
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of O–H⋯O/S conventional hydrogen bonds in considerable Csp2–H blue-shift in the binary systems of acetaldehyde and thioacetaldehyde with substituted carboxylic and thiocarboxylic acids†
Nguyen Truong An a,
Nguyen Thi Duonga,
Nguyen Ngoc Triab and
Nguyen Tien Trung*ab
a,
Nguyen Thi Duonga,
Nguyen Ngoc Triab and
Nguyen Tien Trung*ab
aFaculty of Natural Sciences, Quy Nhon University, Quy Nhon, Vietnam. E-mail: nguyentientrung@qnu.edu.vn
bLaboratory of Computational Chemistry and Modelling (LCCM), Quy Nhon University, Quy Nhon, Vietnam
First published on 9th December 2022
Abstract
Stable binary complexes of RCZOH⋯CH3CHZ (R = CH3, H, F; Z = O, S) are due to contributions from the O–H⋯O/S and Csp2–H⋯O/S hydrogen bonds. The strength of Csp2/O–H⋯O is 1.5 to 2 times greater than that of the Csp2/O–H⋯S bond. The substitution of H(Csp2) of HCZOH by CH3 causes a decrease in complex stability, while the opposite trend occurs for the F atom. A very large red shift of the O–H stretching frequency in O–H⋯O/S bonds was observed. A surprising Csp2–H blue shift up to 104.5 cm−1 was observed for the first time. It is found that the presence of O–H⋯O/S hydrogen bonds and a decisive role of intramolecular hyperconjugation interactions in the complex cause a significant blue shift of the Csp2–H covalent bonds. A striking role of O compared to the S atom in determining the blue shift of Csp2–H stretching vibration and stability of binary complexes is proposed. The obtained results show that the ratio of deprotonation enthalpy and proton affinity could be considered as an index for the classification of the non-conventional hydrogen bond. SAPT2+ results show that the strength of RCSOH⋯CH3CHS complexes is dominated by electrostatic and induction energies, while a larger contribution to the stability of remaining complexes is detected for the electrostatic component.
1. Introduction
Despite exhibiting much lower energy and directionality in comparison to covalent bonds, non-covalent interactions still demonstrate a dominant role owing to their ubiquity in nature and the contribution to the cohesion of chemical systems. The most significant type in non-covalent interactions is the hydrogen bond, which is of great importance in chemistry, biochemistry, and especially in biological systems: DNA, RNA, and proteins.1–3 The A–H⋯B conventional hydrogen bond is a weak non-covalent interaction, in which A and B are highly electronegative elements, and B usually contains a region of high electron density (a lone pair, a negative charge, or a π-system). Conventional hydrogen bonds (so-called red-shifting hydrogen bonds) characterized by A–H bond elongation along with a decrease in its stretching vibration are well-known, and this frequency shift is due to electrostatic attraction between the H atom and B atom.4 The red shift of the A–H stretching vibrational frequency of the A–H bond is proposed from the n(B) → σ*(A–H) intermolecular electron density transfer overcoming the increase in s-character and the A–H bond polarization.5 Nonconventional hydrogen bonds with an A–H contraction associated with an increase in its stretching vibration were discovered in complexes of fluoroparaffins with pyridine, acetone, and dioxane by Trudeau et al. in 1980.6 Such a nonconventional hydrogen bond is named a blue-shifting hydrogen bond.Several hypotheses from both theoretical and experimental research works have been proposed to unravel the phenomenon of blue-shifting hydrogen bonds,2,7–13 however, it has not been deeply understood yet. For instance, Wu et al. indicated that the reason for the shift of A–H stretching vibration arises from the short-range hyperconjugative interaction of n(B) → σ*(A–H) electron density transfer in competition with the long-range electrostatic interaction of the A–H bond and B atom.14 In another report, based on different physical components of interaction energy, Mao et al. suggested that an enhancement of A–H stretching vibration is encouraged by Pauli repulsion, whereas a combination of electrostatic and dispersion forces causes diminishing of A–H stretching vibration.7 The electrostatic and Pauli repulsion energies dominating the sum of polarization and charge transfer interaction define an A–H bond contraction and an increase in its stretching frequency.13 An interesting approach that notices the inherent characteristics of proton donor and acceptor monomers to unfold the origin of blue-shifting hydrogen bonds has been proposed.15–18
The blue-shifting hydrogen bond is mainly discovered in complexes involving C–H bond acting as a proton donor, such as in C–H⋯O/N/halogen/π hydrogen bonds, which are significant due to their abundance and diversity in nature.19–29 Studies were then further expanded in characterizing the blue-shifting hydrogen bond based on the different hybridization types of the C atom in the C–H bond.30–32 It is noteworthy that for the demand for elucidating the characteristics and nature of the blue-shifting hydrogen bond, the non-conventional hydrogen bond involving the Csp3–H bond as a proton donor has been experimentally and theoretically examined much more than the Csp2–H and Csp–H one. A few publications suggested that for the same proton acceptor, the lower the polarity of the proton donor, the larger the blue-shift of stretching frequency, and vice versa.33–35 Consequently, most reports have focused on the Csp3–H bond with its lowest polarity as compared to both the Csp2–H and Csp–H ones. Indeed, in 1999, Hobza et al. used double-resonance infrared ion-depletion (IR-R2PI) spectroscopy to give a predicted blue shift of the Csp3–H stretching frequency of 14 cm−1 for the chloroform⋯fluorobenzene complex.36 A slight Csp3–H stretching frequency blue-shift of 8.7 cm−1 in Csp3–H⋯N nonconventional hydrogen bond was recently observed in the complex between chloroform with acetonitrile in the gas phase using Fourier transform IR (FTIR) spectroscopy by Behera et al.27 Remarkably, a similar change of the Csp3–H stretching frequency from blue shift in the gas phase to red shift in the argon matrix was also observed in complexes of deuterated chloroform with acetone and cyclohexanone.37
Recently, the blue shift of the stretching frequency was found in the Csp2–H bond and the extent of stretching frequency was even much higher than that of the Csp3–H bond as a proton donor, in spite of the larger polarity of the former with respect to the latter one. Indeed, a Csp2–H significant blue shift by 81 ÷ 96 cm−1 in the Csp2–H⋯O hydrogen bond formed in the complexes of formic acid with formaldehyde and thioformaldehyde has been recently discovered.33 An investigation at the B3LYP/6-311++G(d,p) level in the interaction between CH3CHO with two molecules of water predicted a significant blue shift of the Csp2–H vibration amounting to 93 cm−1 in the Csp2–H⋯O hydrogen bond.38 Our group's further reports of the considerable blue shift of Csp2–H stretching vibration in the Csp2–H⋯O/S nonconventional hydrogen bonds in XCHO⋯2H2Z (X = H, F, Cl, Br; Z = O, S, Se, Te) complexes were given in ref. 39. Following complexation, a decrease in the stretching frequency of the O–H bond in the dimers of carboxylic acids40,41 and an increase in Csp2–H stretching vibrational frequency in the complexes of acetaldehyde42–44 were reported. Kaur et al. suggested that the blue shift of Csp2–H stretching vibrational frequency results from the presence of the O–H⋯O bond in the complexes formed between carbonyl compounds and hydrogen bond donors HOR (R = H, Cl, CH3, NH2, C(O)H, C6H5).45
Although some publications related to the considerable blue shifts of Csp2–H stretching vibrational frequencies in the complexes containing carbonyl (Csp2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group have been reported,33,35,46–48 a systematic investigation of binary complexes stabilized by hydrogen bonds with the Csp2–H bond acting as the proton donor for having an insight into the nature of blue-shifting hydrogen bonds is desirable. As a consequence, a study of the complexes formed by interactions of acetaldehyde (CH3CHO) and thioacetaldehyde (CH3CHS) with the substituted carboxylic and thiocarboxylic acids (RCZOH, with R = F, H, CH3, and Z = O, S) is performed in this work to acquire a better understanding of the characteristics of conventional and non-conventional hydrogen bonds along with their mutual impact. The impact of the presence of the O–H⋯O/S hydrogen bond on the stability of Csp2–H⋯O/S nonconventional hydrogen bonds and the blue shift of Csp2–H stretching vibration is estimated. In addition, the effect of the R substituents of RCZOH (F and CH3) acting as electron-withdrawing and electron-donating groups on the strength of binary complexes as well as the characteristics and stability of O–H⋯O/S and Csp2–H⋯O/S hydrogen bonds are evaluated. Furthermore, the role of O compared to S in CH3CHZ in affecting the characteristics of non-conventional and conventional hydrogen bonds is investigated thoroughly.
O) group have been reported,33,35,46–48 a systematic investigation of binary complexes stabilized by hydrogen bonds with the Csp2–H bond acting as the proton donor for having an insight into the nature of blue-shifting hydrogen bonds is desirable. As a consequence, a study of the complexes formed by interactions of acetaldehyde (CH3CHO) and thioacetaldehyde (CH3CHS) with the substituted carboxylic and thiocarboxylic acids (RCZOH, with R = F, H, CH3, and Z = O, S) is performed in this work to acquire a better understanding of the characteristics of conventional and non-conventional hydrogen bonds along with their mutual impact. The impact of the presence of the O–H⋯O/S hydrogen bond on the stability of Csp2–H⋯O/S nonconventional hydrogen bonds and the blue shift of Csp2–H stretching vibration is estimated. In addition, the effect of the R substituents of RCZOH (F and CH3) acting as electron-withdrawing and electron-donating groups on the strength of binary complexes as well as the characteristics and stability of O–H⋯O/S and Csp2–H⋯O/S hydrogen bonds are evaluated. Furthermore, the role of O compared to S in CH3CHZ in affecting the characteristics of non-conventional and conventional hydrogen bonds is investigated thoroughly.
2. Computational methods
The optimized geometries of the monomer, the complexes, and their stretching vibrations are determined on the potential energy surface using the Gaussian 09 package49 using MP2 (second-order Moller–Plesset perturbation theory)50 combined with 6-311++G(3df,2pd).51,52 Proton affinity (PA) at the O and S sites and deprotonation enthalpy (DPE) of C–H and O–H bonds in the isolated monomers were calculated at the CCSD(T)/6-311++G(3df,2pd) level. Interaction energy (ΔE*) corrected by both zero-point vibrational energy (ZPE) and basis set superposition error (BSSE)53 was carried out at the CCSD(T)/6-311++G(3df,2pd) based on the optimizing geometries at the MP2/6-311++G(3df,2pd) level by the supramolecular method, in which, ΔE* is defined as the difference in total electronic energy between the stable complexes (Ecomplex) and the sum of two optimized monomers (Emonomer-1, Emonomer-2) given by| ΔE* = Ecomplex − (Emonomer-1 + Emonomer-2) |
AIM (Atom In Molecule) analysis at MP2/6-311++G(3df,2pd) was performed to determine the bond critical points (BCP), electron density (ρ(rC)), the Laplacian of electron density (∇2ρ(rC)) using AIMAll package.54 The local electron energy density (H(rC)) at BCP was computed by H(rC) = G(rC) + V(rC), in which V(rC) and G(rC) are the electron potential energy density and the electron kinetic energy density, respectively. The individual energy of each hydrogen bond (EHB)55–58 was calculated based on the empirical expression revealed by topological analyses of the electron density: EHB = 0.5 V(rC).58 A natural bond orbital (NBO) analysis at MP2/6-311++G(3df,2pd) using the GenNBO5.G program59 for a comprehensive understanding of the nature of hydrogen bond was carried out by calculating the hyperconjugation energies for intramolecular and intermolecular electron density transfer and orbital occupancies. NCI plot description is further utilized to visualize the weak hydrogen bonds.60,61 Moreover, the Molecular Electrostatic Potential (MEP) analysis24,62 of the monomers is investigated at the MP2/6-311++G(3df,2pd) level. In particular, electrostatic potential, V(r) has been considered to be an important property for analyzing the nature and predicting the strength of several non-covalent interactions. Herein, we used the maximum and minimum surface electrostatic potentials denoted by Vs,max and Vs,min, respectively, in order to characterize hydrogen-bonded complexes. Generally, V(r) is created by the nuclei and electrons of a molecule at any point r in the surrounding space, given by,
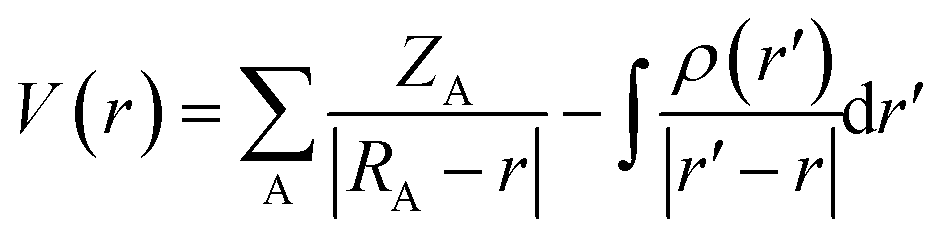
In which, ZA is the charge on nucleus A, located at RA, and ρ(r′) is the molecule's total electronic density with r′ as a dummy integration variable.
To evaluate the physically meaningful components contributing to the stability of investigated complexes, the SAPT (Symmetry-Adapted Perturbation Theory) analysis63 using Psi4 program64 at the MP2/6-311++G(3df,2pd) level was employed. The total interaction energy (ΔESAPT2+) can be divided into the four basic components: exchange-repulsion (Eexch), electrostatic (Eelst), induction (Eind), dispersion (Edisp) terms, and is defined by the following:
| ΔESAPT2+ = Eelst + Eexch + Eind + Edisp + δEHFint,r |
3. Results and discussion
3.1. Structural pattern and stability of complexes
Stable structures of binary complexes of CH3CHZ and RCZOH (with Z = O, S; R = CH3, H, F) at the MP2/6-311++G(3df,2pd) level are shown in Fig. 1a. Twelve stable complexes located on potential energy surfaces are presented in detail in Fig. S1 in the ESI.† Some typical parameters in the observed complexes are tabulated in Table S1.† Briefly, investigated complexes are named RZ–Z, in which, R is CH3, H, and F substitution groups. The O atom or S atom is represented by Z in RCZOH and CH3CHZ, denoted as Z2 and Z7 in the complexes. The sp2-hybridized carbon atom in CH3CHZ is labelled hereafter as Csp2. It can be seen that all structures contain a ring formed by two intermolecular interactions: one Csp2–H⋯Z2 with an obtuse angle (<180°) and one O–H⋯Z7 with an angle of ∼180° (Table S1†). The chalcogen atoms (Z) act as proton-accepting centers and Csp2–H or O–H bonds in the isolated monomers are proton-donating sites.40,65,66 Remarkably, a rotational spectrum study of formic acid – isopropylformate by Spada et al. suggested a similar geometry with hydrogen bonds including O–H⋯O and Csp2–H⋯O.67 Besides, as shown in Table S1,† the distances of H⋯O7, H⋯S7, H⋯O2, and H⋯S2 contacts are close to the sum of the van der Waals radii of relevant atoms (this sum is 3.00 Å and 2.72 Å for the corresponding pair of S and H, O, and H atoms). The lengths of H⋯O distances in Csp2–H⋯O33,45 and O–H⋯O33,45,68 interactions are comparable to those in the complexes of this work. Additionally, the H⋯S distances of O–H⋯S7 hydrogen bond in HO–S are close to those obtained at the MP2/aug-cc-pVDZ level.33 These results roughly suggest the presence of O–H⋯Z7 conventional and Csp2–H⋯Z2 nonconventional hydrogen bonds in the binary complexes.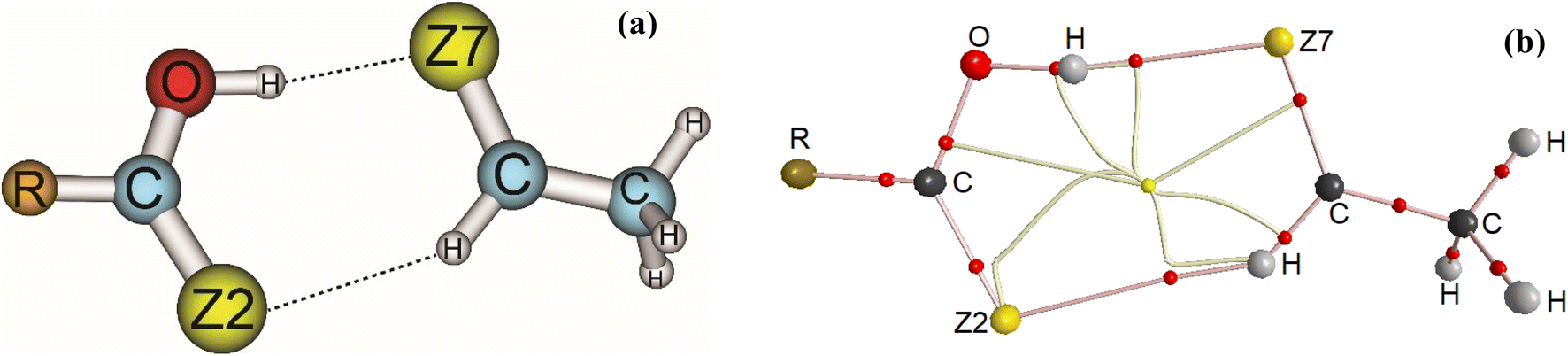 | ||
| Fig. 1 Geometries (a) and topological features (b) of RCZOH⋯CH3CHZ complexes (with R = CH3, H, F; Z = O, S) at MP2/6-311++G(3df,2pd). | ||
Concerning the Csp2–H⋯Z2 bonds, the H⋯Z2 distance is increased when R is an electron-withdrawing group (F) while the opposite is true for an electron-donating group (CH3). Meanwhile, a converse trend is obtained with O–H⋯Z7 bonds. To explain this observation, the effect of the proton-donating or -accepting ability of the isolated monomers on the strength of hydrogen bonds is evaluated through deprotonation enthalpy (DPE) of O/Csp2–H bonds and proton affinity (PA) at the Z sites of CH3CHZ and RCZOH (Table 1). By changing the R substitution in RCZOH, the O–H⋯Z7 intermolecular distance in complexes increases in the order of F < H < CH3. This change accompanies a decrease in the polarity of the O–H bond in RCZOH as one goes from F to H to CH3 substitution (Table 1). Besides, the Csp2–H⋯Z2 intermolecular distance decreases in the substitution order: CH3 > H > F, which relates directly to the increasing proton affinity of the Z2 site in RCZOH.
| Monomer | CH3CHO | CH3CHS | CH3COOH | CH3CSOH | HCOOH | HCSOH | FCOOH | FCSOH |
|---|---|---|---|---|---|---|---|---|
| a Experimental values in parentheses are taken from the NIST webpage (https://webbook.nist.gov/chemistry/). | ||||||||
| PA (Z), kJ mol−1 | 767.2 (768.5) | 807.1 | 768.2 (783.7) | 809.3 | 722.3 (742.0) | 770.3 | 698.5 | 744.0 |
| DPE (Csp2/O–H), kJ mol−1 | 1641.0 (1645.1 ± 4.0) | 1610.0 (1461.0 ± 17.0) | 1454.8 (1457.0 ± 5.9) | 1392.7 | 1440.1 (1449.0 ± 5.0) | 1381.9 | 1366.5 | 1312.4 |
| (1) DPE of Csp2–H in CH3CHZ | DPE(1)/PA(2) | CH3O–O | CH3S–O | HO–O | HS–O | FO–O | FS–O |
| 2.14 | 2.03 | 2.27 | 2.13 | 2.35 | 2.21 | ||
| (2) PA of Z = O/S in RCZOH | DPE(1)/PA(2) | CH3O–S | CH3S–S | HO–S | HS–S | FO–S | FS–S |
| 2.10 | 1.99 | 2.23 | 2.09 | 2.30 | 2.16 |
To evaluate the role of conventional and nonconventional hydrogen bonds in stabilizing the complexes, the second-order perturbative energies Einter from NBO calculations at the MP2/6-311++G(3df,2pd) level are given in Table 2. The large positive values of electron density transfer (EDT) of ca. 0.039–0.079 e show that the electron density is considerably transferred from CH3CHZ to RCZOH following complexation. These EDT values are taken from a stronger electron density transfer from n(Z7) to σ*(O–H) orbital versus n(Z2) to σ*(Csp2–H) one in each binary complex. The values of Einter[n(Z7) → σ*(O–H)] (ca. 22.6–37.4 kJ mol−1) are indeed 10 ÷ 17 times larger than Einter[n(Z2) → σ*(Csp2–H)] (ca. 1.4–3.1 kJ mol−1). It could be thus drawn that the charge-transfer interaction of n(Z7) → σ*(O–H) in the conventional hydrogen bond plays a dominant role in stabilizing each complex, as compared to those of the n(Z2) → σ*(Csp2–H) electron transfer in the non-conventional ones.
| Complex | EDT (e) | Δ% s (%) | Δσ* (e) | Einter (kJ mol−1) | ΔEintra (kJ mol−1) | |||
|---|---|---|---|---|---|---|---|---|
| O(H) | Csp2(H) | O–H | Csp2–H | n(Z7) → σ*(O–H) | n(Z2) → σ*(Csp2–H) | n(Z7) → σ*(Csp2–H) | ||
| a EDT: electron density transfer; Δ% s: change in s-character percentage; Δσ*: change in electron density of anti-bonding orbital; Einter: intermolecular hyperconjugation interaction energy; ΔEintra: difference of intramolecular hyperconjugation interaction energy in isolated monomer and complex. | ||||||||
| CH3O–O | 0.039 | 3.71 | 1.37 | 0.041 | −0.012 | 23.1 | 1.7 | −7.6 |
| CH3S–O | 0.040 | 3.99 | 1.16 | 0.043 | −0.007 | 26.2 | 2.5 | −7.2 |
| HO–O | 0.042 | 3.81 | 1.31 | 0.043 | −0.012 | 25.6 | 1.7 | −7.8 |
| HS–O | 0.041 | 4.14 | 1.12 | 0.044 | −0.007 | 27.8 | 2.4 | −7.4 |
| FO–O | 0.055 | 3.82 | 1.26 | 0.055 | −0.015 | 35.6 | 1.4 | −8.7 |
| FS–O | 0.056 | 3.99 | 1.15 | 0.060 | −0.011 | 37.4 | 2.2 | −8.6 |
| CH3O–S | 0.050 | 3.52 | 1.62 | 0.054 | −0.004 | 22.6 | 2.6 | −3.9 |
| CH3S–S | 0.054 | 3.82 | 1.41 | 0.061 | 0.002 | 26.0 | 3.1 | −3.2 |
| HO–S | 0.056 | 3.73 | 1.55 | 0.059 | −0.004 | 25.1 | 2.6 | −3.8 |
| HS–S | 0.057 | 4.03 | 1.39 | 0.064 | 0.002 | 28.4 | 3.1 | −3.4 |
| FO–S | 0.076 | 3.99 | 1.50 | 0.074 | −0.006 | 33.0 | 2.1 | −4.1 |
| FS–S | 0.079 | 4.18 | 1.39 | 0.083 | −0.001 | 37.0 | 2.7 | −3.7 |
Interaction energies (ΔE*) including ZPE and BSSE corrections of the studied complexes are listed in Table 3. Generally, the values of the interaction energy range from −27.1 to −46.5 kJ mol−1, implying that the considered complexes are stable. The stability of CH3CHZ⋯RCZOH complexes lies between that of (CH3CHZ)2 and (RCZOH)2 dimers, with R = CH3, H, F, and Z = O, S. Particularly, the values of the interaction energy of (HCOOH)2 and (CH3COOH)2 are calculated to be in turn −61.9 and −66.6 kJ mol−1 at the MP2/aug-cc-pVTZ level40 or −56.9 and −60.7 kJ mol−1 at the MP2/6-311++G(d,p),41 respectively. The less negative interaction energy found for (CH3CHO)2 dimer was −14.8 kJ mol−1 at the M062X/6-311++G(3df,3pd) level42 or −11.6 kJ mol−1 at the MP2/6-311++G(d,p) level.44 The investigated complexes are more stable than CH3CHS⋯HOBr (ΔE* is −22.8 kJ mol−1 at the MP2/aug-cc-pVTZ level)69 and CH3CHO⋯H2O and CH3CHS⋯H2O (ΔE* are −15.3 and −11.7 kJ mol−1, respectively, at CCSD(T)/6-311++G(3df,2pd)//MP2/6-311++G(3df,2pd)).70
| Complex | Eelst | Eind | Edisp | δEHF | Eexch | ESAPT2+ | ΔE* | BSSE |
|---|---|---|---|---|---|---|---|---|
| a The values in parentheses for the contribution percentage (%) of energy component on the stability of complexes. | ||||||||
| CH3O–O | −70.6(46) | −44.0(29) | −25.4(17) | −12.5(8) | 98.9 | −53.6 | −35.7 | 5.13 |
| CH3S–O | −72.5(43) | −52.8(31) | −27.8(17) | −15.1(9) | 117.8 | −50.3 | −35.0 | 5.86 |
| HO–O | −73.3(46) | −46.6(29) | −25.3(16) | −13.5(9) | 102.9 | −55.9 | −36.5 | 5.14 |
| HS–O | −74.5(43) | −54.6(32) | −27.7(16) | −16.0(9) | 120.3 | −52.5 | −35.3 | 5.85 |
| FO–O | −81.4(46) | −53.4(30) | −26.0(15) | −16.0(9) | 112.3 | −64.5 | −46.5 | 5.90 |
| FS–O | −83.3(43) | −60.6(32) | −28.7(15) | −18.8(10) | 129.1 | −62.3 | −46.4 | 6.50 |
| CH3O–S | −53.0(40) | −43.8(33) | −25.0(19) | −11.0(8) | 95.8 | −37.0 | −28.7 | 5.42 |
| CH3S–S | −52.8(36) | −52.7(36) | −27.5(19) | −12.8(9) | 114.6 | −31.2 | −27.1 | 6.00 |
| HO–S | −54.3(40) | −46.5(34) | −24.8(18) | −11.9(9) | 99.4 | −38.1 | −28.8 | 5.34 |
| HS–S | −54.1(36) | −54.7(36) | −27.4(18) | −13.6(9) | 117.3 | −32.6 | −27.5 | 6.00 |
| FO–S | −58.5(38) | −54.7(36) | −25.2(17) | −13.9(9) | 106.5 | −45.8 | −36.2 | 6.01 |
| FS–S | −58.1(36) | −60.7(37) | −27.9(17) | −15.7(10) | 124.7 | −37.7 | −34.7 | 6.04 |
With the same atoms Z2 and Z7, the interaction energies of complexes tend to decrease from CH3Z–Z to HZ–Z and finally to FZ–Z. As a result, the substitution of H by F attached to the carboxyl group in HCZOH gives rise to an enhancement of the stabilization energy, while the attachment of the CH3 group diminishes this value. The Einter[n(Z7) → σ*(O–H)] values thus increase in the order of CH3 < H < F substitutions, which are in line with the tendency of enhancement in the polarity of O–H covalent bonds in RCZOH (cf. Table 1). Following this substitution, as observed in Table 2, the Einter[n(Z2) → σ*(Csp2–H)] values decrease slightly, which is due to a lower proton affinity at the Z2 site in the isolated isomers. This indicates a dominant role of the O–H⋯O/S compared to Csp2–H⋯O/S hydrogen bonds in stabilizing the complexes. More negative values of the interaction energy were obtained for CH3CHO⋯HCOOH (−39.8 kJ mol−1) in comparison with CH3CHO⋯CH3COOH (−38.0 kJ mol−1) at the B3LYP-D3/aug-cc-pVTZ level,68 which shows that the H substitution of the Csp2–H group in HCOOH by the CH3 group causes a decrease in the stability of hydrogen-bonded complexes. This observation is also similar to the obtained result in this work and the complexes of alcohol derivatives and amine.71
The values of the maximum surface electrostatic potential (Vs,max) at the H atom of –OH groups in substituted carboxylic and thiocarboxylic acids (RCZOH) are displayed in Fig. S2.† As expected, Vs,max of the RCZOH molecule at the H atom decreases with the substitution of the CH3 group, whereas, it increases in the presence of the F atom, corresponding to the changing trend of the interaction energy in the complexes. Indeed, the F substituted RCZOH having the highest Vs,max is accompanied by the largest negative interaction energy, whereas the CH3 substitution having the lowest Vs,max is associated with the least negative interaction energy. The linear correlations (R2 ≈ 0.9) between the interaction energy of the complexes and the magnitude of Vs,max value are observed in this study, as given in Fig. S3 of ESI.† It shows that the stability of the investigated complexes increases when there is an enhancement of the positive electrostatic potential of the H atom in RCZOH monomers and vice versa.
With the same substituent groups R and Z7, the interaction energy of RO–Z is slightly more negative than that of RS–Z, around 0.1–1.6 kJ mol−1, while Einter[n(S2) → σ*(Csp2–H)] is larger than Einter[n(O2) → σ*(Csp2–H)]. This indicates that the larger strength of Csp2–H⋯O2 versus Csp2–H⋯S2 is caused by the electrostatic attraction between H and Z2 overcoming the charge transfer from n(Z2) to σ*(Csp2–H) orbital. Indeed, the NBO charges given in Table S1 of ESI† show a larger positive charge of the H atom in the Csp2–H⋯O2 bond in RO–Z, as compared to that in Csp2–H⋯S2 in RS–Z. Besides the charge of the O2 atom is also more negative than that of the S2 atom in the complexes. This signifies a stronger electrostatic attraction between O2 and H in the Csp2–H⋯O2 bond than between S2 and H in the Csp2–H⋯S2 bond.
With the same R and Z2, the RZ–O is ca. 7.0–11.7 kJ mol−1 in energy, which is more stable than RZ–S, although electron transfer energy from n(S7) to σ*(O–H) orbital is stronger than that from n(O7) to σ*(O–H) (cf. Tables 2 and 3). Consequently, the stability of the complexes is governed by the electrostatic interaction of Z7 and H atoms as opposed to the energy for the charge transfer from n(Z7) to σ*(O–H) orbital. In short, it is apparent that the larger stability of RO–Z versus RS–Z or RZ–O versus RZ–S comes from the contribution of electrostatic attraction between O and H atoms, overwhelming that between S and H atoms. Indeed, as seen from Table S1,† the NBO charges on O7 atoms have large negative values, whereas positive charges are found on S7 atoms. In addition, more positive charges of H of the –OH group in RZ–O, as compared to those in RZ–S are also observed (cf. Table S1†). Accordingly, the electrostatic attraction between O7 and H in O–H⋯O7 bond is stronger than that between S7 and H in O–H⋯S7 bond. Similarly, the larger stability of HCOOH⋯CH3CHO, as compared to that of HCOOH⋯CH3CHS at the MP2/aug-cc-pVTZ level studied by Trung et al.33 indicates a more important role of the O atom in comparison with the S atom in stabilizing hydrogen-bonded complexes.
The energy components72 were evaluated by employing the SAPT2+ procedures at the MP2/6-311++G(3df,2pd) basis set (Table 3). The calculated results indicate that the interactions in the investigated complexes are mainly contributed by Eelst (36–46%) and Eind (29–37%) components, while Edisp and δEHF only contribute less, half of those, by 15–19% and 8–10%, respectively. As a consequence, the electrostatic and induction components play an important role in stabilizing the complexes, especially, a considerable role of electrostatic energy in complex stabilization. An approximation of the contributions in the percentage of physical energy terms to the stability of the complexes for both electron-donating (CH3) and electron-withdrawing (F) substituent groups was observed (cf. Fig S4†). A great contribution of the electrostatic term to the complex stability occurs in RZ–O in comparison to RZ–S, making the former to be about 1.3–1.4 times more stable than the latter (Table 3). The high values of Eexch seem to be the additional reason for the less stable RS–Z versus RO–Z. Besides, there are approximate contributions of Eelst and Eind components, about 36–37% in RS–S.
3.2. AIM and NCI analyses
In order to clarify the existence and stability of conventional and nonconventional hydrogen bonds in the binary complexes, an AIM analysis was performed and its results showed the bond critical points (BCP) in red between the lines connecting H atoms with Z7 and H atoms with Z2 for all complexes (cf. Fig. 1b and S1 in ESI†). These BCPs again indicate that the O–H⋯Z7 and Csp2–H⋯Z2 contacts in the binary complexes of RCZOH and CH3CHZ are hydrogen bonds. Besides, their existence was confirmed by the values of electron density ρ(rC) and Laplacian at BCPs ∇2ρ(rC), which fall within the criteria for the formation of hydrogen bonds.73 All values of ρ(rC) and ∇2ρ(rC) at these BCPs (cf. Table S2, ESI†) are indeed in the range of 0.0096–0.0521 au and 0.027–0.105 a.u., respectively. The characteristics of conventional and nonconventional hydrogen bonds in the complexes are emphasized by values of local electron energy densities (H(rC)) at BCPs.74,75 The positive values of ∇2ρ(rC) and negative values of H(rC) at BCPs of O–H⋯Z7 contacts in the complexes suggest a partly covalent character of the conventional hydrogen bonds.45 Meanwhile, the positive ∇2ρ(rC) and H(rC) values indicate that the Csp2–H⋯Z2 nonconventional hydrogen bonds are considered to be weak hydrogen bonds.The EHB values from Table S2† suggest that the O–H⋯O7 is twice stabler than O–H⋯S7 and Csp2–H⋯S2 is about 1.5 times less stable than Csp2–H⋯O2. This implies that the O–H⋯Z7 hydrogen bonds keep a substantial part in stabilizing further the complexes, in addition to the stabilization by Csp2–H⋯Z2 hydrogen bonds. This observation is also reported in the hydrogen-bonded complexes between carbonyl derivatives and water.38,46,70 It is found that the contribution to the RZ–Z stabilization is larger for Csp2–H⋯O2/O–H⋯O7 hydrogen bonds than that for Csp2–H⋯S2/O–H⋯S7. With the different substitution of Z2 or Z7 atoms, AIM data shows a larger strength of the Csp2–H⋯O2 hydrogen bond compared to Csp2–H⋯S2,76,77 and a larger strength of the O–H⋯O7 bond in comparison with O–H⋯S7 one.78,79 The second-order correlations between the individual energy of hydrogen bond (EHB) with the value of electron density (ρ(rC)) at the BCP and the distance H⋯Z(r(H⋯Z)) are plotted in Fig. 2. It shows that the more negative the EHB values are, the larger the ρ(rC) values and the shorter the r(H⋯Z) at BCPs. In other words, the ρ(rC) at the BCP of the hydrogen bond is inversely proportional to r(H⋯Z). This result affirms a dominant role of the O–H⋯Z7 bond compared to the Csp2–H⋯Z2 bond in contributing to complexation.
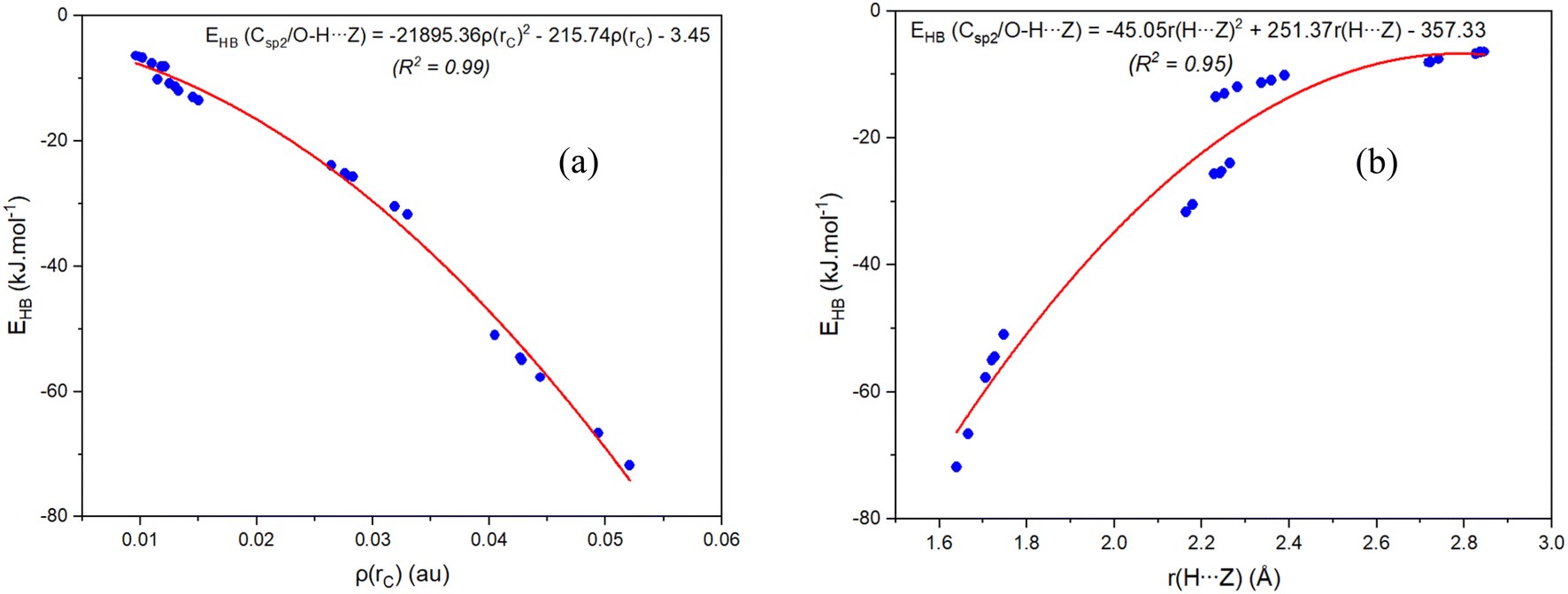 | ||
| Fig. 2 The second-order correlations of individual hydrogen bond energies (EHB) with respect to electron density ρ(rC) at BCPs (a) and intermolecular distances (b) for RCZOH⋯CH3CHZ complexes (Z = O, S; R = CH3, H, F). | ||
For a more obvious understanding of the characteristics of conventional and nonconventional hydrogen bonds in the complexes, NCI (Non-Covalent Interaction) calculations were performed. The reduced density gradient (RDG) isosurfaces and their 2D plots for all complexes are displayed in Fig. S5a and b (in ESI†). In the 2D plots, the low-density and low reduced gradients in the region are where the second eigenvalue (λ2) of the electron-density Hessian matrix is negative, indicating weak and non-covalent attractive interactions between CH3CHZ and RCZOH molecules. It can be seen from Fig. S5a and b† that the volume and site of the tail on the horizontal axis of the blue spike and the green spike in all complexes are not the same. However, in all complexes, the O–H⋯Z7 bond is recognized by the blue spike lying at the higher negative region in comparison with the green spike of the Csp2–H⋯Z2 bond. In RZ–O complexes, the difference in the distance between the tail of the blue spike and the tail of the green spike is larger than that in RZ–S complexes. For instance, with the same site of the tail of the green spike (about −0.01 a.u.), the blue spike for the O–H⋯O7 bond in FS–S lies at about −0.03 a.u., while it lies at −0.05 a.u. for FO–O.
Here, the FO–O complex is chosen as a good example for analysis (Fig. 3). In particular, two spikes (blue and green) are detected in the negative region, confirming again the attendance of O–H⋯Z7 and Csp2–H⋯Z2 hydrogen bonds in the complexes. The O–H⋯Z7 bond is recognized by the blue spike lying at the high negative region (∼0.05 au). This indicates that the O–H⋯Z7 hydrogen bonds are stronger compared to Csp2–H⋯Z2 ones in the complexes, in agreement with the results reported by Lei et al.80 and Cuc et al.39,70 Moreover, the spike found at the positive region with only about 0.01 a.u. in the electron density implies a repulsive non-covalent interaction at the RCP interior to the ring.
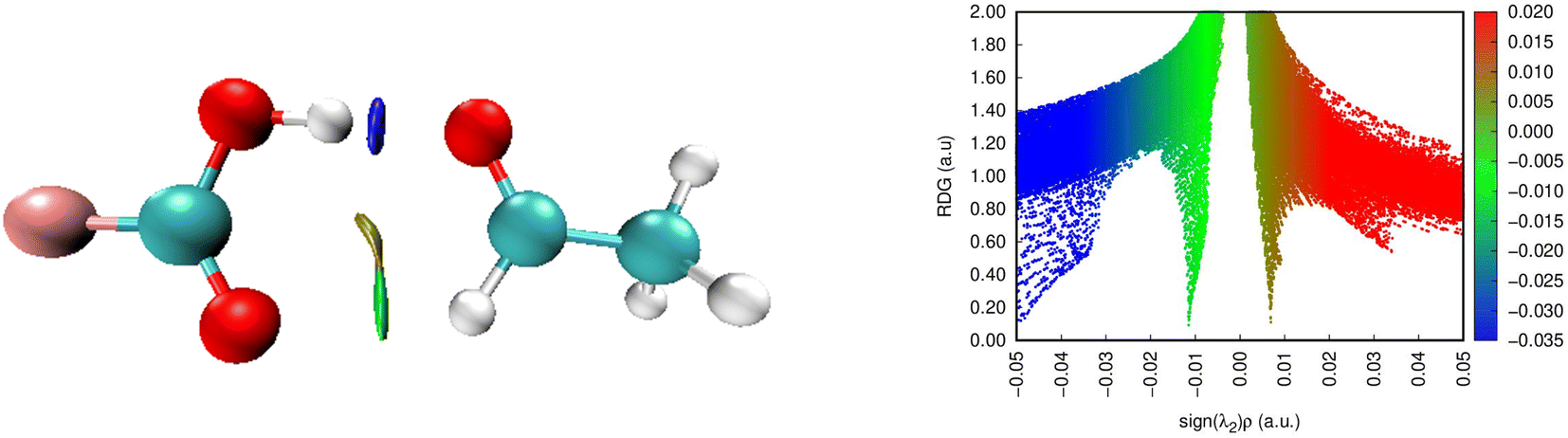 | ||
| Fig. 3 NCI isosurface (3D) and plots of RDG as a function of sign(λ2)ρ(rC) (2D) for FCOOH⋯CH3CHO (the isosurfaces are colored according to the values of sign(λ2)ρ(rC) from −0.05 to 0.05 a.u.). | ||
3.3. Changes in lengths and stretching frequencies of Csp2–H and O–H bonds
The results obtained for the changes in the values of lengths (Δr, mÅ) and stretching frequencies (Δν, cm−1) of O–H and Csp2–H bonds at MP2/6-311++G(3df,2pd) level are tabulated in Table 4 and displayed in Fig. 4. The Csp2–H bond length decreases by 0.5 to 7.6 mÅ, accompanied by an increase in its stretching vibration by 11.0 cm−1 to 104.5 cm−1 in comparison with the corresponding values in the isolated monomers. This observation is remarkable, given that, as introduced in Section 1, a considerable contraction of a Csp2–H bond length has rarely been observed owing to its polarity. The changes in the Csp2–H stretching frequency indicate that all Csp2–H⋯Z2 hydrogen bonds in the studied complexes are blue-shifted. For comparison, a concise summary of the theoretical and experimental publications on the changes in stretching frequencies of the Csp–H, Csp2–H, Csp3–H bonds upon complex formation is given in ESI (Table S3).† It is seen that the red shift in the stretching frequency of the Csp–H bond was found at high levels of theory, while both red or blue shifts of Csp3–H stretching frequencies with weak and moderate magnitude were observed.| Complex | CH3O–O | CH3S–O | HO–O | HS–O | FO–O | FS–O |
|---|---|---|---|---|---|---|
| Δr(O–H) | 19.6 | 20.3 | 21.7 | 21.8 | 28.1 | 30.3 |
| Δν(O–H) | −401.1 | −416.0 | −441.4 | −445.6 | −569.7 | −622.8 |
| Δr(Csp2–H) | −7.0 | −6.5 | −6.8 | −6.4 | −7.6 | −7.5 |
| Δν(Csp2–H) | 96.6 | 83.8 | 95.3 | 82.5 | 104.5 | 96.7 |
| Complex | CH3O–S | CH3S–S | HO–S | HS–S | FO–S | FS–S |
|---|---|---|---|---|---|---|
| Δr(O–H) | 16.3 | 16.9 | 18.1 | 18.4 | 23.4 | 24.8 |
| Δν(O–H) | −344.9 | −358.3 | −382.7 | −389.5 | −494.0 | −528.3 |
| Δr(Csp2–H) | −1.0 | −0.6 | −1.0 | −0.5 | −2.1 | −1.8 |
| Δν(Csp2–H) | 28.1 | 11.9 | 28.3 | 11.0 | 42.3 | 27.7 |
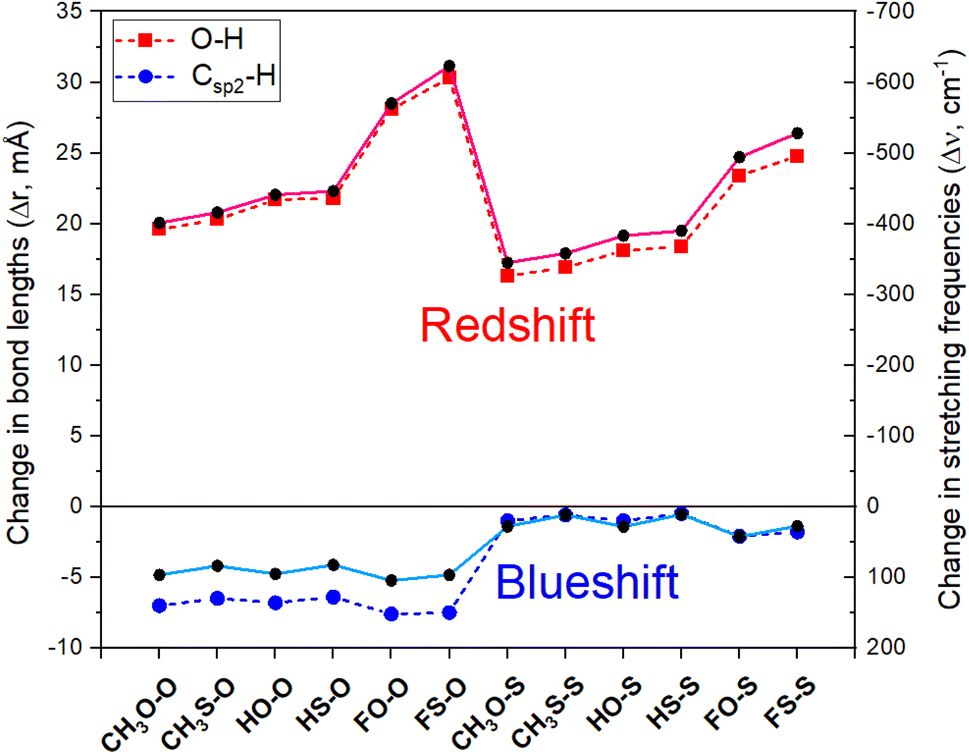 | ||
| Fig. 4 The relationship of changes in Csp2/O–H bond length (Δr in mÅ, dash line, and symbol) and corresponding stretching frequency (Δν in cm−1, solid line and symbol) with different R, Z substitutions for investigated complexes. | ||
The extent of changes in Csp2–H stretching frequencies have been observed in several binary hydrogen-bonded systems (Table S3 in ESI†).33,38,39,46,81 Moderate and strong Csp2–H blue shifts were reported in some complexes between carbonyl/thiocarbonyl compounds with water.38,46,81 Indeed, in complexes between aldehydes and hydrogen chalcogenides (H2O, H2S),39 the Csp2–H stretching frequencies undergo a significant blue shift, amounting to 92 cm−1 for CH3CHO⋯2H2O. The strong blue shift of the Csp2–H stretching frequency (93 cm−1) in the CH3CHO⋯2H2O complex was also found by Chandra et al.38 Remarkably, a strong blue shift of the Csp2–H stretching vibration up to 96 cm−1 in binary complexes between formic acid and acetaldehyde was reported in ref. 33. At the MP2/cc-pVTZ level, Damanjit et al. calculated a significant blue shift of the Csp2–H bond in NH2CHO⋯HCOOH to about 93 cm−1.45 As seen from Table S3† that the blue-shift of the Csp2–H stretching frequency in the dimer of (CH3CHO)2 (including two Csp2–H⋯Z hydrogen bonds) is smaller than that in the HCOOH⋯CH3CHO binary complex (including one Csp2–H⋯Z bond and one O–H⋯O bond). This result emphasizes the important role of O–H⋯O interaction to the large blue-shift of Csp2–H stretching vibration.
Comparing Csp2–H⋯Z types, it is found that the magnitude of the blue shift in the Csp2–H stretching vibrational frequency in O7 = Csp2–H⋯Z2 (RZ–O) is larger than that in S7 = Csp2–H⋯Z2 (RZ–S). This likely relates to the stronger polarity of the Csp2–H bond in CH3CHS with respect to CH3CHO (cf. Table 1). As seen in Fig. 4, a larger contraction of the Csp2–H bond length accompanied by a larger increase in its stretching vibrational frequency was observed for the Csp2–H⋯O2 hydrogen bonds in comparison to the Csp2–H⋯S2. This difference is owing to the larger gas phase Lewis basicity at the S site in RCSOH as compared to that at the O site in RCOOH (cf. Table 1), which denotes a more prominent role of O versus S atom in the larger magnitude of the blue shift of Csp2–H stretching frequency involving hydrogen bond. Importantly, a good correlation between Δν(Csp2–H) and DPE(Csp2–H) along with PA(Z2) is found with R2 ≈ 0.98 (illustrated in Fig. S6†). Hence, the results of the present work demonstrate the fact that the blue shift of stretching vibrational frequency is dependent on the inherent property of the monomer, as discussed in some recent works.18,33–35,39 As seen from Tables 1 and 4, it is found that the higher the DPE/PA ratio for the Csp2–H⋯Z2, the larger blue-shift of the Csp2–H stretching vibration will be, and vice versa. It is also found that the DPE/PA ratio for the Csp2–H⋯Z2 bonds in the complexes with CH3CHZ is larger for Z = O than for Z = S. The highest DPE/PA was estimated in FO–O, and the lowest value was obtained in HS–S. Therefore, it could be proposed that the DPE/PA ratio can be used for the evaluation of the category of hydrogen bonds and the extent of the blue shift of stretching frequency.
The Csp2–H stretching frequency in Csp2–H⋯Z2 bond experiences a strong enhancement upon F substitution in RCZOH compared to H and CH3 substitutions, which has been rarely reported before (cf. Table S3†). It is suggested that a contraction in the length of the Csp2–H bond and a remarkable increase of its stretching vibrational frequencies are affected by the presence of O–H⋯Z7 hydrogen bonds in complexes, especially O–H⋯O hydrogen bonds.45 For instance, the highest extent of the blue shift in Csp2–H⋯Z2 bonds belongs to FO–O (connected by Csp2–H⋯O and O–H⋯O bonds) with a Csp2–H contraction of 7.6 mÅ and a strong increase in its stretching vibration of 104.5 cm−1. This result arises from the largest decrease in the electron density of the σ*(Csp2–H) antibonding orbital (0.015 e), which strengthens the Csp2–H bond and thereby contributes to a blue shift of the Csp2–H stretching vibration.
Table 2 points out that the trend in the change of electron density in the σ*(Csp2–H) orbitals is quite comparable to the change in Csp2–H stretching frequency. Meanwhile, the increase in the percentage of the s-character of Csp2 in the Csp2–H covalent bond is not too much different in the complexes. As a consequence, a decrease of electron density in the σ*(Csp2–H) orbitals, which overwhelms an increase in s character percentage in the hybridization of Csp2(H), governs the magnitude of the blue shift of Csp2–H stretching frequency in the complexes. All complexes including CH3CHO exhibit a large decrease in electron densities in σ*(Csp2–H) (0.007–0.015 e), while a slight increase of 0.002 e or a slight decrease of 0.001 e in the electron density of the σ*(Csp2–H) is estimated for RS–S complexes. Besides, the decreases of the electron density in σ*(Csp2–H) for H or CH3 substituted complexes are comparable and are much smaller than that for F substitutions, which confirms again the strong blue shift of the Csp2–H stretching frequency with the F substituent.
Considering the second-order stabilization energy of interaction11 from Table 2, it shows that a larger decrease in intramolecular hyperconjugation energy of the n(Z7) → σ*(Csp2–H) electron density transfer versus intermolecular hyperconjugation energies of n(Z2) → σ*(Csp2–H) transfer determines the decrease in σ*(Csp2–H) orbital population. It is also noted that a decrease in the hyperconjugation energies associated with intramolecular electron density transfer from n(O7) to σ*(Csp2–H) is twice as large as that from n(S7) to σ*(Csp2–H). A good linear correlation of the changes in the electron density at σ*(Csp2–H) with both Einter[n(Z2) → σ*(Csp2–H)] and ΔEintra[n(Z7) → σ*(Csp2–H)] with R2 ≈ 0.98 is found in Fig S7.† The results show a crucial role in decreasing the hyperconjugation interaction of intramolecular electron transfer from the (Z7) to σ*(Csp2–H) orbital, overcoming the increase in the electron density transfer from the n(Z2) → σ*(Csp2–H) orbital. This result leads to a large decrease in the σ*(Csp2–H) occupation in the complexes, which governs a Csp2–H bond contraction and the associated blue shift. In short, there are two features leading to a significant blue shift of C–H stretching frequency: (i) the presence of O–H⋯Z7 hydrogen bonds in the cyclic structure; and (ii) the decrease in the electron density of σ*(Csp2–H) orbital.
On the other hand, the changes of Δr(O–H) and Δν(O–H), as shown in Fig. 4, denote a strong red shift of O–H stretching frequencies along with its bond elongation in O–H⋯Z7 hydrogen bonds upon complexation. This observation is accompanied by a large enhancement of σ*(O–H) occupation (ca. 0.041–0.083 e) overcoming an increase in the percentage of s-character at O(H) atom (ca. 3.52–4.18%), which does not favor an O–H bond elongation that accompanies an associated red shift of the stretching vibration. From Table 2, the strong hyperconjugation energy for the intermolecular n(Z7) → σ*(O–H) transfer (amounting to 22.6 ÷ 37.4 kJ mol−1) is the reason for the increase in the occupation of σ*(O–H) orbital in complexes. The remarkable red shift of O–H stretching vibration in the O–H⋯O7 hydrogen bond was also observed in the complexes between HCOOH and HCHO derivatives (363 ÷ 604 cm−1)45,68,82,83 or between acetic acid and acetaldehyde (436 cm−1).68 Similar results were also reported in a recent investigation on HCOOH⋯CH3CHS and HCOOH⋯CH3CHO complexes.33
The red shift of O–H stretching frequency of the O–H bond in O–H⋯Z7 hydrogen bonds follows the ordering of CH3 < H < F substitutions, in good agreement with the enhancement of the polarity of the O–H bond in RCZOH in going from CH3 to H and finally to F (Table 1). The red shift of the O–H stretching frequency is greater for RS–Z than for RO–Z, which is in accordance with the higher polarity of the O–H bond in RCSOH, as compared to its value in RCOOH (cf. Table 1). The obtained results indicate that the red shift of O–H stretching frequency in the O–H⋯Z7 hydrogen bond is determined by an increase in σ*(O–H) electron density. From Fig. 4, it is seen that for the same Z2, this O–H stretching frequency red shift in RZ–O is greater than that in RZ–S. Thus, the O–H red-shifting stretching frequency is mainly due to a larger attractive electrostatic force between Z7 and H, which overcomes the n(Z7) → σ*(O–H) charge transfer.
4. Concluding remarks
In this study, twelve stable ring structures of CH3CHZ⋯RCZOH (R = H, F, CH3; Z = O, S) stabilized by O–H⋯O/S and Csp2–H⋯O/S hydrogen bonds, with the interaction energies from −27.1 to −46.5 kJ mol−1 were thoroughly investigated using quantum chemical methods. The substitution of H by the F atom in RCZOH induces an increase in the thermodynamic stability of complexes, while the opposite is true for CH3, which is due to the overwhelming contribution to complex stability of O–H⋯O/S bonds compared to their Csp2–H⋯O/S counterparts. Besides, arising from the electrostatic interaction, a larger role of the O atom in comparison with the S atom in affecting the characteristics and strength of hydrogen bonds as well as the stability of complexes is elucidated in these systems. The strength of Csp2/O–H⋯O is 1.5÷2 times larger than Csp2/O–H⋯S counterparts. SAPT2+ analysis indicates that the attractive electrostatic term plays a crucial role in stabilizing the complexes containing Csp2/O–H⋯O hydrogen bonds. Meanwhile, stabilizing the complexes by Csp2/O–H⋯S hydrogen bonds is due to both electrostatic and induction terms.The Csp2–H stretching frequency in the Csp2–H⋯O/S nonconventional hydrogen bond increases when substituting one H of the –CH group in HCZOH with both, a halogen atom and CH3 group. The largest blue shift of the Csp2–H bond up to 104.5 cm−1 in the FO–O binary complex was observed for the first time, which corresponds to the smallest basicity at the O atom in FCOOH, as compared to that in the other monomers. It is realized that with the presence of O–H⋯O/S bonds, a large decrease in the electron density in σ*(Csp2–H) orbital predominating an increase in the percentage of s-character, causes an abnormal blue-shift of the Csp2–H stretching vibration by ca. 82.5–104.5 cm−1. Besides, the ratio of DPE/PA can be considered a factor in identifying the type of hydrogen bond and the magnitude of blue shift observed upon complexation. NBO results show that the considerable blue shift of Csp2–H stretching vibration is governed by an intramolecular hyperconjugation interaction overcoming the intermolecular one. Moreover, the shift of Csp2–H stretching frequency as a function of net second hyperconjugative energy for the σ*(Csp2–H) orbital was observed.
All the O–H bonds in the O–H⋯O/S conventional hydrogen bonds are red-shifted. The significant elongation of O–H bonds in O–H⋯O/S hydrogen bonds and the strong decrease of their stretching vibrational frequency come from considerable increases in the σ*(O–H) electron density overcoming an increase in the percentage of the s-character of the O/S hybridized atom. It was found that the red shifting of the O–H⋯O bond is larger than that of the O–H⋯S one, and increases in the ordering of CH3Z–Z < HZ–Z < FZ–Z binary complexes.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This research was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.06-2020.28. We are sincerely thankful to Professor Thanh-Tung Nguyen-Dang, Université Laval – Canada for valuable discussions.References
- J. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley, 1st edn, 1995 Search PubMed.
- X. Li, L. Liu and H. B. Schlegel, J. Am. Chem. Soc., 2002, 124, 9639–9647 CrossRef CAS PubMed.
- S. Scheiner, J. Phys. Chem. B, 2006, 110, 18670–18679 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 1367–1400 CrossRef CAS.
- I. V. Alabugin, M. Manoharan, S. Peabody and F. Weinhold, J. Am. Chem. Soc., 2003, 125, 5973–5987 CrossRef CAS.
- G. Trudeau, J. M. Dumas, P. Dupuis, M. Guérin and C. Sandorfy, Top. Curr. Chem., 1980, 93, 91–125 CrossRef CAS PubMed.
- Y. Mao and M. Head-Gordon, J. Phys. Chem. Lett., 2019, 10, 3899–3905 CrossRef CAS.
- A. Vashchenko and A. Afonin, J. Struct. Chem., 2014, 55, 636–643 CrossRef CAS.
- M. Domagała and S. J. Grabowski, J. Phys. Chem. A, 2005, 109, 5683–5688 CrossRef.
- J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620–4632 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
- O. Donoso-Tauda, P. Jaque and J. C. Santos, Phys. Chem. Chem. Phys., 2011, 13, 1552–1559 RSC.
- C. Wang, D. Danovich, S. Shaik and Y. Mo, J. Chem. Theory Comput., 2017, 13, 1626–1637 CrossRef CAS PubMed.
- Y. Mo, C. Wang, L. Guan, B. Braïda, P. C. Hiberty and W. Wu, Chem.–Eur. J., 2014, 20, 8444–8452 CrossRef CAS PubMed.
- S. Michielssens, N. T. Trung, M. Froeyen, P. Herdewijn, M. T. Nguyen and A. Ceulemans, Phys. Chem. Chem. Phys., 2009, 11, 7274–7285 RSC.
- N. T. Trung, T. T. Hue, M. T. Nguyen and T. Zeegers-Huyskens, Phys. Chem. Chem. Phys., 2008, 10, 5105–5113 RSC.
- N. T. Trung, T. T. Hue and M. T. Nguyen, Can. J. Chem., 2010, 88, 849–857 CrossRef CAS.
- N. T. Trung, T. T. Hue and M. T. Nguyen, Phys. Chem. Chem. Phys., 2009, 11, 926–933 RSC.
- Y. Gu, T. Kar and S. Scheiner, J. Am. Chem. Soc., 1999, 121, 9411–9422 CrossRef CAS.
- A. C. Pierce, E. ter Haar, H. M. Binch, D. P. Kay, S. R. Patel and P. Li, J. Med. Chem., 2005, 48, 1278–1281 CrossRef CAS.
- S. Pingali, J. P. Donahue and F. Payton-Stewart, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 388–391 CrossRef CAS.
- L. Spada, Q. Gou, M. Vallejo-López, A. Lesarri, E. J. Cocinero and W. Caminati, Phys. Chem. Chem. Phys., 2014, 16, 2149–2153 RSC.
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS.
- S. R. Garde and R. N. Shirsat, Electrostatics of Atoms and Molecules, Sangam Books Ltd, Hyderabad, 2001 Search PubMed.
- P. Hobza, V. Špirko, H. L. Selzle and E. W. Schlag, J. Phys. Chem. A, 1998, 102, 2501–2504 CrossRef CAS.
- R. Gopi, N. Ramanathan and K. Sundararajan, J. Phys. Chem. A, 2014, 118, 5529–5539 CrossRef CAS.
- B. Behera and P. K. Das, J. Phys. Chem. A, 2019, 123, 1830–1839 CrossRef CAS.
- K. S. Rutkowski, S. M. Melikova, M. Rospenk and A. Koll, Phys. Chem. Chem. Phys., 2011, 13, 14223–14234 RSC.
- I. S. Sosulin, E. S. Shiryaeva, D. A. Tyurin and V. I. Feldman, J. Phys. Chem. A, 2018, 122, 4042–4047 CrossRef CAS.
- X. Chang, Y. Zhang, X. Weng, P. Su, W. Wu and Y. Mo, J. Phys. Chem. A, 2016, 120, 2749–2756 CrossRef CAS.
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed.
- P. Khanh, V. Ngan, N. Man, N. T. Ai Nhung, A. Chandra and N. Trung, RSC Adv., 2016, 6, 106662–106670 RSC.
- N. T. Trung, P. N. Khanh, A. J. P. Carvalho and M. T. Nguyen, J. Comput. Chem., 2019, 40, 1387–1400 CrossRef CAS PubMed.
- N. Thi Hong Man, P. Le Nhan, V. Vo, D. Tuan Quang and N. Tien Trung, Int. J. Quantum Chem., 2017, 117, e25338 CrossRef.
- N. T. Trung, N. P. Hung, T. T. Hue and M. T. Nguyen, Phys. Chem. Chem. Phys., 2011, 13, 14033–14042 RSC.
- P. Hobza, V. Špirko, Z. Havlas, K. Buchhold, B. Reimann, H.-D. Barth and B. Brutschy, Chem. Phys. Lett., 1999, 299, 180–186 CrossRef CAS.
- I. Bhattacharya, J. Sadhukhan, S. Biswas and T. Chakraborty, J. Phys. Chem. A, 2020, 124, 7259–7270 CrossRef CAS PubMed.
- A. K. Chandra and T. Zeegers-Huyskens, J. At., Mol., Opt. Phys., 2012, 2012, e754879 Search PubMed.
- N. T. T. Cuc, C.-T. D. Phan, N. T. A. Nhung, M. T. Nguyen, N. T. Trung and V. T. Ngan, J. Phys. Chem. A, 2021, 125, 10291–10302 CrossRef CAS PubMed.
- K. Tabayashi, O. Takahashi, H. Namatame and M. Taniguchi, Chem. Phys. Lett., 2013, 557, 1–9 CrossRef CAS.
- R. W. Gora, S. J. Grabowski and J. Leszczynski, J. Phys. Chem. A, 2005, 109, 6397–6405 CrossRef CAS.
- T. S. Thakur, M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, Phys. Chem. Chem. Phys., 2011, 13, 14076–14091 RSC.
- Y. Yang and W. Zhang, Acta Chim. Sin., 2009, 67, 599–606 CAS.
- A. Kovács, A. Szabó, D. Nemcsok and I. Hargittai, J. Phys. Chem. A, 2002, 106, 5671–5678 CrossRef.
- D. Kaur and R. Kaur, J. Chem. Sci., 2015, 127, 1299–1313 CrossRef CAS.
- A. K. Chandra and T. Zeegers-Huyskens, J. Comput. Chem., 2012, 33, 1131–1141 CrossRef CAS PubMed.
- N. T. Trung, T. T. Hue and M. T. Nguyen, J. Phys. Chem. A, 2009, 113, 3245–3253 CrossRef CAS.
- D. Voet, J. G. Voet and C. W. Pratt, Fundamentals of Biochemistry, Wiley, New York, Upgrade edn, 2001 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman and J. A. Pople, Gaussian 09 (Revision B.01), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- K. E. Riley, J. A. Platts, J. Řezáč, P. Hobza and J. G. Hill, J. Phys. Chem. A, 2012, 116, 4159–4169 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, J. Phys. Chem. A, 2007, 111, 8257–8263 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. A. Keith and T. K. Gristmill, AIMAll (Version 19.10.12) Software, Overland Park, KS, USA, 2019 Search PubMed.
- P. L. Popelier, Atoms in Molecules: An Introduction, Prentice Hall, Harlow, 1 edn, 2000 Search PubMed.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, New York, 1990 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- E. D. Glemdeming, J. K. Baderhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, GenNBO5.G, Theoretical Chemistry Institute, University of Wisconsin Madison, WI, 2001 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- P. A. Kollman, in Chemical Applications of Atomic and Molecular Electrostatic Potentials: Reactivity, Structure, Scattering, and Energetics of Organic, Inorganic, and Biological Systems, ed. P. Politzer and D. G. Truhlar, Springer US, Boston, MA, 1981, pp. 243–255 Search PubMed.
- R. Bukowski, W. Cencek, M. Jeziorska, B. Jeziorski, V. F. Lotrich, A. J. Misquitta, R. Moszyński, K. Patkowski, R. Podeszwa, F. Rob, S. Rybak, K. Szalewicz, R. J. Wheatley, E. S. Paul, P. E. S. Wormer and P. S. Żuchowski, SAPT2012.2, Warsaw, Poland, 2013 Search PubMed.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Yu. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- R. Kalescky, E. Kraka and D. Cremer, Mol. Phys., 2013, 111, 1497–1510 CrossRef CAS.
- W. Qian and S. Krimm, J. Phys. Chem. A, 2002, 106, 11663–11671 CrossRef CAS.
- L. Spada, L. Evangelisti, W. Li, R. Orlacchio and W. Caminati, J. Phys. Chem. A, 2019, 123, 1785–1789 CrossRef CAS PubMed.
- Q. Zhang and L. Du, Comput. Theor. Chem., 2016, 1078, 123–128 CrossRef CAS.
- Q.-Z. Li, B. Jing, R. Li, Z.-B. Liu, W.-Z. Li, F. Luan, J.-B. Cheng, B.-A. Gong and J.-Z. Sun, Phys. Chem. Chem. Phys., 2011, 13, 2266–2271 RSC.
- N. T. T. Cuc, N. T. An, V. T. Ngan, A. K. Chandra and N. T. Trung, RSC Adv., 2022, 12, 1998–2008 RSC.
- A. S. Hansen, L. Du and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2014, 16, 22882–22891 RSC.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- M. Guin, K. Rautela, R. A. Roopa, C. S. Shantharam and S. Begam Elavarasi, Comput. Theor. Chem., 2021, 1196, 113134 CrossRef CAS.
- B. G. Oliveira, J. Chil. Chem. Soc., 2011, 56, 601–605 CrossRef CAS.
- S. Bedoura, H.-W. Xi and K. Lim, J. Phys. Org. Chem., 2014, 27, 226–236 CrossRef CAS.
- D. Kaur, D. Aulakh, S. Khanna and H. Singh, J. Sulfur Chem., 2014, 35, 290–303 CrossRef CAS.
- J. Lei, S. Alessandrini, J. Chen, Y. Zheng, L. Spada, Q. Gou, C. Puzzarini and V. Barone, Molecules, 2020, 25, 4899 CrossRef CAS PubMed.
- N. T. T. Cuc, H. Q. Dai, N. T. A. Nhung, N. P. Hung and N. T. Trung, Vietnam J. Chem., 2019, 57, 425–432 CrossRef.
- Q. Gou, L. B. Favero, S. S. Bahamyirou, Z. Xia and W. Caminati, J. Phys. Chem. A, 2014, 118, 10738–10741 CrossRef CAS.
- E. Sánchez-García, L. A. Montero and W. Sander, J. Phys. Chem. A, 2006, 110, 12613–12622 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables and figures list of contact distances, the AIM analysis, MEP analysis, SAPT analysis, NCI plot, length changes, and stretching frequency shifts of the Csp2–H and O–H bonds in the complexes. See DOI: https://doi.org/10.1039/d2ra05391h |
| This journal is © The Royal Society of Chemistry 2022 |