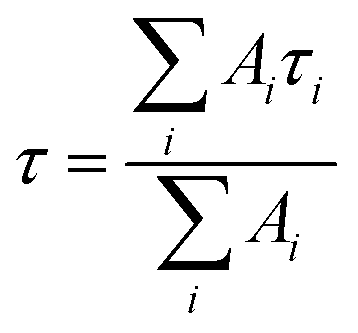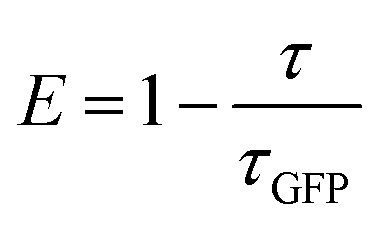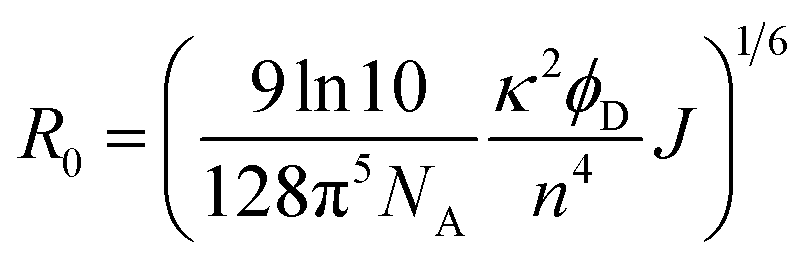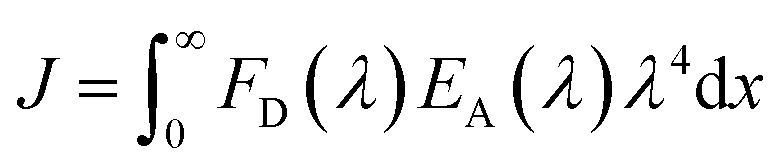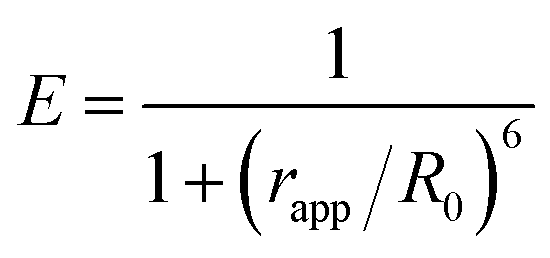DOI:
10.1039/D2RA05249K
(Paper)
RSC Adv., 2022,
12, 28519-28524
A disulphide bond-mediated hetero-dimer of a hemoprotein and a fluorescent protein exhibiting efficient energy transfer†
Received
21st August 2022
, Accepted 22nd September 2022
First published on 6th October 2022
Abstract
Artificial protein hetero-dimerization is one of the promising strategies to construct protein-based chemical tools. In this work, cytochrome b562, an electron transfer hemoprotein, and green fluorescent protein (GFP) mutants with cysteine residues added to their surfaces were conjugated via a pyridyl disulphide-based thiol–disulfide exchange reaction. The eight hetero-dimers, which have cysteine residues at different positions to form the disulphide bonds, were obtained and characterized by gel-electrophoresis, mass spectrometry and size exclusion chromatography. The fluorescence properties of the hetero-dimers were evaluated by fluorescence spectroscopy and fluorescence lifetime measurements. Efficient photoinduced energy transfer from the GFP chromophore to the heme cofactor was observed in each of the hetero-dimers. The energy transfer efficiency is strongly dependent on the cross-linking residues, reaching 96%. Furthermore, the estimated Förster distance and the structure-based maximum possible distances of the donor and acceptor suggest that one of the hetero-dimers has a rigid protein–protein structure with favourable properties for energy transfer. The disulphide bond-mediated protein hetero-dimerization is useful for screening functional protein systems towards further developments.
Introduction
Proteins in their functional forms frequently exist as dimers and higher-order oligomers.1 To gain insights into and develop bio-inspired applications based on these natural complex structures, artificial protein assemblies have been prepared by various approaches utilizing protein–protein interactions formed by metal coordination, covalent-linking, and non-covalent interactions among others.2 Such known applications include drug delivery,3 catalysis,4 and biosensors.5 Among the strategies used, site-specific disulphide bond formation has been reported for covalent linkage of supramolecular protein assemblies in protein crystals6 and a metal coordinated unique cryptand structure.7 Additionally, a disulphide bond cross-linkage is now commonly used in polymerization by dynamic covalent bonds.8 The use of the disulphide bond as the linkage structure provides stability,9 reversibility,10 and stimuli responsiveness11 in dynamic smart materials. Although the disulphide linkage can be formed spontaneously under aerobic conditions,12 this type of random air-oxidation reaction can lead to mixed disulphide aggregations.13 It tends to be a very slow reaction in vivo and is catalysed by protein disulphide isomerase,14 thus leading to the employment of alternative catalysts and various oxidation methods in synthetic routes.15 Due to these complications, we employ a pyridyl disulphide moiety which is known for its high selectivity and reactivity towards thiol groups to form a new disulphide bond.16 The pyridyl disulphide group will activate a thiol group on one protein which will selectively react with a free thiol group on another protein to form a disulphide linkage.17 However, protein hetero-dimerization using the pyridyl disulphide active species is quite limited.18
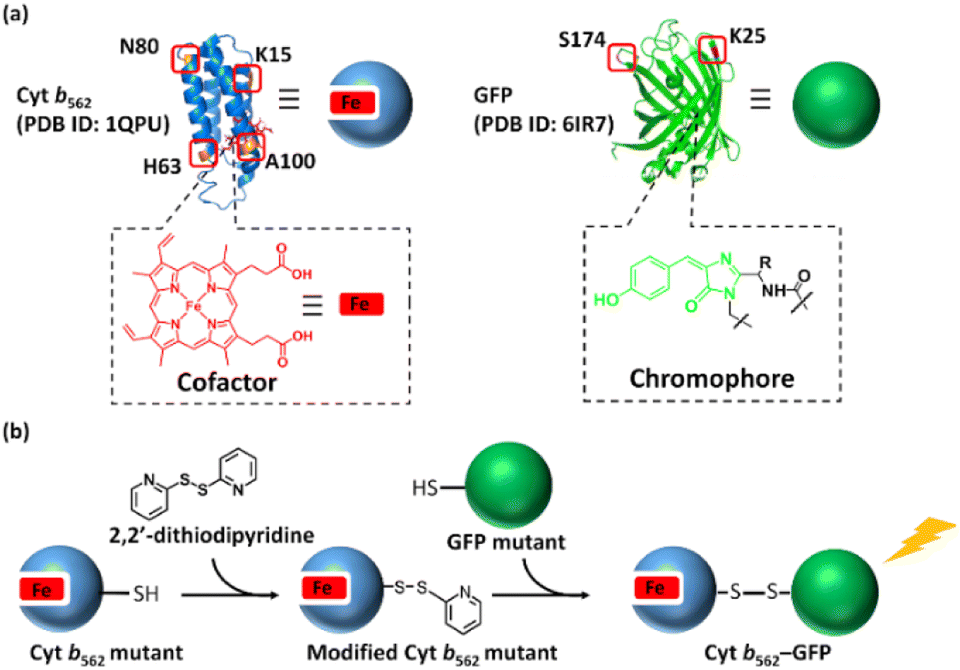
Herein, we employ green fluorescent protein (GFP) and cytochrome b562 (Cyt b562) (Fig. 1) for disulphide hetero-dimerization. Cyt b562 is a small electron transfer protein containing a non-covalently bound heme as a co-factor. Previous reports19 have used Cyt b562 and a GFP mutant to construct a chimera via a conventional recombinant protein fusion method leading to bio-inspired tools.20 Nagamune and co-workers utilized a Gly–Ser linker to prepare a fusion protein displaying 65% of energy transfer from the GFP mutant towards heme, while Jones and co-workers inserted Cyt b562 into the GFP sequence to prepare a di-domain protein scaffold demonstrating the importance of inter-domain interactions in the energy transfer process. Although these previous efforts were successful in expression of the fusion protein, conjugation sites of the components were limited, and optimization of an appropriate linker is necessary. In this work, disulphide bond-mediated hetero-dimerization of Cyt b562 and GFP using the pyridyl disulphide moiety is demonstrated and the energy transfer efficiency in the Cyt b562–GFP hetero-dimers is evaluated (Scheme 1).
