
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrosion behavior of 316 stainless steel arc parts in liquid lead at 650 °C under high oxygen concentrations
Ruizhong Wang ad,
Xi Qiubc,
Shixin Gaobc,
Weihua Liu*d,
Wenjie Li*bc,
Yuanming Libc and
Zhongfeng Tang*ad
ad,
Xi Qiubc,
Shixin Gaobc,
Weihua Liu*d,
Wenjie Li*bc,
Yuanming Libc and
Zhongfeng Tang*ad
aQinghai University, Xining 810016, China
bScience and Technology on Reactor System Design Technology Laboratory, Chengdu 610213, China. E-mail: lwj280@163.com
cNuclear Power Institute of China, Chengdu 610213, China
dShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: liuweihua@sinap.ac.cn; tangzhongfeng@sinap.ac.cn; Fax: +86-21-39194681; Tel: +86-21-39194681
First published on 15th November 2022
Abstract
The corrosion of 316SS in contact with lead complicates the realization of high coolant temperature. To explore the corrosion behavior at high temperature, the corrosion test of 316SS was performed in liquid lead at a high temperature of 650 °C under Ar with oxygen levels of 10−2 wt% and 10−5 wt% by the static corrosion method. The mass changes after corrosion were determined; then, the corrosion depth and the oxide product formed were further characterized. A multi-oxide layer was formed on the 316SS alloy surface, and the thickness reached 17.5 μm over a period of 100 h at the oxygen level of ∼10−2 wt%. Fe oxide was the main product in the outer layer; the dense Fe–Cr oxide was formed in the inner layer and lead was isolated from the metal substrate. When the oxygen content was 10−5 wt%, corrosion by dissolution at a rather high rate was dominant, and the corrosion depth was as high as 50 μm for 100 h. It is speculated that the oxide layer is also formed at the initial stage but gets dissociated when there is no oxygen supply to sustain the oxide layer with prolonged exposure time. The oxygen content in the cover gas greatly influences the corrosion behavior of 316SS, thus directly affecting the application of 316SS immersed in liquid lead at high temperature.
1. Introduction
Lead is a liquid coolant candidate for Generation IV reactors due to its favorable thermal, physical and chemical properties.1,2 It has the advantages of low vapor pressure, natural gamma radiation-shielding ability, efficient retention of fission products and allowing natural circulation in the event of stationary blackouts.3,4 However, lead becomes very corrosive toward containment and structural materials at higher operating temperatures, and this presents a critical challenge in the use of lead in advanced nuclear reactors.5–7 It is for this fundamental reason, the current operating temperature of lead-cooled fast reactors is limited to 550 °C.8 Increasing the maximum operating temperature beyond 600 °C can increase the thermal efficiency of power conversion. However, the corrosivity of liquid metals against structure materials increases sharply with temperature due to the increase in solubility of their constituents at higher temperatures.In the past, many studies on the corrosion behavior of steel materials in contact with liquid metal have been reported. The corrosion of materials in liquid metal is a physical or physio-chemical process, involving the dissolution of material constituents, the migration and penetration of heavy metals and reactions between corrosion products and impurities.9 In the static state, corrosion is mainly divided into oxidation and dissolution corrosion. Compared with oxidation corrosion, dissolution corrosion in liquid metals is deadly and is particularly important for steel alloying elements, such as Ni, Mn, and Cr, which have high solubility in liquid lead or LBE; for example, Ni has a solubility of up to 10 at% in liquid LBE at 700 °C.10 This kind of corrosion significantly changes the surface morphology, microstructure and composition of materials, leading to the deterioration of mechanical properties and seriously threatening the safety of reactor operation.11,12 Therefore, it is of great significance to control the dissolution corrosion of structural materials. A way to mitigate this issue is to establish controlled oxidative passivation through the formation of oxide scales on structural materials by maintaining a desired oxygen concentration in the liquid metal.13 The oxide layer would separate the base from the liquid metal because the diffusion rate of the alloy components in oxides is rather small.14 At different oxygen contents, structural materials undergo different corrosion processes.15 This was confirmed in the study by Oksana Klok et al.16 At oxygen concentrations below 10−7 at% in lead at 550 °C, the corrosion of austenitic steels was dominated by the dissolution of alloy components because no effective oxide layer was formed. At oxygen concentrations above 10−6 at%, oxide films formed on steel surfaces and prevented the direct dissolution of alloy components. In a certain range of oxygen concentration, an oxide film is formed on the surface of the structural material, slowing down the dissolution and erosion of the structural material by the liquid metal and thereby reducing the corrosion rate of the structural material.10
The desired oxygen concentration to form a stable oxide layer can vary and is influenced by many factors, such as temperature, the type of liquid metal and the structural materials. In different liquid metals, the diffusion rate, solubility and partial pressure of oxygen required for the oxidation of structural materials change with temperature.14 In liquid Pb45Bi55, a martensitic-type steel metal was protected from corrosion by the formation of a magnetite and spinel layer containing 10−6 wt% oxygen at 550 °C or less, while the formation of the protective layer failed at higher temperatures.17 At present, most researchers focus on the corrosion behavior on alloying with LBE at temperatures lower than 600 °C while a few studies have been published on the corrosion behavior of LBE at 700 and 800 °C.18 At an average oxygen concentration of 10−5 wt%, a continuous 35 μm thick oxide scale duplex layer was observed on 316 L alloy at 700 °C in LBE, while extensive cracking and localized delamination of the oxide layer were observed at 800 °C. Compared with LBE, lead shows lower activity and oxygen solubility, and thus its corrosion behavior under different oxygen concentrations should be different.
So far, only a few studies have reported the corrosion behavior and mechanism of pure lead under different oxygen contents at temperatures higher than 600 °C.19,20 Complex oxide layers had formed on 20Kh13 chromium steel alloy and Fe–10Cr–4Al alloy in liquid lead under different oxygen contents from 10−6 to 10−3 wt% at 600–700 °C. These layers effectively inhibited the permeation of lead. As a typical structural material, 316SS exhibits high technological maturity and is low in price. When it is used as the structural material in lead-cooled fast reactors, it can significantly improve the overall reactor economy. However, its compatibility with pure lead and corrosion mechanism under high temperatures (≥600 °C) and different oxygen concentrations are still unclear. The purpose of this work is to investigate the difference in the corrosion behavior of molten lead on 316SS arc parts at temperatures above 650 °C in a short period under different oxygen concentrations up to 10−2 wt%.
2. Materials and experimental
2.1 Materials
The chemical composition of the 316SS alloy used in this experiment is summarized in Table 1. The lead particles used in the experiment were obtained from the Sinopharm Group (>99.9 wt% purity), and the impurities present in lead are listed in Table 2.| Alloy | Fe | Cr | Ni | Mn | Si | P | N | Cu | Mo | C |
| 316SS | 69.04 | 16.84 | 10.12 | 1.284 | 0.472 | 0.021 | 0.0082 | 0.09 | 2.08 | 0.0375 |
| Bi | Sn | Sb | Ag | Ni | As | Cu | Fe | Pb |
| 5 | 2 | 3 | 2 | 10 | 1 | 5 | 10 | Bal |
2.2. Experimental methods
As shown in Fig. 1, the 316 SS alloy pipe with a diameter of 1.60 cm was cut into four pieces with an average width of 5 mm each. Then, the samples were laser marked to facilitate their identification after corrosion. The 316SS alloy samples were grinded with 240–3000 grits of SiC abrasive paper. The area and mass of the samples were then measured after drying in a vacuum oven, and the samples were stored in an Ar-filled glove box.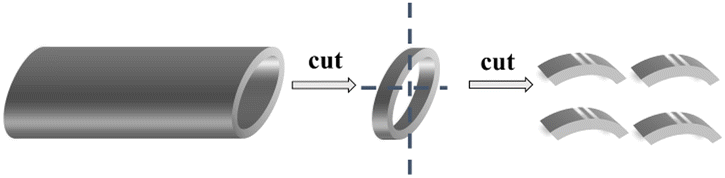 | ||
| Fig. 1 Schematic of the cutting of the 316 stainless steel pipe. | ||
The static immersion corrosion test was used in this work. The schematic of the experimental device is shown in Fig. 2-A. An alumina crucible (inner diameter 42 mm and height 125 mm) was used as the container. An alumina sheet and rod were used to press the 316SS alloy sample into liquid lead due to the higher density of lead than that of the 316SS alloy. To ensure the sample was completely immersed in lead, the crucible was sealed with high-temperature glue and capped. Then, the samples were transferred to the furnace, which was connected to a glove box filled with argon gas. The oxygen concentration in the glove box was ∼10−2 wt% (as determined by a zirconia detector).
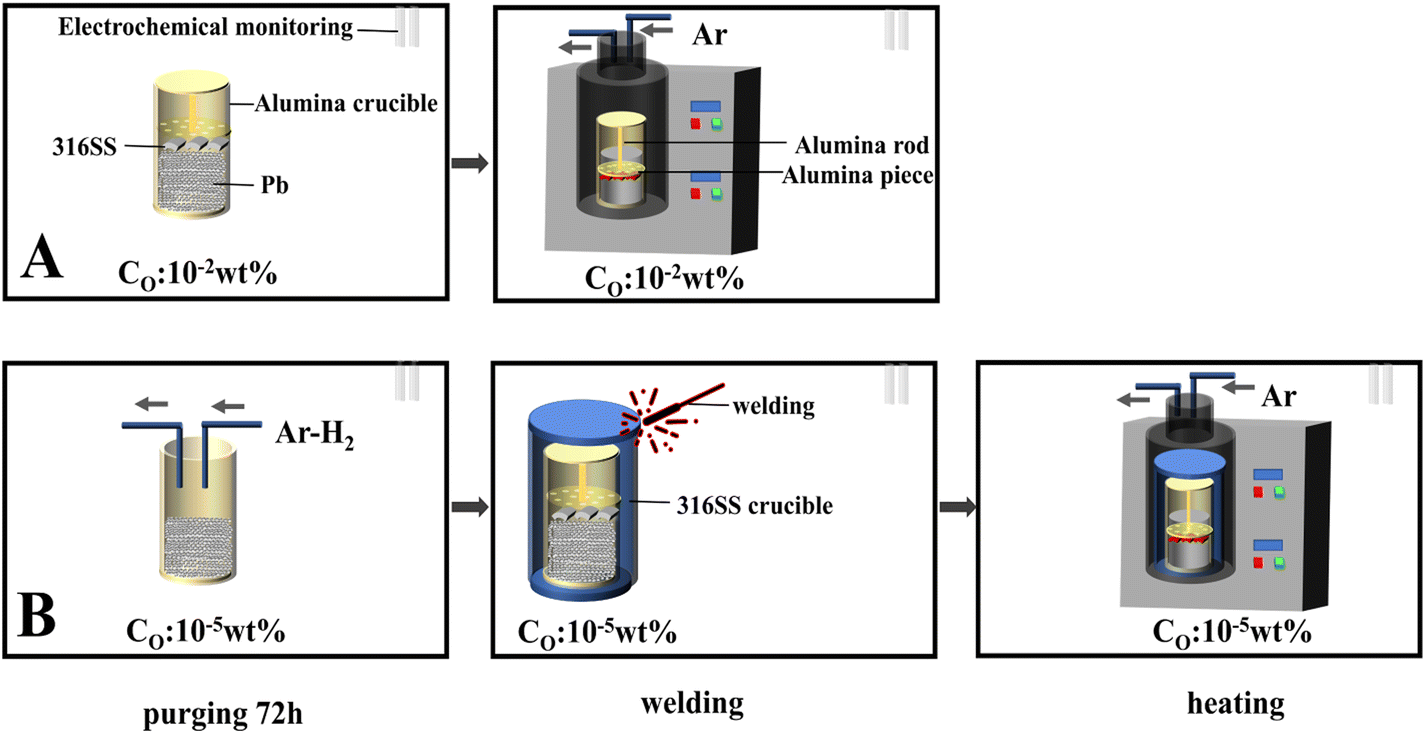 | ||
| Fig. 2 The flow diagram of the corrosion test. | ||
As shown in Fig. 2-B, the entire oxygen-controlled test was also carried out in a glove box with an oxygen content of 10−5 wt%. Firstly, lead was purified by Ar–H2 at 650 °C for 72 h. Then, it was transferred to an alumina crucible, which was further encapsulated in a 316SS crucible by oxygen-free welding. The heating system was connected to a glove box containing the same oxygen content.
The samples were kept at 200 °C for 3 h, then heated to 650 °C at a heating rate of 5 °C min−1 and maintained for 100 h. After corrosion, the samples were cleaned using a mixture of acetic acid: ethanol: hydrogen peroxide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) to completely remove lead from the surface of 316SS.21 The samples were then further washed with deionized water and dried.
:1:1) to completely remove lead from the surface of 316SS.21 The samples were then further washed with deionized water and dried.
2.3. Characterization
The mass of the samples before and after corrosion was measured using an electronic analytical balance (Mettler Toledo ME204E). Scanning electron microscopy (SEM; Zeiss Merlin FE) combined with energy dispersive spectroscopy (EDS; Oxford Instruments X-Max, 80 mm2) was used to characterize the morphology and elemental distribution of the samples before and after corrosion. Electron probe microanalysis (EPMA; Tecnai G2 F20 S-TWIN) was used for the microscopic morphological and compositional analyses of the cross-sections of corroded samples. The microstructures of the pre- and post-corrosion samples were analyzed using X-ray diffraction (XRD; Bruker D8 Advance). The ion concentrations in lead after the corrosion experiment were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES).3. Results
3.1. Mass changes
The mass changes of the 316SS alloy after 100 h of corrosion in liquid lead at 650 °C are shown in Fig. 3. It can be seen that the mass changes were quite different under the two different oxygen concentrations. After 100 h of high-temperature corrosion, the weight gain of the alloy was 0.78 mg cm−2 at the oxygen level of 10−2 wt%. In contrast, a significant mass loss of 17.54 mg cm−2 occurred at the limited oxygen concentration of 10−5 wt%. This indicates that the corrosion behavior of liquid lead toward the 316SS alloy at the two different oxygen concentrations was completely different. In the static state, the corrosion of an alloy in a liquid metal mainly involves oxidation and dissolution.22–24 Obviously, oxidation results in the formation of oxide scales on the material surface, inducing mass gain in the samples, while dissolution induces mass loss. The mass increase after corrosion indicated that an oxide layer may have formed under the condition of ∼10−2 wt% oxygen (CO) and the corrosion was dominated by oxidation. The quality of the sample decreased significantly when CO ∼ 10−5 wt%, which indicated that the corrosion process was dominated by the dissolution of alloy elements. Moreover, the great weight loss in the structural material also confirms the fatality of dissolution corrosion.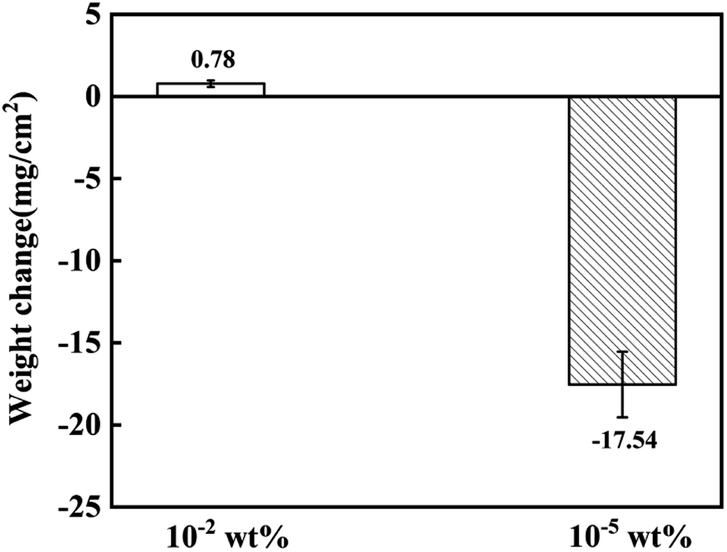 | ||
| Fig. 3 The weight change of 316SS immersed in liquid lead at 650 °C for 100 h under different oxygen concentrations. | ||
3.2 Surface morphology and element analysis
Fig. 4 shows the surface morphology of the alloy after exposure to liquid lead for 100 h at 650 °C under different oxygen levels. In sample A, dense oxide products were produced on the surface of the alloy matrix. The matrix turned non-uniform, but some areas were not covered by the oxide layer. It showed different morphologies, including corrosion zone P1, matrix zone P2 and oxidation product zone P3. Area P1 was rough with small pits, indicating slight corrosion of the matrix. The matrix of the 316SS alloy could be clearly observed in the P2 area, and the surface still maintained a certain level of flatness. Slight corrosion was seen at the scratches caused by the surface treatment of the alloy. Fig. 4 P3 shows an enlarged view of the corrosion product area. Relatively dense and compactly arranged piles of corrosion products were observed on the surface of the alloy in the form of tiles. The corrosion products covered the majority of the alloy surface. This is the reason for the mass increase of the alloy in liquid Pb saturated with oxygen at 650 °C after 100 h exposure. Based on the elemental analysis of the corrosion products, as shown in Fig. 5, the corrosion products formed were mainly Fe oxides.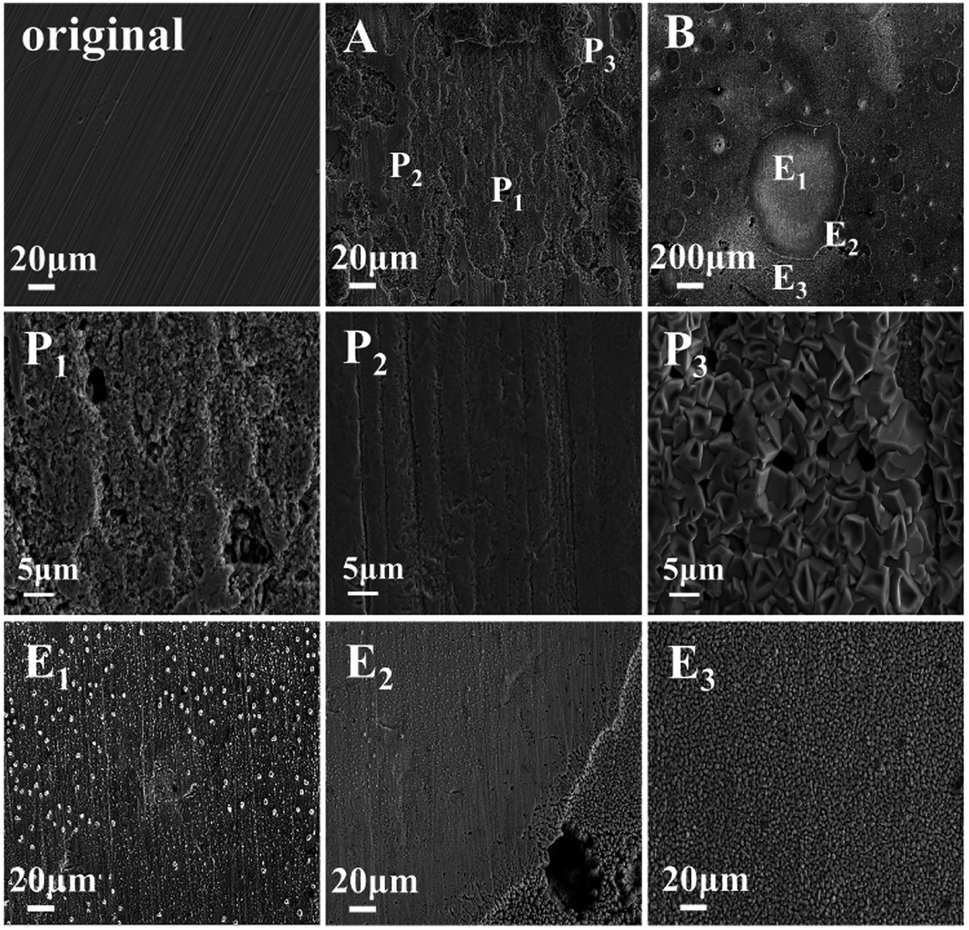 | ||
| Fig. 4 The SEM image of the 316SS alloy surface after immersion in liquid lead for 100 hours at 650 °C under different oxygen contents ((A): CO ∼ 10−2 wt%; (B): CO ∼ 10−5 wt%). | ||
 | ||
| Fig. 5 The surface elemental distribution of the 316SS alloy after corrosion in liquid lead solution at 650 °C for 100 h under different oxygen concentrations ((A): CO ∼ 10−2 wt%; (B): CO ∼ 10−5 wt%). | ||
Fig. 4 E1–E3 correspond to the enlarged images of regions 1, 2 and 3 in B, respectively. On the whole, two types of morphology were observed on the surface of the alloy after etching for 100 h. One showed mainly the matrix with small pitting corrosion, and the other was the porous area produced by etching. According to the elemental analysis shown in Fig. 5, the two types of morphologies exhibit different element distributions. It can be seen that the Fe oxides were not obvious on the surface of the matrix, while Cr and Ni depletion was observed on the surface of the sample. This may be the reason for the significant loss of quality after corrosion in liquid lead with low oxygen content.
3.3 Cross-sectional morphology and elemental analysis
Fig. 6 shows the cross-sectional morphology and the elemental distribution of the 316SS alloy after exposure to liquid lead for 100 h at 650 °C. At 10−2 wt% oxygen, an obvious corrosion layer with a maximum thickness of 17.53 μm was observed. It can be seen from the elemental map that the layer mainly consisted of multiple oxide layers. In the outer layer, Fe oxide was the main product; the oxide of Fe–Cr spinel was mainly formed in the inner layer for 316SS. Fe element was depleted in the inner layer due to the outward diffusion of Fe, while the Cr element diffused into this layer from the matrix and was enriched. However, Ni enrichment, as well as Cr depletion, was observed at the steel-inner oxide interface. The Cr and Ni elements, which have high solubility in molten liquid lead, continue to dissolve in lead, resulting in the lack of Cr element. During the whole dissolution process, oxygen element also continuously penetrates the base metal. The affinity of Fe and Cr elements to O is significantly stronger than that of Ni; thus, when the matrix elements diffuse into the liquid metal-substrate interface, Fe and Cr are oxidized first, while Ni is left behind.25 No corrosion attack was observed in 316SS underneath the oxide layers. Under limited oxygen content (10−5 wt%), the cross-section of 316SS was completely different, and the maximum corrosion depth was 50.04 μm. It can be seen from the elemental distribution map that no obvious oxide layer was formed. Only small amounts of Fe and O were accumulated at the lead/alloy interface, while there was a significant depletion of Cr and Ni in the corresponding corrosion zone. | ||
| Fig. 6 The SEM images of the cross-sectional morphology of the 316SS alloy immersed in a liquid lead solution for 100 h at 650 °C under different oxygen concentrations ((A): CO ∼ 10−2 wt%; (B): CO ∼ 10−5 wt%). | ||
To elucidate the corrosion behavior mechanism under limited oxygen content, the element contribution map across the cross-section was further characterized by EPMA, as shown in Fig. 7. It was obvious that corrosion had occurred in the alloy matrix, as the layer seemed quite different from the morphology of the matrix. The content of Cr and Ni obviously reduced in the corrosion layer, especially Ni, which was almost exhausted in the whole erosion layer. This may be due to the greater solubility of Ni in lead.10 Lead had diffused into the inner part of the samples, leading to a Cr and Ni depletion zone. In the elemental distribution analysis, it was found that the oxygen map was almost the same as that of lead. Oxygen dissolved in lead diffused to the inner region of the samples. However, the Fe element showed a state of enrichment in the erosion layer, which may be due to its low solubility in lead. When 316SS is exposed to lead, the Fe element also dissolves non-selectively, but this is followed by the re-deposition of Fe.26 Therefore, the formation of the protective oxide layer fails due to the decrease in dissolved oxygen content in liquid lead under limited oxygen conditions. Thus, the corrosion process is dominated by the serious dissolution behavior of the alloy. Further, based on the quantitative analysis of elements, it could be concluded that the dissolution of Ni is the main cause for the voids in the alloy section and the obvious mass loss of 316SS.
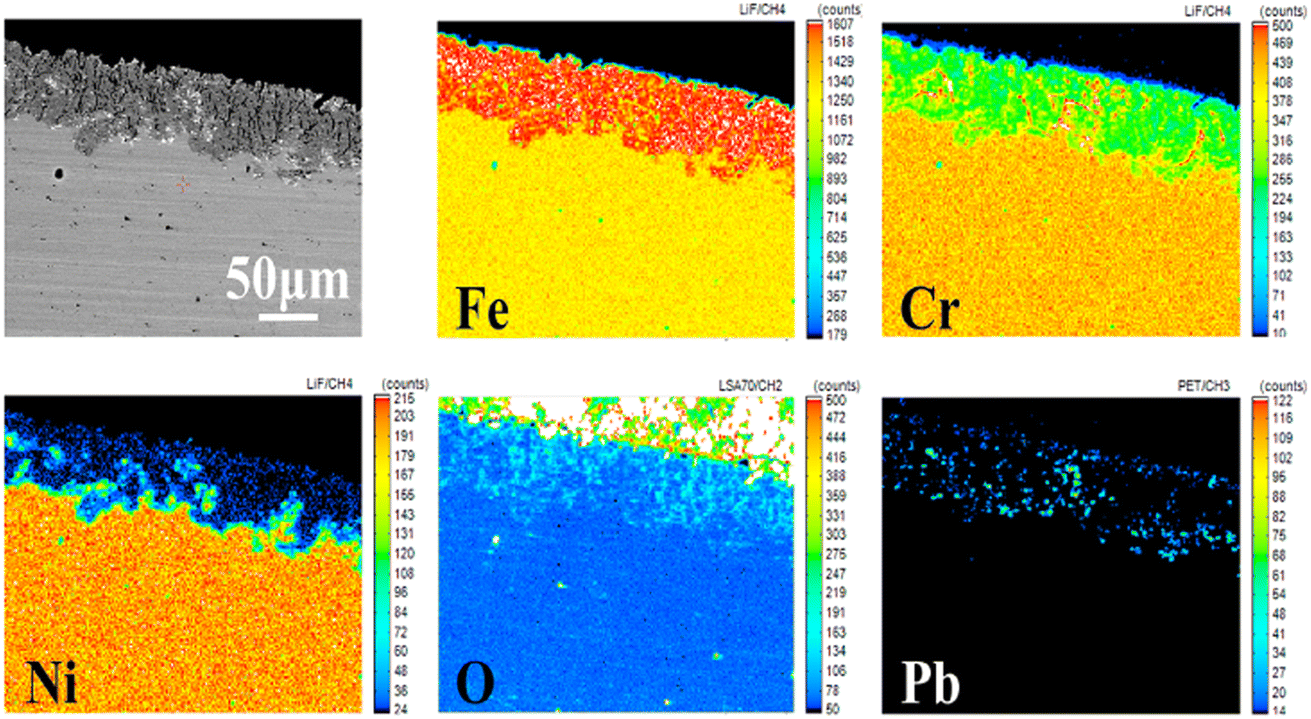 | ||
| Fig. 7 The EPMA images and elemental contribution diagrams of the samples immersed in liquid lead for 100 h at 650 °C under the oxygen content of 10−5 wt%. | ||
3.4 Changes in the crystal phase structure
Fig. 8 displays the XRD patterns of the original 316SS alloy and that after corrosion in liquid lead containing different oxygen contents at 650 °C for 100 h. In sample A, oxygen dissolved in liquid Pb reaches saturation, and the surface of 316SS is covered with the Fe3O4 magnetite phase, while the related Fe–Cr oxide was not detected. This may be attributed to the thick oxide layer of Fe3O4, which was densely distributed over the entire 316SS alloy matrix surface. In B, no new oxidation product was detected on the surface of 316SS, which is consistent with the above SEM and EPMA results. | ||
| Fig. 8 The XRD patterns of the 316SS alloy before and after corrosion in liquid lead containing different oxygen contents at 650 °C for 100 h ((A): CO ∼ 10−2 wt%; (B): CO ∼ 10−5 wt%). | ||
3.5 Changes in the impurities
The content of Fe, Cr, and Ni in lead after corrosion was determined to further understand the corrosion behavior of 316SS under different oxygen concentrations. As seen in Table 3, the concentrations of Fe, Cr and Ni did not change in A after corrosion. This indicates that almost none of these elements was dissolved in liquid lead at 650 °C at this oxygen concentration. This should be due to the protective effect of the oxide layer formed. Meanwhile, the concentration of Fe and Ni in lead after corrosion increased obviously in sample B. The content of Ni in lead reached 106.3 ppm. However, the concentration of Fe after corrosion was unusual due to its low solubility in liquid lead.10 This may be due to the solubility of the Fe oxide product in lead rather than the dissolution of the pure alloying element Fe, which has been confirmed in previous work.26 This indicates that Fe oxide must also have been formed at the beginning under the low oxygen concentration of ∼10−5 wt%.| Sample | Fe/ppm | Cr/ppm | Ni/ppm |
|---|---|---|---|
| A | 12.7 | — | — |
| B | 120.7 | 1.4 | 106.3 |
4. Discussion
The corrosion of structural materials immersed in liquid heavy metals can usually be divided into four types, namely oxidation corrosion, dissolution corrosion, rinse corrosion and wear corrosion.9 These corrosion phenomena may act alone or in combination on structural materials. Oxidation and dissolution corrosion are considered here due to the static corrosion test method used in this work. In this experiment, the corrosion behavior of 316SS in liquid metal was completely different under different oxygen concentrations, and the fundamental reason is the competition between oxidation corrosion and dissolution corrosion. The process is dominated by oxidation corrosion under high oxygen content.27–29The surface of 316SS alloy was rapidly oxidized, and multi-oxide layers containing an outer layer of Fe3O4 and an inner layer of Fe–Cr oxide were formed under the oxygen content of 10−2 wt%. This is also the fundamental reason for the quality increase. The oxide layer of Fe–Cr spinel was found to be dense, and the internal matrix was protected against Pb corrosion due to the much smaller diffusion rate of oxygen in the oxide layer. This is in agreement with previous research, which demonstrated that Fe–Cr spinel is protective against LBE penetration and avoids the dissolution of alloy elements.30,31 The oxygen content of 10−2 wt% has been estimated to be the upper oxygen limit in liquid lead at 650 °C.10 With sufficient oxygen supply, continuous oxidation occurred, the oxidation products gradually accumulated, and the thickness of the oxidation layer reached 17.53 μm in a short period of 100 h. The oxide layer could separate the matrix from the liquid metal, and dissolution corrosion was inhibited. Therefore, neither Ni nor Cr was detected in the liquid lead although they show high solubility in Pb, especially Ni. In previous research by Seung Gi Lee et al.,30 a biphase oxide layer with a thickness of 20 μm was produced on 316SS corroded for 1000 h in a lead–bismuth solution at 600 °C with an oxygen content of 10−5 wt%, protecting the matrix from lead penetration. Compared with the results of Seung Gi Lee et al., the thickness of the oxide layer found in our work is similar but was formed in a shorter time because the relatively high oxygen concentration allows the oxide layer to form quickly.
Oxidation corrosion may involve the following processes: when the alloy material comes in contact with the oxygen-containing liquid metal, an oxide layer of ferrochrome spinel is formed at the interface between lead and the base metal.32,33 With the increase in corrosion time, Fe in the alloy diffuses from the metal base to the oxide layer, supporting the growth of external magnetite and producing nano-level vacancies. At this time, the oxide layer presents a double-layer structure,34 and lead also penetrates the magnetite layer. The oxygen dissolved in lead diffuses to the ferrochrome spinel oxide layer through the nanoscale vacancies to further support the growth of the protective layer.35 Due to the continuous outward diffusion of Fe, the outer magnetite layer grows continuously along the lead interface. Therefore, when the corrosion time is long enough, with the continuous growth of external magnetite, tensile stress accumulates between the magnetite layer and the ferrochrome spinel layer. Moreover, when the stress is large enough, the outer magnetite layer would fall off, causing the chromium oxide layer to be re-exposed to liquid lead.36 Thus, the protective effect of the layer fails, and the oxidation corrosion process would restart.17,37
With limited oxygen when the concentration drops to 10−5 wt%, dissolution corrosion dominated. A significant weight loss, 22 times higher than the weight gain under the oxygen concentration of 10−2 wt%, occurred in this case. The SEM and XRD results certified that no oxidation layer was formed. The dissolution corrosion depth had attained 50 μm with a corrosion rate of 0.5 μm h−1 for 316SS alloy in liquid lead. After corrosion, a higher Ni content in lead was determined by the ICP-OES technique. Moreover, a high Fe concentration was also determined, and this should be due to the dissolution of Fe oxides since Fe has limited solubility in liquid lead. It can be reasonably inferred that the iron oxide formed at the initial stage must have dissociated with time when the oxygen at the sample surface is lower than that for forming iron oxide, and no more iron oxide is formed.10 Then, the rapid dissolution of alloy elements occurs, leading to a significant weight loss.38 Among the dissolved alloy elements, the solubility of Ni element in lead is the highest,39 and its dissolution occurs quickly, which is an important reason for the infiltration of lead into the basement. Yuji Kurata et al. obtained similar results under 2.8 × 10−5 wt% oxygen concentration, and the dissolution reaction of Ni in LBE was the key to controlling the penetration rate of Pb–Bi.7 Seung Gi Lee et al.30 reported that a 20 μm-thick dual-phase oxide layer was formed on the alloy surface in the lead–bismuth solution under the oxygen content of 10−5 wt% at 600 °C. In our work, the completely closed system was used. Therefore, in the process of oxide layer formation and falling off, oxygen is consumed and cannot be replenished in time, causing serious dissolution of the alloy elements in 316SS. Due to low oxygen content and the limited solubility of Fe, the re-deposition of dissolved Fe led to its enrichment in the corrosion area, as confirmed by EMPA. At the same time, Ni and Cr diffused to the lead interface and were partially dissolved. Therefore, from the point of view of material protection, dissolution corrosion will cause greater damage to the material. The corrosion process under different oxygen concentrations is illustrated in Fig. 9. Thus, the oxygen concentration in lead must be actively controlled to avoid heavy dissolution and oxidation, as well as to sustain the protective oxide layer.
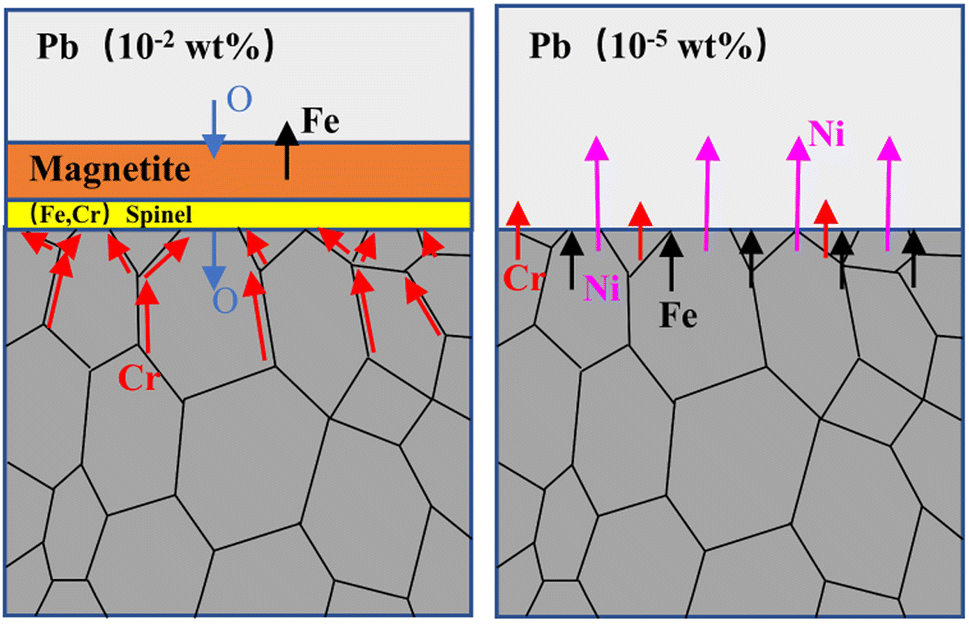 | ||
| Fig. 9 The corrosion mechanism on 316 stainless steel immersed in liquid lead under different oxygen concentrations. | ||
5. Conclusion
Liquid lead with oxygen concentrations of ∼10−2 wt% and ∼10−5 wt% shows significantly different corrosion behaviors on 316SS at 650 °C. Fast oxidation occurs under the high oxygen condition, and in situ oxide scales with a thickness of 17.51 μm are formed on the surface. The oxide scale consisted of Fe3O4 in the outer layer and the oxide of Fe–Cr in the inner layer, which further isolated the metal substrate from lead corrosion. No corrosion attack was observed on 316SS underneath the oxide layers. Due to its low affinity to oxygen, Ni was enriched at the steel-inner oxide interface. At the low oxygen condition, the corrosion was dominated by the fast dissolution of Ni without the protection of the oxide layer. The maximum corrosion depth attained was 50 μm in a short period of 100 h. The corrosion process was aggravated at the high temperature of 650 °C and low oxygen content. The active control of oxygen concentration and the evolution of the oxide scales are to be investigated in the future.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Supported by National Natural Science Foundation of China (52076006).References
- L. Luo, Z. Jiang, Z. Xiao and Q. Huang, Corros. Sci., 2021, 183, 109324 CrossRef.
- H. Tafrishi, S. Sadeghzadeh and R. Ahmadi, RSC Adv., 2022, 12, 14776–14807 RSC.
- J. Wallenius, S. Qvist, I. Mickus, S. Bortot, P. Szakalos and J. Ejenstam, Nucl. Eng. Des., 2018, 338, 23–33 CrossRef CAS.
- J. Wallenius, E. Suvdantsetseg and A. Fokau, Nucl. Technol., 2017, 177, 303–313 CrossRef.
- J. Zhang, Corros. Sci., 2009, 51, 1207–1227 CrossRef CAS.
- J. Ejenstam and P. Szakálos, J. Nucl. Mater., 2015, 461, 164–170 CrossRef CAS.
- Y. Kurata, J. Nucl. Mater., 2014, 448, 239–249 CrossRef CAS.
- P. Dömstedt, M. Lundberg and P. Szakálos, J. Nucl. Mater., 2020, 531, 152022 CrossRef.
- X. Gong, M. P. Short, T. Auger, E. Charalampopoulou and K. Lambrinou, Prog. Mater. Sci., 2022, 126, 100920 CrossRef CAS.
- J. Zhang and N. Li, J. Nucl. Mater., 2008, 373, 351–377 CrossRef CAS.
- F. J. Martín, L. Soler, F. Hernández and D. Gómez-Briceño, J. Nucl. Mater., 2004, 335, 194–198 CrossRef.
- P. Hosemann, D. Frazer, E. Stergar and K. Lambrinou, Scr. Mater., 2016, 118, 37–40 CrossRef CAS.
- N. Arrousse, Y. Fernine, N. Al-Zaqri, A. Boshaala, E. Ech-Chihbi, R. Salim, F. El Hajjaji, A. Alami, M. E. Touhami and M. Taleb, RSC Adv., 2022, 12, 10321–10335 RSC.
- N. Li, J. Nucl. Mater., 2002, 300, 73–81 CrossRef CAS.
- S. Tian, Y. Pang, Z. Jiang, L. Luo and Z. Xiao, Mater. Corros., 2021, 73, 134–142 CrossRef.
- O. Klok, K. Lambrinou, S. Gavrilov, J. Lim and I. De Graeve, J. Nucl. Eng. Radiat. Sci., 2018, 4, 2332–2898 Search PubMed.
- T. Furukawa, G. Müller, G. Schumacher, A. Weisenburger, A. Heinzel and K. Aoto, J. Nucl. Mater., 2004, 335, 189–193 CrossRef CAS.
- C. Cionea, M. D. Abad, Y. Aussat, D. Frazer, A. J. Gubser and P. Hosemann, Sol. Energy Mater. Sol. Cells, 2016, 144, 235–246 CrossRef CAS.
- O. I. Eliseeva and V. P. Tsisar, Mater. Sci., 2005, 41, 603–608 CrossRef CAS.
- V. P. Tsisar, O. I. Eliseeva, V. M. Fedirko and V. A. Lopushans'Kyi, Mater. Sci., 2003, 39, 539–544 CrossRef.
- O. Yeliseyeva, V. Tsisar and Z. Zhou, J. Nucl. Mater., 2013, 442, 434–443 CrossRef.
- C. Schroer, V. Koch, O. Wedemeyer, A. Skrypnik and J. Konys, J. Nucl. Mater., 2016, 469, 162–176 CrossRef.
- C. Schroer, O. Wedemeyer, J. Novotny, A. Skrypnik and J. Konys, Nucl. Eng. Des., 2014, 280, 661–672 CrossRef.
- F. J. Martín-Muñoz, L. Soler-Crespo and D. Gómez-Briceño, J. Nucl. Mater., 2011, 416, 87–93 CrossRef.
- X. Chen, J. F. Stubbins, P. Hosemann and A. M. Bolind, J. Nucl. Mater., 2010, 398, 172–179 CrossRef CAS.
- C. Schroer, O. Wedemeyer, J. Novotny, A. Skrypnik and J. Konys, Corros. Sci., 2014, 84, 113–124 CrossRef CAS.
- E. Charalampopoulou, K. Lambrinou, T. Van der Donck, B. Paladino, F. Di Fonzo, C. Azina, P. Eklund, S. Mráz, J. M. Schneider, D. Schryvers and R. Delville, Mater. Charact., 2021, 178, 111234 CrossRef CAS.
- V. Tsisar, C. Schroer, O. Wedemeyer, A. Skrypnik and J. Konys, J. Nucl. Mater., 2014, 454, 332–342 CrossRef CAS.
- A. Weisenburger, G. Müller, A. Heinzel, A. Jianu, H. Muscher and M. Kieser, Nucl. Eng. Des., 2011, 241, 1329–1334 CrossRef.
- S. G. Lee, Y.-H. Shin, J. Park and I. S. Hwang, Appl. Sci., 2021, 11, 2349–2369 CrossRef.
- Y. S. Zhang, B. S. Chu, H. L. Yu, K. Li, W. H. Wang and W. Yang, RSC Adv., 2022, 12, 9501–9511 RSC.
- L. Martinelli, F. Balbaud-Célérier, G. Picard and G. Santarini, Corros. Sci., 2008, 50, 2549–2559 CrossRef.
- C. Schroer, Z. Voß, O. Wedemeyer, J. Novotny and J. Konys, J. Nucl. Mater., 2006, 356, 189–197 CrossRef CAS.
- J. Zhang, N. Li, Y. Chen and A. E. Rusanov, J. Nucl. Mater., 2005, 336, 1–10 CrossRef CAS.
- L. Martinelli and F. Balbaud-Célérier, Mater. Corros., 2011, 62, 531–542 CrossRef CAS.
- L. Luo, J. Liu, S. Tian, Z. Jiang, Z. Xiao, M. Zhang, W. Huang, L. Sun and W. Luo, J. Nucl. Mater., 2022, 562, 153579 CrossRef CAS.
- A. Heinzel, A. Weisenburger and G. Müller, J. Nucl. Mater., 2014, 448, 163–171 CrossRef CAS.
- J. Zhang and N. Li, Corros. Sci., 2007, 49, 4154–4184 CrossRef CAS.
- E. Yamaki, K. Ginestar and L. Martinelli, Corros. Sci., 2011, 53, 3075–3085 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |