
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on the characterization of metal active sites over Cu-based and Fe-based zeolites for NH3-SCR
Jialing Chen *a,
Wei Huanga,
Sizhuo Baoa,
Wenbo Zhanga,
Tingyu Liang*b,
Shenke Zhengc,
Lan Yia,
Li Guoa and
Xiaoqin Wu*a
*a,
Wei Huanga,
Sizhuo Baoa,
Wenbo Zhanga,
Tingyu Liang*b,
Shenke Zhengc,
Lan Yia,
Li Guoa and
Xiaoqin Wu*a
aKey Laboratory of Hubei Province for Coal Conversion and New Carbon Materials, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan, 430081, China. E-mail: chenjiialing@wust.edu.cn; wuxiaoqin@wust.edu.cn; Tel: +86 027 68862335
bSchool of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan, 430205, China. E-mail: ltingyu2006@126.com
cHubei Key Laboratory for Processing and Application of Catalytic Materials, School of Chemistry and Chemical Engineering, Huanggang Normal University, Huanggang, 438000, China
First published on 28th September 2022
Abstract
Cu-based and Fe-based zeolites are promising catalysts for NH3-SCR due to their high catalytic activity, wide temperature window and good hydrothermal stability, while the detailed investigation of NH3-SCR mechanism should be based on the accurate determination of active metal sites. This review systematically summarizes the qualitative and quantitative determination of metal active sites in Cu-based or Fe-based zeolites for NH3-SCR reactions based on advanced characterization methods such as UV-vis absorption (UV-vis), temperature-programmed reduction with H2 (H2-TPR), X-ray photoelectron spectroscopy (XPS), X-ray absorption fine structure spectroscopy (XAFS), Infrared spectroscopy (IR), Electron paramagnetic resonance (EPR), Mössbauer spectroscopy and DFT calculations. The application and limitations of different characterization methods are also discussed to provide insights for further study of the NH3-SCR reaction mechanism over metal-based zeolites.
1 Introduction
NH3-SCR technology, the selective catalytic reduction of NOx (NO and NO2) to N2 and H2O by NH3 with the aid of catalyst and oxygen, has become one of the main industrial deNOx technologies due to its high efficiency and low cost.1–4 The WO3–V2O5/TiO2 catalyst is the first commercialized NH3-SCR catalyst since the 1970s.5 However, WO3–V2O5/TiO2 catalysts are gradually abandoned in many countries due to their narrow temperature window (320–450 °C), poor hydrothermal stability in NH3-SCR, and more specifically, their high biological toxicity due to the need for vanadium species.6–8Cu-based and Fe-based zeolites are two potential NH3-SCR catalysts with high catalytic activity, wide temperature window and good hydrothermal stability.8–11 By delicately adjusting the content and distribution of metal species,12,13 and by delicately selecting the type of support zeolites,10,14 a variety of highly efficient Cu or Fe-based NH3-SCR zeolite catalysts have been developed, such as Fe/SSZ-13, Fe/Beta, Fe/ZSM-5, Cu/SSZ-13, Cu/SAPO-34, and Cu/LTA zeolites.15,16 Especially, small-pore Cu/SSZ-13 has been successfully applied in exhaust removal for diesel vehicles, though it still suffers from poor hydrothermal stability and poor sulfur resistance.16,17
Generally, the excellent redox properties and strong acidity are two crucial factors deciding the catalytic performance of a catalyst in NH3-SCR.15,16,18,19 The acid sites in catalysts facilitate the adsorption and activation of NH3 molecules, while the redox ability of catalysts originating from metal species mainly catalyze the redox cycle in NH3-SCR.2 According to previous researches,13,20–24 various Fe or Cu species existed in Fe-based or Cu-based zeolites due to the similar formation energy of metal species or the easy interconversion of different metal species under SCR reaction conditions. Therefore, it is important to reveal the catalytic roles of different active metal species in zeolites, so as to clarify the reaction mechanism of NH3-SCR over metal-based zeolites.
According to Gao et al.,13 there were mainly four kinds of iron species on Fe/SSZ-13 zeolites: isolated Fe2+ species, isolated Fe3+ species, dinuclear Fe3+ species ([HO–Fe–O–Fe–OH]2+), and multinuclear FexOy species which consist of trinuclear and highly aggregated FexOy species or nanoparticles. Based on detailed investigation on Fe/SSZ-13 zeolites, Gao et al.13,24 found that isolated Fe3+ species and dinuclear [HO–Fe–O–Fe–OH]2+ species were the dominant active sites at low-temperature (<300 °C) and high-temperature (≥300 °C) ranges for standard NH3-SCR, respectively, though the [HO–Fe–O–Fe–OH]2+ species could also catalyze the undesirable ammonia oxidation side reaction. Besides, isolated Fe2+ species had almost no NH3-SCR activity because of their low redox ability. In addition, the highly aggregated FexOy species in Fe/SSZ-13 zeolites, which catalyzed the ammonia oxidation side reactions, showed limited NH3-SCR activity even at high temperatures. As a summary, isolated Fe3+ and dinuclear [HO–Fe–O–Fe–OH]2+ possess excellent catalytic activity in NH3-SCR, while isolated Fe2+ species, and highly aggregated FexOy species are undesirable for NH3-SCR.13,24
Similar to Fe-based zeolites, several types of Cu species such as isolated Cu+ species, isolated Cu2+ species (including Cu2+–2Z and [Cu(OH)]+–Z species, where Z represents zeolite), Cu2+ dimer species (including single O-bridged dicopper [Cu–O–Cu]2+, double O-bridged dicopper or even bis(μ-hydroxo)-dicopper species), and multinuclear CuOx species or highly aggregated CuOx nanoparticles, can also be observed in Cu-based zeolites, according to previous researches.4,12,25–28
In general, researchers believed that isolated Cu2+ species were the main active sites for NH3-SCR reactions over Cu-based zeolites.12,20,25,26 Xue et al.29 had observed a positive correlation between the concentration of isolated Cu2+ species in Cu/SAPO-34 and the NOx conversion, and further proved that the turnover frequency (TOF) of isolated Cu2+ species in Cu/SAPO-34 remained almost unchanged with Cu loadings at 100–200 °C in NH3-SCR, which strongly proved that isolated Cu2+ species were the main active sites in NH3-SCR. Moreover, Gao et al.27 and Paolucci et al.30 found that both Cu2+–2Z and [Cu(OH)]+–Z species were the main active sites for standard NH3-SCR reactions in Cu/SSZ-13, but Cu2+–2Z species was more stable than [Cu(OH)]+–Z species, as the latter could be easily transformed into CuOx species under hydrothermal aging conditions.
Furthermore, researchers found that dimeric Cu2+ species could also be the main active species for NH3-SCR. Gao et al.12 revealed that at low temperatures (<250 °C), the standard NH3-SCR reaction rate was positively correlated with the square of Cu loadings over Cu/SSZ-13 zeolites, which confirmed the high NH3-SCR activity of dimeric Cu2+ species in NH3-SCR at low temperatures. However, the unstable dimeric Cu2+ species could be converted to isolated Cu2+ species at high temperatures, which became the main active sites for NH3-SCR at high temperatures. Recently, many studies revealed that Cu+ species in Cu-based zeolites also played important roles in NH3-SCR, especially at low temperatures.4,29,31,32 McEwen et al.28 found that both Cu2+ and Cu+ species existed and participated in Cu/SSZ-13 zeolites during the NH3-SCR reactions based on in situ X-ray absorption near edge spectra (XANES). Zhao et al.33 also found that the active species of NH3-SCR over Cu–Mn/SAPO-34 zeolites were a mixture of Cu+ and Cu2+ species. Chen et al.31 revealed that the formation of highly stable Cu+ species in Cu/SSZ-13 was favorable for low-temperature (<200 °C) NH3-SCR reactions.
In conclusion, different kinds of metal species had different redox ability, stability and coordination interactions with zeolite framework, which made them function differently in catalyzing NH3-SCR reactions. However, the coexistence of various Fe species over Fe-based zeolites (or various Cu species over Cu-based zeolites) made the investigation of NH3-SCR reactions mechanism difficult.12 In order to suppress the occurrence of side reactions so as to investigate the detailed reaction pathway of NH3-SCR, the total metal loadings in Cu-based or Fe-based zeolites are usually lower than 5 wt%, normally at about 2 wt%, to suppress the formation of undesired highly aggregated metal oxides or nanoparticles.13,34,35 This poses a problem that, the qualitative and quantitative determination of various metal species over zeolites became difficult due to their low content and high dispersion over zeolites. Therefore, the detailed study on the type, content and distribution of active metal species in metal-based zeolites and their catalytic roles in NH3-SCR with advanced characterization methods are necessary for a better understanding of NH3-SCR reaction mechanism.
In the past 3 years, the studies of NH3-SCR over metal oxides or metal-based zeolites were comprehensively reviewed, which mainly emphasized on the design of catalysts, reaction mechanism and deactivation mechanism. In 2019, Han et al.2 published a comprehensive review on the application of metal oxide catalysts, acidic compound catalysts, metal-based zeolite catalysts, monolith catalysts and their reaction mechanism in NH3-SCR. In addition, because of superior activity and hydrothermal stability in NH3-SCR, many reviews were concentrated on the application of Cu-based zeolite catalysts,36 especially in Cu-SAPO-34,37 Cu-CHA38,39 and Cu-based small-pore zeolites.3,15,40 Besides, several reviews also gave detail information about the design and reaction mechanism of Fe-based zeolites for NH3-SCR.14,18,41,42 Besides, Andana et al.43 summarized the recent research progress on the hybrid metal oxide-zeolite catalysts for low-temperature NH3-SCR, which could enable the in situ NO oxidation over metal oxide and subsequently fast SCR over zeolite component through the “bifunctional mechanism”.
For metal-based catalysts, many reviews focused on Mn-based oxides catalysts,44–47 Ce-based oxides catalysts,48 Fe-based oxides49 and the CeOx–MnOx mixture catalysts50 were published as those catalysts exhibited excellent low-temperature (<100 °C) in NH3-SCR. In addition, the latest progress on the vanadia-based and vanadia-free metal oxides catalysts had also been summarized.49,51 The deactivation mechanism of catalysts in NH3-SCR and corresponding strategies to enhance the poison-resistance of catalysts were also been summarized by researchers.19,52–55 The application of Density functional theory (DFT) in NH3-SCR were reviewed by Guan et al.56 to give clues on the mechanism studies. The influences of spatial confined structure on the catalytic performances of porous metal oxides, metal-based zeolite and metal organic framework catalysts were reviewed by Li et al.57 to give new insights for the designing of future NH3-SCR catalysts.
Though many reviews were published for NH3-SCR, reviews about the characterization methods dealing with the determination of active metal species in Cu-based or Fe-based zeolites for NH3-SCR are scare. Therefore, this review systematically introduced several applicable characterization methods for the accurate determination of active metal sites over Cu-based and Fe-based zeolites in NH3-SCR, such as UV-vis absorption spectra (UV-vis), temperature-programmed reduction with H2 (H2-TPR), X-ray photoelectron spectroscopy (XPS), X-ray absorption fine structure spectrum (XAFS), infrared spectroscopy (IR), electron paramagnetic resonance (EPR), Mössbauer spectroscopy and DFT calculations. In addition, the applications and limitations of different characterization methods in determining Cu-based or Fe-based zeolites are also compared and summarized. The investigation of NH3-SCR reaction mechanism based on the above characterization methods are also introduced, hoping to shed some light on the study of NH3-SCR mechanism over metal-based zeolites.
2 Characterization method of Cu-based or Fe-based zeolites for NH3-SCR
2.1 UV-vis
UV-vis absorption spectroscopy (UV-vis) is one of the powerful methods to determine the chemical state and content of metal species in zeolites.20 For example, the existence of isolated Fe3+ species in tetrahedral or octahedral coordination, oligomeric FexOy species, and hematite-type Fe2O3 species can be qualitatively determined by the characteristic absorption band of UV-vis spectra.58 According to literature,34,59,60 for Fe-based zeolites, the absorption band with wavelength lower than 300 nm in UV-vis spectra can be attributed to the isolated Fe3+ species, among which the UV absorption peak at 220–250 nm belongs to the four-coordinated isolated Fe3+ species while those at 250–300 nm are related to isolated Fe3+ species with higher coordination number such as octahedral-coordinated Fe3+ species. In addition, the absorption band at 300–400 nm is usually assigned to the charge transition peak of octahedral coordination aggregated Fe3+ species such as small FexOy species, while those with wavelength larger than 400 nm belongs to Fe2O3 nanoparticles. Based on the above assignments, the relative content of various Fe3+ species in zeolites can be estimated by the deconvolution of UV bands.34Generally, for the UV-vis spectra of Cu-based zeolites, the absorption peak at about 210 and 280 nm can be attributed to charge transfer from lattice O2− to Cu2+, the UV band at ca. 750 nm is related to the d–d transitions of Cu2+ species with distorted octahedral coordination, all of those bands are characteristic bands of isolated Cu2+ species.61–64 In addition, the absorption bands at ca. 250 nm and 450 nm can be assigned to CuOx species which are caused by the charge transfer and d–d transition of octahedral coordinated Cu2+ in CuOx species.25,61,62,65 Similarly, the quantitative estimation of various Cu species can be achieved based on the above assignments, though the wavelengths of the same type of Cu species over different zeolites slightly change due to different interactions between Cu cations with zeolite framework.62–64
UV-vis spectroscopy conducting in situ is a powerful tool to provide information on the NH3-SCR mechanism of catalysts. Zhang et al.20 studied the NH3-SCR mechanism of two active Cu species, i.e., isolated Cu2+ and [Cu(OH)]+ species, in Cu/SSZ-13 by in situ UV-vis with the aid of DFT calculations. They assigned the UV-vis band at 215 nm, 240 nm and 355 nm to [Cu(OH)]+ species, isolated Cu2+ and dimer [Cu2O2]2+ species, respectively, and found that two isolated [Cu(OH)]+ species could bridge to form a transient [Cu2O2]2+ species upon O2 activation, while isolated Cu2+ species remained unchanged. In addition, the [Cu(OH)]+ species exhibited stronger activity than isolated Cu2+ species during both reduction by NH3 and NO oxidation reactions. Moreover, the [Cu2O2]2+ intermediates could be detected under low-temperature SCR conditions. Those results indicated that [Cu(OH)]+ species might play a more important role than isolated Cu2+ species in Cu/SSZ-13 for NH3-SCR at low temperatures.