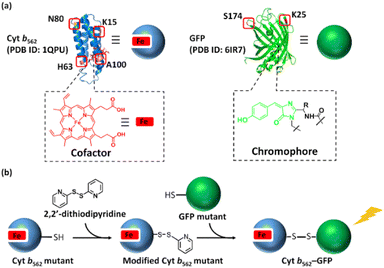 |
| | Scheme 1 (a) Mutation points of Cyt b562 and GFP. (b) Schematic representation of the hetero-dimerization of Cyt b562 and GFP mutants. | |
Experimental
Preparation of modified Cyt b562 mutants
Cyt b562 mutants (500 μM, 900 μL) were reduced upon addition of a 1 M DTTaq (100 μL) solution and incubated for 1 h at 4 °C. DTT was removed using a HiTrap desalting column (eluent: 100 mM potassium phosphate buffer at pH 7.0) and the obtained protein solution was modified upon addition of 2,2′-dithiodipyridine (1 mM, 100 μL) in DMSO and incubated at 4 °C for 1 h. The modified protein was passed through a 5 mL HiTrap desalting column pre-equilibrated by 25 mL of 100 mM potassium phosphate buffer.
Preparation and purification of hetero-dimer
Purified GFP mutants in 100 mM potassium phosphate buffer were reduced upon addition of 1 M DTTaq (100 μL) solution and incubated at 37 °C for 1 h before passing through HiTrap desalting column and diluted to 100 μM. Reduced GFP mutants were incubated with modified Cyt b562 (3 eq.) at 25 °C for 2 h. The crude protein mixtures were purified straight away via Superdex 75 Increase 10/300 GL column using ÄKTA Purifier System (GE Healthcare) eluting by 100 mM potassium phosphate buffer containing 0.3 M NaCl at 0.5 mL min−1 elution rate at 4 °C.
Fluorescence lifetime analysis
Fluorescence lifetime measurements were measured at 25 °C in a constant temperature circulating water bath. The instrument response function (IRF) was determined from scattered light signals in the measurements of a LUDOX SM colloidal silica solution (30 wt% suspension in water, Sigma-Aldrich). The IRF showed a width of 60 ps measured as the full width at half maximum intensity. The IRF was employed in order to fit the fluorescence decay curves using a bi- or tri-exponential decay model. The quality of the fit was assessed from the χ2 values and distribution of the residuals with fixed τ values for the respective GFP mutants to achieve closest χ2 = 1.00 by bi- or tri-decay fitting. All fluorescence lifetime measurement values are an average of at least three sample measurements.
The intensity average lifetime, τav, was calculated by eqn (1):
| |
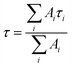 | (1) |
where
Ai and
τi represent the amplitude and the fluorescence lifetime respectively of the individual components.
21,22| |
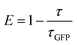 | (2) |
where
E is the energy transfer efficiency,
τGFP value is lifetime obtained by the fluorescence decay for a corresponding GFP mutant, and
τav is the average fluorescence lifetime calculated from
eqn (1).
The averaged Förster distance (R0), in which energy transfer from donor to acceptor occurs with a 50% probability, was calculated by eqn (3).23
| |
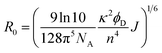 | (3) |
where
κ represents the orientation factor for the transition dipoles. Assuming that the dipole orientation factor (
κ2) was a limiting value (
κ2 is taken as 2/3 if the orientation of the donor and acceptor is assumed to be random),
n is the refractive index of water (1.333),
ϕD is the quantum yield of GFP (0.68),
24 J is the integral of the overlap of the emission from GFP
K25C and the absorption from Cyt
b562N80C given by:
| |
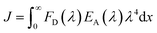 | (4) |
where
J was calculated using the experimental spectra giving
J = 5.67 × 10
14 M
−1 cm
−1 nm
4.
25
The apparent distance (rapp) is estimated with the value of energy efficiency E derived from time-resolved fluorescence measurement and eqn (5).
| |
 | (5) |
Distance estimation in protein structure
For the computational design of the modification of Cyt b562 and GFP, the protein structural data of 1QPU and 6IR7 from the Protein Data Bank were utilized in PyMol, respectively. The distances between Cα and the heme centre of Cyt b562 and between Cα and the fluorophore centre of GFP were estimated by the distance option in the measurement tool while highlighting the two specified residues (Cα to the heme centre of Cyt b562 or GFP fluorophore).
Results and discussion
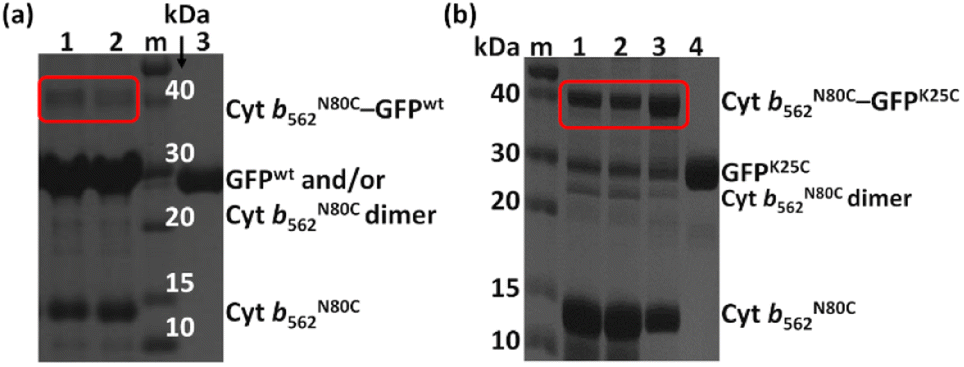
Initially, the modification of a Cyt b562 mutant having a cysteine residue at the N80 position located on the protein surface (Cyt b562N80C) with 2,2′-dithiodipyridine was carried out. The modified Cyt b562N80C bearing the pyridyl disulphide moiety was characterized by MALDI-TOF MS: found m/z = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880, calcd m/z = 11879 (Fig. S1a†). The hetero-dimerization was first attempted using the wild-type GFP as it is known to have one exposed cysteine residue at the 47th position. The reactivity was first tested by conjugating the wild-type GFP (GFPwt) with an excess amount of 2,2′-dithiodipyridine modified Cyt b562N80C. The analysis of the crude reaction mixture by non-reducing SDS-PAGE shows that GFPwt does not provide an efficient hetero-conjugation product with Cyt b562N80C (Fig. 1a). Furthermore, an excess amount of homo-dimerization of Cyt b562N80C was detected, although the band of the Cyt b562N80C dimer overlaps with the GFPwt monomer band at ca. 30 kDa. This is plausibly caused by steric inaccessibility of the inherent cysteine residue at the 47th position.26 Therefore, the exposed 25th position of GFP was selected as a conjugation site for the hetero-dimerization with Cyt b562N80C after mutation of the 47th cysteine residue to alanine (GFPK25C).27 In comparison, GFPK25C readily conjugates to modified Cyt b562N80C under the same conditions with high conjugation efficiency to afford the hetero-dimer (Cyt b562N80C–GFPK25C) with 82% yield (Fig. 1b). Control experiments of air-oxidation of reduced Cyt b562N80C and GFPK25C under the same conditions and analysed by non-reducing SDS-PAGE did not show hetero-dimerization product (Fig. S3†).
880, calcd m/z = 11879 (Fig. S1a†). The hetero-dimerization was first attempted using the wild-type GFP as it is known to have one exposed cysteine residue at the 47th position. The reactivity was first tested by conjugating the wild-type GFP (GFPwt) with an excess amount of 2,2′-dithiodipyridine modified Cyt b562N80C. The analysis of the crude reaction mixture by non-reducing SDS-PAGE shows that GFPwt does not provide an efficient hetero-conjugation product with Cyt b562N80C (Fig. 1a). Furthermore, an excess amount of homo-dimerization of Cyt b562N80C was detected, although the band of the Cyt b562N80C dimer overlaps with the GFPwt monomer band at ca. 30 kDa. This is plausibly caused by steric inaccessibility of the inherent cysteine residue at the 47th position.26 Therefore, the exposed 25th position of GFP was selected as a conjugation site for the hetero-dimerization with Cyt b562N80C after mutation of the 47th cysteine residue to alanine (GFPK25C).27 In comparison, GFPK25C readily conjugates to modified Cyt b562N80C under the same conditions with high conjugation efficiency to afford the hetero-dimer (Cyt b562N80C–GFPK25C) with 82% yield (Fig. 1b). Control experiments of air-oxidation of reduced Cyt b562N80C and GFPK25C under the same conditions and analysed by non-reducing SDS-PAGE did not show hetero-dimerization product (Fig. S3†).
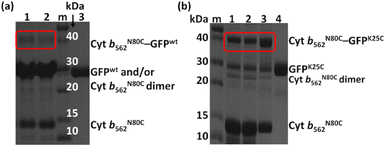 |
| | Fig. 1 (a) Non-reducing SDS-PAGE for samples in lanes 1 and 2 from GFPwt (100 μM) conjugation with an excess amount of pyridyl disulphide-modified Cyt b562N80C. Conditions: modified Cyt b562N80C: reduced GFPwt = 2:1 in lane 1 and 3:1 in lane 2, and control GFPwt in lane 3. Lane m shows protein markers. (b) Non-reducing SDS-PAGE for samples in lanes 1, 2, and 3. Conditions: modified Cyt b562N80C: reduced GFPK25C 1:1 in lane 1, 2:1 in lane 2, 3:1 in lane 3, and GFPK25C control in lane 4. Lane m shows protein markers. | |
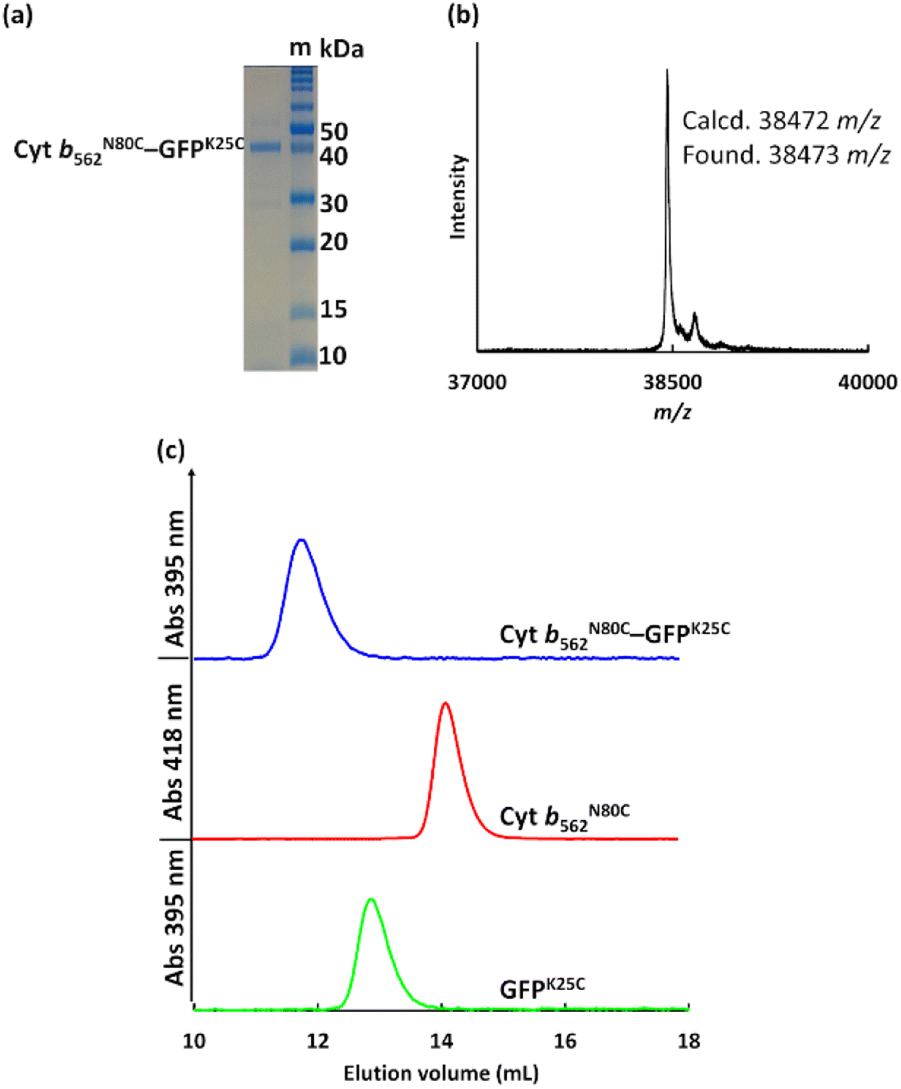
The crude Cyt b562N80C–GFPK25C mixtures purified using size exclusion chromatography (SEC, Fig. S2†) and ultrafiltration (30 kDa cut-off) showed a single distinct band at ca. 40 kDa in non-reducing SDS-PAGE consistent with the MALDI-TOF MS result: found m/z = 38473, calcd m/z = 38472 (Fig. 2a and b). In the SEC analyses, purified Cyt b562N80C–GFPK25C provides a single peak at 11.7 mL (Fig. 2c) which is a smaller elution volume compared to monomeric proteins of Cyt b562N80C and GFPK25C eluted at 14.6 mL and 13.1 mL, respectively. These results indicate that the designed protein hetero-dimer can be successfully obtained with high purity. To verify the hetero-dimers linked by disulphide bonds, analysis by reducing SDS-PAGE with dithiothreitol (DTT) as a reductant was carried out. The analysed samples indicate cleavage of the disulphide bond resulting in the separation of the two components, Cyt b562 and GFP by the appearance of their monomeric bands at ca. 15 kDa and 30 kDa, respectively (Fig. S4†).
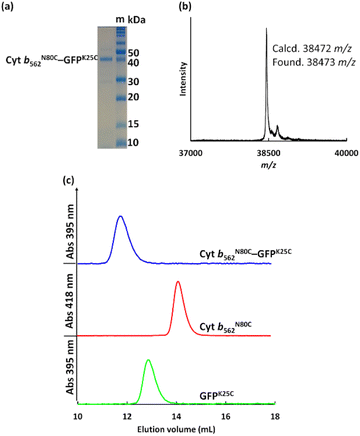 |
| | Fig. 2 (a) Non-reducing SDS-PAGE of purified Cyt b562N80C–GFPK25C with protein markers, m, (b) MALDI-TOF MS of Cyt b562N80C–GFPK25C, and (c) SEC profiles of purified Cyt b562N80C–GFPK25C, Cyt b562N80C, and GFPK25C. Conditions: eluent = 100 mM potassium phosphate buffer containing 300 mM NaCl, pH 7.0 at 4 °C, column = Superdex 75 Increase 10/300 GL, elution rate = 0.5 mL min−1. | |
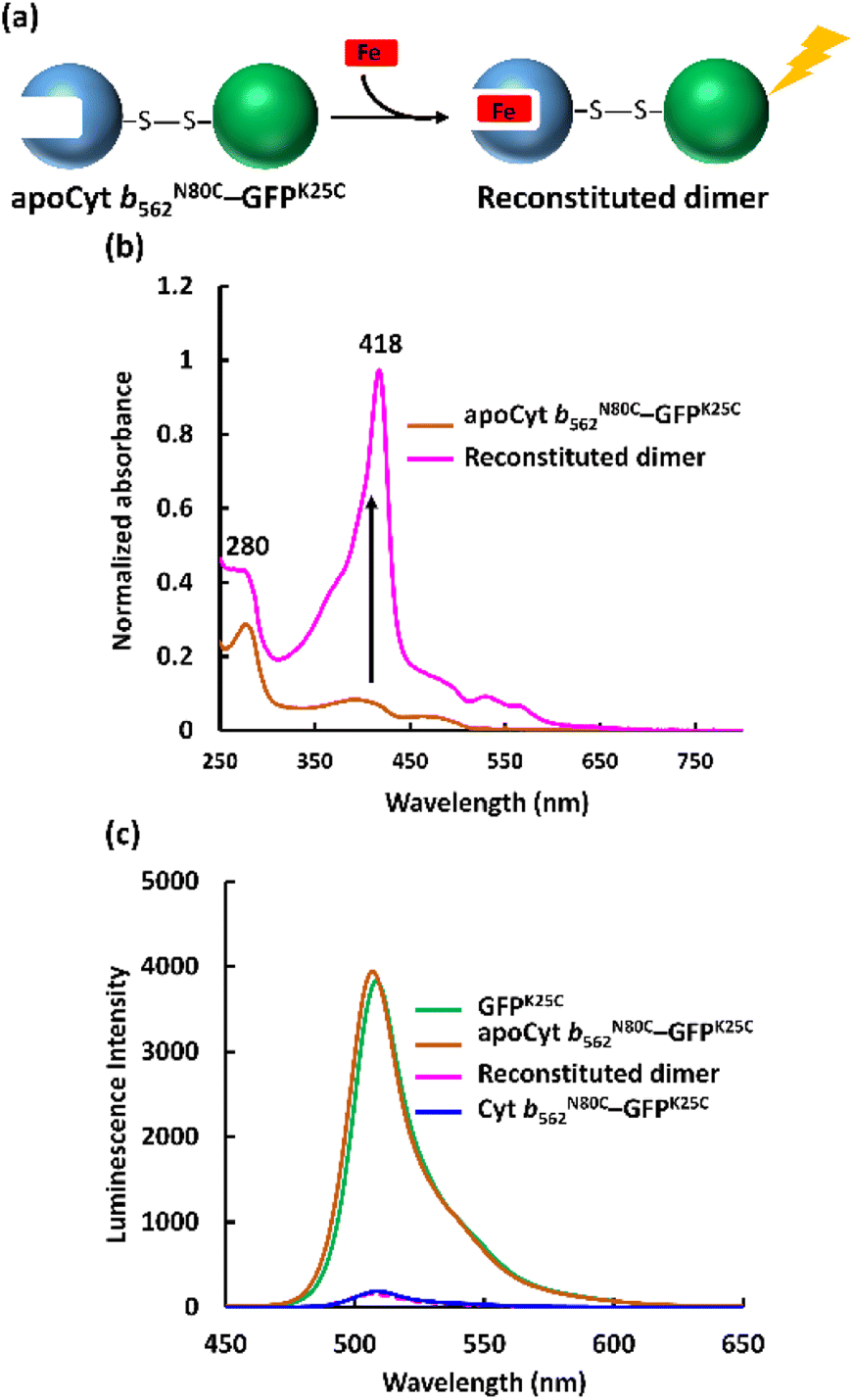
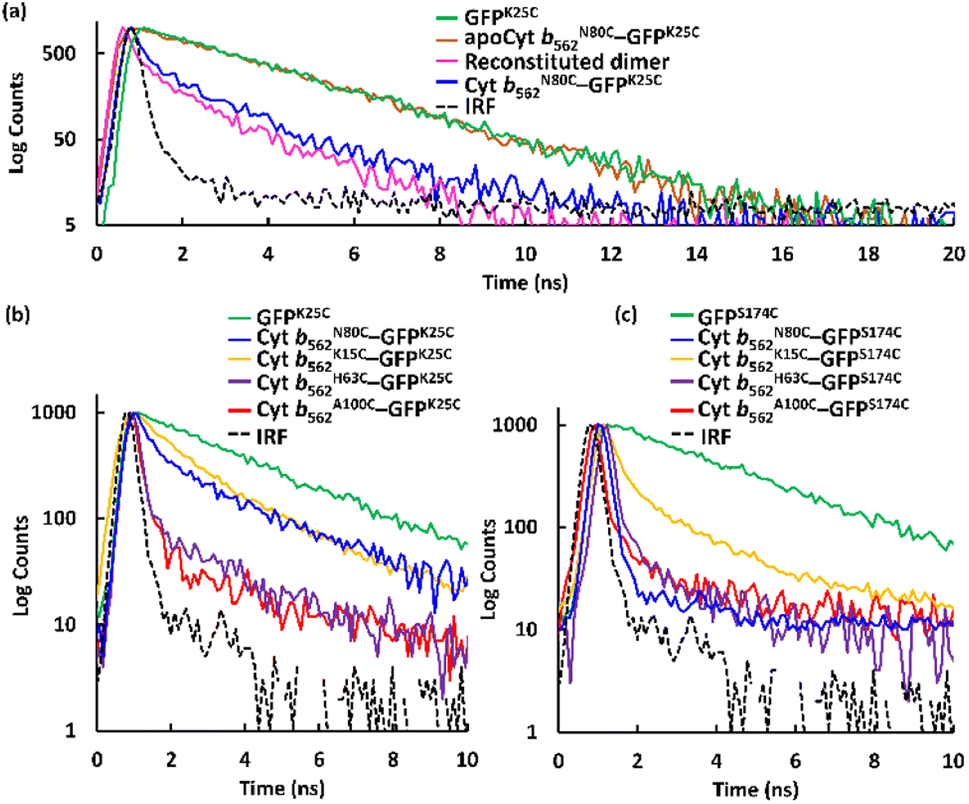
The apo form of modified Cyt b562 was obtained by the conventional method28 (Fig. S5†), and was subsequently conjugated with GFPK25C. The reaction mixture was purified using the same method, and SEC analysis shows that apoCyt b562N80C–GFPK25C is generated with an elution volume similar to that of Cyt b562N80C–GFPK25C (Fig. S6†). The reconstitution of the apoCyt b562N80C–GFPK25C with heme illustrated in Fig. 3a results in the re-appearance of the characteristic Soret band absorption at 418 nm as shown in Fig. 3b, confirming successful heme binding. The fluorescence of the reconstituted hetero-dimer is ca. 90% quenched compared to apoCyt b562N80C–GFPK25C which exhibits fluorescence similar to that of GFPK25C (Fig. 3c). This indicates that the heme-dependent quenching occurs in the hetero-dimer. Moreover, the fluorescence lifetime measurements exhibit fast decay from both Cyt b562N80C–GFPK25C and the reconstituted hetero-dimer: τ1 = 0.31 ns and 0.28 ns, respectively (Fig. 4a and Table 1).29,30 In contrast, apoCyt b562N80C–GFPK25C has τ = 2.93 ns which is similar to that of GFPK25C. The identical fluorescence lifetimes of GFPK25C and apoCyt b562N80C–GFPK25C indicate that there is no quenching behaviour within the apoCyt b562N80C–GFPK25C due to the absence of heme. Thus, the rapid decay observed in the reconstituted dimer demonstrates efficient static quenching behaviour of the GFPK25C by the heme cofactor (Fig. 3c and 4a).
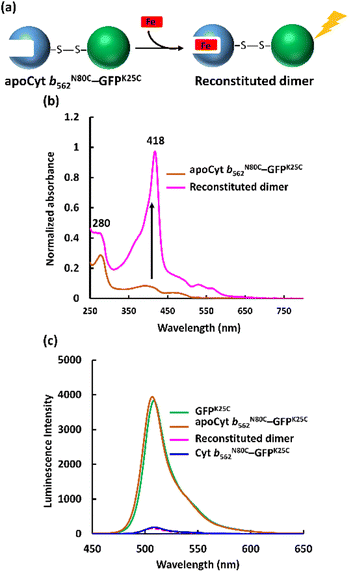 |
| | Fig. 3 (a) Schematic illustration of the hetero-dimer reconstitution with heme. (b) UV-vis spectra of apoCyt b562N80C–GFPK25C (brown) and reconstituted sample (pink). Conditions: [protein] = 50 μM in 100 mM potassium phosphate buffer, pH 7.0. (c) Fluorescence spectra of the hetero-dimers and GFPK25C. Conditions: [protein] = 5 μM, in 100 mM potassium phosphate buffer, pH 7.0 at 25 °C, λex = 395 nm. | |
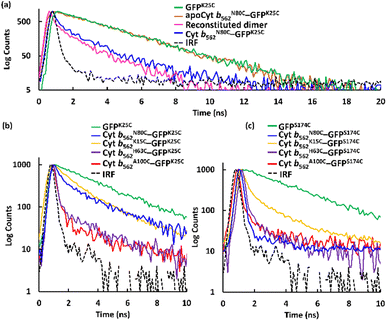 |
| | Fig. 4 Fluorescence lifetime measurements fitted taking the instrument response function (IRF) into account by a bi- or tri-exponential decay model for the respective samples. (a) GFPK25C, apoCyt b562N80C–GFPK25C, reconstituted dimer obtained by incorporation of heme into apoCyt b562N80C–GFPK25C, and Cyt b562N80C–GFPK25C. (b) GFPK25C and hetero-dimers with GFPK25C. (c) GFPS174C and hetero-dimers with GFPS174C. Conditions: [protein] = 20 μM, 1 mL, prepared in 0.1 M potassium phosphate buffer, pH 7.0 at 25 °C. Full scale fluorescence lifetime measurements are shown in Fig. S10.† | |
Table 1 Fluorescence lifetime values of purified hetero-dimera
| Protein |
τ1 (ns) |
τ2 (ns) |
τ3 (ns) |
A1 (%) |
A2 (%) |
A3 (%) |
χ2 |
τavb (ns) |
| Conditions: [protein] = 20 μM, 1 mL, prepared in 0.1 M potassium phosphate buffer pH 7.0 at 25 °C. Fluorescence lifetimes were evaluated by bi- or tri-exponential decay model. Values expressed are means ± SD of three parallel measurements. The intensity average lifetimes were calculated using eqn (1). Hetero-dimer formed by apoCyt b562N80C–GFPK25C and then reconstituted by heme. |
| GFPK25C |
2.92 ± 0.06 |
— |
— |
100 |
— |
— |
1.05 ± 0.00 |
|
| apoCyt b562N80C–GFPK25C |
2.93 ± 0.01 |
99.9 ± 0.05 |
1.00 ± 0.03 |
|
| Reconstituted dimerc |
0.28 ± 0.08 |
2.92 |
— |
93.7 ± 0.26 |
6.3 ± 0.16 |
— |
0.99 ± 0.01 |
|
| Cyt b562A100C–GFPK25C |
0.16 ± 0.04 |
1.35 ± 0.01 |
2.92 |
91.7 ± 0.05 |
4.90 ± 0.12 |
3.78 ± 0.52 |
1.09 ± 0.01 |
0.32 |
| Cyt b562H63C–GFPK25C |
0.20 ± 0.01 |
2.92 |
— |
93.7 ± 5.42 |
6.31 ± 5.42 |
6.31 ± 5.42 |
0.97 ± 0.00 |
0.37 |
| Cyt b562N80C–GFPK25C |
0.31 ± 0.03 |
1.53 ± 0.26 |
2.92 |
97.5 ± 0.79 |
2.50 ± 0.44 |
— |
1.07 ± 0.05 |
0.49 |
| Cyt b562K15C–GFPK25C |
0.56 ± 0.03 |
2.17 ± 0.33 |
2.92 |
92.9 ± 1.76 |
5.44 ± 0.88 |
1.68 ± 0.09 |
1.10 ± 0.07 |
0.70 |
| GFPS174C |
2.88 ± 0.02 |
|
|
100 |
|
|
1.02 ± 0.00 |
| Cyt b562A100C–GFPS174C |
0.15 ± 0.00 |
2.88 |
— |
99.8 ± 0.01 |
0.22 ± 0.02 |
— |
1.06 ± 0.15 |
0.15 |
| Cyt b562H63C–GFPS174C |
0.12 ± 0.02 |
1.67 ± 0.10 |
2.88 |
89.8 ± 6.56 |
6.60 ± 4.25 |
3.59 ± 2.37 |
0.98 ± 0.12 |
0.32 |
| Cyt b562N80C–GFPS174C |
0.10 ± 0.00 |
0.75 ± 0.39 |
2.88 |
99.2 ± 0.07 |
0.77 ± 0.07 |
— |
1.06 ± 0.02 |
0.11 |
| Cyt b562K15C–GFPS174C |
0.23 ± 0.01 |
2.82 ± 0.05 |
2.88 |
80.5 ± 1.57 |
16.7 ± 1.50 |
2.76 ± 0.15 |
0.97 ± 0.02 |
0.74 |
Three additional Cyt b562 single mutants containing cysteine at positions K15 (Cyt b562K15C), H63 (Cyt b562H63C) and A100 (Cyt b562A100C) (Scheme 1a) and one GFP mutant containing cysteine at position S174 (GFPS174C)26 (Scheme 1a) were employed for the hetero-dimerization. The mutants were chosen due to their high expression levels, modification and hetero-conjugation yields. Similar to the Cyt b562N80C–GFPK25C hetero-dimerization, each Cyt b562 mutant was modified with 2,2′-dithiodipyridine (Fig. S1†) and conjugated with GFP, yielding a total of seven purified hetero-dimers characterized by non-reducing SDS-PAGE (Fig. S7†) and MALDI-TOF MS (Fig. S8 and S9†).
Fig. 4b, c, and Table 1 show the results in the fluorescence lifetime measurements of a series of the hetero-dimers of Cyt b562 and GFP mutants. Interestingly, different lifetime values were obtained and GFPK25C exhibits the shortest lifetime τ1 = 0.16 ns among the conjugations with Cyt b562A100C, while GFPS174C provides the shortest lifetime of τ1 = 0.10 ns among the conjugations with Cyt b562N80C.29 The highest energy efficiencies obtained from Cyt b562A100C–GFPK25C and Cyt b562N80C–GFPS174C, were calculated (eqn (2)) to be 89% and 96% respectively (Table 2).
Table 2 Distances between acceptor and donor
| Protein |
E (%) |
dhc (Å) |
dcd (Å) |
dmaxe (Å) |
rappf (Å) |
| Energy efficiency for hetero-dimers conjugated with GFPK25C was calculated using eqn (2) and τ1 value of GFPK25C. Energy efficiency for hetero-dimers conjugated with GFPS174C was calculated using eqn (2) and τ1 value of GFPS174C. Distances from the Cα of the mutated residue in the crystal structure of wild type Cyt b562 to the Fe center. Distances from the Cα of the mutated residue in the crystal structure of wild type GFP to the GFP fluorophore. The maximum possible distance was estimated as the sum of dh and dc with the typical length of the disulfide bond (2.04 Å).32 The apparent distances from heme to the GFP chromophore calculated using eqn (5), experimental R0 and E. |
| GFPK25C |
|
|
22.7 |
|
|
| Cyt b562A100C–GFPK25C |
89a |
9.6 |
|
34.3 |
39 |
| Cyt b562H63C–GFPK25C |
87a |
12.0 |
|
36.7 |
40 |
| Cyt b562N80C–GFPK25C |
83a |
23.9 |
|
48.6 |
42 |
| Cyt b562K15C–GFPK25C |
76a |
15.4 |
|
40.1 |
46 |
| GFPS174C |
|
|
21.0 |
|
|
| Cyt b562A100C–GFPS174C |
95b |
9.6 |
|
34.3 |
34 |
| Cyt b562H63C–GFPS174C |
89b |
12.0 |
|
36.7 |
39 |
| Cyt b562N80C–GFPS174C |
96b |
23.9 |
|
48.6 |
32 |
| Cyt b562K15C–GFPS174C |
74b |
15.4 |
|
40.1 |
46 |
Since the Cyt b562 and GFP mutants have similar spectra to Cyt b562N80C and GFPK25C, respectively, superposition of the GFPK25C emission spectrum with the Cyt b562N80C absorption spectrum shows overlap, suggesting the possible FRET of the hetero-dimer (Fig. S11c†). The Förster distance (R0) was estimated to be 56 Å using the reported quantum yield of GFP,24 assuming random donor and acceptor orientation.29 From the calculated R0 values and experimental results of energy transfer efficiencies (E), the apparent distances (rapp) between heme and GFP chromophore are found to be between 32 Å and 46 Å (Table 2). The distances from each cysteine mutation point to the chromophore or to the heme were estimated.30 These estimations indicate that the distances between the Cα atom of the mutated residues and the heme centre in Cyt b562 range from 9.6 Å to 23.9 Å, while in GFP the Cα atom of the K25 and S174 residues are situated away from the chromophore with a distance of 22.7 Å and 21.0 Å, respectively (Fig. S12 and S13†).31 The maximum possible distances (dmax) between the chromophore and heme were also determined from these structural data and the typical length of the disulphide bond (2.04 Å),32 showing that most hetero-dimers exhibit values similar to rapp with differences of less than 7 Å (Table 2). The only exception is the Cyt b562N80C–GFPS174C hetero-dimer which exhibits a significantly shorter rapp of 32 Å relative to dmax of 49 Å. This result suggests a favourable conformation with a short distance and/or proper orientation of the heme cofactor and chromophore in Cyt b562N80C–GFPS174C. Although the single disulphide bond in the hetero-dimer is generally flexible with free rotation of the protein units, this finding indicates that a unique disulphide bond at appropriate mutation points triggers the induced protein–protein interaction33 to strain the rotation achieving the favourable conformation for energy transfer.
Conclusions
In conclusion, we successfully achieved hetero-dimerization of the Cyt b562 and GFP mutants by employing a pyridyl disulphide moiety to rapidly react with thiols under mild conditions forming a disulphide bond. The purified hetero-dimers demonstrate efficient energy transfer in a heme-dependent quenching manner as foreseen. Simple protein linkage by disulphide bond formation is also useful to construct an interprotein energy transfer system as well as reported genetic protein fusion.19 Our work presents a rapid and efficient site-selective bio-conjugation approach which allows for a much broader sampling and screening process to determine efficient energy transfer pairs. Further detailed investigations of the specific protein–protein interactions and hetero-dimer conformations are expected to contribute to the refinement of a useful process for hetero-dimerization of proteins.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by Grants-in-Aid for Scientific Research provided by JSPS KAKENHI Grant Numbers JP15H05804, JP18K19099, JP18KK0156, JP20H02755, JP20H00403 and JP20KK0315.
Notes and references
-
(a) M. H. Ali and B. Imperiali, Bioorg. Med. Chem., 2005, 13, 5013–5020 Search PubMed;
(b) D. S. Goodsell and A. J. Olson, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 105–153 Search PubMed.
-
(a) J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217–2221 Search PubMed;
(b) F. Sakai, G. Yang, M. S. Weiss, Y. Liu, G. Chen and M. Jiang, Nat. Commun., 2014, 5, 4634–4642 Search PubMed;
(c) S. Abe, B. Maity and T. Ueno, Chem. Commun., 2016, 52, 6496–6512 Search PubMed;
(d) Y. Suzuki, G. Cardone, D. Restrepo, P. D. Zavattieri, T. S. Baker and F. A. Tezcan, Nature, 2016, 533, 369–373 Search PubMed;
(e) D. A. Uhlenheuer, D. Wasserberg, H. Nguyen, L. Zhang, C. Blum, V. Subramaniam and L. Brunsveld, Chem.–Eur. J., 2009, 15, 8779–8790 Search PubMed.
-
(a) E. J. Lee, N. K. Lee and I. Kim, Adv. Drug Delivery Rev., 2016, 106, 157–171 Search PubMed;
(b) M. Zdanowicz and J. Chroboczek, Acta Biochim. Pol., 2016, 63, 469–473 Search PubMed;
(c) J. A. Mackay, M. Chen, J. R. McDaniel, W. G. Liu, A. J. Simnick and A. Chilkoti, Nat. Mater., 2009, 8, 993–999 Search PubMed.
-
(a) W. Kang, J. Liu, J. Wang, Y. Nie, Z. Guo and J. Xia, Bioconjugate Chem., 2014, 25, 1387–1394 Search PubMed;
(b) T. Tong, S. Schoffelen, S. F. M. van Dongen, T. A. van Beek, H. Zuilhof and J. C. M. van Hest, Chem. Sci., 2011, 2, 1278–1285 Search PubMed.
-
(a) J. Byeon, F. T. Llimpoco and R. C. Bailey, Langmuir, 2010, 26, 15430–15435 Search PubMed;
(b) Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2013, 52, 4088–4106 Search PubMed.
-
(a) S. Abe, T. T. Pham, H. Negishi, K. Yamashita, K. Hirata and T. Ueno, Angew. Chem., Int. Ed., 2021, 60, 12341–12345 Search PubMed;
(b) H. Negishi, S. Abe, K. Yamashita, K. Hirata, K. Niwase, M. Boudes, F. Coulibaly, H. Mori and T. Ueno, Chem. Commun., 2018, 54, 1988–1991 Search PubMed;
(c) T. K. Nguyen, H. Negishi, S. Abe and T. Ueno, Chem. Sci., 2019, 10, 1046–1051 Search PubMed.
- A. Medina-Morales, A. Perez, J. D. Brodin and F. A. Tezcan, J. Am. Chem. Soc., 2013, 135, 12013–12022 Search PubMed.
-
(a) Q. Zhang, D. H. Qu, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2022, 144, 2022–2033 Search PubMed;
(b) A. H. J. Engwerda and S. P. Fletcher, Nat. Commun., 2020, 11, 4156–4163 Search PubMed.
-
(a) J. Wang, L. Chen, J. Wu, W. Li, K. Liu, T. Masuda and A. Zhang, Asian J. Chem., 2018, 13, 3647–3652 Search PubMed;
(b) B. Li, J. Rozas and D. T. Haynie, Biotechnol. Prog., 2006, 22, 111–117 Search PubMed.
-
(a) X. Lin, G. Godeau and M. W. Grinstaff, New J. Chem., 2014, 38, 5186–5189 Search PubMed;
(b) A. Fava, A. Iliceto and E. Camera, J. Am. Chem. Soc., 1957, 79, 833–838 Search PubMed;
(c) J. W. Sadownik and R. V. Ulijn, Curr. Opin. Biotechnol., 2010, 21, 401–411 Search PubMed.
-
(a) Y. Z. You, C. Y. Hone and C. Y. Pan, J. Phys. Chem. C, 2007, 111, 16161–16166 Search PubMed;
(b) O. Hayashida, K. Ichimura, D. Sato and T. Yasunaga, J. Org. Chem., 2013, 78, 5463–5469 Search PubMed;
(c) H. Wang, P. Wang, H. Xing, N. Li and X. Ji, Polym. Chem., 2015, 53, 2079–2084 Search PubMed.
- H. Nakamoto and J. C. A. Bardwell, Biochim. Biophys. Acta, 2004, 1694, 111–119 Search PubMed.
- A. Zapun, J. C. A. Bardwell and T. E. Creighton, Biochemistry, 1993, 32, 5083–5092 Search PubMed.
-
(a) J. C. A. Bardwell and J. Beckwith, Cell, 1993, 74, 769–771 Search PubMed;
(b) A. S. Patel and W. J. Lees, Bioorg. Med. Chem., 2012, 20, 1020–1028 Search PubMed.
-
(a) T. Shi and D. L. Rabenstein, Tetrahedron Lett., 2001, 42, 7203–7206 Search PubMed;
(b) H. F. Gilbert, J. Biol. Chem., 1997, 272, 29399–29402 Search PubMed;
(c) J. D. Gough, R. H. Williams, A. E. Donofrio and W. J. Lees, J. Am. Chem. Soc., 2002, 124, 3885–3892 Search PubMed;
(d) T. E. Creighton and D. P. Goldenberg, J. Mol. Biol., 1984, 179, 497–526 Search PubMed.
-
(a) J. D. Thomas and T. R. Burke, Tetrahedron Lett., 2011, 52, 4316–4319 Search PubMed;
(b) Y. Akiyama, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2004, 15, 424–427 Search PubMed;
(c) D. R. Grassetti and J. F. Murray Jr, Arch. Biochem. Biophys., 1967, 119, 41–49 Search PubMed.
-
(a) T. Ishii, M. Yamada, T. Hirase and Y. Nagasaki, Polym. J., 2005, 37, 221–228 Search PubMed;
(b) J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Polym. Chem., 2009, 47, 4302–4313 Search PubMed.
- A. J. William, R. D. Ovadia and G. S. Giese, Ind. Eng. Chem. Res., 2017, 56, 1713–1722 Search PubMed.
-
(a) S. Takeda, N. Kamiya, R. Arai and T. Nagamune, Biochem. Biophys. Res. Commun., 2001, 289, 299–304 Search PubMed;
(b) J. A. J. Arpino, H. Czapinska, A. Piasecka, W. R. Edwards, P. Barker, M. J. Gajda, M. Bochtler and D. D. Jones, J. Am. Chem. Soc., 2012, 134, 13632–13640 Search PubMed.
-
(a) E. A. Dolgopolova, D. E. Williams, A. B. Greytak, A. M. Rice, M. D. Smith, J. A. Krause and N. B. Shustova, Angew. Chem., Int. Ed., 2015, 54, 13639–13643 Search PubMed;
(b) D. A. Hanna, R. M. Harvey, O. Martinez-Guzman, X. Yuan, B. Chandrasekharan, G. Raju, F. W. Outeen and A. R. Reddi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7539–7544 Search PubMed;
(c) S. Basak, N. Saikia, L. Dougherty, Z. Guo, F. Wu, F. Mindlin, J. W. Lary, J. L. Cole, F. Ding and M. E. Bowen, J. Mol. Biol., 2021, 433, 166793 Search PubMed;
(d) M. Sadoine, M. Reger, K. M. Wong and W. B. Frommer, ACS Sens., 2021, 6, 1779–1784 Search PubMed;
(e) N. Soleja, O. Manzoor, P. Nandal and M. Mohsin, Org. Biomol. Chem., 2019, 17, 2413–2422 Search PubMed.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 Search PubMed.
- S. E. Webber, Photochem. Photobiol., 1997, 65, 33–38 Search PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815–829 Search PubMed.
- R. Heim, A. B. Cubitt and R. Y. Tsien, Nature, 1995, 373, 663–664 Search PubMed.
- M. J. Patel, G. Yilmaz, L. Bhatia, E. E. Biswas-Fiss and S. B. Biswas, MethodsX, 2018, 5, 419–430 Search PubMed.
- L. M. Constantini, M. Baloban, M. L. Markwardt, M. A. Rizzo, F. Guo, V. V. Verkhusha and E. L. Snapp, Nat. Commun., 2015, 6, 7670–7683 Search PubMed.
- GFPK25C and GFPS174C contain cysteine residues at the 25th and 174th positions respectively, after mutating the inherent 47th cysteine to alanine..
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- The fluorescence decay of GFPK25C was fitted by a first-order exponential equation suggesting a single lifetime,23 while a multi-exponential equation was used for the hetero-dimers due to more complex decays possibly showing characteristics of conformational heterogeneity (Table 1)..
- The distances from the Fe centre to Cα of the mutated residue were estimated by the NMR structure of wild type Cyt b562 (1QPU), while the distances from the chromophore centre to the α carbon of the mutated residue were estimated by the crystal structure of wild type GFP (6IR7)..
- M. Sun, Y. Wang, Q. Zhang, Y. Xia, W. Ge and D. Guo, BMC Genomics, 2017, 18, 279–289 Search PubMed.
- K. Oohora, N. Fujimaki, R. Kajihara, H. Watanabe, T. Uchihashi and T. Hayashi, J. Am. Chem. Soc., 2018, 140, 10145–10148 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Takashi Hayashi
* and
Takashi Hayashi