
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative bridgehead functionalization of (4 + 3) cycloadducts obtained from oxidopyridinium ions†
Wanna Sungnoi and
Michael Harmata *
*
Department of Chemistry, University of Missouri-Columbia, Columbia, Missouri 65211, USA. E-mail: harmatam@missouri.edu
First published on 6th October 2022
Abstract
Treatment of selected (4 + 3) cycloadducts derived from oxidopyridinium ions with N-iodosuccinimide (NIS) in hexafluoroisopropanol (HFIP) resulted in the formation of bridgehead ethers via a net oxidative C–H activation.
Since their beginnings in the mid-1970s, the (4 + 3) cycloaddition reactions of N-substituted oxidopyridinium ions have provided an attractive and facile method for the construction of nitrogenous, heterocyclic seven-membered rings (Scheme 1).1,2 While N-aryl and N-alkenyl substitution of the pyridinium nitrogen have largely dominated the literature, to the best of our knowledge, there had been only one example of a (4 + 3) cycloaddition reaction of N-alkyl oxidopyridinium ions reported prior to 2017.2a Our recent reports of the reaction of N-methyl oxidopyridinium ions with conjugated dienes expanded the scope of the (4 + 3) process, due to the incorporation of an ester functional group at the 5-position of the hydroxypyridine starting material.3 Cycloadducts are formed in good to excellent yields, and the reactions of select dienes proceed in high regioselectivity. Furthermore, the cycloaddition process can be steered towards a high preference for the endo diastereomer when the starting diene bears a bulky trialkylsilyl groups at C2, as we have recently reported (Scheme 2).4
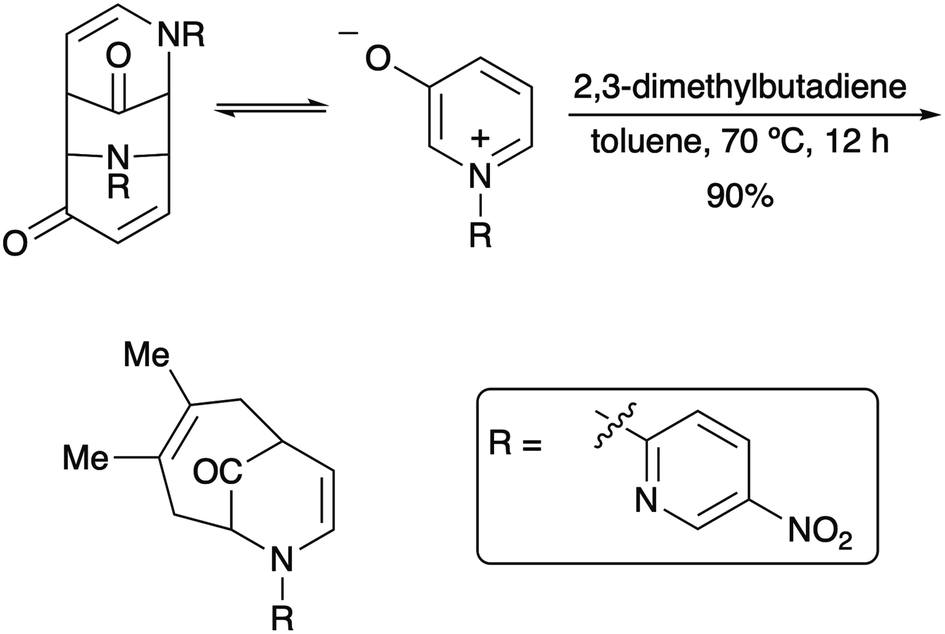 | ||
| Scheme 1 Example of a (4 + 3) cycloaddition of an oxidopyridinium ion. | ||
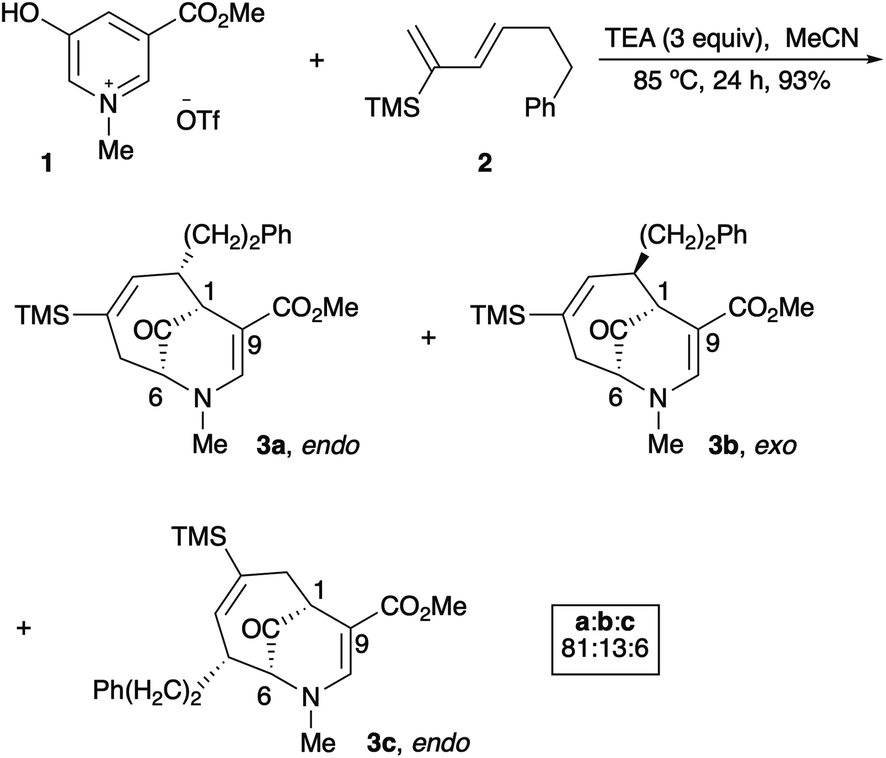 | ||
| Scheme 2 Endo selective (4 + 3) cycloaddition of an oxidopyridinium ion. | ||
Upon our initial exploration into the chemistry of the latter (4 + 3) cycloadduct products, our intentions were the replacement of the trialkylsilyl group with a halogen atom such as iodine or bromine. We were particularly inspired by a report from the Zakarian group, who showed that vinylsilanes react with N-iodosuccinimide in hexafluoroisopropanol to afford iodoalkenes stereospecifically and in good yield (Scheme 3).5 However, when our (4 + 3) cycloadducts were exposed to such reaction conditions, the replacement of the C–Si bond by a C–I bond was not observed. Thus, when a mixture of 3a–c was treated with NIS in hexafluoroisopropanol (HFIP),6 a vinyl iodide was not formed. Instead, functionalization of the bridgehead carbon as a hexafluoroisopropyl ether was observed, a formal oxidative C–H activation process, affording 4a (Scheme 4).4 This communication reports further examples of this process and other results that provide some insights for future studies.
 | ||
| Scheme 3 Zakarian's vinyl iodide synthesis. | ||
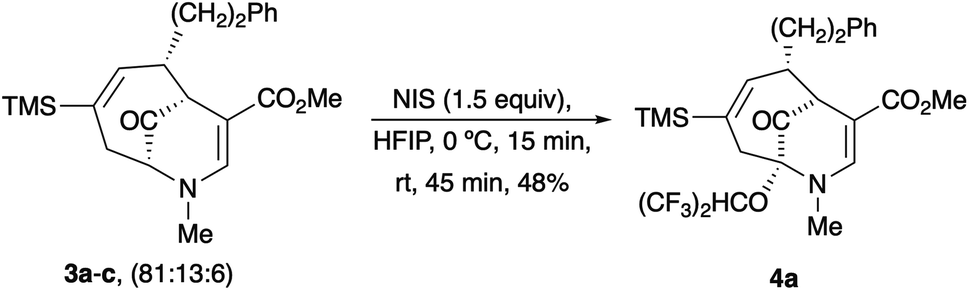 | ||
| Scheme 4 First example of the bridgehead oxidation of a (4 + 3) cycloadduct. | ||
The substrates for the process are known compounds, produced in our laboratories using methodology we developed for the (4 + 3) cycloaddition of oxidopyridinium ions with dienes.4 From the very first example of the process, we noted an interesting phenomenon. For example, entry 1 of Table 1 (see also Scheme 4) makes use of an inseparable mixture of diastereomers as the starting material. All three of the diastereomers are detectable by 1H NMR in the mixture. Nevertheless, the product of the oxidation is a single diastereomer. The structure of 4a was supported by 1H NMR data. The fate of the minor diastereomers of the starting material is unknown at this time, as nothing else could be isolated from the reaction mixture. This is also true for similar entries in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Substratea | Timea (h) | Product | Yieldb (%) |
| a The major diastereomer/regioisomer of the mixture is shown.b Yields are based on the entire mass of the starting material, including isomers that may not have given rise to the product observed.c The substitution pattern of the major isomer is given in parentheses. | ||||
| 1 | 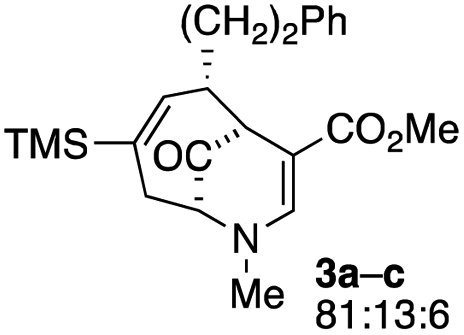 |
1 | 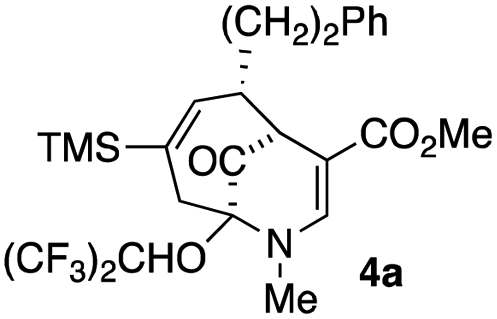 |
52 |
| 2 | 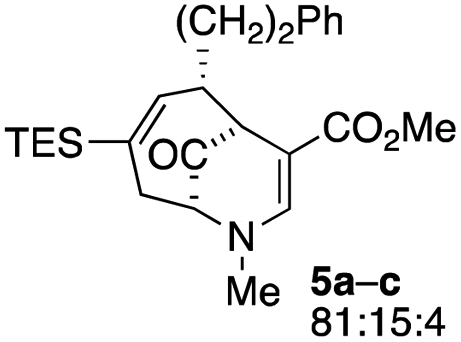 |
23 | 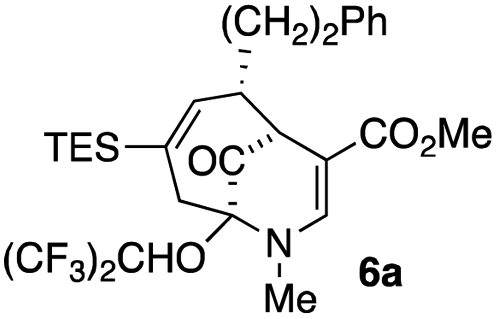 |
54 |
| 3 | 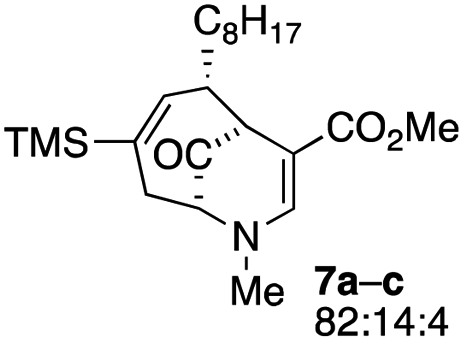 |
1.5 | 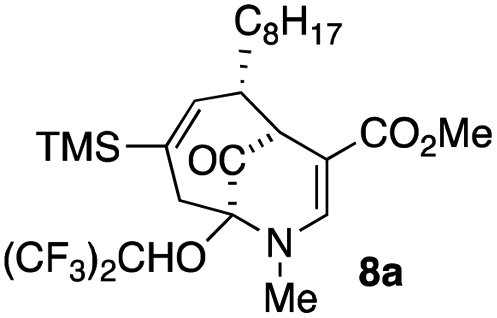 |
52 |
| 4 | 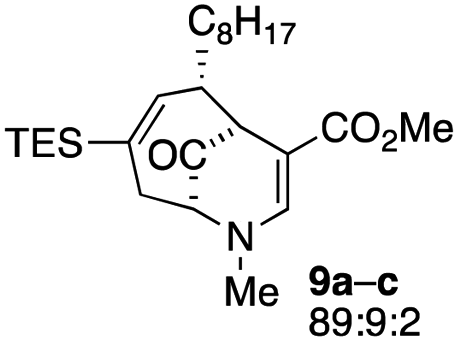 |
22 | 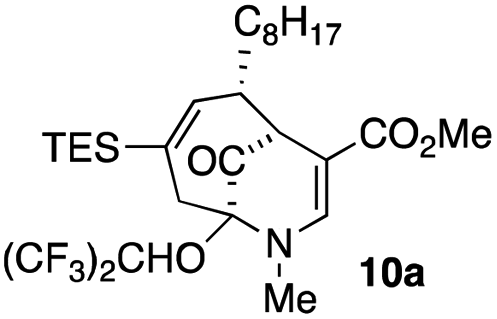 |
51 |
| 5 | 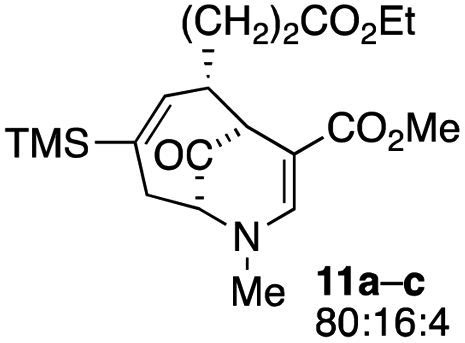 |
21 | 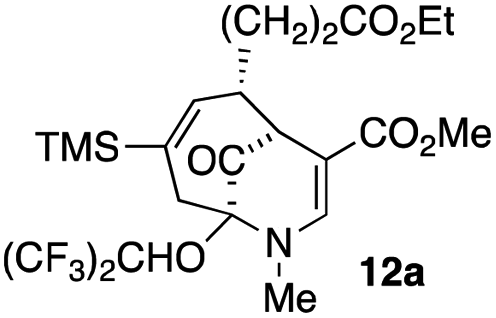 |
44 |
| 6 | 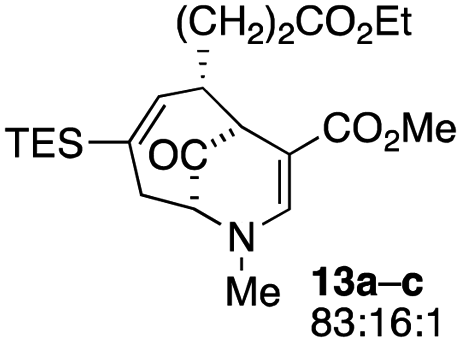 |
23 | 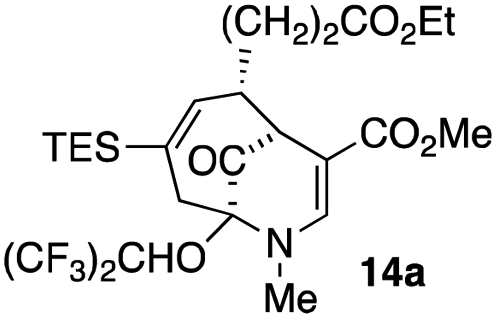 |
38 |
| 7 | 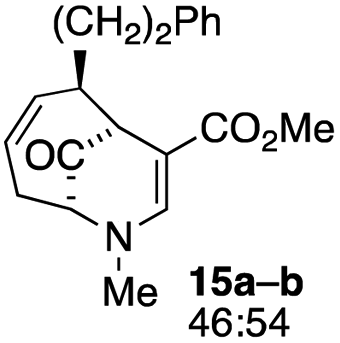 |
23 | 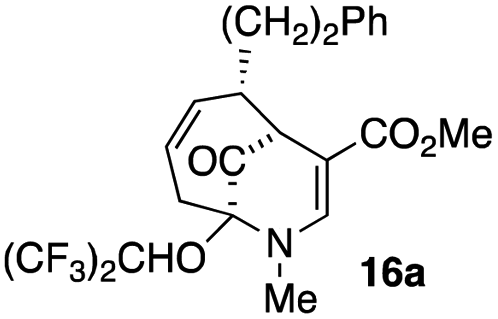 |
16 |
| 8 |  |
5.5 | 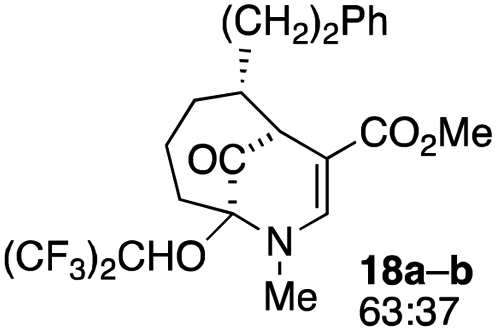 |
33 |
| 9c | 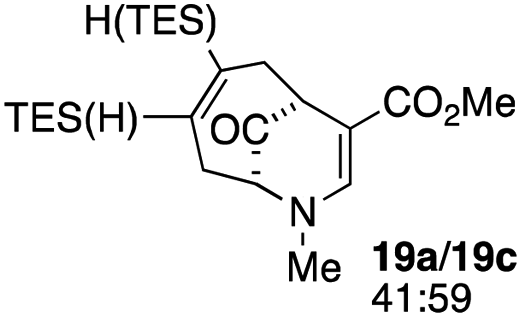 |
2 | 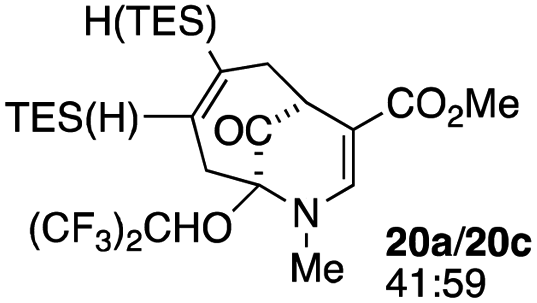 |
35 |
| 10c | 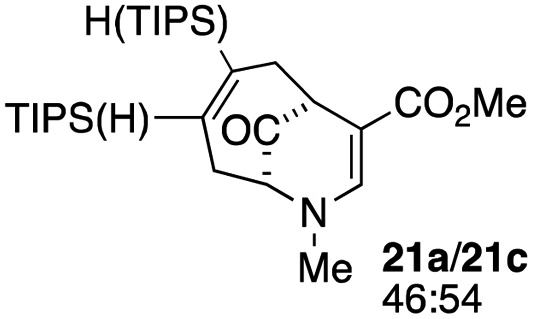 |
3 | 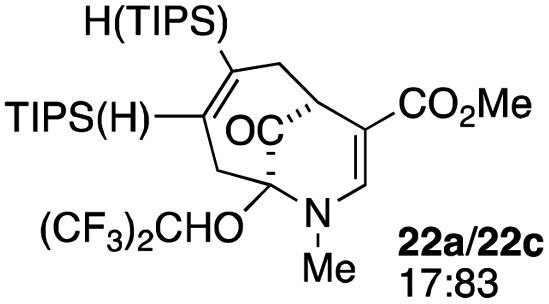 |
59 |
| 11 | 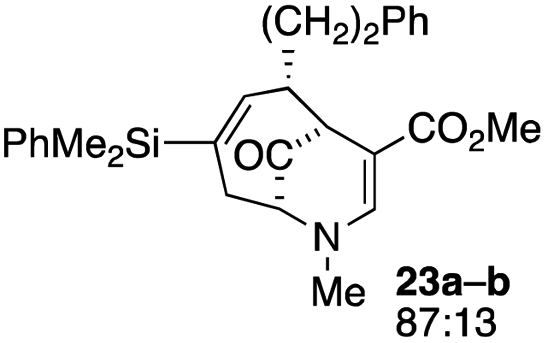 |
4 | 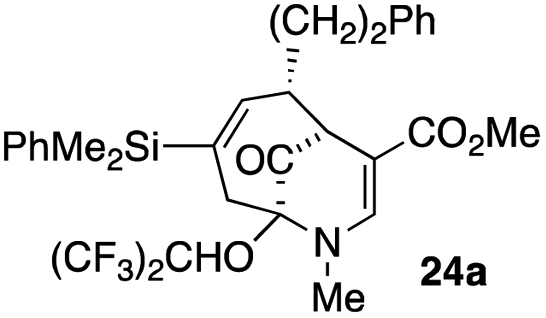 |
31 |
| 12 | 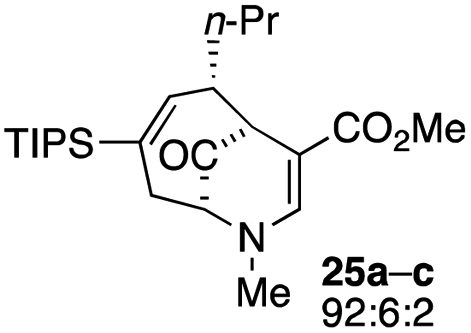 |
23 | 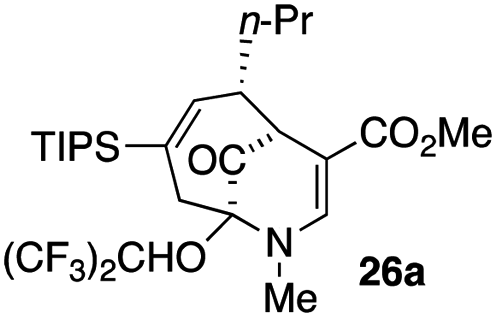 |
64 |
In general, the reactions were conducted with 0.25 mmol of substrate dissolved in 4 mL of hexafluoroisopropanol (HFIP). The stirred solution was cooled to 0 °C in an ice bath. After addition of N-iodosuccinimide (1.5 equiv.), the mixture was allowed to slowly warm to the room temperature. The reaction was monitored by TLC until starting material was completely consumed.
The substrates that contained an alkyl substituent at C-2 and a trialkylsilyl group at C-4 afforded product in about 50% yield (entries 1–4, Table 1). The assignment of structure using NMR was supported by an X-ray crystal structure of 8a.8
Not all substrates bearing an alkyl substituent at C-2 and a trialkylsilyl group at C-4 afforded product in the 50% range. The lower yield of 24a from 23a–b, is perhaps due to the phenyl group on the silicon being sufficiently reactive to cause side product formation (entry 11, Table 1). In the case of 25a–c, the TIPS group at C-4 may have afforded protection against side product formation, resulting in a relatively high yield for the formation of 26a (entry 12, Table 1).
The substrates lacking a trialkylsilyl group (15a–b) gave a very low yield of product, indicating that the trialkylsilyl group affords some protection against degradation in the oxidation process (entry 7, Table 1). There is improvement in yield when the double bond of 15 removed, indeed the yield is doubled (entry 8, Table 1). Interestingly, this is the only case in which a substituent is located at C-2 in which both diastereomers lead to product.
When no substituent appears at C-2 but a trialkylsilyl group appears at C-3 or C-4, both regioisomers appear to participate in the oxidation reaction (entries 8–9, Table 1). However, some inconsistencies are apparent. When the trialkylsilyl group is TES, the yield is low but both isomers seem to react at equal rates, as the ratio does not change in going from substrate to product (entry 9, Table 1). This is not the case for the substrate with a TIPS group, in which it is clear that one isomer behaves better than the other (entry 10, Table 1). The reasons for this are not known.
Fig. 1 shows substrates that did not afford bridgehead oxidation product and led to complex reaction mixtures when treated with NIS. It does appear that the silyl group provides protection to the alkene, but the effect is clearly not universal.
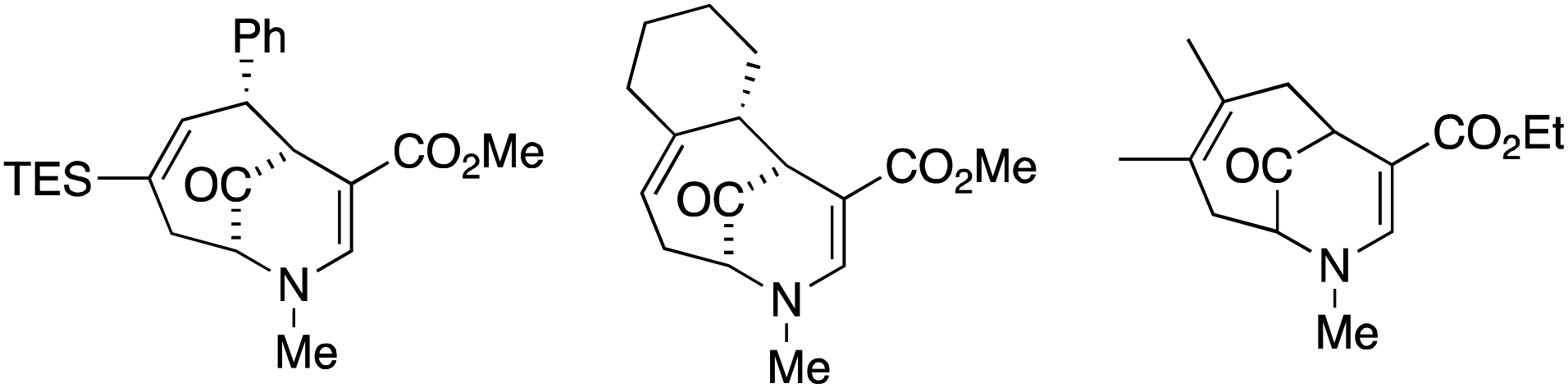 | ||
| Fig. 1 Substrates that did not lead to oxidation. | ||
We attempted to modify the reaction conditions in the hope of obtaining different products or limit the need for HFIP as solvent using 3a–c as a model substrate. When LiI, NaI, or NaBr was added to the reaction mixture, oxidation products were obtained in low yield. No halogen incorporation was observed, though such products might be expected to be labile in any case, as they would likely be readily solvolyzed. Attempts to reduce the amount of HFIP used in the process (2–30 equiv. in MeCN) gave bridgehead substitution products in 5–46% yield with recovered starting material isolated in yields ranging from 29–64%. Addition of small amounts of trifluoroacetic acid to the standard reaction mixture led to decomposition. Excess NIS (15 equiv.) gave decomposition and NBS was not effective at all in producing the oxidation product under the standard reaction conditions.
Our working mechanism for this reaction is shown in Scheme 5. Reaction of 3a with NIS activated by HFIP through hydrogen bonding produces the iminium ion 27a.7 Deprotonation of this intermediate with succinimide anion affords the bridgehead iminium species 28a, which is trapped with HFIP to produce the product 4a (Scheme 5). Inclusion of allyltrimethylsilane in the reaction to trap 28a led to a complex product mixture.
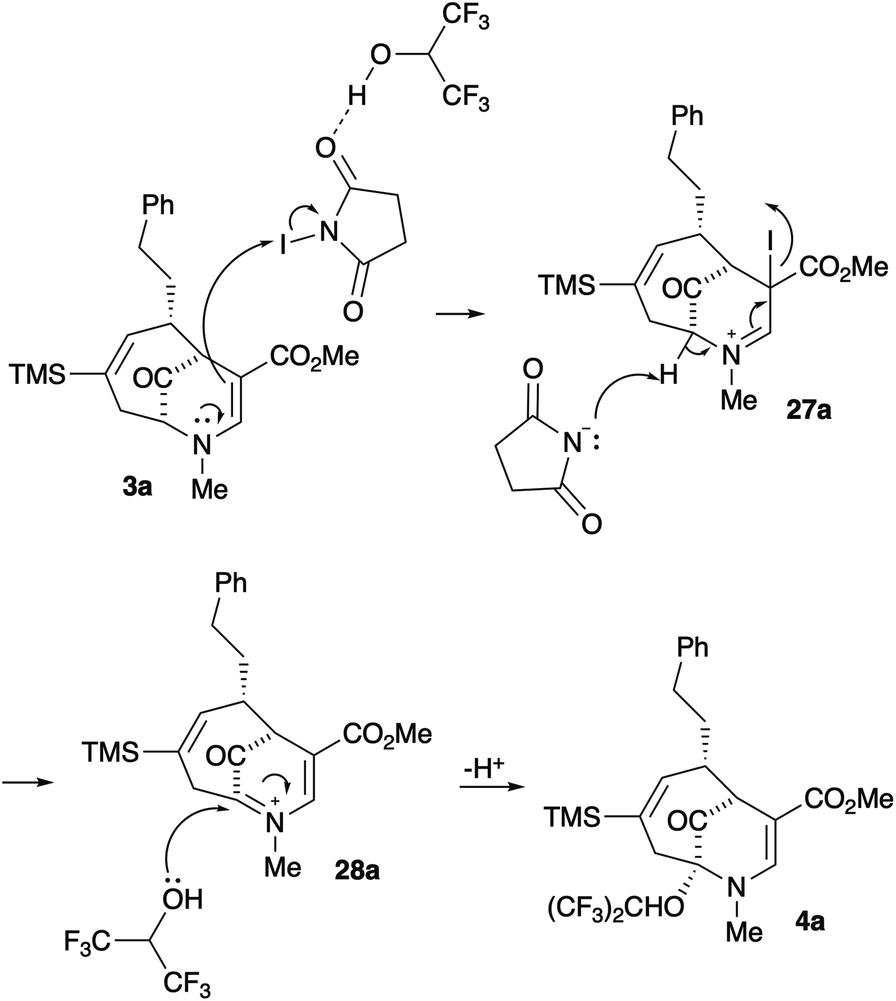 | ||
| Scheme 5 Proposed mechanism of the oxidation. | ||
In conclusion, we report a unique bridgehead oxidation process of selected (4 + 3) cycloadducts derived from oxidopyridinium ions. While this process will certainly possess limitations, it raises questions about mechanism and what other reagents might be used to effect such an oxidation in a more general way, and whether more general bridgehead functionalization might be possible through such a mechanism. We plan on addressing these questions. Results will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation of the United States for their continuing financial support of our program (CHE-1764437). We thank Ms Madison Clark (University of Missouri-Columbia) and Mr Alexander S. Harmata (University of Michigan) for assistance in the preparation of the manuscript. We thank Dr Steven P. Kelley for acquisition of the X-ray data.Notes and references
- Selected reviews of the (4 + 3) cycloaddition reaction: (a) M. Harmata, Chem. Commun., 2010, 46, 8886 RSC; (b) M. Harmata, Chem. Commun., 2010, 46, 8904 RSC; (c) J. L. Mascareñas, M. Gulias and F. Lopez, in Comprehensive Organic Synthesis, ed. P. Knochel and G. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 5, pp. 595–655 Search PubMed; (d) S. Y. Y. Lam and P. Chiu, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, John Wiley & Sons, Hoboken, NJ, 2014, pp. 565–598 Search PubMed; (e) D. E. Jones and M. Harmata, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, John Wiley & Sons, Hoboken, NJ, 2014, pp. 599–630 Search PubMed.
- (a) N. Dennis, A. R. Katritzky and R. Rittner, J. Chem. Soc., Perkin Trans. 1, 1976, 1, 2329 RSC; (b) A. R. Katritzky, S. Rahimi-Rastgoo, G. J. Sabongi and G. W. Fischer, J. Chem. Soc., Perkin Trans. 1, 1980, 1, 362 RSC; (c) A. R. Katritzky and N. Dennis, Chem. Rev., 1989, 89, 827 CrossRef CAS; (d) M. J. Sung, H. I. Lee, Y. Chong and J. K. Cha, Org. Lett., 1999, 1, 2017 CrossRef CAS PubMed; (e) H. I. Lee, M. J. Sung, H. B. Lee and J. K. Cha, Heterocycles, 2004, 62, 407 CrossRef CAS.
- (a) C. Fu, N. Lora, P. L. Kirchhoefer, D. R. Lee, E. Altenhofer, C. L. Barnes, N. L. Hungerford, E. H. Krenske and M. Harmata, Angew. Chem., Int. Ed., 2017, 56, 14682 CrossRef CAS PubMed; (b) C. Fu, S. P. Kelley, J. Tu and M. Harmata, J. Org. Chem., 2021, 86, 7028 CrossRef CAS.
- W. Sungnoi, A. B. Keto, R. B. Roseli, J. Liu, H. Wang, C. Fu, E. L. Regalado, E. H. Krenske and M. Harmata, Org. Lett., 2021, 23, 8302 CrossRef CAS PubMed.
- E. A. Liardi, C. E. Stivala and A. Zakarian, Org. Lett., 2008, 10, 1727 CrossRef.
- For chemistry demonstrating the special properties of HFIP, see: (a) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé,Chem. Rev., 2022, 122, 12544 Search PubMed; (b) N. Zeidan, S. Bicic, R. J. Mayer, D. Leboeuf and J. Moran, Chem. Sci., 2022, 13, 8436 RSC; (c) T. Bhattacharya, A. Ghosh and D. Maiti, Chem. Sci., 2021, 12, 3857 RSC; (d) V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Leboeuf, Chem. Commun., 2020, 56, 11548 RSC; (e) A. M. Arnold, A. Pöthig, M. Drees and T. Gulder, J. Am. Chem. Soc., 2018, 140, 4344 CrossRef CAS; (f) A. D. Salvo, M. David, B. Crousse and D. Bonnet-Delpon, Adv. Synth. Catal., 2006, 348, 118 CrossRef.
- For leading references to bridgehead iminium ions, see: (a) K. M. Brummond and S.-P. Hong, J. Org. Chem., 2005, 70, 907 CrossRef CAS; (b) M. H. Franz, S. Roeper, R. Wartchow and H. M. R. Hoffmann, J. Org. Chem., 2004, 69, 2983 CrossRef CAS PubMed; (c) B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS PubMed.
- Crystal structure of compound 8a (CCDC 2171958†) has been deposited at the Cambridge Crystallographic Data Center.
Footnote |
| † Electronic supplementary information (ESI) available: General information, table of trapping studies, synthesis procedures and characterization. CCDC 2171958 of compound 8a. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05189c |
| This journal is © The Royal Society of Chemistry 2022 |