UV-vis spectra in combination with other techniques such as EPR, ICP-AES (inductively coupled plasma atomic emission spectroscopy) is an effective way to estimate the distribution of different metal species in zeolites.20,35,66 However, the estimation of the fraction for various Fe species or Cu species based on the deconvolution of UV-vis spectra is only a semiquantitative method due to the unknown extinction coefficients of different adsorption band currently.59 Meanwhile, only Fe3+ and Cu2+ species can be observed by UV-vis spectra, Fe2+ and Cu+ species existing in zeolites are invisible in the wavelength range of 200–800 nm in UV-vis experiments (for example, the adsorption band of Fe2+ species is located in the near infrared range around 1000 nm (ref. 66)). Moreover, only the coordination state of metal species can be obtained from UV-vis spectra, the detailed chemical structure, for example, whether the tetrahedral coordinated Cu2+ or Fe3+ species are in the framework or extra-framework of zeolites cannot be distinguished.64,67 Therefore, the accurate determination of metal species in zeolites by UV-vis should be available with the aid of other methods, such as EPR, ICP-AES, H2-TPR and IR experiments. In conclusion, the application and determination of Cu or Fe species in zeolites for NH3-SCR by UV-vis are summarized in Fig. 1.
 | ||
| Fig. 1 Summary on the determination of Cu or Fe species in zeolites by UV-vis spectra. | ||
2.2 H2-TPR
Temperature-programmed reduction with H2 (H2-TPR) is a widely used method to differentiate chemical valence of Cu or Fe species in zeolites by the reduction reaction between hydrogen and metal cations. The reduction temperature can be used to distinguish the chemical valence of metal species, while the deconvoluted reduction peak area in the H2-TPR profiles can be used to estimate the content of metal species.27,68The reduction process of Fe-based zeolites are quite complex, as various kinds of Fe species such as isolated Fe3+ species, Fe2+ species, FexOy clusters, large Fe2O3 nanoparticles can be easily formed in zeolites due to their similar energy of formation.13 However, by summarizing previous literature,24,69,70 the reduction peak of different Fe species can be roughly divided into several temperature ranges. The reduction of isolated Fe3+ at the ion-exchange sites of zeolites to Fe2+ are usually occurred at 380–430 °C, while the further reduction of these Fe2+ species to Fe0 can only take place at about 900–1000 °C due to the strong electrostatic interactions between Fe cations and the O–Al sites of zeolites.69,71,72 In addition, the reduction temperature of iron oxide clusters increases with their particle size, according to Brandenberger et al.69 Generally, the reduction of small FexOy clusters and large Fe2O3 nanoparticles are in the temperature range of 500–560 °C and 680–750 °C, respectively. In addition, some highly aggregated Fe2O3 or Fe3O4 particles are hard to be reduced during the H2-TPR experiments. Those Fe species can be reduced only by increasing the reduction temperatures to about 1000 °C, which usually causes the collapse of zeolite framework.34,73
For Cu-based zeolites, the reduction of isolated Cu2+ species at the ion-exchange sites of zeolites usually undergo two steps:27,29,74–76 the reduction of Cu2+ to Cu+, and further to Cu0. The reduction of two kinds of isolated Cu2+ species, i.e., Cu2+–2Z and [Cu(OH)]+–Z (Z represents zeolite) happen at different temperatures due to their different stability in zeolites:27,74,77 the reduction of more stable Cu2+–2Z to Cu+ happens at about 400 °C, while that of less stable [Cu(OH)]+–Z species at about 250 °C with a further reduction peak of Cu+ to Cu0 at about 360 °C. The reduction of CuO nanoparticles in Cu-based zeolites are much easier than Cu cations. CuO nanoparticles could be directly reduced to Cu0 by hydrogen at around 300 °C.27,29,74 Occasionally, reduction peak at around 700–900 °C emerged at the H2-TPR profiles of Cu-based zeolites, which could be attributed to the reduction of Cu+ at the ion-exchange sites of zeolites to Cu0 process, as the strong interactions between zeolite framework and Cu+ species hindered the reduction process.27,29 Table 1 summarizes the reduction steps of Cu-based or Fe-based zeolites by H2-TPR experiments.
| Zeolite | Reduction steps | Ref. |
|---|---|---|
| Cu-based zeolites | Two step reduction of isolated Cu2+ species: Cu2+ to Cu+: ∼400 °C; Cu+ to Cu0: 700–900 °C | 27, 29, 74 and 77 |
| Two step reduction of [Cu(OH)]+ species: [Cu(OH)]+ to Cu+: ∼250 °C; Cu+ to Cu0: 360 °C | 27, 74 and 77 | |
| Directly reduction of CuO to Cu0: ∼300 °C | 27, 29 and 74 | |
| Fe-based zeolites (reduction temperature increases with FexOy particle size) | Two step reduction of isolated Fe3+ species: Fe3+ to Fe2+: 380–430 °C; Fe2+ to Fe0: 900–1000 °C | 69, 71 and 72 |
| Reduction of small FexOy species: 500–560 °C | 24, 69 and 70–72 | |
| Reduction of large Fe2O3 nanoparticles: 680–750 °C | 24, 69 and 70–72 | |
| Reduction of aggregated Fe2O3 or Fe3O4 particles: >1000 °C | 34 and 73 |
Calculating the H2 consumption of each reduction peak by deconvolution the H2-TPR profiles is an effective way to quantitatively estimate the content of various metal species in zeolites. The reduction process of different kinds of Cu species in Cu-based zeolites are shown in formula (1)–(4). Combining the reduction temperatures and H2 consumption of various Cu species in H2-TPR profiles, the content of various Cu species in Cu-based zeolites could be roughly estimated, according to previous literature.77–79
 | (1) |
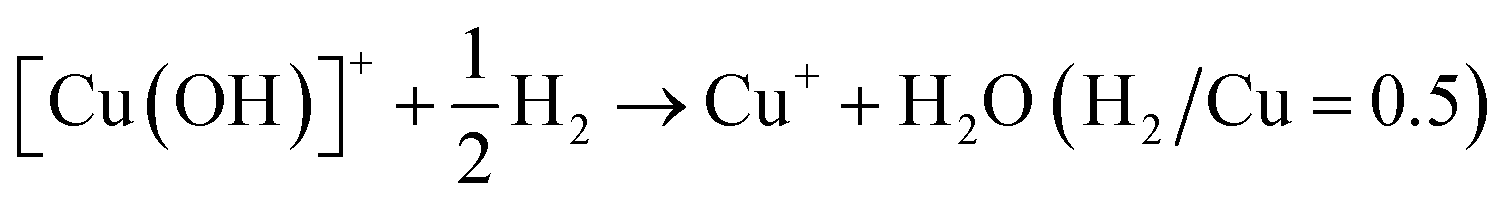 | (2) |
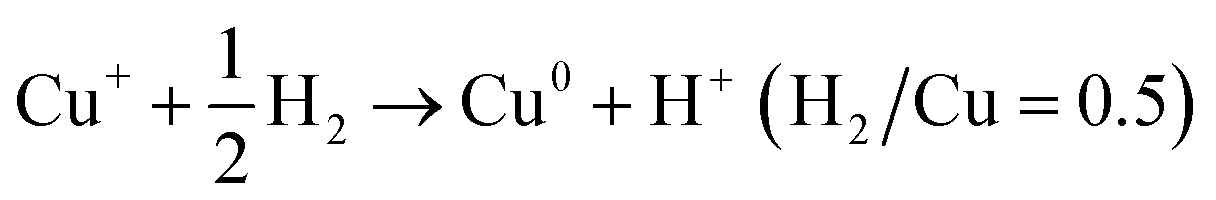 | (3) |
| Cu2+ + H2 → Cu0 + 2H+ (H2/Cu = 1) | (4) |
By conducting H2-TPR experiments, Gao et al.77 had semi-quantitively determined the isolated Cu2+ and [Cu(OH)]+ species and excluded the existence of CuO aggregated species in Cu/SSZ-13. Further, by combing with EPR and NH3-SCR kinetic experiments, they investigated the influences of Si/Al ratio and Cu content in Cu/SSZ-13 on the NH3-SCR mechanism and found that the six-membered rings (6MR) faces with 2 Al atoms in the CHA structure were the most favorable site in stabilizing Cu2+ ions, while Cu+ and [Cu(OH)]+ became the most stable Cu species in the absence of 2 Al sites. Song et al.27 also distinguished Cu2+ and [Cu(OH)]+ in Cu/SSZ-13 by H2-TPR and further studied the hydrothermal stability of them in NH3-SCR by combining EPR, DFT and kinetic reactions. They found that isolated Cu2+ species exhibited higher stability than [Cu(OH)]+ under hydrothermal aging conditions, which provided the atomic-level understanding of transformation of Cu species in NH3-SCR. Those literature indicate that H2-TPR is also an effective tool for the investigation of NH3-SCR mechanism.
However, when H2-TPR is used for the determination of metal species in zeolites, there are certain limitations: the reduction temperature of H2-TPR is usually below 1000 °C, so it is incapable to effectively detect the metal species that are extremely difficult to reduce.34 Moreover, the H2-TPR experiments cannot detect the zero valent metals in the zeolites. Furthermore, though H2-TPR can be used to determine both Cu+ and Cu2+ or Fe2+ and Fe3+ species in Cu-based or Fe-based zeolites, the reduction peak could be affected by many factors.
Firstly, the differentiation of reduction peaks for various metal species in zeolites are sometimes difficult, as the reduction peak of the same type of metal species usually shift with the zeolite supports and preparation method. For example, the reduction temperature of Cu species in zeolites slightly changed when using different zeolites as support, as the reduction behaviors of metal species were sensitive to the zeolite support and local chemical environment of metal species.62,75,78,80 Secondly, the reduction temperature of metal species may also shift with the increasing of metal content, as the gradually formation of aggregation metal species may be undetectable in H2-TPR or the overlapped reduction peaks may form a large broad peak.35 Thirdly, the H2O molecules adsorbed on the metal-based zeolites also affect the determination of H2-TPR, as the dehydration process of metal-based zeolites before the H2-TPR experiments can cause the auto-reduction of metal species which eventually decrease the H2 consumption in the H2-TPR profiles.77,81 Lastly, the shape and area of reduction peak in the H2-TPR experiments could also be affected by the heating rate of temperature-programmed process.82 Therefore, when H2-TPR is used to detect active metals on zeolites, it is necessary to select reasonable and repeatable experimental conditions, be careful in qualitative and quantitative analysis of H2-TPR results, and combine with other characterization methods to ensure the accuracy of the results.
2.3 XPS
X-ray photoelectron spectroscopy (XPS) based on photoelectric effect equation is often used to determine the composition and chemical valence of elements on zeolite catalysts by exciting the inner electrons and measuring the binding energy of elements.83The XPS signal of Cu 2p in Cu-based zeolites are usually separated into Cu 2p3/2 and Cu 2p1/2 doublet structures (two peaks) due to the spin–orbit coupling. Generally, the Cu+ 2p3/2 and 2p1/2 signals are located at binding energy of ca. 932.5 ± 0.2 eV and 952.3 ± 0.2 eV, respectively, while Cu2+ 2p3/2 and 2p1/2 signals are located at about 933.7 ± 0.2 eV and 953.6 ± 0.2 eV, respectively.84,85 In addition, the satellite peak at about 944 eV is also characteristic of Cu2+ species. However, the shift of binding energy for the same kind of Cu species are usually observed by many researchers, as the coordination structure and chemical state of Cu species can be significantly affected by the Cu loadings, preparation method and the zeolite supported of Cu-based zeolites. Xu and co-workers86 had observed the Cu2+ 2p2/3 and 2p1/2 signals at about 935.2 eV and 955.0 eV on Cu/Beta zeolites, which were related to the special local structure of Cu2+ species between Cu species and BEA framework. Moreover, previous researchers had different assignments for the binding energy at 928–940 eV of Cu 2p3/2 structure in Cu-based zeolites. Han and co-workers87 assigned the two asymmetric signals at 933.5 eV and 936.5 eV to CuO species and isolated Cu2+ species in Cu/SSZ-13, which were supported by the Auger electron spectra and many other researchers.88–90 Therefore, the distinguish of Cu+ and Cu2+ species are not easy by XPS method, as the chemical state of Cu species are related to the support, which should be analyzed by combining several types of characterization methods.91
Similar to Cu 2p spectra, the XPS signal of Fe 2p spectra are also split into Fe 2p3/2 and Fe 2p1/2 doublet structures due to the spin–orbit coupling.84,92–94 Unlike Cu 2p spectra, the assignment of Fe 2p spectra are relatively clear, though slight shift of binding energy for different catalysts.92,94,95 Generally, the Fe2+ 2p3/2 and 2p1/2 signals are located at binding energy of ca. 710 eV and 723 eV with satellite peaks at ca. 715 eV and 729 eV, respectively, while Fe3+ 2p3/2 and 2p1/2 signals are located at about 711 eV and 724 eV with satellite peaks at ca. 719 eV and 733 eV, respectively.92,96 According to the above assignments, researcher had estimated the relative content of Fe3+ and Fe2+ species over Fe/Beta, Fe/ZSM-5 and Fe/MOR zeolites, which was in accordance with 57Fe Mössbauer spectra results.96
Based on XPS results of catalysts before and after hydrothermal aging treatment, Lee et al.83 confirmed the formation of CuAl2O4 species (935 eV) in Cu/SSZ-13, Cu/UZM-35, Cu/Beta, Cu/ZSM-5 zeolites during the hydrothermal aging process, by combing with H2-TPR, UV-vis and XANES experiments. They found that the extent of transformation of isolated Cu2+ species into CuOx and CuAl2O4 species during hydrothermal aging process was higher in the order of Cu/SSZ-13 < Cu/UZM-35 < Cu/Beta < Cu/ZSM-5, which was in line with the higher stability of Cu/SSZ-13 and Cu/UZM-35 than Cu/Beta and Cu/ZSM-5 in NH3-SCR. Xu et al.86 had compared the NH3-SCR activity of copper and iron bimetal modified Beta zeolite (Cu–Fe/Beta) with that of Cu/Beta and Fe/Beta mono-component Beta zeolites and found that Cu–Fe/Beta exhibited higher low-temperature activity and wider temperature window than the rest two zeolites. Based on XPS measurement, they revealed that the dispersion state of active components and the ratios of Cu2+/Cu+ and Fe3+/Fe2+ in the Cu–Fe/Beta zeolite were increased when comparing with the other two zeolites due to the synergistic effect of copper and iron species, which was also supported by XRD, UV-vis and EPR results. Based on above researches, Table 2 summarizes the attribution of XPS signals in Cu-based or Fe-based zeolites.
| Attribution of XPS signals | Ref. | |
|---|---|---|
| Cu-based zeolites | Characteristic of Cu+ species: Cu+ 2p3/2: ∼932.5 eV; Cu+ 2p1/2: ∼952.3 eV, shift to 935.2 eV and 955.0 eV in Cu/Beta | 84–86 |
| Characteristic of Cu2+ species: Cu2+ 2p3/2: ∼933.7 eV; Cu2+ 2p1/2: ∼953.6 eV; satellite peak: ∼944 eV | 84 and 85 | |
| Different attributions in Cu/SSZ-13: CuO: 933.5 eV; isolated Cu2+ species: 936.5 eV | 87 | |
| Cu2Al2O4: ∼935 eV | 83 | |
| Fe-based zeolites | Characteristic of Fe2+ species: Fe2+ 2p3/2: ∼710 eV; Fe2+ 2p1/2: ∼723 eV; satellite peaks: ∼715 eV and 729 eV | 92 and 94–96 |
| Characteristic of Fe3+ species: Fe3+ 2p3/2: ∼711 eV; Fe3+ 2p1/2: ∼724 eV satellite peaks: ∼719 eV and 733 eV | 92 and 94–96 |
Thought XPS can distinguish various Fe or Cu species by nondestructive testing which only need small amounts of sample (10–50 mg for zeolites), it can only detect elements of nanoscale thickness on the surface of zeolites, while the accurate determination of composition of the whole zeolites required element analysis by ICP-AES method.84,97,98 In addition, the automatic reduction of Cu2+ to Cu+ (or Fe3+ to Fe2+) species was sometimes inevitable due to the ultra-high vacuum conditions of XPS determination, which would cause the deviation of XPS testing results from the actual state of zeolites.99 Moreover, the binding energy of metal species may shift with zeolite supports due to the different interactions between metal species and zeolite framework.89,96 Therefore, the accurate analysis of various metal species by XPS should take the above factors into account and often needs to be carried out by combining other characterization methods such as ICP-AES, EPR, Auger electron spectra and UV-vis spectra.83,90,93,97,100
2.4 XAFS
The X-ray absorption fine structure spectroscopy (XAFS) is an atomic-scale tool to study the local structure and chemical state of metal species in zeolites.101–103 When the energy of X-ray resonates with the ionization energy of the inner layer electron of atoms, the electron is excited to form a continuous spectrum (XAFS). The X-ray absorption near-edge structure (XANES) consists of the regime from −10 eV below to ca. 50 eV above the edge energy E0, which can be used to determine the electronic state of the absorbing atom, such as oxidation number and the geometric structure.104 The extended X-ray absorption fine structures (EXAFS) comprises the regime from about 50 eV to about 1000 eV above E0, which can be used to determine the local structure of atoms, as EXAFS can provide the interatomic distances and coordination numbers for several coordination shells around the absorbing atom.104In recent years, XAFS has been widely used to study the active sites of metal-based zeolites in NH3-SCR reactions. Deka et al.26 had studied the local environment of copper species in Cu/SSZ-13 under realistic NH3-SCR conditions by in situ XAFS and in situ XRD experiments, and confirmed that the isolated Cu2+ species located in the double-six-ring (D6R) subunit of CHA structure were the main active sites for NH3-SCR. In addition, they found that the isolated Cu2+ species suffered from a conformational change in the local geometry from a planar form to a distorted tetrahedron due to a preferential interaction with NH3 at low temperatures, which process resulted in the stymieing of activity. In contrast, due to the weak interactions between the isolated Cu2+ species and NH3, the local structure of isolated Cu2+ species remained unchanged at high temperatures, which results in the high activity of Cu/SSZ-13 in NH3-SCR. Korhonen et al.25 also found that isolated Cu2+ species were the main active sites in NH3-SCR over Cu/SSZ-13 zeolites based on EXAFS experiments.
McEwen et al.28 had explored the Cu oxidation state and coordination environment in Cu/SSZ-13 zeolites during the NH3-SCR reactions by operando XAFS experiments and found that, tetrahedral coordinated Cu2+ species dominated in Cu/SSZ-13 under fast NH3-SCR (NO2/NOx = 0.5) and NO2-SCR (NO2/NOx = 1) reaction conditions, while both Cu+ and Cu2+ species existed in Cu/SSZ-13 zeolite under standard NH3-SCR (NO2/NOx = 0) reaction conditions, which indicated that partial reduction of copper species occurred under the standard NH3-SCR atmosphere. As shown in Fig. 2, Lomachenko et al.105 had monitored the oxidation state, mobility of Cu species over Cu-CHA zeolites during NH3-SCR reactions at 150–400 °C range by operando XAFS experiments and unambiguously identified two distinct regimes for the catalytic mechanism of Cu active sites. In the low temperature range (<200 °C), Cu+ (m-Cu(I) complexes) and Cu2+ species (Z–Cu(II) and m-Cu(II) complexes) were the main catalytic active centers, which were solvated by NH3 due to the strong coordination between NH3 and copper species. In the middle and high temperature range (250–400 °C), the main catalytic active centers were isolated Cu2+ species (Z–Cu(II) complexes) which were coordinated with zeolite framework.
 | ||
| Fig. 2 The relationship between the relative percentages of Z–Cu(II), m-Cu(II) and m-Cu(I) species and the NH3-SCR activity in different temperatures. Z–Cu(II) stands for the sum of Z–[Cu(II)OH−] and Z–[Cu(II)NO3−] complexes, while m-Cu(II) represents the sum of mobile [Cu(II)(NH3)4]2+ and [Cu(II)(H2O)6]2+ complexes. m-Cu(I) represents the mobile [Cu(I)(NH3)2]+ complexes in Cu/SSZ-13 zeolites (atoms color code: Cu: green, O: red, Al: yellow, Si: gray, N: blue, H: white).105 (with permission from ACS publications). | ||
The detail understanding of the redox cycle of active metal species is important for the revealing of NH3-SCR reaction mechanism, as NH3-SCR is a redox reaction. Ueda et al.106 had investigated the redox cycle of Cu species in Cu/ZSM-5 for NH3-SCR based on in situ XAFS experiments. They found that Cu2+ species could be slowly reduced to Cu+ species by NH3 flow, which process could be accelerated by adding NO to the NH3 flow. However, for the oxidation half-cycle, the complete oxidization of Cu+ to Cu2+ species could only be achieved by the NO and O2 mixture flow but oxygen alone. Those results indicated that the oxidation half-cycle of Cu+ to Cu2+ process was more difficult than the reduction half-cycle, which was the rate-limiting step for the whole NH3-SCR reactions. Further, Gao et al.32 had studied the detailed pathway of the oxidation half-cycle on Cu/SSZ-13 by in situ EXAFS experiments combined with DFT calculations, and confirmed that for NH3-SCR reactions at low temperatures, the oxidation half-cycle of Cu(I) to Cu(II) requires the participation of two isolated Cu+ species by forming [CuI(NH3)2]+–O2–[CuI(NH3)2]+ as intermediates.
Lercher et al.107 had studied the redox process of Fe/Beta in NH3-SCR by determining the fraction of Fe2+ and Fe3+ species through the combination of XANES and Mössbauer spectroscopy, they found that the distribution of Fe2+ and Fe3+ in Fe/BEA zeolites depended mainly on both the Fe content in zeolites and the conditions of the thermal treatment. In addition, they also revealed that the ion exchanged isolated Fe2+/Fe3+ species are reversibly oxidized and reduced under real NH3-SCR conditions, which were the main active sites for NH3-SCR. Doronkin et al.108 had investigated the structure of iron sites in Fe/Beta and copper sites in Cu-SAPO-34 zeolites during the real NH3-SCR conditions by operando spatially-resolved and time-resolved XAFS experiments, respectively. They found strong gradients of Fe and Cu oxidation state along the Fe/Beta and Cu-SAPO-34 catalyst bed, respectively. By detailed studies on the change tendency of relative percentages of Fe and Cu oxidation state in NH3-SCR reactions over Fe/Beta and Cu-SAPO-34 catalyst, respectively, they concluded that the re-oxidation of Cu or Fe in zeolites was the rate-limiting step in NH3-SCR. Dahl et al.109 had studied the NH3-SCR reaction mechanism over Fe-Beta zeolites by in situ XANES and EXAFS experiments. They also found a relation between the oxidation state of iron and the NH3-SCR catalytic activity of Fe/Beta zeolites and concluded that isolated iron species were the active sites while the re-oxidation of iron species was one of the rate-limiting steps in NH3-SCR.
It is worthy to note that the chemical state of Cu or Fe species in zeolites during the NH3-SCR reactions will undergo successive reduction and oxidation steps so as to catalyze the redox process. Therefore, in most of the literature, the investigation of chemical state of the metal species in zeolites were conducted by in situ or operando XAFS under NH3-SCR reaction conditions, so as to reveal the detail reaction mechanism of NH3-SCR. However, the ex situ XAFS experiment of metal-based zeolites can still provide valuable information to NH3-SCR mechanism. Firstly, the ex situ XAFS experiment can be used to determine the state of metal species in the as-prepared catalysts, supported by other characterization methods such as UV-vis, H2-TPR, etc.110 Korhonen et al.25 compared the EXAFS spectra of Cu/SSZ-13 zeolites before and after calcination and found the coordination number of Cu2+ ions in SSZ-13 reduced from 4 to 3, along with the decrease of average distance of Cu–O distance from 2.02 Å to 1.93 Å, which indicated that the state of Cu in SSZ-13 is sensitive to reaction conditions. In addition, the good fitness of experimental EXAFS and calculated ones confirmed the validity of the proposed local structure of the isolated Cu2+ species in SSZ-13, which is important for the further studying of NH3-SCR mechanism. Secondly, by comparing the ex situ XAFS results of catalysts treated at different gas and temperature conditions, it is possible to speculate the reaction mechanism of catalysts in NH3-SCR. Deka et al.26 had studied the Cu K-edge XANES spectra of Cu-SSZ-13 after calcination and under NH3-SCR conditions at different temperatures (125–300 °C). They found that the local environment of isolated Cu2+ species (located on the plane and slightly distorted from the center of the D6R subunits of CHA) under NH3-SCR reaction at 300 °C was similar to that seen after calcination, whereas a conformational change from a square planar to a distorted tetrahedral type structure was observed for isolated Cu2+ species at 125 °C in NH3-SCR conditions due to a direct interaction of NH3 with copper, according to XANES and EXAFS spectra results. Those results indicated that the reaction mechanism of Cu-SSZ-13 in NH3-SCR changed with reaction temperatures, which was further confirmed by Lomachenko et al.111 through operando XAFS and Emission Spectroscopies. In conclusion, the application of XAFS over Cu-based zeolites for investigating NH3-SCR mechanism is summarized in Fig. 3.
 | ||
| Fig. 3 Summary on the studying of NH3-SCR mechanism in Cu/zeolite by XAFS. | ||
The fine structures at X-ray absorption edges contain information about the geometrical and electronic structure of absorbing atoms, which has been widely used in the study of NH3-SCR reaction mechanism over metal-based zeolite catalysts.101,112–114 However, the major disadvantage of XAFS experiment is that the signals of all absorbing atoms of one type in the sample may overlap at the edge, which made it difficult to distinguish the individual signals belonging to different species when the sample contains an element in several different atomic environments.102 Another disadvantage of XAFS is that the accessibility of synchrotrons of X-ray is not easy, as beam times are scarce which need to be scheduled months ahead.104
2.5 FTIR spectroscopy
Infrared (IR) spectroscopy can detect the framework vibration of zeolites, the chemical bond vibration of metal cationic in zeolites and the vibration of bonds between adsorbent and zeolite framework, so as to analyze the structure of metal-based zeolites, or capture the surface adsorption groups and active intermediates on catalysts.115–118 In NH3-SCR, the Fourier transform infrared spectra (FTIR) after in situ adsorption of NO or CO (FTIR of adsorption NO or CO) are often used to explore the status of metal active species on the zeolite catalysts,119–122 as CO and NO molecules can form coordination complexes with metal cations which can be detected by infrared spectroscopy.The FTIR of adsorption NO or CO experiments can be used to selectively determine the Fe2+ species in zeolites, as the adsorption of NO or CO on Fe3+ are too weak to be detected.24 Though the pre-treatments of Fe-based zeolites for FTIR experiments under high temperature and ultra-high vacuum (about 300–500 °C and 10−5 Pa) may cause the automatic reduction of Fe3+ species to Fe2+ species, thus bringing about the deviation of FTIR spectra from the actual one, the FTIR spectra of adsorption NO or CO can still provide key information about the catalysts.115,123,124
Gao et al.24,125 showed that NO could coordinate with Fe2+ species to form a variety of Fe complexes on Fe/SSZ-13 zeolites: the mononitrosyl Fe2+–NO species (1885 cm−1) formed in the six-membered rings (6MR) of CHA structure, the dinitrosyl Fe2+–(NO)2 (1850 and 1772 cm−1) and trinitrosyl Fe2+–(NO)3 (1916, 1810 and 1797 cm−1) species formed in the eight-membered ring (8MR) windows of CHA cage. By comparing the chemical statues and content of Fe2+ species before and after hydrothermal aging treatments, the migration and transformation behaviors of Fe2+ species in Fe/SSZ-13 zeolites were also clarified by Gao et al.,24 which suggested that even hydrothermal aging at 600 °C could result in the aggregation of Fe2+ species and the formation of FeAlOx clusters with low reducibility in NH3-SCR. Szanyi and co-workers125 found two kinds of mononitrosyl Fe2+–NO species existed in Fe/SSZ-13 zeolites by FTIR of NO adsorption experiments, one was the Fe2+–NO species in the restricted environment (at the ion-exchange sites in six-membered rings of CHA structure, with FTIR signal at 1880 cm−1) which were thermodynamically stable, the other was the unstable Fe2+–NO species in the open environment (in the CHA cages, with FTIR signal at 1900 cm−1), which would gradually transform into trinitrosyl Fe2+–(NO)3 complex, as supported by the shift of infrared absorption band to 1800–1830 cm−1. According to Szanyi et al.,125 the assignment of FTIR bands after NO adsorption over Fe-based H-SSZ-13 zeolites are summarized in Table 3. The Fe2+ species located in the eight-membered ring (8MR) of the large CHA cage could form three types of Fe2+–(NO)x species with NO (i.e., Fe2+–NO, Fe2+–(NO)2 and Fe2+–(NO)3) by increasing the adsorption pressure of NO, whereas only mononitrosyl Fe2+–NO species could be formed in Fe2+ species located in the six-membered rings (6MR) of double six-member (D6R) prisms due to the strong electrostatic interactions between Fe2+ ions and zeolite framework.
| Adsorption center | PNO | Low | Medium | High |
|---|---|---|---|---|
| Fe2+ in 8 MR of CHA | Ads. complex | Fe2+(NO) | Fe2+(NO)2 | Fe2+(NO)3 |
| IR peak position | 1900 | 1771 | 1801 | |
| 1810 | ||||
| 1852 | 1917 | |||
| Fe2+ in 6 MR of CHA | Ads. complex | Fe2+(NO) | Fe2+(NO) | Fe2+(NO) |
| IR peak position | 1884 | 1884 | 1884 |
CO is a sensitive probe for the characterization of Fe2+ species over zeolites. Kim and co-workers58 had investigated the distribution of Fe species on Fe/Beta zeolites by IR spectroscopy of adsorbed CO, they found that the primary bands at 2190, 2175 and 2157 cm−1 were attributed to Fe2+–CO species, the acidic OH–CO adducts and to CO interacting with Fe3+–OH species, respectively, as the direct interactions between Fe3+ and CO were too weak to form Fe3+–CO species.126 In addition, the weak bands at about 2225 cm−1 and 2130 cm−1 were attributed to Al3+–CO complex and physiosorbed CO, respectively. Moreover, by introducing O2 onto the surface of Fe/Beta zeolites, the fraction of Fe2+ species (reflecting by the Fe2+–CO bands at 2190 cm−1) apparently decreased correspondingly with the dramatically increasing of Fe3+ species (representing by the Fe3+–OH⋯CO band at 2157 cm−1), which suggested the oxidation of Fe2+ species to Fe3+ species in the presence of O2 over Fe/Beta zeolites. Malpartida and co-workers122 also found that CO could distinguish two kinds of Fe2+ species in Fe/FER zeolites by IR spectroscopy of adsorbed CO under dynamic vacuum: one is the mono-carbonyl Fe2+–CO species (with strong bands at 2196 cm−1) formed by CO with the most abundant iron sites at large cavities of ferrierite zeolites, which could be transformed to di-carbonyl species by increasing CO pressure; the other is the mono-carbonyl Fe2+–CO species (with weak IR bands at 2187 cm−1) formed by CO coordinating with less abundant iron sites located in more confined sites, which were more stable than the former one and couldn't be converted to di-carbonyl species. Szanyi et al.125 also investigated the FTIR spectra after CO adsorption over Fe/SSZ-13 zeolites, in which several IR bands were different from the above-mentioned Fe-based zeolites, which was reasonable as the coordination environments of Fe2+ species in different zeolites were slightly changed. By summarizing the above literature, the assignments of IR bands after CO adsorption over several Fe-based zeolites are summarized in Table 4.
| FTIR bands after CO adsorption over Fe-based zeolites (cm−1) | Ref. | ||||||
|---|---|---|---|---|---|---|---|
| Fe2+–CO | Fe2+–(CO)2 | OH–CO adducts | Fe3+–OH⋯CO | Al3+–CO complex | FeOx–(CO) | ||
| Fe/Beta | 2190 | — | 2175 | 2157 | 2225 | — | 58 |
| Fe/FER | 2187, 2195 (in small cavities); 2196 (in large cavities) | 2188 | 2173 | — | — | — | 122 |
| Fe/SSZ-13 | 2198 (in 6 MR) | — | 2175 | — | 2221 | 2129 | 125 |
| 2207 (in 8 MR) | 2231 | 2148 | |||||
| 2177 | |||||||
Similar to Fe-based zeolites, IR spectroscopy of adsorbed CO can be used to selectively determine the Cu+ species in Cu-based zeolites, while the adsorption of CO on Cu2+ species are much weak.127,128 According to Corma and co-workers,127 the IR absorption band of the mono-carbonyl Cu+–CO complex formed by CO interacting with Cu+ at the ion exchange sites of Cu/CHA zeolites appeared at about 2155 cm−1, while the absorption peak at about 2180 cm−1 could be attributed to the asymmetric stretching of the bicarbonyl Cu+–(CO)2 complex formed by two CO molecules with one Cu+ species. Szanyi et al.129 had studied the distribution of Cu species on Cu/SSZ-13 by IR spectroscopy and found that the adsorbed CO at room temperature primarily formed Cu+–CO species, but Cu+–(CO)2 and Cu2+–CO species could also be formed when excessive CO were dosed.
In contrast to IR spectroscopy of CO adsorption, IR spectroscopy of NO adsorption can detect both Cu2+ and Cu+ species in Cu-based zeolites, due to the strong interactions between NO and copper cations.127,129,130 Concepcion et al.127 found that only the mono-nitroso complex Cu2+–NO could be formed on Cu/SSZ-13 and Cu/SAPO-34 zeolites due to the strong coordination between isolated Cu2+ and framework oxygen of zeolites, which exhibited two IR bands at around 1925 cm−1 and 1950 cm−1, attributing to two kinds of Cu2+–NO complexes formed by NO reacting with Cu2+ species located in the six-membered and eight-membered rings of CHA structures, respectively. In contrast, various nitro complexes could be formed between NO and Cu+ species over Cu/SSZ-13 and Cu/SAPO-34 zeolites: the infrared absorption peak at 1805–1820 cm−1 and 1660–1720 cm−1 could be assigned to mono-nitroso Cu+–NO complex and di-nitroso Cu+–(NO)2 complex, respectively. In addition, NO could also coordinate with hydrated Cu2+ species ([Cu(OH)+]) to form the IR band at around 1790 cm−1, which was confirmed by a newly formed hydroxyl vibrational absorption peak at 3668 cm−1. Moreover, NO could also interact with dimeric Cu2+ species ([Cu–O–Cu]2+) to form two kinds of IR absorption bands at 1712 cm−1 and 1887 cm−1, which were attributed to the adsorption of NO on the CuII–O bridge bonds, and the direct adsorption of NO on CuII in [Cu–O–Cu]2+ dimers, respectively. Giordanino et al.131 summarized the FTIR bands after CO adsorption or NO adsorption over different Cu-based zeolites and revealed that the preparation method of catalysts and testing conditions (temperature, NO pressure, oxidation or reduction atmosphere) of FTIR experiments could resulted in slightly shift of FTIR bands for Cu–NO or Cu–CO complexes over different Cu-based zeolites. Base on above literature, the typical assignments of FTIR bands after CO adsorption or NO adsorption over different Cu-based zeolites are summarized in Tables 5 and 6, respectively.
| Zeolite | FTIR bands after CO adsorption over Cu-based zeolites (cm−1) | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cu+–CO | Cu+–(CO)2 | Cu+–(CO)3 | Cu2+–CO | OH−–CO | [Cu(OH)]+⋯CO | Al3+⋯CO | CuOx⋯CO | ||
| Cu-SSZ-13 | 2155 | 2150, 2178 | 2134, 2169, 2194 | — | — | 2207 | 2220 | — | 131 |
| Cu-SSZ-13 | 2158 | 2148 | — | — | 2177 | — | — | — | 127 |
| Cu-SSZ-13 | 2135, 2154 | 2178 | — | 2220 | — | — | — | — | 129 |
| Cu/SAPO-34 | 2154 | 2147, 2178 | 2139, 2163, 2187 | — | 2171 | — | — | 2131 | 127 |
| Cu/ZSM-5 | 2158 | 2150, 2178 | 2134, 2166, 2192 | — | — | — | — | — | 131 and 132 |
| Cu/Beta | 2158, 2153 | 2152, 2180 | 2134, 2168, 2193 | — | — | — | — | — | 131 |
| Cu/MOR | 2159 | 2152, 2180 | 2146, 2167, 2193 | — | — | — | — | — | 133 |
| Cu/FER | 2157 | 2149, 2178 | 2135, 2170, 2191 | — | — | — | — | — | 134 |
| Zeolite | FTIR bands after NO adsorption over Cu-based zeolites (cm−1) | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Cu+–NO | Cu+–(NO)2 | Cu2+–NO | Cu2+–(NO)2 | [Cu(OH)]+⋯NO | [Cu–O–Cu]2+⋯(NO)2 | ||
| Cu/SSZ-13 | 1809 | 1826, 1728 | 1890 | — | — | — | 131 |
| Cu/SSZ-13 | 1803, 1816 | 1663, 1800, 1654, 1783 | 1925, 1947, 1960, 1965, 1977 | 1801, 1892, 1869, 1813, 1874 | 1795, 1788 | — | 127 |
| Cu/SAPO-34 | 1811, 1821 | 1720, 1831, 1714, 1828 | 1907, 1940, 1943, 1968 | 1809, 1888, 1802, 1868 | 1790, 1798 | 1712, 1887 | 127 |
| Cu/ZSM-5 | 1813 | 1730, 1825 | 1921, 1912, 1905, 1895 | — | — | — | 131 and 135 |
| Cu/Beta | 1802, 1815 | 1828, 1734 | 1912, 1903, 1895 | — | — | — | 131 and 136 |
| Cu/MOR | 1813 | 1730, 1828 (main channel); 1785, 1870 (side pocket) | 1960, 1938, 1921, 1909, 1895 | — | — | — | 135 and 137 |
Infrared spectroscopy of NO or CO adsorption is a highly valuable tool to provide information about the chemical nature of metal species in zeolites, such as the oxidation state, coordination environment, which are important in NH3-SCR reactions.115,138,139 Through in situ adsorption of CO and NO infrared experiments, researchers had detailly investigated the distribution of Cu species on Cu/SSZ-13 zeolites under different pretreatment conditions, which provided valuable information for the understanding of detailed NH3-SCR reaction mechanism.7,14,127,129 However, the quantitative of various metal species by IR bands are difficult, as the unknown extinction coefficients of different adsorption band for metal species. In addition, the assignments of IR bands for NO or CO adsorption over different zeolites should be careful, as the interactions between zeolites with different framework and metal species were different, which would cause the shift of IR bands.115 Moreover, the overlap of absorption peaks may occur at high adsorption pressure of CO or NO or when high loading of metal species was achieved at zeolites, which would make the assignment of IR bands difficult.1,79 Therefore, care must be taken when general conclusions are drawn about the adsorption and reactive properties of metal species based on infrared spectroscopy of NO or CO adsorption in zeolites, which may should be supported by other characterization methods.
2.6 EPR
Electron paramagnetic resonance (EPR) is a magnetic resonance technology based on the magnetic moment of unpaired electrons which move around the nucleus and spin at the same time, resulting in electric current and magnetic moment.140 Under an external electromagnetic field, the electrons with low energy levels can be excited to high energy levels by absorbing microwave energy, resulting in electron paramagnetic resonance. The g factor (spectral splitting factor) in paramagnetic resonance spectra which reflecting the local magnetic field information can be used to analyze the chemical environments of metal atoms.21,43EPR spectroscopy has been successfully used to characterize the coordination environment and local structure of copper species in Cu-based zeolites.22 As Cu+ lacks paramagnetic electrons, [Cu(OH)]+ has the pseudo Jahn–Teller effect, CuOx clusters and [Cu–O–Cu]2+ have antiferromagnetic interactions, these copper species are EPR silent species.27 However, under hydration, [Cu(OH)]+ species can be converted into [Cu(OH)(H2O)5]+ species with EPR activity and thus be detected by EPR. In contrast, when dehydrated, [Cu(OH)(H2O)5]+ species can be transformed into [Cu(OH)]+ species, or be automatically reduced to Cu+ species or be condensed to form [Cu–O–Cu]2+ or CuOx species, which are all EPR silent species. On the other hand, isolated Cu2+ species, either in the hydrated form ([Cu(H2O)6]2+ or [Cu(OH)(H2O)5]+) or dehydrated form (Cu2+), are paramagnetic and can be detected by EPR.27,77 Therefore, the relative content of [Cu(OH)]+ and isolated Cu2+ species on Cu-based zeolites can be quantitatively calculated by EPR spectroscopy through determining the Cu-based zeolites under dehydration and hydration conditions, as described in Fig. 4.
 | ||
| Fig. 4 Summary on the EPR determination and quantification of Cu species in Cu/zeolite. | ||
Gao et al.27 had quantitatively determining the content of various Cu species on Cu/SSZ-13 zeolites by combing EPR experiments under hydrated and dehydrated conditions with ICP-AES method. As summarized in Fig. 5, firstly, the content of isolated Cu2+ species can be calculated by EPR spectra of dehydrated Cu/SSZ-13 zeolites. Secondly, the total content of isolated Cu2+ species and [Cu(OH)]+ species can be measured by EPR spectra of hydrated Cu/SSZ-13 zeolites; then the content of [Cu(OH)]+ species on Cu/SSZ-13 zeolites can be calculated by the difference between the above two EPR spectra. Thirdly, the total content of all the Cu species on Cu/SSZ-13 zeolites can be measured by ICP-AES analysis. Therefore, the total content of copper species without EPR activity (such as Cu+, [Cu–O–Cu]2+ and CuOx) can be calculated by combining the quantitative results of ICP-ASE and EPR experiments. Based on the quantitative determining of various Cu species on Cu/SSZ-13 zeolites, Gao et al.27 found that the isolated Cu2+ species showed good hydrothermal stability in NH3-SCR. In contrast, during the hydrothermal aging process, the [Cu(OH)]+ species in Cu/SSZ-13 zeolites would gradually transform into CuOx species, which had almost no NH3-SCR activity but high activity to the side reactions of ammonia oxidation, and eventually cause the decrease of NH3-SCR activity.
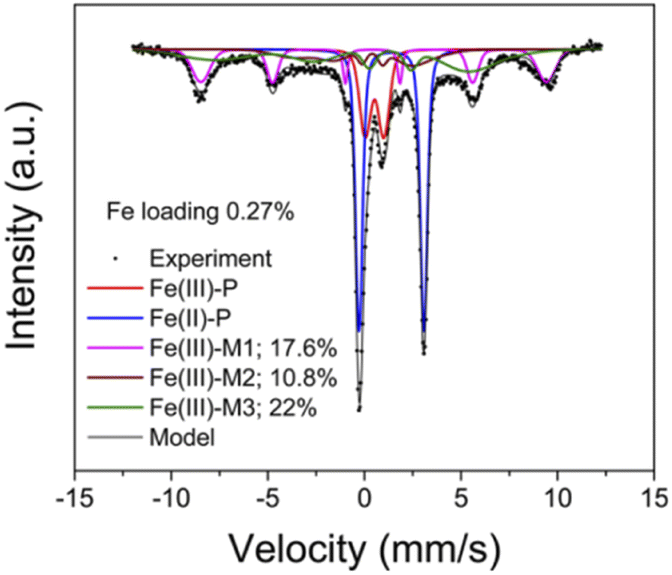 | ||
| Fig. 5 Mössbauer spectroscopy of the ambient 57Fe/SSZ-13 sample measured at 8 K. Peak fitting results and percentages of different components, Fe(III)-P, Fe(II)-P, Fe(III)-M are also displayed144 (with permission from ACS publications). | ||
EPR spectra can also be used to distinguish the location and distance of isolated Cu2+ species. Gao et al.78 had estimated the Cu–Cu distance of isolated Cu2+ ions in Cu/SSZ-13 zeolite based on the line broadening of EPR spectra. They found that the Cu–Cu distance was greater than 20 Å at low copper loadings, indicating that there was allowed only one Cu2+ ions within one hexagonal unit cell. As the Cu loading increased, the Cu–Cu distance decreased significantly, indicating that several Cu2+ ions were located in the large CHA cages and were close to the eight-membered rings.
In Fe-based zeolites, EPR spectra can be used to selectively detect Fe3+ species, as Fe2+ was silent in EPR spectra due to the lack of paramagnetic electrons. According to literature,73,141,142 the EPR signal at g = 4.3 can be attributed to the tetrahedral Fe3+ species in the framework of zeolites, while the signa at g = 6.0 and 8.8 are attributed to the distorted tetrahedral Fe3+ species and octahedral coordinated Fe3+ species, respectively. In addition, the highly symmetric octahedral coordinated Fe3+, which are the predominant active species in NH3-SCR, has characteristic EPR signals at g = 2.0; while the aggregated FexOy species show EPR characteristic signals at g = 2.3. Li et al.73 had prepared Fe-ZSM-5 zeolites with different iron content by one-pot method, and found that the NH3-SCR catalytic activity of Fe-ZSM-5 were positively correlated with the EPR iron species at g = 2.0 in the EPR spectra, which supported the above assignments. However, Shen et al.141 had investigated the distribution of Fe3+ species in Fe/Beta zeolites by UV-vis and in situ EPR spectra and found that the isolated Fe3+ species in distorted tetrahedral (g = 6) and octahedral (g = 8.8) environments showed higher NH3-SCR activity, while those in tetrahedral framework environments (g = 4.3) exhibited lower activity. The controversial results of EPR spectra over different Fe-based zeolites demonstrates that the differences in zeolite support, in the type and content of iron species and in the different experimental conditions could bring in discrepancies in the EPR spectra, which should be carefully analyzed considering the detailed experimental conditions of EPR spectra or by combining with other characterizations such as UV-vis, FTIR or XAFS.13
EPR is a powerful tool for determining metal species in zeolites with high sensitivity, which can provide qualitative and quantitative information on the oxidation state and chemical environment of paramagnetic metal ions by combining with other characterization methods such as ICP and UV-vis.29 However, there are still some limitations in the application of EPR spectra. Firstly, the EPR spectra with high resolution can only be obtained under ultra-low temperature of about 155 K for Cu-based zeolites78 and about 7 K for Fe-based zeolites,24 in order to avoid the signal broadening due to the migration of metal species and the anti-ferromagnetic interactions. Secondly, EPR spectra of metal-based zeolites are highly sensitive to water and paramagnetic species, so the testing conditions of EPR spectra should be strictly controlled.21 Thirdly, when the structure of zeolite support changes, the g value of EPR signal for the same kind of metal species may shift due to different interactions between metal species and zeolites, which made the assignment of EPR signal difficult.109 Therefore, the EPR spectra should be analyzed carefully according to the preparation method, zeolite support and metal content of catalysts, or by combining with other characterizations.
2.7 Mössbauer spectroscopy
Mössbauer spectroscopy is a kind of gamma ray spectrum based on “Mössbauer effect”, i.e., “the emission and subsequent resonant absorption of nuclear gamma rays by the nuclei of certain atoms embedded in a solid material”.143 When the energy of the gamma ray equal to the energy of nuclear energy level transition, the resonant absorption phenomenon can be observed in Mössbauer spectroscopy. Mössbauer spectroscopy can be used to study the chemical valence or oxidation state, magnetic properties, coordination numbers and electron density of specific atoms. However, though the “Mössbauer effect” of more than 80 isotopes is measurable, only two of them (57Fe, 119Sn) are applied in practice due to the restrict testing conditions and equipment.143 As the obtaining of Mössbauer spectrum require the gamma ray source with high energy at low temperature, the further application of Mössbauer spectrum is greatly limited.57Fe Mössbauer spectroscopy conducting at cryogenic temperatures (8 K) can prevent signal loss caused by recoil of mobile species, and are proved to be more accurate than UV-vis and H2-TPR in quantification of Fe species in zeolites.68 According to previous studies,13,23,24,68 four key parameters, i.e. average center shift (CS, mm s−1), average quadrupole splitting (QS, mm s−1), standard deviation of QS (ε, mm s−1) and average magnetic hyperfine field (HF, Tesla) could be used to distinguish different Fe species in the Mössbauer spectroscopy of Fe/SSZ-13 zeolites.57 Generally, the component with a CS value of ∼1.4 mm s−1 is attributed to Fe(II) species, while those with CS value of ∼0.5 mm s−1 can be assigned to Fe(III) species. Also, the paramagnetic Fe species give doublet features in Mössbauer spectroscopy (designated as P), while the magnetic Fe species display sextet signals (designated as M).13,23,24,68 Based on above features, the Fe species in Fe/SSZ-13 zeolites can be distinguished and divided into three types:13 Fe(III)-P representing Fe3+ dimers (HO–Fe(III)–O–Fe(III)–OH), Fe(II)-P assigning to isolated Fe2+ species, and Fe(III)-M to a mixture of isolated Fe3+ species ([Fe(OH)2]+) and Fe-oxide clusters/particles. By spectra deconvolution, the relative percentages of three types of Fe species in Fa/SSZ-13 zeolites can be estimated, as shown in Fig. 5, according to Gao and co-workers.13
Based on the quantification of 57Fe Mössbauer spectroscopy and kinetic experiments, Gao and co-workers13 had detailly investigated the active Fe sites in NH3-SCR reactions over a series of Fe/SSZ-13 zeolites with differentiate iron contents. They found that the isolated Fe3+ species were the main active sites for standard NH3-SCR at low temperatures (≤260 °C), while the [HO–Fe–O–Fe–OH]2+ species dominated the NH3-SCR reactions in the high temperature range (260–550 °C). Moreover, the migration and aggregation of Fe species in Fe/SSZ-13 zeolites during the hydrothermal aging treatments had also been clearly clarified by 57Fe Mössbauer spectroscopy:24 the isolated Fe3+ species would gradually transform to [HO–Fe–O–Fe–OH]2+ under mild hydrothermal conditions (600–700 °C), which was beneficial for the enhancement of NH3-SCR activity. However, once the hydrothermal aging temperature exceeding 800 °C, the NH3-SCR activity of aged Fe/SSZ-13 zeolites dramatically decreased due to serious framework dealumination, accompanying by the agglomeration of active isolated Fe3+ species to FexOy clusters or nanoparticles (without or with very low NH3-SCR activity) and even the incorporation of Al into FexOy species (almost no SCR activity).
As the intensity of the Mössbauer signal is strongly temperature dependent (usually, the ultra-low testing temperature of 8 K is required for high resolution), the quantitative determination of the oxidation of Fe species in zeolites by in situ Mössbauer spectroscopy is not possible under real NH3-SCR reactions at elevated temperatures.145 However, Maier et al.107 has developed an effective method to quantitative determining the oxidation state of Fe species in Fe/Bata zeolites under real NH3-SCR conditions by combining in situ XANES and Mössbauer spectroscopy. They had obtained a linear correlation between the edge energy of the XANES and the oxidation state determined by Mössbauer spectroscopy, which in turn allowed them to determining the relative concentrations of Fe3+ and Fe2+ species under real NH3-SCR conditions through in situ XANES with the aid of Mössbauer spectroscopy. In conclusion, the application and determination of Fe species in zeolites for NH3-SCR are summarized in Fig. 6.
 | ||
| Fig. 6 Summary on the determination of Fe species in Fe/zeolite by Mössbauer spectroscopy. | ||
Compared with UV-vis, H2-TPR and EPR experiments, Mössbauer spectroscopy is the most accurate method for the simultaneously determination of both Fe2+ and Fe3+ species in zeolites with high resolution and sensitivity.13,24 In addition, only several type of nucleuses (57Fe and 119Sn) have the resonance absorption of Mössbauer effect, the determination of Mössbauer spectroscopy is not disturbed by other elements, which guarantees the high accuracy of Mössbauer spectroscopy but also limits its application. Moreover, the high cost of equipment with γ-ray sources and the low testing temperature (8 K) for high resolution also limit its application.143
2.8 DFT calculation
The density functional theory (DFT) calculation is another powerful tool to study the chemical state of metal species, the key reaction intermediates and the reaction mechanism of NH3-SCR, which could be used to study the interactions between atoms or molecules with the catalysts from atomic-scale.As a gas–solid multiphase reaction, the gas molecules adsorption process is important for the study of NH3-SCR mechanism, which is usually investigated by experimental method such as FTIR spectra, XAFS, and reaction kinetics. DFT can be used to predict and verify the spectral results of gas molecules adsorption (CO, NO, N2, NH3, etc.) so as to investigate the key metal species and reaction mechanism of NH3-SCR. Zhang et al.124 had investigated the NO adsorption process over Cu-SSZ-13 zeolite by FTIR spectra and DFT calculations and found a perfect agreement between FTIR bands and DFT calculation results for the NO vibration frequencies, as shown in Table 7. Base on FTIR results and DFT calculations, they concluded that NO molecules did bind stronger on [Cu(OH)]+ located in 8MR than isolated Cu2+ species in the 6MR of CHA structure in Cu/SSZ-13 zeolites, which indicated that Cu2+ ions were stabilized with ligands in 8MR of CHA cages in NH3-SCR. Similarly, Corma et al.127 had also clearly identified different kinds of Cu–NO complexes in Cu/SAPO-34 and Cu/SSZ-13 zeolites by combining FTIR spectra of NO adsorption and DFT calculations, which could be used to indirectly determining the distribution of Cu species in zeolites.
| NO frequency (cm−1) | Experimental results | Computational results |
|---|---|---|
| Cu2+–NO | 1850–1950 | 1895–1932 |
| [Cu(II)OH]+–NO | 1870–1915 | 1874, 1907 |
| Cu+–NO | 1770–1808 | 1794, 1788, 1795 |
| Cu–N2O | ∼2250 | 2367, 2339, 2362 |
| Cu+–NO+ | 2160–2170 | — |
DFT can also be used to determine the distribution and spatial location of metal active sites, the key intermediate species and to reaction pathway in NH3-SCR. Li et al.146 had explored the possible locations of Cu2+ species in Cu/SAPO-18 by DFT calculations and EPR experiments. They calculated and found two of seven possible locations of Cu2+ species were the most stable energy state, and then revealed the NH3-SCR reaction mechanism over Cu/SAPO-18 based on that. Mao et al.147 calculated the binding energy of Cu2+ species on five type of ion exchange sites in SAPO-34 and found that Cu2+ species located in the 6MR plane with slightly off-center position had the lowest binding energy, which were the most stable exchange sites for Cu2+ in SAPO-34. Further research indicated that those stable Cu2+ species were the main active sites for Cu/SAPO-34 zeolites in NH3-SCR.
McEwen et al.28 investigated the oxidation state and coordination environment of Cu species in Cu/SSZ-13 under standard SCR or rapid SCR conditions by operando XAS with the aid of DFT calculations. They found that the oxidation state of Cu species changed with reaction conditions: four-fold-coordinated Cu2+ species dominated the Cu/SSZ-13 zeolites under fast and slow SCR conditions, in which the NO2/NOx ratios were 0.5 and 1 in the feed gas, respectively. In contrast, mixed Cu+ and Cu2+ oxidation states were observed under standard SCR conditions without NO2 in the feed gas, which indicated a reduction in the average Cu coordination and highlighted the role of Cu redox chemistry in NH3-SCR process. Based on DFT calculation and kinetic experiments, Gao et al.32 studied the oxidation half-cycle of Cu+ to Cu2+ process over Cu/SSZ-13 in NH3-SCR and found that the isolated Cu+ species were unable to complete oxidation half-cycle at low temperatures, which process could only occur with the participation of two isolated Cu+ ions by forming [CuI(NH3)2]+–O2–[CuI(NH3)2]+ intermediates. In addition, the above oxidation half-cycle was the rate-determining step for NH3-SCR at low temperatures, which was confirmed by DFT calculations.
Recently, Guan et al.56 reviewed the application of DFT in NH3-SCR research from the aspects of surface adsorption, metal active sites characteristics, reaction mechanism, hydrothermal aging mechanism and poisoning mechanism in catalysts. DFT calculations can obtain a lot of information of catalysts in NH3-SCR, which can not only verify the experimental characterization results to make them more reliable, but also reveal the underlying causes of experimental results from microscopic atomic level. The current use of DFT in NH3-SCR included: determining the location and distribution of active sites on the catalysts, calculating the activation energy of the elementary reaction to reveal the rate-determining step for NH3-SCR, determining the reaction pathway by calculating the intermediates species and transition state energy of NH3-SCR reactions, etc.
Though DFT is a universal method to investigate the physicochemical and reactivity properties of catalysts which is not restricted by the research project or the research process, there are still shortcomings. Firstly, when constructing models, saturation of the boundary of the cluster model with hydrogen atoms may cause deviations from the actual structures. Secondly, zeolite with complex spatial structures would result in huge calculations and the loss of accuracy of calculation results sometimes. Thirdly, the DFT calculation results of some special systems were heavily dependent on the selection of calculation methods. However, the development of calculation software and methods may help to solve those problem. By combining with advanced experimental methods, DFT calculation has become one of the most important tool to investigate the reaction mechanism of NH3-SCR.
3 Conclusions and outlook
Cu-based and Fe-based zeolites are promising catalysts for NH3-SCR due to the high catalytic activity, wide temperature window and good hydrothermal stability. The detailed investigation of NH3-SCR mechanism based on Cu-based and Fe-based zeolites are important for further development of high-efficiency NH3-SCR catalysts, which should be based on the accurate determining of active metal sites. Fig. 7 summarizing the effective characterization methods of various Cu or Fe species in zeolites for NH3-SCR. As also summarized in Table 8, UV-vis, H2-TPR, XPS, XAFS, FTIR (adsorption of CO or NO), EPR, Mössbauer spectroscopy and DFT calculations introduced in this review are characteristic methods to determine the type, content, distribution or even local structure of various Cu or Fe species in zeolites, though they have different applicability and limitations. In addition, the accurate qualitative and quantitative determination of various metal active sites in Cu-based or Fe-based zeolites usually requires a combination of several characterization methods by considering the preparation method of catalysts and by careful analyzing the characterization results. Moreover, off-line characterization methods often cannot precisely reveal the chemical environment of metal active species in real NH3-SCR conditions, as well as the interactions between active sites with reactants. Therefore, the in situ or operando characterization methods such as in situ IR, in situ UV-vis, in situ EPR, operando XAFS, etc., as well as DFT calculations should also be applied to more accurately reveal the activation and reaction process of active species on the catalysts so as to reveal the detailed reaction mechanism of NH3-SCR. | ||
| Fig. 7 The characterization methods of various Cu or Fe species in zeolites for NH3-SCR. | ||
| Characterization methods | Fe species | Cu species | Limitation |
|---|---|---|---|
| UV-vis | (1) 200–300 nm: 220–250 nm belongs to the four-coordinated isolated Fe3+ species, 250–300 nm are related to isolated Fe3+ species with higher coordination number | (1) 210 and 280 nm: isolated Cu2+ species (charge transfer from lattice O2− to Cu2+) | (1) Only Fe3+ and Cu2+ species can be detected, Fe2+ and Cu+ species are invisible in 200–800 nm of UV-vis spectra62–64,66 |
| (2) 300–400 nm: charge transition peak of octahedral coordination aggregated Fe3+ species such as small FexOy species | (2) ca. 750 nm: isolated Cu2+ species (d–d transitions of Cu2+ species with distorted octahedral coordination)61–64 | (2) Only a semiquantitative method due to the unknown extinction coefficients of different adsorption band of various metal species59,66 | |
| (3) >400 nm: Fe2O3 nanoparticles34,59,60 | (3) 250 nm and 450 nm: CuOx species (the charge transfer (250 nm) and d–d transition (450 nm) of octahedral coordinated Cu2+ in CuOx species25,61,62,65 | (3) Only the coordination state of metal species can be obtained, the detailed chemical structure of metal species cannot be determined64,67 | |
| H2-TPR | (1) 380–430 °C: the reduction of isolated Fe3+ to Fe2+ species24,69,70 | (1) 400 °C: the reduction of Cu2+–2Z to Cu+ species | (1) Unable to determine irreducible metal species such as Cu0 and Fe0 in zeolites |
| (2) 500–560 °C: the reduction of aggregated FexOy species24,69,70 | (2) The reduction of [Cu(OH)]+–Z to Cu+ at 250 °C, then Cu+ to Cu0 at 360 °C (ref. 27, 74 and 77) | (2) The distinguish of reduction peak for various metal species are difficult as they may shift and overlap with each other35 | |
| (3) 680–750 °C: the reduction of Fe2O3 nanoparticles69 | (3) 300 °C: the reduction of CuO to Cu0 species27,29,74 | (3) The assignment of reduction peak should consider the zeolite support, metal content, preparation method of catalysts62,75,78,80 | |
| (4) 900–1000 °C: the reduction of Fe2+ species in ion-exchange sites and high aggregated Fe2O3 or Fe3O4 particles34,69,71–73 | (4) 700–900 °C: Cu+ at the ion-exchange site of zeolite be reduced to Cu0 species27,29 | (4) The automatic reduction of metal species in the dehydration pre-treatment process caused by adsorbed H2O in zeolites may decrease the H2 consumption77,81 | |
| (5) The shape and area of reduction peak in the H2-TPR experiments could be affected by the heating rate of temperature-programmed process82 | |||
| XPS | (1) Fe2+ 2p3/2 and 2p1/2 signals are those located at binding energy of ca. 710 eV and 723 eV with satellite peaks at ca. 715 eV and 729 eV, respectively74,84,92–94 | (1) Cu 2p1/2 peak splits into two signals at 952.3 ± 0.2 eV for Cu+ and at 953.6 ± 0.2 eV for Cu2+ species84,85 | (1) Only elements with nano-scale thickness on the surface of zeolites can be detected84,97,98 |
| (2) Fe3+ 2p3/2 and 2p1/2 signals are those located at about 711 eV and 724 eV with satellite peaks at ca. 719 eV and 733 eV, respectively92,96 | (2) The assignment of Cu 2p3/2 signals are controversial. Some researchers attributed the Cu 2p3/2 signals at 932.5 ± 0.2 eV for Cu+ and those at 933.7 ± 0.2 eV for Cu2+ species,84–86 while others attributing them to CuO species and isolated Cu2+ species, respectively87 | (2) The automatic reduction of Cu2+ or Fe3+ species under pretreatment conditions of ultra-high vacuum99 | |
| (3) The assignment of XPS peak are sometimes difficult due to shift and overlapping of binding energy signals for metal species,89,96 better be carefully taken and with the aid of ICP-AES, EPR, Auger electron spectra and UV-vis spectra88–92,94,95 | |||
| XAFS | (1) Distinguish and quantification of Fe2+ and Fe3+ species in Fe/zeolites107 | (1) Distinguish and quantification of Cu+ and Cu2+ species in Cu/zeolites28,105 | (1) The signals of all absorbing atoms of one type in the sample may overlap at the edge, which made it difficult to distinguish different kind of species102 |
| (2) Obtain the local structure of Fe species such as coordination number and bond lengths for Fe–O and Fe–Fe coordination spheres108 | (2) Obtain the local structure of Cu species such as coordination number and bond lengths for Cu–O and Cu–Cu coordination spheres25,26 | (2) The accessibility of synchrotrons of X-ray is not easy, as beam times are scarce and need to be scheduled months ahead104 | |
| (3) Operando XAS can be used to monitor the oxidation and reduction half-cycle of Fe species in real NH3-SCR reaction conditions109 | (3) Operando XAS can be used to monitor the oxidation and reduction half-cycle of Cu species under real NH3-SCR reaction conditions28,32,105,106 | ||
| FTIR spectroscopy | (1) FTIR of adsorption CO: selectively detecting Fe2+ species by weak adsorption to form Fe2+–CO species, while Fe3+–OH could coordinated with CO to form Fe3+–OH⋯CO species58,122,126 | (1) FTIR of adsorption CO: selectively detecting Cu+ species by forming Cu+–CO, Cu+–(CO)2 species; Cu2+–CO can only be formed under excessive CO condition127–129 | (1) The automatic reduction of Cu2+ or Fe3+ species under pretreatment conditions of ultra-high vacuum115,123,124 |
| (2) FTIR of adsorption NO: selectively detecting Fe2+ species by strong adsorption to form Fe2+–NO, Fe2+–(NO)2 and Fe2+–(NO)3 species24,125 | (2) FTIR of adsorption NO: both Cu+ and Cu2+ species can be detected by strong adsorption to form Cu2+–NO, Cu+–NO, Cu+–(NO)2 complexes; [Cu(OH)]+ and [Cu–O–Cu]2+ can also coordinated with NO to form IR signal127,129,130 | (2) The assignment of IR bands should be careful due to shifting and overlapping of signals for metal species when catalysts with high metal content or complex composition1,7,14,79,115,127,129 | |
| (3) Only a semiquantitative method due to the unknown extinction coefficients of different adsorption band of various metal species148 | |||
| EPR | (1) EPR silent: Fe2+ species13 | (1) EPR silent: Cu+, [Cu(OH)]+, CuOx and [Cu–O–Cu]2+ species27 | (1) Low testing temperature (155 K for Cu-based zeolites and 7 K for Fe-based zeolites) is required for taking high-resolution EPR spectra so as to relieve the peak broadening resulting from metal mobility and antiferromagnetic interactions24,78 |
| (2) EPR active: Fe3+ species, including framework, extra-framework Fe3+ in zeolites and Fe3+ in FexOy clusters73,141,142 | (2) EPR active: isolated Cu2+, [Cu(H2O)6]2+ and [Cu(OH)(H2O)5]+ species27,77 | (2) High sensitivity of EPR signal to H2O and paramagnetic species21 | |
| (3) The quantitative measuring of different Cu species in Cu/SSZ-13 can be achieved by combining EPR and ICP-AES method27 | (3) The deviation of g values for metal species with different zeolite supports made the assignments of EPR signal difficult109 | ||
| (4) The Cu–Cu distance of isolated Cu2+ ions in Cu/SSZ-13 can be estimated by the line broadening of EPR spectra78 | |||
| Mössbauer spectroscopy | (1) Determination of Fe2+ and Fe3+ at the same time | Unavailable | (1) Only applicable to several nucleus with Mossbauer effect, such as 57Fe and 119Sn143 |
| (2) Distinguishing of different types of Fe species: Fe(III)-P representing Fe3+ dimers (HO–Fe(III)–O–Fe(III)–OH)), Fe(II)-P assigning to isolated Fe2+ species, and Fe(III)-M to a mixture of isolated Fe3+ species ([Fe(OH)2]+) and Fe-oxide clusters/particles13,23,24,68 | (2) Super-low testing temperature (8 K) for high resolution13,24,68 | ||
| (3) High cost of equipment with γ-ray sources which are not easy to be available143 | |||
| DFT | (1) Verify the spectral results of gas molecules adsorption (CO, NO, etc.)124,127 | (1) Calculation results may deviate from the actual structures due to the saturation of the boundary of the cluster model with hydrogen atoms56 | |
| (2) Determining the location and distribution of active sites on the catalysts146,147 | (2) Zeolite with complex spatial structures would result in huge calculations and the loss of accuracy of calculation results sometimes56 | ||
| (3) Calculating the activation energy of the elementary reaction to reveal the rate-determining step for NH3-SCR146,147 | (3) Calculation results of some special systems were heavily dependent on the selection of calculation methods56 | ||
| (4) Determining the reaction pathway by calculating the intermediates species and transition state energy of NH3-SCR reactions, etc.28,32,56 | |||
Author contributions
Jialing Chen: conception, investigation and writing-review & editing; Wei Huang: investigation and methodology; Sizhuo Bao: investigation and methodology; Wenbo Zhang: investigation and methodology; Tingyu Liang: methodology, and review & editing; Shenke Zheng: IR spectroscopy analysis; Lan Yi: UV-vis analysis; Li Guo: H2-TPR analysis; Xiaoqin Wu: review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is devoted to the 60th birthday of our beloved Prof. Jianguo Wang (Shanxi Institute of Coal Chemistry, University of Chinese Academy of Sciences). We are grateful for the financial supports of the National Natural Science Foundation of China (22002114 and 22102124), the National Innovation and Entrepreneurship Training Program for College Students (201810488016); the Education Department of Hubei Province (Q20211501), the Open Fund of Key Laboratory of Hubei Province for Coal Conversion and New Carbon Materials (WKDM202208). We would like to thank Mr Guohong Zhang, Jinhui Zhou and Feizhou Wang at the Analytical & Testing Center of Wuhan University of Science and Technology for the help on XPS and FTIR experimental method.References
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J.-y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS.
- L. Zhang, Q. Wu, X. Meng, U. Müller, M. Feyen, D. Dai, S. Maurer, R. McGuire, A. Moini, A.-N. Parvulescu, W. Zhang, C. Shi, T. Yokoi, X. Pan, X. Bao, H. Gies, B. Marler, D. E. De Vos, U. Kolb and F.-S. Xiao, React. Chem. Eng., 2019, 4, 975–985 RSC.
- F. Liu, Y. Yu and H. He, Chem. Commun., 2014, 45, 8445–8463 RSC.
- Y. Yue, B. Liu, P. Qin, N. Lv, T. Wang, X. Bi, H. Zhu, P. Yuan, Z. Bai, Q. Cui and X. Bao, Chem. Eng. J., 2020, 398, 125515–125527 CrossRef CAS.
- A. M. Beale, I. Lezcano-Gonzalez, T. Maunula and R. G. Palgrave, Catal., Struct. React., 2015, 1, 25–34 Search PubMed.
- T. Jiang and R. F. Lobo, in Structure and Reactivity of Metals in Zeolite Materials, ed. J. Pérez Pariente and M. Sánchez-Sánchez, Springer International Publishing, Cham, 2018, DOI:10.1007/430_2018_23, pp. 155–178.
- J. Wang, H. Zhao, G. Haller and Y. Li, Appl. Catal., B, 2017, 202, 346–354 CrossRef CAS.
- C. Paolucci, J. R. Di Iorio, F. H. Ribeiro, R. Gounder and W. F. Schneider, in Advances in Catalysis, ed. C. Song, 2016, vol. 59, pp. 1–107 Search PubMed.
- R. Zhang, N. Liu, Z. Lei and B. Chen, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS PubMed.
- T. J. Toops, J. A. Pihl and W. P. Partridge, in Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, ed. I. Nova and E. Tronconi, Springer New York, New York, 2014, DOI:10.1007/978-1-4899-8071-7_4, pp. 97–121.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- F. Gao, Y. Zheng, R. K. Kukkadapu, Y. L. Wang, E. D. Walter, B. Schwenzer, J. Szanyi and C. H. F. Peden, ACS Catal., 2016, 6, 2939–2954 CrossRef CAS.
- F. Gao, ChemCatChem, 2020, 10, 1324 CAS.
- Y. Shan, J. Du, Y. Zhang, W. Shan, X. Shi, Y. Yu, R. Zhang, X. Meng, F. S. Xiao and H. He, Natl. Sci. Rev., 2021, 8, 134–153 CrossRef PubMed.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- A. Wang, Y. Chen, E. D. Walter, N. M. Washton, D. Mei, T. Varga, Y. Wang, J. Szanyi, Y. Wang, C. H. F. Peden and F. Gao, Nat. Commun., 2019, 10, 1137–1147 CrossRef PubMed.
- W. Zhang, J. Chen, L. Guo, W. Zheng, G. Wang, S. Zheng and X. Wu, J. Fuel Chem. Technol., 2021, 49, 1294–1315 CrossRef.
- W. Zheng, J. Chen, L. Guo, W. Zhang, H. Zhao and X. Wu, J. Fuel Chem. Technol., 2020, 48, 1193–1210 CrossRef CAS.
- Y. Zhang, J. Zhang, H. Wang, W. Yang, C. Wang, Y. Peng, J. Chen, J. Li and F. Gao, J. Phys. Chem. C, 2022, 126, 8720–8733 CrossRef CAS.
- Y. Wu, Y. Ma, Y. Wang, K. G. Rappé, N. M. Washton, Y. Wang, E. D. Walter and F. Gao, J. Am. Chem. Soc., 2022, 144, 9734–9746 CrossRef CAS PubMed.
- Y. Zhang, Y. Peng, J. Li, K. Groden, J.-S. McEwen, E. D. Walter, Y. Chen, Y. Wang and F. Gao, ACS Catal., 2020, 10, 9410–9419 CrossRef CAS.
- F. Gao, M. Kollár, R. K. Kukkadapu, N. M. Washton, Y. Wang, J. Szanyi and C. H. Peden, Appl. Catal., B, 2015, 164, 407–419 CrossRef CAS.
- L. Kovarik, N. M. Washton, R. K. Kukkadapu, A. Devaraj, A. Wang, Y. Wang, J. Szanyi, C. H. F. Peden and F. Gao, ACS Catal., 2017, 7, 2458–2470 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS.
- J. Song, Y. L. Wang, E. D. Walter, N. M. Washton, D. H. Mei, L. Kovarik, M. H. Engelhard, S. Prodinger, Y. Wang, C. H. F. Peden and F. Gao, ACS Catal., 2017, 7, 8214–8227 CrossRef CAS.
- J. S. McEwen, T. Anggara, W. F. Schneider, V. F. Kispersky, J. T. Miller, W. N. Delgass and F. H. Ribeiro, Catal. Today, 2012, 184, 129–144 CrossRef CAS.
- J. Xue, X. Wang, G. Qi, J. Wang, M. Shen and W. Li, J. Catal., 2013, 297, 56–64 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. A. Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- B. Chen, R. Xu, R. Zhang and N. Liu, Environ. Sci. Technol., 2014, 48, 13909–13916 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- S. Zhao, L. Huang, B. Jiang, M. Cheng, J. Zhang and Y. Hu, Chin. J. Catal., 2018, 39, 800–809 CrossRef CAS.
- J. Chen, G. Peng, W. Zheng, W. Zhang, L. Guo and X. Wu, Catal. Sci. Technol., 2020, 10, 6583–6598 RSC.
- J. Chen, G. Peng, T. Liang, W. Zhang, W. Zheng, H. Zhao, L. Guo and X. Wu, Nanomaterials, 2020, 10, 2170 CrossRef CAS PubMed.
- S. Mohan, P. Dinesha and S. Kumar, Chem. Eng. J., 2020, 384, 123253–123262 CrossRef CAS.
- M. Jabłońska, Mol. Catal., 2022, 518, 112111 CrossRef.
- H. Lei, V. Rizzotto, A. Guo, D. Ye, U. Simon and P. Chen, Catalysts, 2021, 11, 52 CrossRef CAS.
- S. Zhang, L. Pang, Z. Chen, S. Ming, Y. Dong, Q. Liu, P. Liu, W. Cai and T. Li, Appl. Catal., A, 2020, 607, 117855 CrossRef CAS.
- C. H. F. Peden, J. Catal., 2019, 373, 384–389 CrossRef CAS.
- Q. Liu, C. Bian, S. Ming, L. Guo, S. Zhang, L. Pang, P. Liu, Z. Chen and T. Li, Appl. Catal., A, 2020, 607, 117865 CrossRef CAS.
- Y. Pu, X. Xie, W. Jiang, L. Yang, X. Jiang and L. Yao, Chin. Chem. Lett., 2020, 31, 2549–2555 CrossRef CAS.
- T. Andana, K. G. Rappé, F. Gao, J. Szanyi, X. Pereira-Hernandez and Y. Wang, Appl. Catal., B, 2021, 291, 120054 CrossRef CAS.
- Z. Zhang, J. Li, J. Tian, Y. Zhong, Z. Zou, R. Dong, S. Gao, W. Xu and D. Tan, Fuel Process. Technol., 2022, 230, 107213 CrossRef CAS.
- J. Yang, Y. Huang, J. Su, L. Chen, M. Zhang, M. Gao, M. Yang, F. Wang, X. Zhang and B. Shen, Sep. Purif. Technol., 2022, 297, 121544 CrossRef CAS.
- R. T. Guo, B. Qin, L. G. Wei, T. Y. Yin, J. Zhou and W. G. Pan, Phys. Chem. Chem. Phys., 2022, 24, 363–6382 Search PubMed.
- N. Zhang, H. He, D. Wang and Y. Li, J. Solid State Chem., 2020, 289, 121464 CrossRef CAS.
- M. Cai, X. Bian, F. Xie, W. Wu and P. Cen, Catalysts, 2021, 11, 361 CrossRef CAS.
- W. Shan, Y. Yu, Y. Zhang, G. He and H. He, Catal. Today, 2020, 376, 292–301 CrossRef.
- S. J. Park, Catalysts, 2021, 11, 1519 CrossRef.
- Z. Lian, Y. Li, W. Shan and H. He, Catalysts, 2020, 10, 1421 CrossRef CAS.
- C. Liu, H. Wang, Z. Zhang and Q. Liu, Catalysts, 2020, 10, 1034 CrossRef CAS.
- J. Xu, Y. Zhang, X. Zou, T. Tang, Q. Zhang, F. Guo and H. Liu, New J. Chem., 2022, 46, 2053–2067 RSC.
- G. Xu, X. Guo, X. Cheng, J. Yu and B. Fang, Nanoscale, 2021, 13, 7052–7080 RSC.
- K. Guo, J. Ji, W. Song, J. Sun, C. Tang and L. Dong, Appl. Catal., B, 2021, 297, 120388 CrossRef CAS.
- B. Guan, H. Jiang, Y. Wei, Z. Liu, X. Wu, H. Lin and Z. Huang, Mol. Catal., 2021, 510, 111704 CrossRef CAS.
- M. Li, Y. Guo and J. Yang, ChemCatChem, 2020, 19, 5599–5610 CrossRef.
- J. Kim, A. Jentys, S. M. Maier and J. A. Lercher, J. Phys. Chem. C, 2013, 117, 986–993 CrossRef CAS.
- J. Pérez-Ramírez, J. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. Debbagh and L. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef.
- R. Nedyalkova, S. Shwan, M. Skoglundh and L. Olsson, Appl. Catal., B, 2013, 138–139, 373–380 CrossRef CAS.
- H. Praliaud, S. Mikhailenko, Z. Chajar and M. Primet, Appl. Catal., B, 1998, 16, 359–374 CrossRef CAS.
- Z. Zhao, R. Yu, R. Zhao, C. Shi, H. Gies, F. S. Xiao, D. D. Vos, T. Yokoi, X. Bao and U. Kolb, Appl. Catal., B, 2017, 217, 421–428 CrossRef CAS.
- A. El-Trass, H. ElShamy, I. El-Mehasseb and M. El-Kemary, Appl. Surf. Sci., 2012, 258, 2997–3001 CrossRef CAS.
- M. H. Groothaert, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS.
- I. Lezcano-Gonzalez, U. Deka, H. E. V. D. Bij, P. Paalanen, B. Arstad, B. M. Weckhuysen and A. M. Beale, Appl. Catal., B, 2014, 154–155, 339–349 CrossRef CAS.
- J. Pérez-Ramírez, M. Santhosh Kumar and A. Brückner, J. Catal., 2004, 223, 13–27 CrossRef.
- B. Ipek, M. J. Wulfers, H. Kim, F. Goltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- A. Wang, Y. Wang, E. D. Walter, R. K. Kukkadapu, Y. Guo, G. Lu, R. S. Weber, Y. Wang, C. H. F. Peden and F. Gao, J. Catal., 2018, 358, 199–210 CrossRef CAS.
- S. Brandenberger, O. Kröcher, M. Casapu, A. Tissler and R. Althoff, Appl. Catal., B, 2011, 101, 649–659 CrossRef CAS.
- K. Niu, G. Li, J. Liu and Y. Wei, J. Solid State Chem., 2020, 287, 121330–1213336 CrossRef CAS.
- X. Shi, H. He and L. Xie, Chin. J. Catal., 2015, 36, 649–656 CrossRef CAS.
- K. Krishna, G. B. F. Seijger, C. M. V. D. Bleek, M. Makkee and H. P. A. Calis, Catal. Lett., 2003, 86, 121–132 CrossRef CAS.
- E. Yuan, G. Wu, W. Dai, N. Guan and L. Li, Catal. Sci. Technol., 2017, 7, 3036–3044 RSC.
- J. H. Kwak, H. Zhu, J. H. Lee, C. H. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- H. Wang, R. Xu, Y. Jin and R. Zhang, Catal. Today, 2019, 327, 295–307 CrossRef CAS.
- Y. Cui, Y. Wang, E. D. Walter, J. Szanyi, Y. Wang and F. Gao, Catal. Today, 2019, 339, 233–240 CrossRef.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- F. Lin, T. Andana, Y. Wu, J. Szanyi, Y. Wang and F. Gao, J. Catal., 2021, 401, 70–80 CrossRef CAS.
- J. Li, N. Wilken, K. Kamasamudram and N. W. Currier, Top. Catal., 2013, 56, 201–204 CrossRef CAS.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. F. Peden, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS.
- J. M. Kanervo and A. O. I. Krause, J. Catal., 2002, 207, 57–65 CrossRef CAS.
- J. H. Lee, Y. J. Kim, T. Ryu, P. S. Kim, H. K. Chang and S. B. Hong, Appl. Catal., B, 2017, 200, 428–438 CrossRef CAS.
- Q. Ye, L. Wang and R. T. Yang, Appl. Catal., A, 2012, 427, 24–34 CrossRef.
- J. Chastain and R. C. King Jr, Handbook of X-ray photoelectron spectroscopy, J Perkin-Elmer Corporation, 1992 Search PubMed.
- L. Xu, C. Shi, B. B. Chen, Q. Zhao, Y. J. Zhu, H. Gies, F. S. Xiao, D. De Vos, T. Yokoi, X. H. Bao, U. Kolb, M. Feyen, S. Maurer, A. Moini, U. Muller and W. P. Zhang, Microporous Mesoporous Mater., 2016, 236, 211–217 CrossRef CAS.
- H. Jiang, B. Guan, H. Lin and Z. Huang, Fuel, 2019, 255, 115587 CrossRef CAS.
- H. Yang, X. Cui, S. Li, Y. Cen, T. Deng, J. Wang, U. Olsbye and W. Fan, Microporous Mesoporous Mater., 2020, 305, 110381 CrossRef CAS.
- H. Yang, Y. Chen, X. Cui, G. Wang, Y. Cen, T. Deng, W. Yan, J. Gao, S. Zhu and U. Olsbye, Angew. Chem., Int. Ed., 2018, 130, 1854–1858 CrossRef.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- S. Velu, K. Suzuki, M. Vijayaraj, S. Barman and C. S. Gopinath, Appl. Catal., B, 2005, 55, 287–299 CrossRef CAS.
- F. Mercier, N. Thromat and C. Beaucaire, Appl. Surf. Sci., 2000, 165, 2880302 Search PubMed.
- J. Gurgul, K. Łątka, I. Hnat, J. Rynkowski and S. Dzwigaj, Microporous Mesoporous Mater., 2013, 168, 1–6 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- D. D. Hawn and B. M. DeKoven, Surf. Interface Anal., 1987, 10, 63–74 CrossRef CAS.
- P. Boron, L. Chmielarz, J. Gurgul, K. Łatka, B. Gil, B. Marszałek and S. Dzwigaj, Microporous Mesoporous Mater., 2015, 203, 73–85 CrossRef CAS.
- Y. Li, X. Han, Y. Hou, Y. Guo, Y. Liu, N. Xiang, Y. Cui and Z. Huang, Chin. J. Catal., 2017, 38, 1831–1841 CrossRef CAS.
- Y. Yue, H. Liu, P. Yuan, C. Yu and X. Bao, Sci. Rep., 2015, 5, 9270–9279 CrossRef CAS PubMed.
- M. Milanesio, G. Croce, D. Viterbo, H. O. Pastore, A. J. dos Santos Mascarenhas, E. C. de Oliveira Munsignatti and L. Meda, J. Phys. Chem. A, 2008, 112, 13745 CrossRef CAS.
- B. Pereda-Ayo, U. D. L. Torre, M. J. Illán-Gómez, A. Bueno-López and J. R. González-Velasco, Appl. Catal., B, 2014, 147, 420–428 CrossRef CAS.
- A. Boubnov, H. W. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J.-D. Grunwaldt, J. Am. Chem. Soc., 2014, 136, 13006–13015 CrossRef CAS.
- A. Williams, Rev. Sci. Instrum., 1983, 54, 193–197 CrossRef CAS.
- H. Maeda, T. Fukunaga, K. Suzuki, K. Osamura, M. Hida, H. Terauchi and N. Kamijo, Jpn. J. Appl. Phys., 1988, 27, L938–L940 CrossRef CAS.
- P. Behrens, Molecular Sieves – Science and Technology, 2004, vol. 4, pp. 427–466 Search PubMed.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- K. Ueda, J. Ohyama and A. Satsuma, Chem. Lett., 2017, 46, 1390–1392 CrossRef CAS.
- S. M. Maier, A. Jentys, E. Metwalli, P. Müller-Buschbaum and J. A. Lercher, J. Phys. Chem. Lett., 2011, 2, 950–955 CrossRef CAS.
- D. E. Doronkin, M. Casapu, T. Günter, O. Müller, R. Frahm and J.-D. Grunwaldt, J. Phys. Chem. C, 2014, 118, 10204–10212 CrossRef CAS.
- M. Hoj, M. J. Beier, J.-D. Grunwaldt and S. r. Dahl, Appl. Catal., B, 2009, 93, 166–176 CrossRef.
- Y. Xin, N. Zhang, X. Wang, Q. Li, X. Ma, Y. Qi, L. Zheng, J. A. Anderson and Z. Zhang, Catal. Today, 2019, 332, 35–41 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- F. Gao and J. Szanyi, Nat. Catal., 2018, 1, 174–175 CrossRef.
- S. M. Maier, A. Jentys, M. Janousch and J. A. van Bekkum, J. Phys. Chem. C, 2012, 116, 5846–5856 CrossRef CAS.
- I. A. Pankin, H. I. Hamoud, K. A. Lomachenko, S. B. Rasmussen, A. Martini, P. Bazin, V. Valtchev, M. Daturi, C. Lamberti and S. Bordiga, Catal. Sci. Technol., 2021, 11, 846–860 RSC.
- Y. Zhang, Y. Peng, K. Li, S. Liu, J. Chen, J. Li, F. Gao and C. H. F. Peden, ACS Catal., 2019, 9, 6137–6145 CrossRef CAS.
- X. Wei, Q. Ke, H. Cheng, Y. Guo, Z. Yuan, S. Zhao, T. Sun and S. Wang, Chem. Eng. J., 2019, 9, 123491–123502 Search PubMed.
- R. P. Vélez, U. Bentrup, W. Grünert and A. Brückner, Top. Catal., 2017, 60, 1641–1652 CrossRef.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC.
- D. Sun, Q. Liu, Z. Liu, G. Gui and Z. Huang, Catal. Today, 2009, 132, 122–126 CAS.
- H. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16–23 CrossRef CAS.
- P. R. Chen and U. Simon, Catalysts, 2016, 6, 204 CrossRef.
- I. Malpartida, E. Ivanova, M. Mihaylov, K. Hadjiivanov, V. Blasin-Aubé, O. Marie and M. Daturi, Catal. Today, 2010, 149, 295–303 CrossRef CAS.
- H.-Y. Chen, M. Kollar, Z. Wei, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, Catal. Today, 2019, 320, 61–71 CrossRef CAS.
- R. Zhang, J.-S. McEwen, M. Kollár, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, ACS Catal., 2014, 4, 4093–4105 CrossRef CAS.
- J. Szanyi, F. Gao, J. H. Kwak, M. Kollár, Y. Wang and C. H. F. Peden, Phys. Chem. Chem. Phys., 2016, 18, 10473–10485 RSC.
- R. Kefirov, E. Ivanova, K. Hadjiivanov, S. Dzwigaj and M. Che, Catal. Lett., 2008, 125, 209–214 CrossRef CAS.
- P. Concepcion, M. Boronat, R. Millan, M. Moliner and A. Corma, Top. Catal., 2017, 60, 1653–1663 CrossRef CAS.
- A. Frache, M. Cadoni, C. Bisio, L. Marchese, A. J. S. Mascarenhas and H. O. Pastore, Langmuir, 2002, 18, 6875–6880 CrossRef CAS.
- J. Szanyi, J. H. Kwak, H. Zhu and C. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368–2380 RSC.
- M.-L. Tarot, E. E. Iojoiu, V. Lauga, D. Duprez, X. Courtois and F. Can, Appl. Catal., B, 2019, 250, 355–368 CrossRef CAS.
- F. Giordanino, P. N. R. Vennestrm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- C. Lamberti, S. Bordiga, M. Salvalaggio, G. Spoto, A. Zecchina, F. Geobaldo, G. Vlaic and M. Bellatreccia, J. Phys. Chem. B, 1997, 101, 344–360 CrossRef CAS.
- C. Lamberti, S. Bordiga, A. Zecchina, M. Salvalaggio, F. Geobaldo and C. O. Areán, J. Chem. Soc., Faraday Trans., 1998, 94, 1519–1525 RSC.
- G. Turnes Palomino, S. Bordiga, C. Lamberti, A. Zecchina and C. Otero Areán, in Studies in Surface Science and Catalysis, ed. R. Aiello, G. Giordano and F. Testa, Elsevier, 2002, vol. 142, pp. 199–206 Search PubMed.
- J. Dedecek, Z. Sobalik, Z. Tvaruazkova, D. Kaucky and B. Wichterlova, J. Phys. Chem., 1995, 99, 16327–16337 CrossRef CAS.
- G. Turnes Palomino, A. Zecchina, E. Giamello, P. Fisicaro, G. Berlier, C. Lamberti and S. Bordiga, in Studies in Surface Science and Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, vol. 130, pp. 2915–2920 Search PubMed.
- C. Lamberti, E. Groppo, G. Spoto, S. Bordiga and A. Zecchina, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Academic Press, 2007, vol. 51, pp. 1–74 Search PubMed.
- M. Lemishka, J. Dedecek, K. Mlekodaj, Z. Sobalik, S. Sklenak and E. Tabor, Pure Appl. Chem., 2019, 91, 1721–1732 CrossRef CAS.
- J. Grzybek, B. Gil, W. J. Roth, M. Skoczek, A. Kowalczyk and L. Chmielarz, Spectrochim. Acta, Part A, 2018, 196, 281–288 CrossRef CAS PubMed.
- G. Lancaster, J. Mater. Sci., 1967, 2, 489–495 CrossRef CAS.
- H. Liu, J. Wang, T. Yu, S. Fan and M. Shen, Catal. Sci. Technol., 2014, 4, 1350–1356 RSC.
- M. Hoj, M. J. Beier, J. D. Grunwaldt and S. R. Dahl, Appl. Catal., B, 2009, 93, 166–176 CrossRef.
- G. Principi, Metal, 2020, 10, 992 CrossRef.
- H.-Y. Chen and W. M. H. Sachtler, Catal. Today, 1998, 42, 73–83 CrossRef CAS.
- A. R. Overweg, M. W. J. Crajé, A. M. van der Kraan, I. W. C. E. Arends, A. Ribera and R. A. Sheldon, J. Catal., 2004, 223, 262–270 CrossRef CAS.
- Y. Li, J. Deng, W. Song, J. Liu, Z. Zhao, M. Gao, Y. Wei and L. Zhao, J. Phys. Chem. C, 2016, 120, 14669–14680 CrossRef CAS.
- Y. Mao, Z. Y. Wang, H. F. Wang and P. Hu, ACS Catal., 2016, 6, 7882–7891 CrossRef CAS.
- S. Bordiga, L. Regli, D. Cocina, C. Lamberti, M. Bjørgen and K. P. Lillerud, J. Phys. Chem. B, 2005, 109, 2779–2784 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |