298 Å in (E′′). In the next step, the formation of Ru–H species, (B′) or (F′), takes place, where Ru–H distances were found to be 1.598 Å in (B′) and (F′), and the formation of this species is found to be exergonic by 90.7 kJ mol−1 for (B′) and 66.0 kJ mol−1 for (F′) from (A′′) and (E′′) respectively. From species (F′) and (B′), elimination of aldehyde (G) and ketone (C) leading to the formation of active species (D′). The formation of (D′) from (1b) is exothermic by 257.3 kJ mol−1.
In the entire potential energy surface, the Ru–Ccarbene bond was found only to alter marginally, and this suggests that the Ru–NHC bond is an anchor for the catalyst offering significant stability during the catalytic cycle. The formation of (E′) and (A′) incurs additional energy costs compare to (E) and (A) and the formation of (D) is exothermic by 214 kJ mol−1 while (D′) is endothermic by 257.3 kJ mol−1. Therefore it is clear that the mechanism proceeds via (E) and (A) to form (D). And then finally, aldehyde (G) and ketone (C) would undergo cross-aldol condensation under basic reaction condition containing NaOiPr and producing the cross-aldol α,β-unsaturated compound PhCOCH = CHPh (H).
Hydrogenation of olefins and ketones
The last step is the hydrogenation of olefin and ketones. The computed energy profile and geometries of this step are given the Fig. 8 and ESI Fig. S141† respectively. Formation of active (I) species form (D) by 42.9 kJ mol−1. For the metal–ligand non-cooperative mechanism, proceeding by a neutral pathway, the computed barrier for hydrogenation of olefin is 86.0 kJ mol−1. The Ru–H bond is elongated from 1.593 Å in (I) to 1.758 Å at the transition state 1,4-TSIJ, and the bond distance of C(β-carbon)–H is found at 1.437 Å, and the same distance for the 1,2 addition is found to be 1.448 Å. The difference in barrier height computed between the 1,4- and 1,2 addition is 5 kJ mol−1, and this is attributed to the greater positive charge found on the β carbon. The 1,4 addition and 1,2-addition result in the formation of intermediates (J) and 1,2-(J) respectively, and the intermediate (J) regenerates the species (D) along with (4′) on alcoholysis. The reaction proceeds further upon hydrogenation of the keto group in (4′) by the Ru–H active species (D). This step has a computed barrier of 85.0 kJ mol−1. The Ru–H is elongated to 2.122 Å in the transition state TSKL compared to 1.593 Å in (K), and the distance between the carbonyl carbon and hydrogen atom of Ru–H is 1.448 Å at the transition state TSKL. This step results in the intermediate (L) which regenerates (D) along with (4) on alcoholysis. This mechanism is consistent with the experimental observation that the alcohol product, PhCH(OH)CH2CH2Ph (4), and the ketone product, PhCOCH2CH2Ph (4′) were isolated from the catalysis mixture (ESI Fig. S31–S42†).For the metal–ligand cooperative mechanism, proceeding by an ionic pathway, the formation of the intermediate (I′) species from (D) is endothermic by 23.2 kJ mol−1. There exists a strong hydrogen bond between the hydrogen of N–H and O atom unsaturated carbonyl compound at the species (I′). (ESI Fig. S143, S146 and S147†). First, the hydride transfer occurs from ruthenium hydride to β carbon of α,β-unsaturated carbonyl compound (1,4 addition) or the hydride transfer to carbonyl carbon (1,2-addition). Our calculations revealed that the 1,2 addition has a 20 kJ mol−1 high barrier than the 1,4 addition. The product (J′) formed upon 1,4 addition is lower in energy by 12.8 kJ mol−1 compared to the 1,2 addition product 1,2-(J′). The hydrogen bond between the oxygen (Ru–O) and hydrogen of N–H is absent in the case of 1,2-(J′) (1, 2-product) but present in (J′) (1, 4-product), and this is one of the reasons for its stability. The distance between Ru and oxygen is found to be 2.096 Å in (J′), and the Ru–H bond is elongated from 1.599 Å in (I′) to 1.806 Å in 1,4-TSI′J′ and also from 1.599 Å in (I′) to 1.829 Å 1,2-TSI′J′ (in the case of 1,2 addition). The distance from carbonyl carbon to the hydrogen atom of Ru–H is 1.430 Å and 1.541 Å in the case of 1,2 addition and 1,4 addition transition states, respectively. This also explains the destabilisation of the 1,2 addition transition state compared to that of the 1,4 addition transition state. The distance from carbonyl carbon to α-carbon is decreased from 1.434 Å in 1,4-TSI′J′ to 1.353 Å in (J′), and the distance between α,β -carbons is elongated by 1.403 Å in 1,4-TSI′J′ to 1.512 Å in (J′). In the next step, the oxygen atom deprotonates the N–H proton through the TSJ′1b′ transition state. This step has a 20 kJ mol−1 barrier. The Ru–O and O–C (carbonyl) bonds are elongated from 2.096 Å in (J′) to 2.170 Å in TSJ′1b′ and 1.355 Å in (J′) to 1.384 Å in TSJ′1b′, respectively. This step results in the formation of a hydrogenated product (4′) and intermediate (1b′) which regenerates the (D′). The reaction proceeds further upon hydrogenation of the keto group in (4′) by the Ru–H active species (D′). Again, Ru–H hydride transfer is favourable and has a computed barrier of 79.0 kJ mol−1. The Ru–H is elongated to 1.770 Å in TSK′L′ as compared to 1.599 Å in (K′), and the distance between the carbonyl carbon and hydrogen atom of Ru–H is 1.450 Å at the transition state TSK′L′. The oxygen atom deprotonated the nitrogen in the next step, and this is found to be very facile with a very small barrier of 7 kJ mol−1. This step results in the formation of (4) and the intermediate (1b′), which regenerates the (D′) on alcoholysis.
If we compare the geometries across the potential energy surface, the Ru–N(amido) and Ru–C(benzene) distances were found to alter during the course of the reaction, but the Ru–Ccarbene distances are more robust, suggesting the carbene ligands act as an anchoring group to avoid decomposition of catalysts.
Thus, the current manuscript combines the experimental and theoretical insights on the alcohol–alcohol coupling reactions by the ruthenium N-heterocyclic carbene complexes. Quite importantly, both the possibilities, (i) metal–ligand non-cooperative mechanism, proceeding by a neutral pathway (Scheme 2) and the (ii) metal–ligand cooperative mechanism, proceeding by an ionic pathway (ESI Scheme S2†) were validated by the DFT studies. Several intermediates namely, the HRMS characterization of the acetophenone bound Ru–H species (B) (Fig. 4) and the benzaldehyde bound Ru–H species (F) (Fig. 5), that were detected in the reaction mixture were also validated by the computational model of the neutral pathway (Fig. 7). Furthermore, the detailed insight into the neutral pathway as obtained from the theoretical studies, suggests a lower activation energy barrier for the CC bond hydrogenation (1,4-TSIJ) than for the CO bond hydrogenation (1,2-TSIJ) (Fig. 8), and which too is in agreement with the experimental findings.
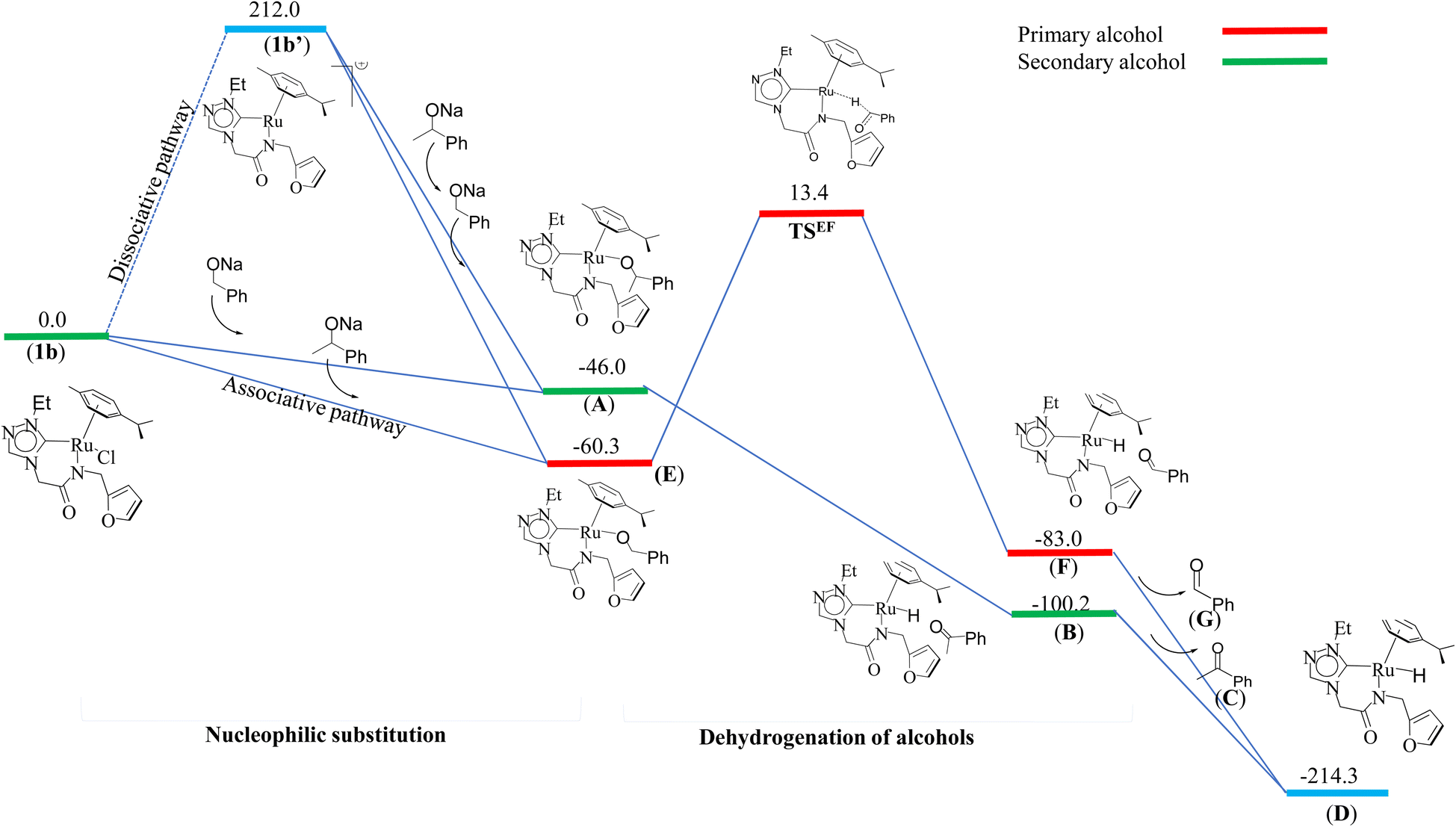 | ||
| Fig. 7 Energy profile diagram of nucleophilic substitution and dehydrogenation of alcohols by (1b) (deprotonated alcohol pathway or the neutral pathway). | ||
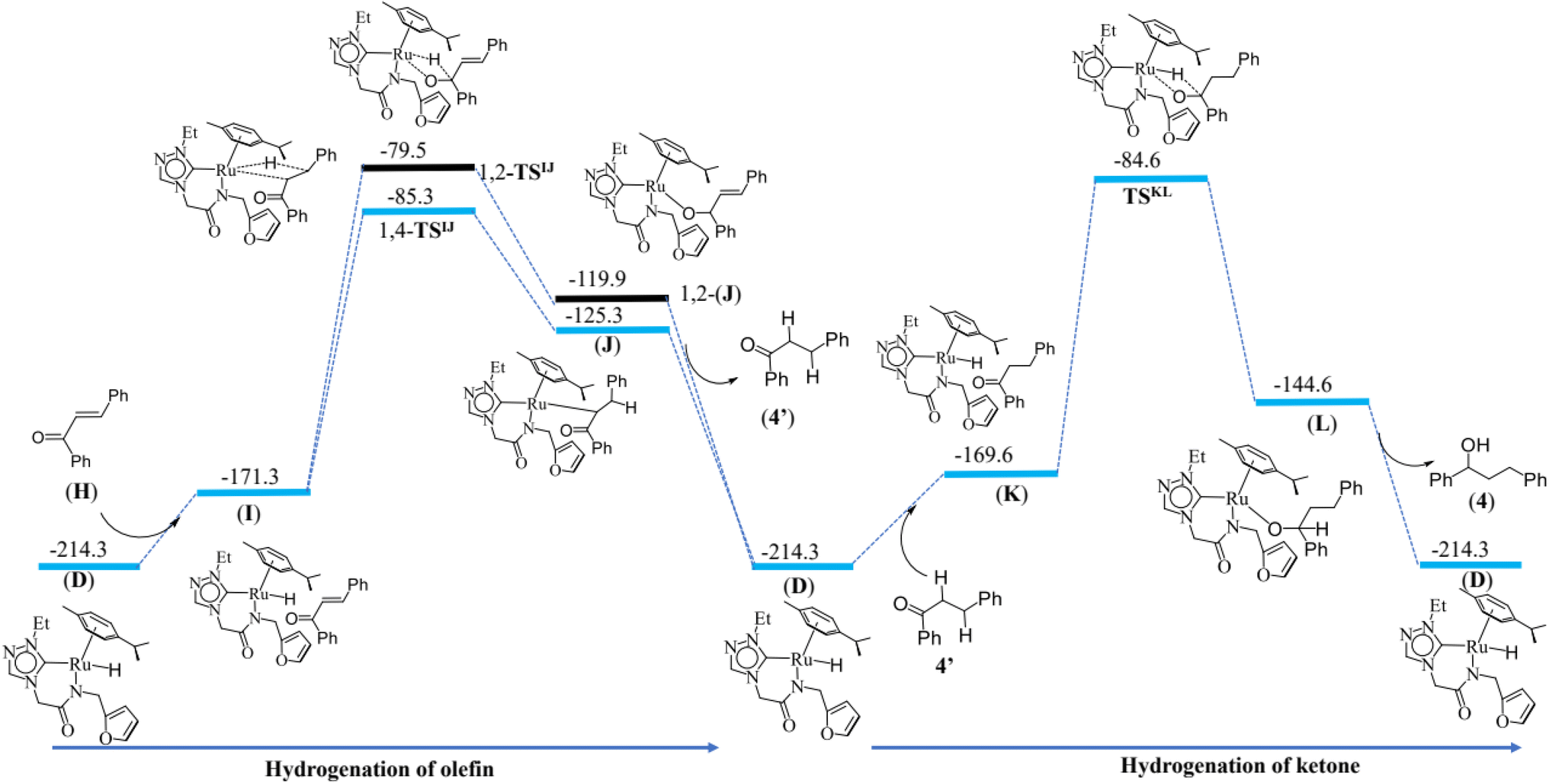 | ||
| Fig. 8 Energy profile diagram for hydrogenation of ketone and olefin by ruthenium hydride active species (D) (deprotonated alcohol pathway or the neutral pathway). | ||
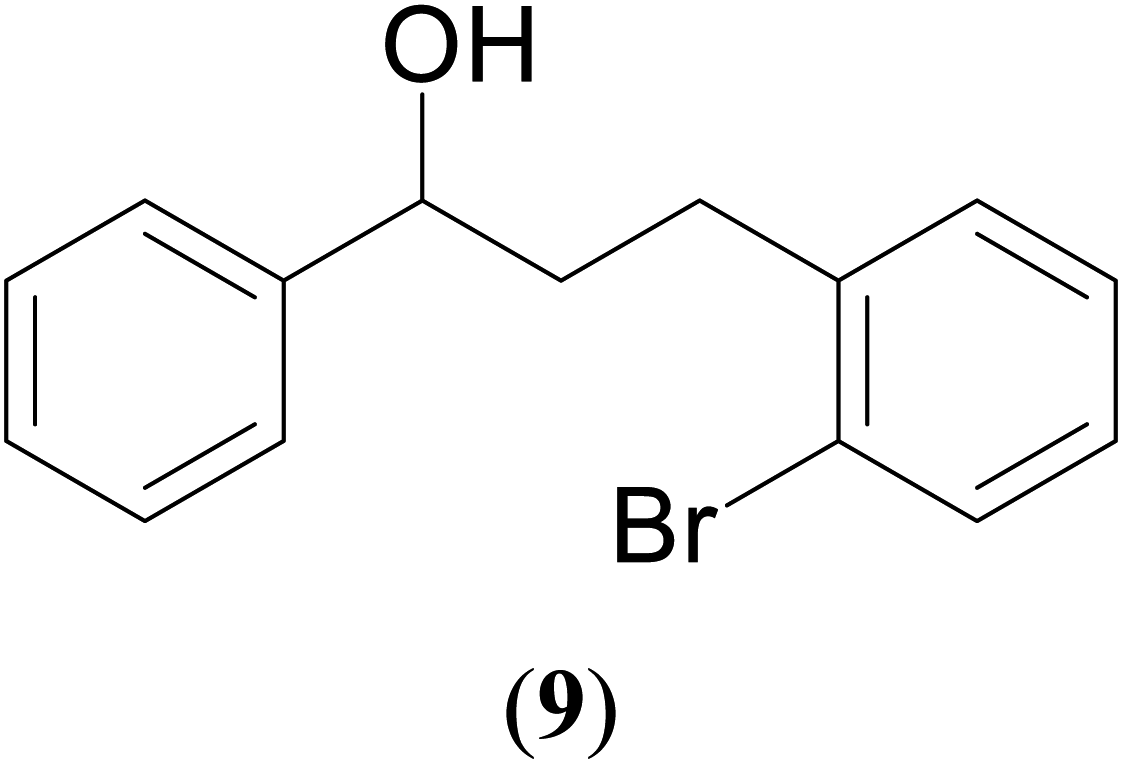
A hands-on utility of these ruthenium (1–3)b complexes in facilitating one-pot synthetic protocol for preparing a variety of bioactive molecules mainly flavan derivatives (16–20), was achieved using the reaction of the corresponding 2-arylethanol with 2-bromobenzyl alcohol (Scheme 3 and see ESI Table S5†).32 Flavonoids are a group of plant metabolites with valuable antioxidant properties having significant health benefits.32 The flavan core structure is ubiquitous in various flavonoid natural products displaying favorable biological and pharmacological properties. Several synthetic routes to the flavan derivatives that exist contain intriguing multi-step sequences46–49 like, tosylhydrazine mediated transformation of 2-hydroxyl chalcones to 2-arylchromans.50 and the three-step strategy for the synthesis of functionalized flavans using combination of Pd and Cu catalysts.51,52 These approaches are plagued with longer reaction times, multistep synthesis, and harsh reaction conditions, making them less attractive. In this context, significantly enough, the ruthenium (1–3)b complexes catalyzed the reaction of 1-phenylethanol with 2-bromobenzyl alcohol to give the corresponding dehydrogenative alcohol–alcohol cross coupled product, which upon subsequent treatment with CuI yielded the desire flavan product, 2-phenylchroman (16), in ca. 26% at 110 °C after 24 hours reaction time. Additionally, four other flavan derivatives (17–20) were obtained under analogous conditions. More interestingly, we succeeded in isolating the intermediate dehydrogenative alcohol–alcohol cross coupled product, PhCH(OH)CH2CH2PhBr (9), in ca. 64% yield of the reaction of the 1-phenylethanol with 2-bromobenzyl alcohol using the ruthenium (1–3)b complexes. (Table 1, entry 6) Further treatment of PhCH(OH)CH2CH2PhBr (9) with CuI and 2,2′-bipyridine produced 2-phenylchroman (16), in ca. 32%.
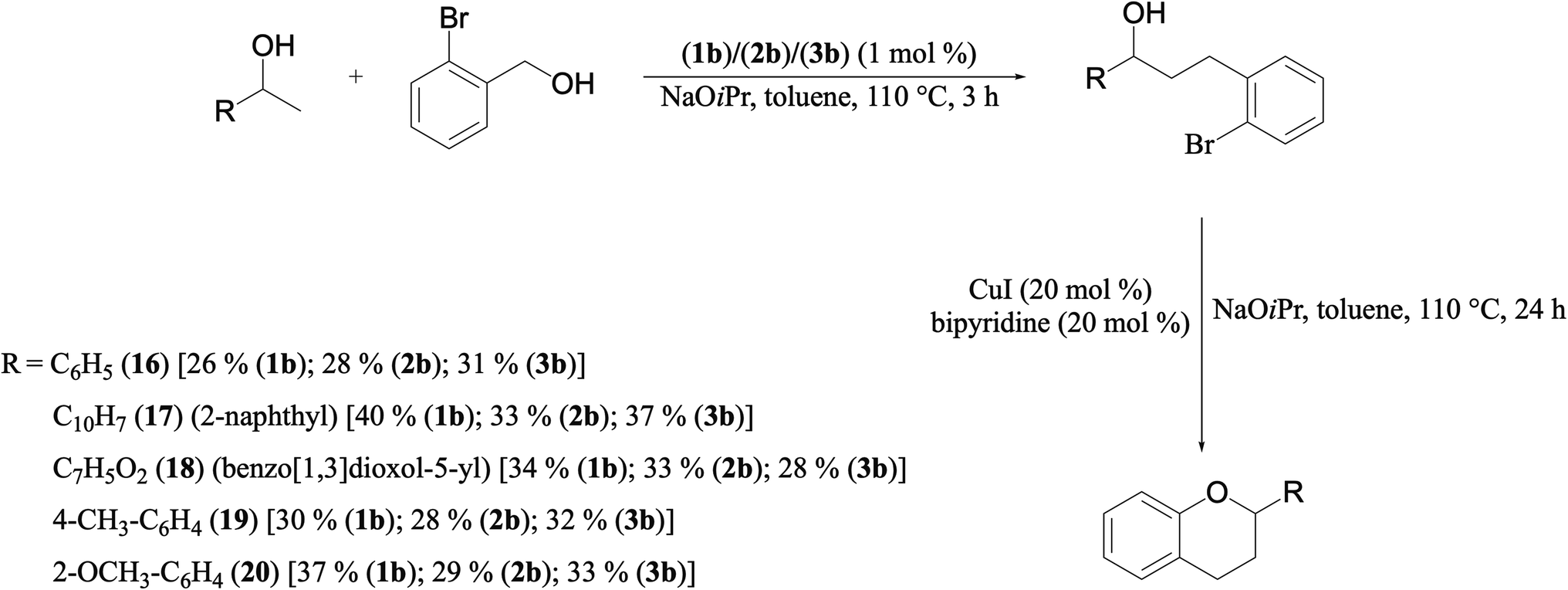 | ||
| Scheme 3 A simple practical synthesis of a variety of flavan derivatives (16–20), by Ru–NHC (1–3)b complexes in a one-pot tandem fashion. | ||
Conclusion
In summary, three neutral amido-functionalized ruthenium complexes of 1,2,4-triazole derived N-heterocyclic carbene (1–3)b were synthesized by transmetallation reaction with [(p-cymene)RuCl2]2, from their respective silver counterparts. The ruthenium (1–3)b complexes efficiently carried out the one-pot tandem reaction for a variety of substrates having electron withdrawing and electron donating substrates in moderate to good isolated yields. A careful scrutiny of the substrate scope study revealed that isopropyl group containing neutral ruthenium (2b) complex gave a good to excellent yield of ca. 59–72%, whereas the ethyl group containing neutral ruthenium (1b) complex and benzyl group containing neutral ruthenium (3b) complex gave a comparable yield of ca. 49–71% and ca. 51–69% respectively. The time dependence study performed on a representative (1b) complex showed the major catalysis product modulating between partially reduced CC hydrogenated carbonyl species PhCOCH2CH2Ph (4′) and fully reduced CO and CC hydrogenated secondary alcohol, PhCH(OH)CH2CH2Ph (4) over extended period of 120 hours. Mechanistic validation of the proposed catalytic cycle were obtained by the mass spectrometric characterization of two key catalytic intermediates namely, the ketone coordinated ruthenium hydride species (B) {[M + H]+ at 591.1913 (calcd 591.1912)} and aldehyde coordinated ruthenium hydride species, (F) {[M + H]+ at 577.1751 (calcd 577.1755)}. Additionally, the CC hydrogenated carbonyl intermediate species PhCOCH2CH2Ph (4′) along with final fully reduced CO and CC hydrogenated species PhCH(OH)CH2CH2Ph (4) were isolated and characterized. The utility of the neutral ruthenium (1–3)b complexes in the alcohol–alcohol coupling reaction was demonstrated through the one-pot synthesis of five different bioactive flavan derivatives (16–20). Employing DFT methods, we have explored two possible mechanistic pathways that were adapted based on the experimental evidence and literature precedents. Our results suggest that the initial step of generating the catalytic precursor involves the substitution of Cl− ion by a primary or secondary alcohol via an associative mechanism. Of the two pathways studied, the barriers involving alcohols in the ionic pathway are relatively smaller compared to the neutral pathway in the dehydrogenation and the hydrogenation steps. However, the energetic cost associated with the generation of catalytic precursors, (A′) and (E′) are extremely high for the ionic pathway compared to the catalytic precursors, (A) and (E) for the neutral pathway. Despite a slightly higher barrier in the second dehydrogenation and the third hydrogenation steps, the neutral pathway has a significant energy advantage in the first nucleophilic substitution step. Hence, reaction is expected to proceed via the neutral pathway. The findings from those study will direct the development of ruthenium-based N-heterocyclic carbene catalysts for the dehydrogenative cross-coupling of primary and secondary alcohols.
Experimental section
General procedures
All manipulations were carried out using a combination of a glovebox and standard Schlenk techniques. Solvents were purified and degassed by standard procedures. 1-(R)-4-N (Furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride [R = Et (1),42 i-Pr (2),42 Bn (3)42], {[1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazole-5-ylidene]2Ag}+Cl− (1a)41 and {[1-(ethyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− (2a)41 were synthesized according to the modified literature procedures. 1H and 13C{1H} NMR spectra were recorded on Bruker 400 MHz and Bruker 500 MHz NMR spectrometer. 1H NMR peaks are labeled as singlet (s), doublet (d), triplet (t), quartet (quat), quintet, broad (br), triplet of triplets (tt), doublet of doublets (dd), doublet of triplets (dt), doublet of quartets (dq), multiplet (m), and septet. Infrared spectra in transmission mode were recorded on a PerkinElmer Spectrum One FT-IR spectrometer. Mass spectrometry measurements were done on a Micromass Q-Tof and Bruker Maxis Impact spectrometer. Elemental analysis was carried out on Thermo Quest FLASH 1112 SERIES (CHNS) Elemental Analyzer. X-ray diffraction data for compounds (1–3)b were collected on a Bruker APEX 2 CCD platform diffractometer (Mo Kα (k = 0.71073 Å)) equipped with an Oxford liquid nitrogen cryostream. Crystals were mounted in a nylon loop with Paratone–N cryoprotectant oil. The structures were solved using direct methods and standard difference map techniques, and were refined by full-matrix least-squares procedures on F2 with SHELXTL (Version 6.14).53 Compound 2b exhibited a disordered isopropyl group on a p-cymene, which was modeled and refined freely. Crystals of 3b were found to be non-merohedrally twinned. CELL_NOW (version 2008/2)54 was used to find two twin domains and both components were integrated with SAINT using the multiple-component orientation matrix produced by CELL_NOW. The data were absorption corrected and scaled with TWINABS (version 2008/4).55 Initial solutions were found and refined with merged and roughly detwinned HKLF 4 format data before final refinement against HKLF 5 format twin data yielding twin ratio (BASF) 0.496(6). For the catalysis runs, the GCMS analyses were done using Agilent Technologies 7890A GC systems with 5975C inert XL EI/CI MSD Triple-Axis detector.:3 v/v) to give the product 1b as an orange solid (0.311 g, 89%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ ppm, 8.07 (s, 1H, NCN), 7.46 (br, 1H, C43O), 6.44 (t, 1H, 3JHH = 3 Hz, C43O), 6.39 (d, 1H, 3JHH = 3 Hz, C43O), 5.29 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 5.07 (d, 1H, 2JHH = 15 Hz, C2), 5.04 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.95 (d, 2H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.60 (d, 1H, 2JHH = 15 Hz, C2), 4.58 (qd, 1H, 3JHH = 7 Hz, C2CH3), 4.48 (qd, 1H, 3JHH = 7 Hz, C2CH3), 4.40 (d, 1H, 2JHH = 16 Hz, C2), 4.35 (d, 1H, 2JHH = 16 Hz, C2), 2.53 (sept, 1H, 3JHH = 7 Hz, p-CH3C6H4C(CH3)2 2.04 (s, 3H, p-C3C6H4CH(CH3)2), 1.60 (t, 3H, 3JHH = 7 Hz, CH2C3), 1.16 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2), 1.01 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2. 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ ppm, 180.5 (Ru–N![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) N), 168.8 (O), 156.4 (4H3O), 142.0 (N–(3)-N), 140.3 (4H3O), 111.3 (4H3O), 108.6 (p-CH36H4CH(CH3(C4H3O)2), 107.6 (4H3O), 100.3, (p-CH36H4CH(CH3)2), 88.6 (p-CH36H4CH(CH3)2), 86.3 (p-CH36H4CH(CH3)2), 84.3 (p-CH36H4CH(CH3)2), 82.8 (p-CH36H4CH(CH3)2), 52.8 (H2), 48.6 (H2), 47.4 (H2CH3), 31.5 (p-CH3C6H4H(CH3)2), 24.3 (p-CH3C6H4CH(H3)2), 20.8 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2), 15.6 (CH2H3). IR data (KBr pellet) cm−1: 1589 (s) (νco). HRMS (ES): m/z 505.0948 [M + H]+, calcd 505.0941. Anal. calcd for C21H27RuClN4O2: C, 50.05; H, 5.40; N, 11.12 found: C, 50.77; H, 5.05; N, 11.85%.:3 v/v) to give product 2b as a orange solid (0.286 g, 52%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ ppm, 8.18 (s, 1H, NCN), 7.38 (br, 1H, C43O), 6.39 (br, 1H, C43O), 6.37 (br, 1H, C43O), 5.22 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 5.18 (sept, 1H, 3JHH = 6 Hz, C(CH3)2, 4.98 (d, 1H, 2JHH = 15 Hz, C2), 4.95 (br, 1H, p-CH3C64CH(CH3)2), 4.90 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.63 (d, 1H, 2JHH = 15 Hz, C2), 4.31 (d, 1H, 2JHH = 15 Hz, C2), 4.30 (d, 1H, 2JHH = 15 Hz, C2), 2.57 (sept, 1H, 3JHH = 7 Hz, p-CH3C6H4C(CH3)2), 2.03 (s, 3H, p-C3C6H4CH(CH3)2, 1.59 (d, 3H, 3JHH = 7 Hz, CH(C3)2), 1.53 (d, 3H, 3JHH = 7 Hz, CH(C3)2), 1.15 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2), 1.00 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ ppm, 179.3 (Ru–NN), 168.8 (O), 156.6 (4H3O), 141.9 (N–(3)-N), 140.2 (4H3O), 111.3 (4H3O), 108.3 (p-CH36H4CH(CH3)2), 107.6 (4H3O), 100.3 (p-CH36H4CH(CH3)2), 88.2 (p-CH36H4CH(CH3)2), 85.9 (p-CH36H4CH(CH3)2), 84.3 (p-CH36H4CH(CH3)2), 83.1 (p-CH36H4CH(CH3)2), 54.1 (H(CH3)2), 52.7 (H2), 48.5 (H2), 31.5 (p-CH3C6H4H(CH3)2), 24.2 (CH(H3)2, 23.4 (CH(H3)2, 23.3 (p-CH3C6H4CH(H3)2), 21.0 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2). IR data (KBr pellet) cm−1: 1584 (s) (νco). HRMS (ES): m/z 519.1096 [M + H]+, calcd. 519.1098. Anal. calcd for C22H29RuClN4O2: C, 51.01; H, 5.64; N, 10.82. Found: C, 50.62; H, 5.52; N, 11.22%.CO), 8.41 (s, 2H, N–C(3)–N), 7.28 (br, 6H, C65), 7.23 (br, 2H, C43O), 7.19 (br, 4H, C65), 6.23 (br, 2H, C43O), 6.22 (br, 2H, C43O), 5.33 (s, 4H, C2), 5.26 (s, 4H, C2), 4.40 (br, 4H, C2NH). 13C{1H} NMR (DMSO-d6, 100 MHz, 25 °C): δ ppm, 183.3 (Ag–NN), 166.1 (O), 151.5 (4H3O), 145.2 (ipso-6H5), 142.3 (N–(3)-N), 142.3 (4H3O), 136.1 (6H5), 128.7 (6H5) 128.1 (6H5), 110.5 (4H3O), 107.3 (4H3O), 56.0 (H2), 50.24 (H2), 35.8 (H2). IR data (KBr pellet cm−1): 1667 (s) (νco). HRMS (ES): m/z 699.1590 [(NHC)2Ag]+, calcd 699.1592. Anal. calcd for C32H32AgClN8O4·0.5H2O: C, 51.59; H, 4.46; N, 15.04. Found: C, 52.23; H, 4.80; N, 14.08%.:3 v/v) to give the product 3b as an yellow solid (0.302 g, 76%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ ppm, 8.20 (s, 1H, NCN), 7.41–7.37 (m, 3H, C43O & C65), 7.34–7.30 (m, 3H, C65), 6.40 (br, 1H, C43O), 6.36 (br, 1H, C43O), 5.99 (d, 1H, 2JHH = 16 Hz, C2), 5.58 (d, 1H, 2JHH = 16 Hz, C2), 5.04 (d, 1H, 2JHH = 16 Hz, C2), 4.92 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.79 (br, 1H, p-CH3C64CH(CH3)2), 4.76 (d, 2H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.64 (d, 1H, 2JHH = 16 Hz, C2), 4 39 (d, 1H, 2JHH = 15 Hz, C2), 4.34 (d, 1H, 2JHH = 15 Hz, C2), 2.45 (sept, 1H, 3JHH = 7 Hz, p-CH3C6H4C(CH3)2), 1.93 (s, 3H, p-C3C6H4CH(CH3)2), 1.03 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2), 0.88 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ ppm, 182.3 (Ru–NN), 168.8 (O), 156.4 (4H3O), 142.3 (N–(3)H–N), 140.2 (4H3O), 137.0 (6H5), 129.0 (6H5), 128.3 (6H5), 127.0 (6H5), 111.2 (4H3O), 108.7 (p-CH36H4CH(CH3)2), 107.5 (4H3O), 100.3 (p-CH36H4CH(CH3)2), 88.1 (p-CH36H4CH(CH3)2), 85.3 (p-CH36H4CH(CH3)2), 84.5 (p-CH36H4CH(CH3)2), 83.4 (p-CH36H4CH(CH3)2), 55.3 (CH2), 52.8 (H2), 48.8 (H2), 31.3 (p-CH3C6H4H(CH3)2), 24.1 (p-CH3C6H4CH(H3)2), 20.9 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2. IR data (KBr pellet) cm−1: 1587 (s) (νco). HRMS (ES): m/z 567.1097 [M + H]+, calcd 567.1099. Anal. calcd for C26H29RuClN4O2: C, 55.17; H, 5.16; N, 9.90. Found: C, 54.38; H, 5.56; N, 10.50%.
N), 168.8 (O), 156.4 (4H3O), 142.0 (N–(3)-N), 140.3 (4H3O), 111.3 (4H3O), 108.6 (p-CH36H4CH(CH3(C4H3O)2), 107.6 (4H3O), 100.3, (p-CH36H4CH(CH3)2), 88.6 (p-CH36H4CH(CH3)2), 86.3 (p-CH36H4CH(CH3)2), 84.3 (p-CH36H4CH(CH3)2), 82.8 (p-CH36H4CH(CH3)2), 52.8 (H2), 48.6 (H2), 47.4 (H2CH3), 31.5 (p-CH3C6H4H(CH3)2), 24.3 (p-CH3C6H4CH(H3)2), 20.8 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2), 15.6 (CH2H3). IR data (KBr pellet) cm−1: 1589 (s) (νco). HRMS (ES): m/z 505.0948 [M + H]+, calcd 505.0941. Anal. calcd for C21H27RuClN4O2: C, 50.05; H, 5.40; N, 11.12 found: C, 50.77; H, 5.05; N, 11.85%.:3 v/v) to give product 2b as a orange solid (0.286 g, 52%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ ppm, 8.18 (s, 1H, NCN), 7.38 (br, 1H, C43O), 6.39 (br, 1H, C43O), 6.37 (br, 1H, C43O), 5.22 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 5.18 (sept, 1H, 3JHH = 6 Hz, C(CH3)2, 4.98 (d, 1H, 2JHH = 15 Hz, C2), 4.95 (br, 1H, p-CH3C64CH(CH3)2), 4.90 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.63 (d, 1H, 2JHH = 15 Hz, C2), 4.31 (d, 1H, 2JHH = 15 Hz, C2), 4.30 (d, 1H, 2JHH = 15 Hz, C2), 2.57 (sept, 1H, 3JHH = 7 Hz, p-CH3C6H4C(CH3)2), 2.03 (s, 3H, p-C3C6H4CH(CH3)2, 1.59 (d, 3H, 3JHH = 7 Hz, CH(C3)2), 1.53 (d, 3H, 3JHH = 7 Hz, CH(C3)2), 1.15 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2), 1.00 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ ppm, 179.3 (Ru–NN), 168.8 (O), 156.6 (4H3O), 141.9 (N–(3)-N), 140.2 (4H3O), 111.3 (4H3O), 108.3 (p-CH36H4CH(CH3)2), 107.6 (4H3O), 100.3 (p-CH36H4CH(CH3)2), 88.2 (p-CH36H4CH(CH3)2), 85.9 (p-CH36H4CH(CH3)2), 84.3 (p-CH36H4CH(CH3)2), 83.1 (p-CH36H4CH(CH3)2), 54.1 (H(CH3)2), 52.7 (H2), 48.5 (H2), 31.5 (p-CH3C6H4H(CH3)2), 24.2 (CH(H3)2, 23.4 (CH(H3)2, 23.3 (p-CH3C6H4CH(H3)2), 21.0 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2). IR data (KBr pellet) cm−1: 1584 (s) (νco). HRMS (ES): m/z 519.1096 [M + H]+, calcd. 519.1098. Anal. calcd for C22H29RuClN4O2: C, 51.01; H, 5.64; N, 10.82. Found: C, 50.62; H, 5.52; N, 11.22%.CO), 8.41 (s, 2H, N–C(3)–N), 7.28 (br, 6H, C65), 7.23 (br, 2H, C43O), 7.19 (br, 4H, C65), 6.23 (br, 2H, C43O), 6.22 (br, 2H, C43O), 5.33 (s, 4H, C2), 5.26 (s, 4H, C2), 4.40 (br, 4H, C2NH). 13C{1H} NMR (DMSO-d6, 100 MHz, 25 °C): δ ppm, 183.3 (Ag–NN), 166.1 (O), 151.5 (4H3O), 145.2 (ipso-6H5), 142.3 (N–(3)-N), 142.3 (4H3O), 136.1 (6H5), 128.7 (6H5) 128.1 (6H5), 110.5 (4H3O), 107.3 (4H3O), 56.0 (H2), 50.24 (H2), 35.8 (H2). IR data (KBr pellet cm−1): 1667 (s) (νco). HRMS (ES): m/z 699.1590 [(NHC)2Ag]+, calcd 699.1592. Anal. calcd for C32H32AgClN8O4·0.5H2O: C, 51.59; H, 4.46; N, 15.04. Found: C, 52.23; H, 4.80; N, 14.08%.:3 v/v) to give the product 3b as an yellow solid (0.302 g, 76%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ ppm, 8.20 (s, 1H, NCN), 7.41–7.37 (m, 3H, C43O & C65), 7.34–7.30 (m, 3H, C65), 6.40 (br, 1H, C43O), 6.36 (br, 1H, C43O), 5.99 (d, 1H, 2JHH = 16 Hz, C2), 5.58 (d, 1H, 2JHH = 16 Hz, C2), 5.04 (d, 1H, 2JHH = 16 Hz, C2), 4.92 (d, 1H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.79 (br, 1H, p-CH3C64CH(CH3)2), 4.76 (d, 2H, 3JHH = 6 Hz, p-CH3C64CH(CH3)2), 4.64 (d, 1H, 2JHH = 16 Hz, C2), 4 39 (d, 1H, 2JHH = 15 Hz, C2), 4.34 (d, 1H, 2JHH = 15 Hz, C2), 2.45 (sept, 1H, 3JHH = 7 Hz, p-CH3C6H4C(CH3)2), 1.93 (s, 3H, p-C3C6H4CH(CH3)2), 1.03 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2), 0.88 (d, 3H, 3JHH = 7 Hz, p-CH3C6H4CH(C3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ ppm, 182.3 (Ru–NN), 168.8 (O), 156.4 (4H3O), 142.3 (N–(3)H–N), 140.2 (4H3O), 137.0 (6H5), 129.0 (6H5), 128.3 (6H5), 127.0 (6H5), 111.2 (4H3O), 108.7 (p-CH36H4CH(CH3)2), 107.5 (4H3O), 100.3 (p-CH36H4CH(CH3)2), 88.1 (p-CH36H4CH(CH3)2), 85.3 (p-CH36H4CH(CH3)2), 84.5 (p-CH36H4CH(CH3)2), 83.4 (p-CH36H4CH(CH3)2), 55.3 (CH2), 52.8 (H2), 48.8 (H2), 31.3 (p-CH3C6H4H(CH3)2), 24.1 (p-CH3C6H4CH(H3)2), 20.9 (p-CH3C6H4CH(H3)2), 18.8 (p-H3C6H4CH(CH3)2. IR data (KBr pellet) cm−1: 1587 (s) (νco). HRMS (ES): m/z 567.1097 [M + H]+, calcd 567.1099. Anal. calcd for C26H29RuClN4O2: C, 55.17; H, 5.16; N, 9.90. Found: C, 54.38; H, 5.56; N, 10.50%.General procedure for β-alkylation of alcohols
In a typical catalysis run, performed in a pressure vial, a mixture of primary alcohol (1.00 mmol), secondary alcohol (1.00 mmol), NaO-i-Pr (1.00 mmol), and the Ru–NHC complex (1b)/(2b)/(3b) (0.01 mmol, 1 mol%) in 2 mL of toluene was heated at 110 °C for 3 hours. The reaction mixture was then cooled to room temperature, volatiles were evaporated thus obtained crude product was further purified by silica gel column chromatography using a mixed medium of petroleum ether and EtOAc to give the desired products (4–15).General procedure for mass experiment for the detection of catalytic intermediates
In a typical mass experiment for (1b), a 1:1:1 mixture of benzyl alcohol (0.1 mmol), 1-phenylethanol (0.1 mmol), NaO-i-Pr (0.1 mmol), and the Ru–NHC complex (1b) (0.01 mmol, 1 mol%) in ca. 2 mL of toluene was refluxed at 110 °C for 5 minutes and aliquot was subjected to mass analysis. ESI-MS data of the acetophenone bound Ru–H species (B) (Fig. 4) and benzaldehyde bound Ru–H species (F) (Fig. 5).
General procedure for the synthesis of the flavan derivatives
A mixture of Ru–NHC complex (1b)/(2b)/(3b) (0.01 mmol, 1 mol%), NaO-i-Pr (1.00 mmol), (2-bromophenyl)methanol (1.00 mmol) and 1-phenylethanol/1-(naphthalen-2-yl)ethanol/1-(benzo[1,3]dioxol-5-yl)ethanol/1-(4-tolyl)ethanol/1-(2-methoxyphenyl)ethanol (1.00 mmol), and ca. 2 mL toluene was stirred at 110 °C for 3 h. After cooled to ambient temperature, CuI (0.1 mmol, 20 mol%), 2,2′-bipyridine (0.1 mmol, 20 mol%), NaO-i-Pr (1.00 mmol), and ca. 1 mL toluene were added to the same reactor, and the resultant mixture was stirred at 110 °C for 24 h. The reaction mixture was cooled and quenched with aqueous NH4Cl solution followed by CH2Cl2 (ca. 20 mL) was added to the reaction mixture and the organic layer was separated. This process was repeated two times and the combined organic phase was dried over anhydrous Na2SO4. Then the solvent was removed under reduced pressure. The corresponding flavan derivatives was isolated through silica gel (230–400 mesh) column chromatography using pet ether/ethyl acetate (200:1, v/v) as eluent. The final products (16–20) were authenticated by NMR, GCMS and CHN.
General procedure for synthesis of one representative flavan intermediate (9)
In a typical catalysis run, performed in a pressure vial, a mixture of 2-bromobenzyl alcohol (1.00 mmol), 1-phenylethanol (1.00 mmol), NaO-i-Pr (1.00 mmol), and the Ru–NHC complex (1b)/(2b)/(3b) (0.01 mmol, 1 mol%) in 2 mL of toluene was heated at 110 °C for 3 hours. The reaction mixture was then cooled to room temperature, volatiles were evaporated thus obtained crude product was further purified by silica gel column chromatography using a mixed medium of petroleum ether and EtOAc to give the desired products (9) with yield ca. 64% (1b), 64% (2b), 62% (3b).General procedure for synthesis of one representative flavan derivative 2-phenylchroman (16) from flavan intermediate (9)
In a typical catalysis run, performed in a pressure vial, a mixture of (9) (1.00 mmol), CuI (0.1 mmol, 20 mol%), 2,2′-bipyridine (0.1 mmol, 20 mol%), and NaO-i-Pr (1.00 mmol), in 2 mL of toluene was heated at 110 °C for 24 hours. The reaction mixture was cooled and quenched with aqueous NH4Cl solution followed by CH2Cl2 (ca. 30 mL) was added to the reaction mixture and the organic layer was separated. This process was repeated two times and the combined organic phase was dried over anhydrous Na2SO4. Then the solvent was removed under reduced pressure. The corresponding flavan derivative (16) was isolated through silica gel (230–400 mesh) column chromatography using pet ether/ethyl acetate (200:1, v/v) as eluent with yield ca. 32%.
Computational methods
All DFT calculations were performed using G09 version D suite of programs.56 All geometry optimization were carried out using B3LYP-D2/LACVP* basis set, which encompasses the LanL2DZ basis set for Ru and 6-31G* for the rest of the atoms.57,58 To confirm the nature of the species, vibrational frequencies were estimated, and all the minima were found to have positive frequencies and transition states were characterized by one imaginary frequency which was further analysed using visualization program to affirm the correct nature of the TS. Single point calculations were then performed on the optimized geometry at B3LYP-D2/def2-TZVP basis set59 level, including solvent correction employing a PCM model with Toluene as solvent.60 Thus, all the quoted energies were free energies incorporating corrections from frequency calculations performed. For the selected transition states, Intrinsic Reaction Coordinate (IRC) calculations were performed to confirm if it connects to the desired reactant and product.61 For visualization of geometries and frequencies, Chemcraft software was used. (Lite version build 2005, 8).62Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DST and SERB (grant no: SR/S1/IC-50/2011, EMR/2014/000254, CRG/2019/000029, CRG/2018/00430, DST/CSA-03/2018-10, and SB/SJF/2019-20/12), New Delhi, India and Council of Scientific & Industrial Research (CSIR) {01(2880)/17/EMR-II} New Delhi, India for financial support of this research. A. K. thanks CSIR, New Delhi. S. T. and C. N. thank IIT Bombay for research fellowships.References
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC.
- J. Yang, X. Liu, D.-L. Meng, H.-Y. Chen, Z.-H. Zong, T.-T. Feng and K. Sun, Adv. Synth. Catal., 2012, 354, 328–334 CrossRef CAS.
- Z. D. Young and R. J. Davis, Catal. Sci. Technol., 2018, 8, 1722–1729 RSC.
- T. Matsu-ura, S. Sakaguchi, Y. Obora and Y. Ishii, J. Org. Chem., 2006, 71, 8306–8308 CrossRef CAS PubMed.
- D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876–3902 RSC.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CAS.
- Q. Wang, K. Wu and Z. Yu, Organometallics, 2016, 35, 1251–1256 CrossRef CAS.
- S. Genç, B. Arslan, S. Gülcemal, S. Günnaz, B. Çetinkaya and D. Gülcemal, J. Org. Chem., 2019, 84, 6286–6297 CrossRef PubMed.
- M. V. Jimenez, J. Fernandez-Tornos, F. J. Modrego, J. J. Perez-Torrente and L. A. Oro, Chem.–Eur. J., 2015, 21, 17877–17889 CrossRef CAS PubMed.
- C. Xu, L. Y. Goh and S. A. Pullarkat, Organometallics, 2011, 30, 6499–6502 CrossRef CAS.
- T. Liu, L. Wang, K. Wu and Z. Yu, ACS Catal., 2018, 8, 7201–7207 CrossRef CAS.
- A. Alanthadka, S. Bera and D. Banerjee, J. Org. Chem., 2019, 84, 11676–11686 CrossRef CAS PubMed.
- B. Pandey, S. Xu and K. Ding, Org. Lett., 2019, 21, 7420–7423 CrossRef CAS PubMed.
- F. Freitag, T. Irrgang and R. Kempe, Chem.–Eur. J., 2017, 23, 12110–12113 CrossRef CAS PubMed.
- T. Miura, O. Kose, F. Li, S. Kai and S. Saito, Chem. - Eur. J., 2011, 17, 11146–11151 CrossRef CAS PubMed.
- S. Liao, K. Yu, Q. Li, H. Tian, Z. Zhang, X. Yu and Q. Xu, Org. Biomol. Chem., 2012, 10, 2973–2978 RSC.
- D.-W. Tan, H.-X. Li, D.-L. Zhu, H.-Y. Li, D. J. Young, J.-L. Yao and J.-P. Lang, Org. Lett., 2018, 20, 608–611 CrossRef CAS PubMed.
- G. Tang and C.-H. Cheng, Adv. Synth. Catal., 2011, 353, 1918–1922 CrossRef CAS.
- M.-J. Zhang, H.-X. Li, D. J. Young, H.-Y. Li and J.-P. Lang, Org. Biomol. Chem., 2019, 17, 3567–3574 RSC.
- H. W. Cheung, T. Y. Lee, H. Y. Lui, C. H. Yeung and C. P. Lau, Adv. Synth. Catal., 2008, 350, 2975–2983 CrossRef CAS.
- S. Shee, B. Paul, D. Panja, B. C. Roy, K. Chakrabarti, K. Ganguli, A. Das, G. K. Das and S. Kundu, Adv. Synth. Catal., 2017, 359, 3888–3893 CrossRef CAS.
- D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321–325 CrossRef CAS.
- J. Shi, B. Hu, P. Ren, S. Shang, X. Yang and D. Chen, Organometallics, 2018, 37, 2795–2806 CrossRef CAS.
- P. Satyanarayana, G. M. Reddy, H. Maheswaran and M. L. Kantam, Adv. Synth. Catal., 2013, 355, 1859–1867 CrossRef CAS.
- A. P. da Costa, M. Sanaú, E. Peris and B. Royo, Dalton Trans., 2009, 6960–6966 RSC.
- S. Genç, S. Gülcemal, S. Günnaz, B. Çetinkaya and D. Gülcemal, J. Org. Chem., 2020, 85, 9139–9152 CrossRef PubMed.
- S. Kumar, A. Narayanan, M. N. Rao, M. M. Shaikh and P. Ghosh, J. Chem. Sci., 2011, 123, 791–798 CrossRef CAS.
- D. Kumar, A. P. Prakasham, S. Das, A. Datta and P. Ghosh, ACS Omega, 2018, 3, 1922–1938 CrossRef CAS PubMed.
- A. P. Prakasham, S. Ta, S. Dey and P. Ghosh, Dalton Trans., 2021, 50, 15640–15654 RSC.
- S. Musa, L. Ackermann and D. Gelman, Adv. Synth. Catal., 2013, 355, 3077–3080 CrossRef CAS.
- K. Chakrabarti, B. Paul, M. Maji, B. C. Roy, S. Shee and S. Kundu, Org. Biomol. Chem., 2016, 14, 10988–10997 RSC.
- A. R. Sahoo, G. Lalitha, V. Murugesh, C. Bruneau, G. V. M. Sharma, S. Suresh and M. Achard, J. Org. Chem., 2017, 82, 10727–10731 CrossRef CAS PubMed.
- B. C. Roy, K. Chakrabarti, S. Shee, S. Paul and S. Kundu, Chem.–Eur. J., 2016, 22, 18147–18155 CrossRef CAS PubMed.
- C. S. Cho, B. T. Kim, H.-S. Kim, T.-J. Kim and S. C. Shim, Organometallics, 2003, 22, 3608–3610 CrossRef CAS.
- X. Yang, ACS Catal., 2013, 3, 2684–2688 CrossRef CAS.
- M. Huang, Y. Li, J. Liu, X.-B. Lan, Y. Liu, C. Zhao and Z. Ke, Green Chem., 2019, 21, 219–224 RSC.
- Y.-Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q.-Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- A. Kumar, M. K. Gangwar, M. M. Shaikh and P. Ghosh, Proc. Natl. Acad. Sci. U. S. A., 2016, 86, 605–609 CAS.
- A. Kumar, L. P. Bheeter, M. K. Gangwar, J.-B. Sortais, C. Darcel and P. Ghosh, J. Organomet. Chem., 2015, 786, 63–70 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1932, 54, 3570–3582 CrossRef CAS.
- M. Viciano, M. Sanaú and E. Peris, Organometallics, 2007, 26, 6050–6054 CrossRef CAS.
- A. Prades, M. Viciano, M. Sanaú and E. Peris, Organometallics, 2008, 27, 4254–4259 CrossRef CAS.
- K.-i. Oyama and T. Kondo, J. Org. Chem., 2004, 69, 5240–5246 CrossRef CAS PubMed.
- O. Mazimba, I. B. Masesane and R. R. Majinda, Tetrahedron Lett., 2011, 52, 6716–6718 CrossRef CAS.
- S. Nakamura, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 5282–5283 CrossRef CAS PubMed.
- K. J. Hodgetts, Tetrahedron, 2005, 61, 6860–6870 CrossRef CAS.
- X. Shang, X. Zhou, W. Zhang, C. Wan and J. Chen, Tetrahedron, 2015, 71, 8187–8193 CrossRef CAS.
- B. V. Ramulu, L. Mahendar, J. Krishna, A. G. K. Reddy, B. Suchand and G. Satyanarayana, Tetrahedron, 2013, 69, 8305–8315 CrossRef CAS.
- A. G. K. Reddy, J. Krishna and G. Satyanarayana, ChemistrySelect, 2016, 1, 1151–1155 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, University of Göttingen, Germany, 2008.
- G. M. Sheldrick, Google Scholar There is no corresponding record for this reference Search PubMed.
- M. J. T. Frisch, G. W., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, 2009.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- K. Ishida, K. Morokuma and A. Komornicki, J. Chem. Phys., 1977, 66, 2153–2156 CrossRef CAS.
- G. A. Zhurko and D. A. Zhurko, https://www.chemcraftprog.com/, 2014.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C{1H} NMR, IR, HRMS, elemental analysis data of silver carbene complexes (3b) and ruthenium carbene complexes (1–3)b, 1H NMR, 13C{1H} NMR, GC-MS chromatograms and elemental analysis data of the catalysis products (4–15), five different flavan derivatives (16–20) and the CIF file giving X-ray crystallographic data of ruthenium carbene complexes (1–3)b. All computational study related coordinates, figures, energy profiles of the intermediates and transition states. CCDC 807541 (1b), 817082 (2b) and 823520 (3b). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05531g |
| ‡ A. K., S. T. and C. N. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |