
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced stability of vanadium-doped Li1.2Ni0.16Co0.08Mn0.56O2 cathode materials for superior Li-ion batteries†
Miaomiao Zhou‡
a,
Jianjun Zhao‡ b,
Xiaodong Wang‡a,
Ji Shena,
Jin-Lin Yangcd,
Wenhao Tanga,
Yirui Denga,
Shi-Xi Zhao*c and
Ruiping Liu*a
b,
Xiaodong Wang‡a,
Ji Shena,
Jin-Lin Yangcd,
Wenhao Tanga,
Yirui Denga,
Shi-Xi Zhao*c and
Ruiping Liu*a
aSchool of Chemical & Environmental Engineering, China University of Mining and Technology (Beijing), Beijing, 100083, China. E-mail: lrp@cumtb.edu.cn
bState Key Laboratory of Chemical Resources Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, College of Chemistry, Beijing University of Chemical Technology, Beijing, 100029, China
cTsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, China. E-mail: zhaosx@sz.tsinghua.edu.cn
dSchool of Materials Science and Engineering, Tsinghua University, Beijing, 100084, China
First published on 16th November 2022
Abstract
Lithium–manganese-based cathode materials have attracted much attention due to its high specific capacity, but the low initial coulomb efficiency, poor rate performance and voltage attenuation during cycling limit its application. In this work, Li1.2Ni0.16Co0.08Mn0.56−xVxO2 samples (x = 0, 0.005, 0.01, 0.02, 0.05) were prepared using the sol–gel method, and the effects of different V5+ contents on the structure, valence state, and electrochemical performance of electrode materials were investigated. The results show that the introduction of high-valence V5+ in cathode materials can reduce partial Mn4+ to active Mn3+ ions for charge conservation, which not only improves the discharge capacity and coulomb efficiency of Li-rich manganese-based cathode materials, but also inhibits the voltage attenuation. The initial discharge capacity of the Li1.2Ni0.16Co0.08Mn0.55V0.01O2 is as high as 280.9 mA h g−1 with coulomb efficiency of 77.7% at 0.05C, which is much higher than that of the undoped pristine sample (236.6 mA h g−1 with coulomb efficiency of 74.0%). After 100 cycles at 0.1C, the capacity retention rate of Li1.2Ni0.16Co0.08Mn0.55V0.01O2 was 92.3% with the median voltage retention rate of 95.6%. This work provides a new idea for high performance of lithium-rich manganese-based cathode materials.
1. Introduction
Lithium-ion batteries (LIBs) have been widely used in different portable electronic devices and electric vehicles due to its high energy density, long cycle life and environment benign, thereby becoming an integral part of our lives.1–4 As is well known, capacity is determined by the ability to store lithium ions, while the energy density is proportional to the capacity and potential of discharge,5,6 and thus the cathode materials with layered structure exhibit great advantages and potential for use in new generation of renewable power sources.7–9 Among them, LiCoO2 with a relative high theoretical specific capacity of 274 mA h g−1 is one of the most popular cathode materials, however, the scarcity of cobalt resources, lower actual specific capacity of 140 mA h g−1, the collapse of the structure and lattice distortion during cycling restrain its widely use.10–12 Therefore, it is highly desired to explore the new cathode materials with high theoretical specific capacity and low cost.13–17Lithium-rich cathode materials with discharge capacities exceeding 250 mA h g−1 and energy densities of reaching 300 W h kg−1 have been attracted more attention.18–20 However, the low diffusion coefficient of lithium ions of lithium-rich cathode materials will lead to poor rate performance, and after the initial loss of oxygen, Li-rich NMC deliver descent capacity and lose energy density due to voltage decay, which is caused by irreversible migration of transition metals into the interlayer space during oxidation, where their redox potential is lower.21–23 Moreover, the Ni4+ ions will migrate to the surface of the materials and undergo severe side reactions with the electrolyte during cycling, which will lead to the reduced redox capacity and instability of interaction between Mn and Ni, and finally result in the voltage attenuation.24,25 To solve these problems, numerous strategies including surface modification and bulk doping have been proposed.26–28 Although the dissolution damage of the cathode materials in electrolyte solution can be inhibited by surface modification to some extent, the crystal structure of lithium-rich cathode materials tends to change during high-voltage charging and discharging.29–31 In contrast, appropriate doping can reinforce the crystal structure to improve the structural stability of materials, meanwhile improve the diffusion coefficient lithium ions in electrode materials,32,33 and thus it is considered to be a more effective modification strategy. Up to now, numerous metal cations have been used for doping, such as Fe,34 Sn,35 Zr,36 Ti, Ru,37 Cr and Al. Fe doping can stabilize the layered structure and improve the rate performance.38,39 Cr doping can increase the area of the inclined region (below 4.5 V), thereby restrain voltage fading, and consequently, the amount of oxygen evolution above the voltage plateau region (4.5 V) can be decreased.40 The cycling stability of Li1.2Mn0.56−xAlxNi0.16CO0.08O2 has been improved by aluminum doping, and in particular, the capacity retention rate of Li1.2Mn0.51Al0.05Ni0.16Co0.08O2 and Li1.2Mn0.48Al0.08Ni0.16Co0.08O2 electrodes are enhanced. However, the lower coulombic efficiency of the untreated material (only 68%),41 the lower median voltage, and the electrolyte corrosion of the cathode materials are the main issues which further need to explore.42,43
Vanadium ions are considered as the most suitable substitute to manganese ions due to their similar ionic radii.44 Appropriate V doping can reduce the mixing degree of Li/Ni, thus stabilize the structure and improve the electrochemical performance.45 Herein, Li1.2Ni0.16Co0.08Mn0.56−xVxO2 samples were prepared by using the sol–gel method. The results show that Li1.2Ni0.16Co0.08Mn0.55V0.01O2 exhibits best electrochemical performance within 2.0–4.8 V at 0.1C rate. After 100 cycles, the discharge specific capacity of 225.66 mA h g−1 and the capacity retention rate of as high as 92.3% can be obtained. The median voltage attenuation is only 0.15 V, and the retention rate is 95.6%. The doping of high-valence vanadium cannot only improve the cyclic performance of the lithium-rich cathode material, but also inhibit the voltage attenuation. This work provides a new way for improving the electrochemical performance of Li-rich cathode materials.
2. Experimental section
2.1. Synthesis of materials
A series of V-doped Li1.2Ni0.16Mn0.56−xCo0.08VxO2 samples (x = 0, 0.005, 0.01, 0.02, 0.05) were prepared by the sol–gel method, which are denoted as pristine, V-0.5%, V-1%, V-2%, and V-5%, respectively. Citric acid was used as a chelating agent and deionized water was used as solvent. Typically, a certain stoichiometric ratio of the solute (lithium acetate, nickel acetate, cobalt acetate, manganese acetate, ammonium metavanadate) was dissolved in water to form transparent solution. Subsequently, a certain amount of citric acid solution was added via continuous stirring until the pH was stable (the molar ratio of citric acid to metal ions is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The solution was transferred to a water bath at 60 °C under stirring for 4 h, and then kept at 95 °C to form the sticky gel after evaporating the water. The Li1.2Ni0.16Co0.08Mn0.56−xVxO2 electrode materials can be obtained after drying the sticky gel at 120 °C for 24 h, pyrolysis at 500 °C for 5 h and calcining at 900 °C for 16 h in air.
:1). The solution was transferred to a water bath at 60 °C under stirring for 4 h, and then kept at 95 °C to form the sticky gel after evaporating the water. The Li1.2Ni0.16Co0.08Mn0.56−xVxO2 electrode materials can be obtained after drying the sticky gel at 120 °C for 24 h, pyrolysis at 500 °C for 5 h and calcining at 900 °C for 16 h in air.
2.2 Characterizations methods
The morphologies of the cathode materials before and after cycling were observed by scanning electron microscopy (JSM-7800F, Tokyo, Japan) and transmission electron microscopy (TEM-2100, JEOL, Tokyo, Japan). The phase composition of the V-doped Li1.2Ni0.16Mn0.56–xCo0.08VxO2 cathode materials (x = 0, 0.005, 0.01, 0.02, 0.05) was analyzed by a powder X-ray diffractometer (XRD) at a scan rate of 10 deg min−1 in the 2θ range of 10°–80° X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Fisher K-Alpha with an Al Kα X-ray source. The specific surface area of the material was evaluated by means of the Brunauer–Emmett–Teller (BET) model, based on nitrogen adsorption–desorption isotherm tests performed on a BeiShiDe 3H-2000 PM1 instrument.2.3 Electrochemical measurements
The 2032 type coin cells were assembled in glove box under argon atmosphere. The cathode was prepared by mixing the Li1.2Ni0.16Co0.08Mn0.56−xVxO2, carbon black and polyvinylidene fluoride (PVDF) with the mass ratio of 8:1:1 in the N-methyl-2-pyrrolidone (NMP) to form a uniform slurry firstly, and then the slurry was coated on an aluminum foil and dried overnight at 60 °C in vacuum oven. Lithium foil was used as the counter, the porous polyethylene was selected as the separator, and LiPF6 in the mixture of EC:EMC:DMC = 1:1:1 by volume was used as the electrolyte. Six cells of each cathode material were prepared for simultaneous testing. The cell performance was evaluated at different rates in the voltage range of 2.0–4.8 V at the room temperature on LAND 2001 battery test system (Wuhan Land, China). CV tests and EIS measurements were carried out on IVIUMnSTAT multichannel electrochemical analyzer in the same voltage range at the scanning rate of 0.1 mV s−1 and in the frequency range of 0.01–10000 Hz, respectively.
3. Result and discussion
The XRD patterns of the samples with different amount of vanadium-doping are shown in Fig. 1(a). The main peaks for all samples correspond to the layered LiCoO2 phase structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group).46 Among them, two pairs of splitting peaks ((006)/(102) and (108)/(110)) are extremely evident, indicating that both the pristine and vanadium substituted samples exhibit a well-formed layered structure.47 In addition, a small diffraction peak appears between 20° and 25°, demonstrating the presence of the monoclinic Li2MnO3 phases.48 Due to the slightly larger ion radius of V5+ (0.54 Å) than that of Mn4+ (0.53 Å), the c/a values of the samples increase with the increase of doping amount (Table S1†). The vanadium ion occupies the TM (Co, Mn and Ni) layer. Due to the slightly larger ion radius of V5+ (0.54 Å) than that of Mn4+ (0.53 Å), and it is possible that it occupies the lattice position of manganese ions.49 Meanwhile, the I003/I104 ratios of the V-1% sample are larger than that of the pristine material, indicating that the layered structure is more stable. The Li3VO4 impurity phase can be clearly observed in the V-2% and V-5% samples, which may result in the poor electrochemical performance of the cathode material (Fig. 1(b)).
m space group).46 Among them, two pairs of splitting peaks ((006)/(102) and (108)/(110)) are extremely evident, indicating that both the pristine and vanadium substituted samples exhibit a well-formed layered structure.47 In addition, a small diffraction peak appears between 20° and 25°, demonstrating the presence of the monoclinic Li2MnO3 phases.48 Due to the slightly larger ion radius of V5+ (0.54 Å) than that of Mn4+ (0.53 Å), the c/a values of the samples increase with the increase of doping amount (Table S1†). The vanadium ion occupies the TM (Co, Mn and Ni) layer. Due to the slightly larger ion radius of V5+ (0.54 Å) than that of Mn4+ (0.53 Å), and it is possible that it occupies the lattice position of manganese ions.49 Meanwhile, the I003/I104 ratios of the V-1% sample are larger than that of the pristine material, indicating that the layered structure is more stable. The Li3VO4 impurity phase can be clearly observed in the V-2% and V-5% samples, which may result in the poor electrochemical performance of the cathode material (Fig. 1(b)).
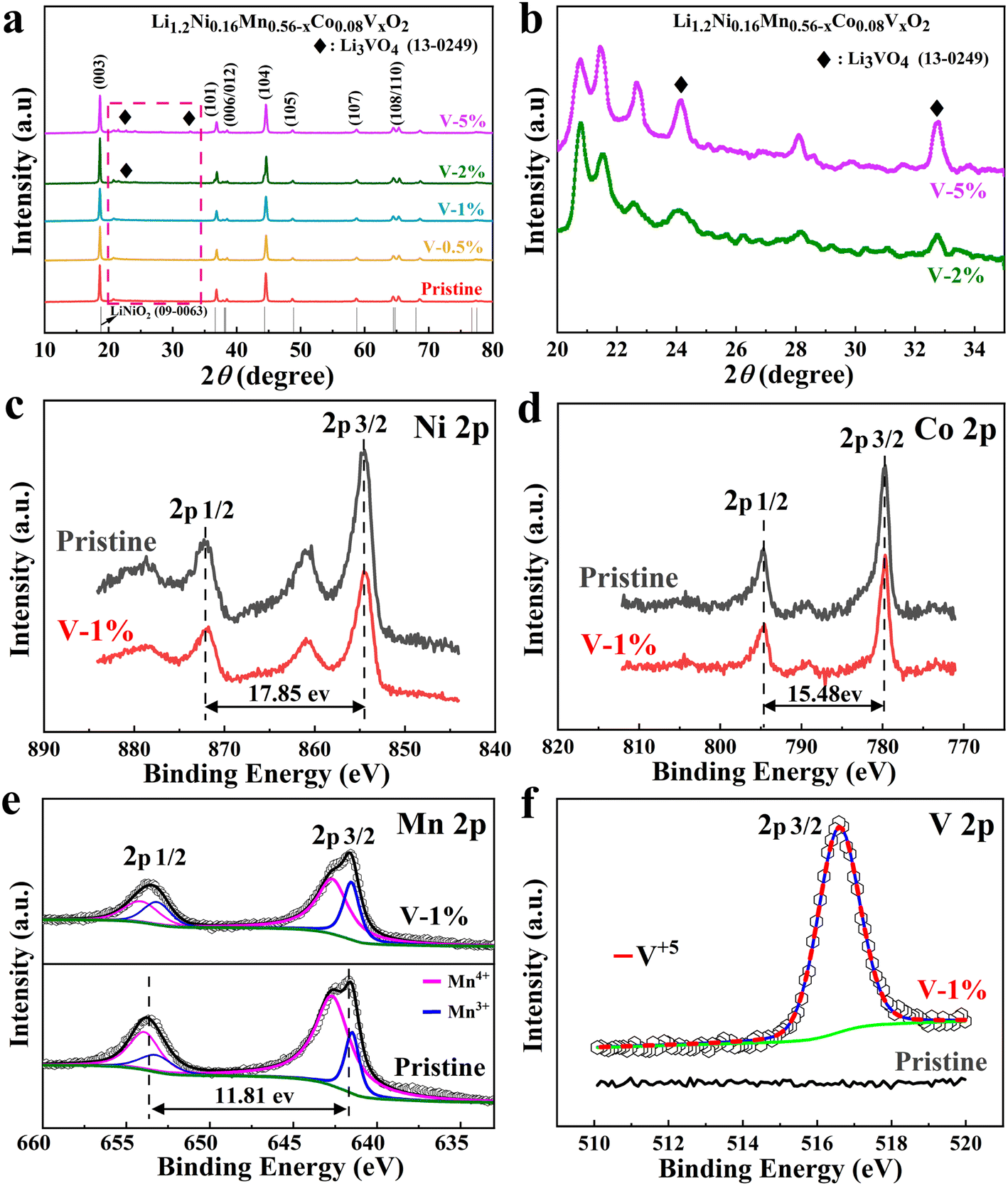 | ||
| Fig. 1 (a) XRD patterns of samples with different vanadium-doping. (b) XRD patterns of V-2% and V-5% samples at 20°–35°. (c–f) X-ray photoelectron spectroscopy (XPS) of elements (Ni, Co, V, Mn) in pristine (x = 0) and V-1% sample. | ||
X-ray photoelectron spectroscopy (XPS) of the pristine and V-1% samples are shown in Fig. 1(c–f) and S1.† The binding energies of Ni 2p2/3 and 2p1/2 for the corresponding electrodes are 854.6 eV and 872.4 eV, respectively,50,51 and the distance between binding energy peaks is 17.85 eV,52 indicates that the valence state of Ni for both samples is +2 (Fig. 1(c)).53,54 The difference of 15.48 eV between two main peaks of Co 2p2/3 (780.06 eV) and Co 2p1/2 (795.54 eV) for the pristine sample are observed, which has been observed previously for Co3+ ions (Fig. 1(d)).55 The V 2p2/3 peak at 516.58 eV for the V-1% sample demonstrates the successful vanadium doping (Fig. 1(e)). It also should be noted that the percent of Mn3+ ions in the V-1% sample is higher (31.67%) than that of pristine material (19.12%), which can be due to the effect of V5+ doping (Table S2†). The large charge disbalance in the compound may be ascribed to the concentration difference between the internal and surface of the material, with the surface concentration being greater and the internal concentration being less, which may result in the substitution of Mn4+ by V5+ having a greater effect on the surface, manifesting as an increase in Mn3+ on the surface. When V5+ partially replaces Mn4+, a part of Mn4+ will transform into Mn3+ to keep the valence balance. However, the difference in Mn3+ content between the V-1% and the pristine samples is sufficient to show that some of the Mn4+ will be converted to Mn3+ to maintain valence equilibrium when V5+ partially replaces Mn4+. Generally, Mn3+ can contribute capacity due to its electrochemically active characteristic. However, excessive Mn3+ will have a negative impact due to the disproportionation reaction of Mn3+, which will produce Mn2+ and Mn4+, and Mn2+ will dissolve in the electrolyte or migrate to adjacent tetrahedral positions to form a spinel phase. Conversely, Mn4+ in the crystal structure is important for ensuring the structural stability of the materials. Therefore, an appropriate ratio of Mn3+/Mn4+ has an important effect on the rate and cycle performance of lithium-rich manganese-based cathode materials.56,57
Fig. 2 and S2–S4† show the SEM and TEM images of different samples. It can be observed that the primary particles of the pristine material are polygonal spheroids, which exhibit a size of approximately 100–150 nm with a slight agglomeration (Fig. 2(a)). The addition of element V reduces the agglomeration of the material and lowers the ionic diffusion impedance, while improving the electrical conductivity of the material and accelerating charge transfer, which provides an opportunity for the material to improve its electrochemical performance. As shown in Fig. 2(b), when the doping amount of vanadium is 0.01, the particle size of the sample grows into approximately 200–300 nm, and the uniform distribution of the lithium (yellow), manganese (blue), nickel (purple), cobalt (green), and vanadium (orange) are observed (Fig. S5†), the particle size of the material with 5% vanadium doping is about 100 μm, indicating a severe agglomerate (Fig. S2†). It should be worth noting that V5+ cannot be evenly distributed into the lattice of lithium–manganese rich materials when the vanadium doping is larger than 2%. The HRTEM images for the pristine and V-1% samples show that the particle size of the pristine and V-1% samples are similar with a interplanar spacing of 0.475 nm and 0.471 nm (003 reflection), respectively, which is typical for the Rm layered structure (Fig. 2(c–f)). No other impurities lattice stripes are observed (Fig. 2(e and f)), which indicates that V is successfully doped into the lattice of the material. The results verify the high ordering state of the crystal structure, which is consistent with the XRD results.
 | ||
| Fig. 2 SEM images of pristine (a) and sample with 1% vanadium doping (b), HRTEM images of pristine (c and d) and sample with 1% vanadium doping (e and f). | ||
The size distribution of secondary particles of the pristine, V-1% and V-5% samples is shown in Fig. S6(a and b),† where D90, D50 and D10 are the equivalent diameters (average particle size) of the largest particles in the distribution curve at a cumulative distribution of 90%, 50% and 10%, respectively. It can be observed that the D10, D50, and D90 of the V-1% sample are smaller compared with those of the other two materials, and the particle size is more uniform. According to BET results (Fig. S6(c)†), the surface areas and average pore size of the pristine and sample with 1% vanadium doping are 3.37 m2 g−1/13.1 nm and 3.21 m2 g−1/14.5 nm, respectively. It can be assumed that the difference between the particle size for undoped and doped samples is due to the agglomeration of particles, as observed for the V-5% sample.
The initial charge–discharge curves of the pristine, V-0.5%, V-1%, V-2% and V-5% samples in the voltage range of 2.0–4.8 V and at 0.05C rate (1C = 250 mA h g−1) were shown in Fig. S7(a).† All samples showed similar charge–discharge curves, existing slopes and large platform areas, which is similar to other lithium rich manganese based materials. Initial stage (<4.5 V), the Li-ions remove from Li2MnO3 phase and accompanied with the oxidation of transition metal Ni2+ to Ni3+, and Co3+ to Co4+, in second stage (above 4.5 V), O2− is oxidized to On− (1 < n< 2), followed by oxidation to O2.58 With the increase of the amount of V doping, the length of the inclined region increases first and then decreases. As observed, the largest discharge capacity of 134.26 mA h g−1 from 2.0 to 4.5 V can be obtained for the V-1% sample, which can be ascribed to the presence of more electrochemically active Mn3+ ions, which is in good agreement with the results of the XPS analysis. The total charge/discharge capacity of pristine, V-0.5%, V-1%, V-2% and V-5% are 319.7/236.6 mA h g−1, 348.6/266.2 mA h g−1, 361.6/280.9 mA h g−1, 305.2/223.8 mA h g−1 and 296/214.6 mA h g−1 respectively, and the initial coulombic efficiency of the materials are 74.1%, 76.3%, 77.7%, 73.3%, and 72.4%, respectively (Table S3†). Furthermore, the discharge voltage platform along the arrow direction decreases with the increase of the number of cycles, and the sample V-1% exhibits the minimum capacity fading, while too much vanadium doping will cause rapid voltage decay.
Cyclic voltammetry curves for Li1.2Ni0.16Co0.08Mn0.56−xVxO2 (x = 0, 0.005, 0.01, 0.02, 0.05) electrode materials are presented in Fig. 3(a and b) and S8.† The oxidation peak at 4.1 V of the all the sample can be ascribed to the oxidation reactions of Ni2+ → Ni4+ and Co3+ → Co4+, and the oxidation peak at 4.72 V corresponds to the oxidization of O2− to On− (1 < n< 2), followed by oxidation to O2. Three reduction peaks, corresponding to the reduction of Ni4+, Co4+ and Mn4+, are observed in the first cycling process for all electrode materials.59,60 Compared to the first cycle, the sharp oxidation peak at 4.72 V disappears in the subsequent cycles, which can be attributed to an excessive Li+ remove from layered Li2MnO3 and an irreversible lattice oxygen escape, and finally resulting in a large irreversible capacity loss in the initial charge/discharge cycle. As the number of cycles increase, a new reduction peak around 3.1 V appears during the negative sweep, which corresponds to the reduction of Mn4+ to Mn3+ and the gradual transformation of the layered phase to the spinel phase.61 It should be noted that the reduction peak of the V-1% sample (at approximately 3.1 V) changes insignificantly from the first cycle to the third cycle, which proves that a proper amount of V doping can inhibit the conversion of the layered structure to the spinel structure and improve the structural stability of the material. Furthermore, appropriate V doping can reduce the polarization of REDOX, and the smallest potential difference of 0.36 V can be obtained in the V-1% sample. Moreover, the cyclic voltammetry curves of the V-1% sample was well overlapped with an increase in the number of cycles, indicating the good reversibility of the materials.
 | ||
| Fig. 3 (a and b) Cyclic voltammetry curves of the pristine, V-0.5%, V-1%, V-2% and V-5% samples at 0.1 mV s−1. (c) Dependence of the average discharge profile on the number of cycles for the Li1.2Ni0.16Co0.08Mn0.56−xVxO2 electrodes. (d) The rate and (e) cycling performance of the electrode materials at 0.1C in the range of 2.0–4.8 V. | ||
Fig. 3(c) shows that the median voltage of the pristine and the V-1% samples decrease as the number of cycles increases. The voltage decay of the pristine sample is evidently faster, especially in the first few cycles, and it can be ascribed to the phase transition of the cathode materials during charging/discharging. The median voltage of the samples with vanadium doping is higher, which can be due to the improvement of the structural stability of the material. The drop in the average discharge voltage is 0.400, 0.390, 0.156, 0.332 and 0.388 V for pristine, V-0.5%, V-1%, V-2% and V-5%, respectively. It can be concluded that an appropriate amount of V doping can improve the structural stability of the lithium–manganese rich layered oxide and inhibit the transformation of the layered structure to the spinel structure.
The rate performance of the pristine sample and the V-0.5%, V-1%, V-0.2%, V-2% and V-5% samples is evaluated at 0.1, 0.2, 0.5, 1 and 2C in the voltage range of 2.0–4.8 V for 5 cycles (Fig. 3(d)). The results show that the rate performance of the sample doped with 1% of V is better than that of the original sample, but excessive doping has the negative effect. The discharge capacity of the pristine, V-1% and V-5% samples is 168.65 mA h g−1, 158.13 mA h g−1 and 137.37 mA h g−1 at 1C, respectively. The highest discharge capacity of 238.5 mA h g−1 for the V-1% sample can be restored when the current density goes back to 0.1C, while the sample with 5% vanadium doping deliver the minimum discharge capacity of 188.27 mA h g−1, and it may be attributed to the formation of the LiV3O4 heterogeneous phase, which will greatly reduce the conductivity of the material. As shown in Fig. 3(e), after 100 charge/discharge cycles at 0.1C, the capacity retention of V-1% electrodes (92.3%) is much higher than that of the pristine (85.1%), V-0.5% (90.8%), V-2% (82.2%) and V-5% (86.1%) electrodes, which further indicates the improved structural stability of the electrode after doping with 1% vanadium.
The electrochemical impedance spectra of all samples after 3rd and 100th cycles are shown in Fig. S9.† The semicircle in the high frequencies region corresponds to the battery's ohmic resistance (Rs) and charge transfer resistance (Rct) at the electrode/electrolyte interface, and the slope in the low-frequency region corresponds to the Warburg impedance associated with Li+ diffusion in the bulk materials, and the CPE is the capacitance of the electrode/electrolyte bilayer. The EIS spectrum after the third cycle showed that the charge transfer resistance decreased linearly with the increase of doping amount of vanadium, and then increased significantly (Table S4†). The charge transfer resistance across the electrolyte/intercalate interface (Rct) of the pristine, V-0.5%, V-1%, V-2% and V-5% samples is 49.85, 45.15, 14.61, 105.9 and 113.5 Ω, respectively. A decrease in Rct indicates a significant effect of vanadium doping on the transport rate of lithium ions through the electrode/electrolyte interface. This behavior is observed when the vanadium content increases up to 1%, which confirms the earlier assumption that the crystal structure is stabilized by doping with vanadium ions. At the same time, excessive doping leads to the formation of an impurity phase and the agglomeration of particles. The increase in particle agglomeration leads to a decrease in the solid phase diffusion coefficient and electron conductivity, an increase in the internal resistance of the cell and an increase in the charge transfer impedance.
To further study the influence of vanadium doping on the morphology of the material, SEM images of the pristine, V-1% and V-5% samples after 100 cycles were obtained (Fig. 4). The particles of the pristine material are evidently damaged with rough surfaces and rounded edges after cycling. The lithium-rich manganese based electrode materials can react with the electrolyte during subsequent cycling processes, which result in the destroyed structure of the material. The sample with 5% vanadium doping also tends to pulverization due to the corrosion of the electrolyte. Compared with that of the pristine and V-5% samples, the morphology of the V-1% material is integrity and smooth, which is benefit from the stabilization of the internal structure of the material by a small amount of vanadium doping.
 | ||
| Fig. 4 SEM images of the pristine (a–c), V-1% (d–f) and V-5% (g–i) samples after 100 cycles at 0.1C (1C = 250 mA g−1). | ||
The crystal structures of the pristine, V-1% and V-5% samples before and after 100 cycles can be clearly observed in Fig. 5. LiMn2O4 phase with F![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) m space group appears for all materials, indicating that the crystal structure of the electrode is changed, and the new spinel phase is formed due to the structural rearrangement of certain layered compositions during charge–discharge process (Fig. 5(a)). Thus, a decrease in the diffraction intensity of the (003) peak occurs due to the transformation of the layered structure to a spinel structure during cycling. As shown in Fig. 5(b), owing to an axis expansion after cycling, the (003) diffraction peak was shifted to the low angle by 0.13°, 0.05° and 0.08° for the pristine, V-1% and V-5% samples, respectively. Among them, the V-1% sample exhibits the least deviation in the diffraction angle after cycling, indicating the improved structure stability of the material. Similar to the results before cycling, as shown in Table S5,† the degree of cation mixing of the V-1% samples remained lower than that of the original samples after cycling, indicating that proper V-doping can enhance the structural stability of the Li-rich cathode material.
m space group appears for all materials, indicating that the crystal structure of the electrode is changed, and the new spinel phase is formed due to the structural rearrangement of certain layered compositions during charge–discharge process (Fig. 5(a)). Thus, a decrease in the diffraction intensity of the (003) peak occurs due to the transformation of the layered structure to a spinel structure during cycling. As shown in Fig. 5(b), owing to an axis expansion after cycling, the (003) diffraction peak was shifted to the low angle by 0.13°, 0.05° and 0.08° for the pristine, V-1% and V-5% samples, respectively. Among them, the V-1% sample exhibits the least deviation in the diffraction angle after cycling, indicating the improved structure stability of the material. Similar to the results before cycling, as shown in Table S5,† the degree of cation mixing of the V-1% samples remained lower than that of the original samples after cycling, indicating that proper V-doping can enhance the structural stability of the Li-rich cathode material.
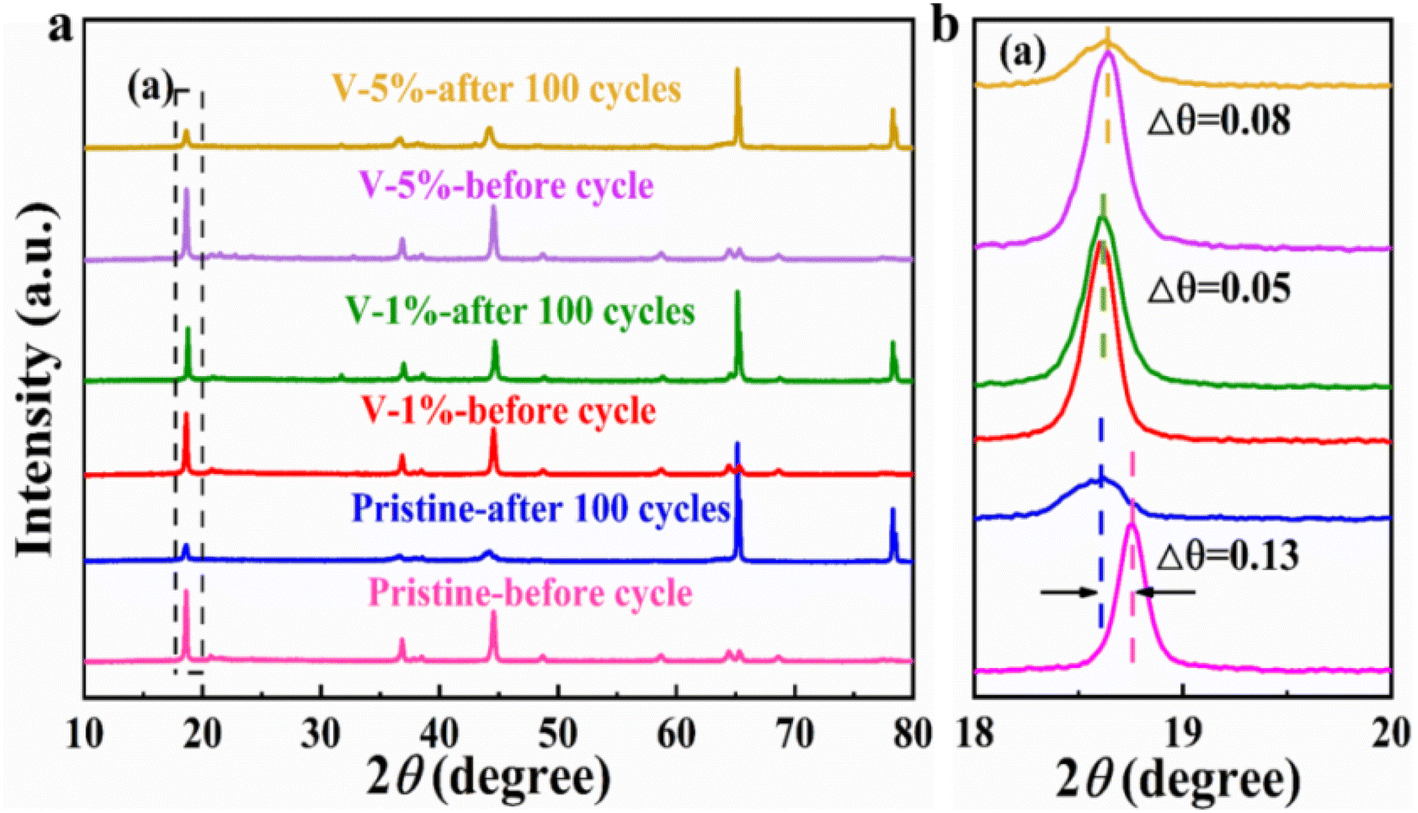 | ||
| Fig. 5 (a) XRD patterns of the pristine, V-1% and V-5% samples after 100 cycles at 0.1C (1C = 250 mA h g−1); (b) magnified view of the XRD patterns at 18°–20°. | ||
The structural transformation diagram of the cathode materials is shown in Fig. 6. The structures of the Li-rich samples are composed of LMO2 and Li2MnO3 phases. During cycling, part of Li ions escape from the LMO2 lattice and enter into the Li2MnO3 lattice, meanwhile, some transition metal ions may occupy the position of Li+ in the Li layers, resulting in local areas TM/Li mixing in the domain, and finally leading to the two-phase coexistence of layered structure (Rm) and spinel structure (Fdm). After vanadium doping with the appropriate content, a large number of transition metal ions is relatively stable and will not entered into the lithium layer structure.62 Thus it can stabilize the layered structure to some extent and improve the electrochemical performance of lithium-rich cathode materials.
 | ||
| Fig. 6 Schematic illustration of structural transformation of the cathode materials during the cycling. | ||
4. Conclusions
In this work, lithium rich Li1.2Ni0.16Co0.08Mn0.56−xVxO2 cathode materials with different amount of vanadium doping were successfully synthesized via sol–gel method. The as-obtained lithium-rich manganese-based materials with 1% of V doping exhibit pure Rm phase layered structure, uniform particle size without agglomeration. The introduction of V5+ into the cathode materials can not only reduce some of the Mn4+ to active Mn3+ ions, but also decrease the particle size of the as-obtained materials, which is very favorable for Li+ transport and accelerating of the electrochemical reaction kinetics, and thus improves the cycling stability and coulombic efficiency of lithium-rich manganese-based cathode materials. The appropriate amount of V doping improves the structural stability of the Li–Mn-rich layered oxide, reduces the degree of cation mixing and inhibits the conversion of the layered structure to a spinel structure. Sample V-1% showed a 5% increase in median voltage retention after 100 cycles at 0.1C, indicating that a moderate amount of V doping can also slow down the decay of the median voltage. This work provides a new way to stabilize the lithium-rich manganese-based cathode material during cycling and makes it a promising candidate for high specific energy LIBs.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the National Natural Science Foundation of China (52272258), the Fundamental Research Funds for the Central Universities (No. 2021JCCXJD01), Key R & D and transformation projects in Qinghai Province (2021-HZ-808) and Hebei Province (21314401D). The author would also like to thank the shiyanjia lab (https://www.shiyanjia.com) for the XRD and XPS tests.References
- Y. Shao-Horn, L. Croguennec, C. Delmas, E. C. Nelson and M. A. O'Keefe, Nat. Mater., 2003, 2, 464–467 CrossRef PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Y. Wang and S. Zhao, J. Wuhan Univ. Technol. Mater. Sci. Ed., 2016, 31, 945–949 CrossRef CAS.
- D. Li, B. Zhang, X. Qu, J. Zhang, K. Meng, G. Ji, P. Li and J. Xu, Chin. Chem. Lett., 2021, 30, 2333–2337 CrossRef.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef CAS PubMed.
- Y. X. Xie, S. Z. Chen, W. Yang, H. B. Zou, Z. Y. Lin and J. C. Zhou, J. Alloy Compd., 2019, 772, 230–239 CrossRef CAS.
- P. Wu, B. Shi, H. Tu, C. Guo, A. Liu, G. Yan and Z. Yu, J. Adv. Ceram., 2021, 10, 1129–1139 CrossRef CAS.
- Y. Ding, D. Mu, B. Wu, R. Wang, Z. Zhao and F. Wu, Appl. Energy, 2017, 195, 586–599 CrossRef CAS.
- Q. Wang, H. Zhao, B. Li, C. Yang, M. Li, Y. Li, P. Han, M. Wu, T. Li and R. Liu, Chin. Chem. Lett., 2021, 32, 1157–1160 CrossRef CAS.
- M. Zhou, Int. J. Electrochem. Sci., 2020, 10759–10771 CrossRef CAS.
- T. Ohzuku and A. Ueda, J. Electrochem. Soc., 1994, 141, 2972–2977 CrossRef CAS.
- C. Y. Cui, X. L. Fan, X. Q. Zhou, J. Chen, Q. C. Wang, L. Ma, C. Y. Yang, E. Y. Hu, X. Q. Yang and C. S. Wang, J. Am. Chem. Soc., 2020, 142, 8918–8927 CrossRef CAS PubMed.
- Q. Zhang, Y. He, G. Lin, X. Ma, Z. Xiao, D. Shi and Y. Yang, J. Mater. Chem. A, 2021, 9, 10652–10660 RSC.
- X. Wu, S. X. Zhao, L. Wei, H. Deng and C. W. Nan, J. Alloy Compd., 2018, 735, 1158–1166 CrossRef CAS.
- F. Yang, X. Feng, Y. S. Liu, L. C. Kao, P. A. Glans, W. Yang and J. Guo, Energy Environ. Mater., 2021, 4, 139–157 CrossRef CAS.
- W. Xu and Y. Wang, Ionics, 2018, 24, 2523–2532 CrossRef CAS.
- L. Zhu, Y. Liu, W. Wu, X. Wu, W. Tang and Y. Wu, J. Mater. Chem. A, 2015, 3, 15156–15162 RSC.
- B. Li, X. Wang, Y. Gao, B. Wang, J. Qiu, X. Cheng and D. Dai, J. Materiom, 2019, 5, 149–155 CrossRef.
- Z. Yang, C. Zheng, Z. Wei, J. Zhong, H. Liu, J. Feng, J. Li and F. Kang, Energy Mater., 2022, 2, 200006 Search PubMed.
- K. Mu, Y. Cao, G. Hu, K. Du, H. Yang, Z. Gan and Z. Peng, Electrochim. Acta, 2018, 273, 88–97 CrossRef CAS.
- T. Liu, S.-X. Zhao, L.-L. Gou, X. Wu and C.-W. Nan, Rare Met., 2019, 38, 189–198 CrossRef CAS.
- K. Bi, S. X. Zhao, C. Huang and C. W. Nan, J. Power Sources, 2018, 389, 240–248 CrossRef CAS.
- Y. Shang, T. Chu, B. Shi and K. Fu, Energy Environ. Mater., 2020, 4, 213–221 CrossRef.
- P. Hou, J. Yin, M. Ding, J. Huang and X. Xu, Small, 2017, 13, 201701802 Search PubMed.
- W. Huang, W. Li, L. Wang, H. Zhu, M. Gao, H. Zhao, J. Zhao, X. Shen, X. Wang, Z. Wang, C. Qi, W. Xiao, L. Yao, J. Wang, W. Zhuang and X. Sun, Small, 2021, 17, 202104282 Search PubMed.
- J. Zhang, H. Zhang, R. Gao, Z. Li, Z. Hu and X. Liu, Phys. Chem. Chem. Phys., 2016, 18, 13322–13331 RSC.
- R. Tatara, Y. Yu, P. Karayaylali, A. K. Chan, Y. Zhang, R. Jung, F. Maglia, L. Giordano and Y. Shao-Horn, ACS Appl. Mater. Interfaces, 2019, 11, 34973–34988 CrossRef CAS PubMed.
- R. Sharpe, R. A. House, M. J. Clarke, D. Forstermann, J. J. Marie, G. Cibin, K. J. Zhou, H. Y. Playford, P. G. Bruce and M. S. Islam, J. Am. Chem. Soc., 2020, 142, 21799–21809 CrossRef CAS PubMed.
- J. Zheng, Y. Xie, S. S. Jiang, Y. Z. Long, X. Ning and Z. G. Lan, Phys. Chem. Chem. Phys., 2019, 21, 26502–26514 RSC.
- Z. Yang, J. J. Zhong, J. L. Li, Y. Y. Liu, B. B. Niu and F. Y. Kang, Ceram. Int., 2019, 45, 439–448 CrossRef CAS.
- Y. L. Heng, Z. Y. Gu, J. Z. Guo, X. T. Wang, X. X. Zhao and X. L. Wu, Energy Mater., 2022, 2, 200017 Search PubMed.
- Y. Zhao, J. Liu, S. Wang, R. Ji, Q. Xia, Z. Ding, W. Wei, Y. Liu, P. Wang and D. G. Ivey, Adv. Funct. Mater., 2016, 26, 4760–4767 CrossRef CAS.
- S. Y. Zheng, K. Zhou, F. Zheng, H. D. Liu, G. M. Zhong, W. H. Zuo, N. B. Xu, G. Zhao, M. Z. Luo, J. Wu, C. Y. Zhang, Z. R. Zhang, S. Q. Wu and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 40347–40354 CrossRef CAS PubMed.
- J. Billaud, D. Sheptyakov, S. Sallard, D. Leanza, M. Talianker, J. Grinblat, H. Sclar, D. Aurbach, P. Novák and C. Villevieille, J. Mater. Chem. A, 2019, 7, 15215–15224 RSC.
- C. S. Yoon, M. J. Choi, D. W. Jun, Q. Zhang, P. Kaghazchi, K. H. Kim and Y. K. Sun, Chem. Mater., 2018, 30, 1808–1814 CrossRef CAS.
- X. Feng, Y. R. Gao, L. B. Ben, Z. Z. Yang, Z. X. Wang and L. Q. Chen, J. Power Sources, 2016, 317, 74–80 CrossRef CAS.
- B. H. Song, M. O. Lai and L. Lu, Electrochim. Acta, 2012, 80, 187–195 CrossRef CAS.
- G. R. Hu, Z. C. Xue, Z. Y. Luo, Z. D. Peng, Y. B. Cao, W. G. Wang, Y. X. Zeng, Y. Huang, Y. Tao, T. F. Li, Z. Y. Zhang and K. Du, Ceram. Int., 2019, 45, 10633–10639 CrossRef CAS.
- X. Liu, T. Huang and A. Yu, Electrochim. Acta, 2014, 133, 555–563 CrossRef CAS.
- B. H. Song, C. F. Zhou, H. L. Wang, H. W. Liu, Z. W. Liu, M. O. Lai and L. Lu, J. Electrochem. Soc., 2014, 161, A1723–A1730 CrossRef CAS.
- P. K. Nayak, J. Grinblat, M. Levi, O. Haik, E. Levi, S. Kim, J. W. Choi and D. Aurbach, ChemElectroChem, 2015, 2, 1957–1965 CrossRef CAS.
- P. M. Gonzalez Puente, S. Song, S. Cao, L. Z. Rannalter, Z. Pan, X. Xiang, Q. Shen and F. Chen, J. Adv. Ceram., 2021, 10, 933–972 CrossRef CAS.
- X. Wu, X. Liang, X. Zhang, L. Lan, S. Li and Q. Gai, J. Adv. Ceram., 2021, 10, 347–354 CrossRef CAS.
- Y. Zang, X. Sun, Z. F. Tang, H. F. Xiang and C. H. Chen, RSC Adv., 2016, 6, 30194–30198 RSC.
- C. Lu, S. Yang, H. Wu, Y. Zhang, X. Yang and T. Liang, Electrochim. Acta, 2016, 209, 448–455 CrossRef CAS.
- L. Y. Bao, L. Wei, N. T. Fu, J. Y. Dong, L. Chen, Y. F. Su, N. Li, Y. Lu, Y. J. Li, S. Chen and F. Wu, J. Energy Chem., 2022, 66, 123–132 CrossRef CAS.
- Y. Y. He, S. Wang, H. Y. Zhang, X. Chen, J. Li, H. Y. Xu, Y. H. Zhang, K. H. Hu, G. P. Lv, Y. Meng and W. Xiang, J. Colloid Interface Sci., 2022, 607, 1333–1342 CrossRef CAS PubMed.
- Y. Xie, Y. C. Jin and L. Xiang, Particuology, 2022, 61, 1–10 CrossRef CAS.
- T. Nakamura, K. Ohta, X. Hou, Y. Kimura, K. Tsuruta, Y. Tamenori, R. Aso, H. Yoshida and K. Amezawa, J. Mater. Chem. A, 2021, 9, 3657–3667 RSC.
- Z.-Y. Kou, Y. Lu, C. Miao, J.-Q. Li, C.-J. Liu and W. Xiao, Rare Met., 2021, 40, 3175–3184 CrossRef CAS.
- E. Y. Zhao, M. H. Zhang, X. L. Wang, E. Y. Hu, J. Liu, X. Q. Yu, M. Olguin, T. A. Wynn, Y. S. Meng, K. Page, F. W. Wang, H. Li, X. Q. Yang, X. J. Huang and L. Q. Chen, Energy Storage Mater., 2020, 24, 384–393 CrossRef.
- P. P. Zhang, X. H. Zhai, H. Huang, J. F. Zhou, X. B. Li, Y. P. He and Z. C. Guo, Ceram. Int., 2020, 46, 24723–24736 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X. Yuan, A. A. Razzaq, Y. Chen, Y. Lian, X. Zhao, Y. Peng and Z. Deng, Chin. Chem. Lett., 2021, 32, 890–894 CrossRef CAS.
- R. A. House, G. J. Rees, M. A. Perez-Osorio, J. J. Marie, E. Boivin, A. W. Robertson, A. Nag, M. Garcia-Fernandez, K. J. Zhou and P. G. Bruce, Nat. Energy, 2020, 5, 777–785 CrossRef CAS.
- S. Ramakrishnan, B. Park, J. Wu, W. L. Yang and B. McCloskey, J. Am. Chem. Soc., 2020, 142, 8522–8531 CrossRef CAS PubMed.
- Z. Wang, H.-Q. Lu, Y.-P. Yin, X.-Y. Sun, X.-T. Bai, X.-L. Shen, W.-D. Zhuang and S.-G. Lu, Rare Met., 2017, 36, 899–904 CrossRef CAS.
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvère and J.-M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed.
- A. J. Naylor, I. Kallquist, D. Peralta, J. F. Martin, A. Boulineau, J. F. Colin, C. Baur, J. Chable, M. Fichtner, K. Edstrom, M. Hahlin and D. Brandell, ACS Appl. Energy Mater., 2020, 3, 5937–5948 CrossRef CAS PubMed.
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 2, 264–279 CrossRef CAS.
- Z. P. Shi, Q. W. Gu, L. Yun, Z. N. Wei, D. Hu, B. Qiu, G. Z. Chen and Z. P. Liu, J. Mater. Chem. A, 2021, 9, 24426–24437 RSC.
- Q. Ma, R. Li, R. Zheng, Y. Liu, H. Huo and C. Dai, J. Power Sources, 2016, 331, 112–121 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05126e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |