
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent effects on the motion of a crown ether/amino rotaxane†
Zhen Wua,
Shuangshuang Wang *ab,
Zilin Zhangb,
Yanjun Zhangb,
Yanzhen Yin*b,
Haixin Shib and
Shufei Jiaob
*ab,
Zilin Zhangb,
Yanjun Zhangb,
Yanzhen Yin*b,
Haixin Shib and
Shufei Jiaob
aSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China. E-mail: doublewang123@163.com
bGuangxi Key Laboratory of Green Chemical Materials and Safety Technology, Beibu Gulf University, Qinzhou 535011, China. E-mail: yinyanzhen2009@163.com
First published on 26th October 2022
Abstract
Solvents have been recognized as a significant factor for modulating the shuttle of rotaxanes and regulating their functions regarding molecular machines by a lot of published studies. The mechanism of the effects of solvents on the motion of crown ether/amino rotaxanes, however, remains unclear. In this work, a rotaxane, formed by dibenzo-24-crown-8 (C[8]) and a dumbbell-shaped axle with two positively charged amino groups, was investigated at the atom level. Two-dimensional free-energy landscapes characterizing the conformational change of C[8] and the shuttling motions in chloroform and water were mapped. The results indicated that the barriers in water were evidently lower than those in chloroform. By analyzing the trajectories, there was no obvious steric effect during shuttling. Instead, the main driving force of shuttling was verified from electrostatic interactions, especially strong hydrogen bonding interactions between the axle and water, which resulted in the fast shuttling rate of the rotaxane. All in all, the polarity and hydrogen bond-forming ability of solvents are the main factors in affecting the shuttling rate of a crown ether/amino rotaxane. In addition, C[8] would adopt S-shaped conformations during shuttling except for situating in the amino sites with C-shaped ones adopted due to π–π stacking interactions. The results of this research improve the comprehension of the solvent modulation ability for shuttling in crown ether-based rotaxanes and illustrate the effects of structural modifications on motions. These new insights are expected to serve the efficient design and construction of molecular machines.
1 Introduction
Mechanical interlocked molecular machines are a kind of molecules with some specific functions, which are composed of two or more molecular elements entangled by mechanical bonds in space. As the fundamental archetypes of these molecules, molecular shuttles have attracted increasing public attention due to their diverse functions and great applications such as information storage,1 molecular logic gates,2 molecular switches,3 and medical transportation4.5,6 Rotaxanes7–9 are a typical kind of molecular shuttle and are composed of a linear molecule flanked with stoppers at both termini and a macrocycle threaded onto the latter. The shuttling motion of a rotaxane, namely, the macrocycle in a rotaxane shuttling between two or more sites, could be triggered by assorted external stimuli,7,10 such as temperature changes,11,12 pH changes,13,14 and solvent changes,15,16 which drive rotaxanes as key factors for the construction of molecular shuttles and motors.17–19Many macrocyclic molecules can be available for the ring components of rotaxanes, for example, crown ethers,20,21 pillararenes,11,22 and cyclodextrins23.24,25 Crown ethers, as the most flexible macrocyclic molecules,26 were synthesized and first used as the guest of rotaxanes by Stoddart and coworkers.27,28 Subsequently, a number of crown ether-based rotaxanes were synthesized and studied such as mushrooms after rain.29–31 According to these research studies, it was readily found that crown ethers could shuttle between some recognition sites,32–37 for example, amino groups, benzimidazolium, and bis(pyridinium). Wherein, the molecular machines with the positively charged amino sites attracted more attention. For this kind of molecular machine, the mechanism of movements in rotaxanes and the corresponding driving forces can be summarized as a function of the environment. However, till now, the majority of published studies about crown ether/amino rotaxanes focus on their synthesis and only a small amount of them marginally considered the mechanisms of the movement. In terms of this, Stoddart et al. synthesized a molecular shuttle28 with bis(pyridinium) and amino recognition sites, and found that the shuttle preferred to stay on the positively charged amino sites and could be reversed by a deacidified treatment. Then, Takata et al. constructed a thermoresponsive rotaxane shuttling system32 with amino recognition sites, and the resulting molecular machine could be driven by the chemoselective thermal decomposition of ammonium trichloroacetate. Nonetheless, the mechanisms underlying the solvent-controlled behavior of crown ether/amino rotaxanes are still lacking in further studies.
Molecular dynamics simulations (MDS) are a widely used strategy for revealing the internal mechanisms of molecular machines. Cai et al.38 investigated a crown ether-based rotaxane with benzimidazolium and bis(pyridinium) using MDS and indicated that solvents have a significant impact on the shuttling rate. In addition, water could act as lubrication in accelerating the motion of the rotaxane as well. Thus, in this research, all-atom MDS were used for investigating solvent effects on the motion of a crown ether/amino rotaxane. One model of the rotaxane in Fig. 1 was built to compare two solvents, chloroform (CHCl3) and water, which are of great polarity difference. We determined the two-dimensional free-energy landscapes characterizing the shuttling in the rotaxane accompanied by the conformational change of dibenzo-24-crown-8 (C[8]) and then gaged the least free-energy pathways, according to which the most likely conformations of C[8] during shuttling were inferred. By parsing trajectories and dividing the total free energy into physically meaningful individual components, the free-energy contributions extracted from the potentials of means force were analyzed to interpret the driving forces of the shuttling motion and conformational transition. The present work provided a reasonable understanding of the characteristics of driving forces and the mechanisms of the effect of the solvents on the shuttling of crown ether/amino-based rotaxanes. These theoretical findings could be helpful for the rational design of crown ether-based molecular shuttles, which could be controlled by solvents.
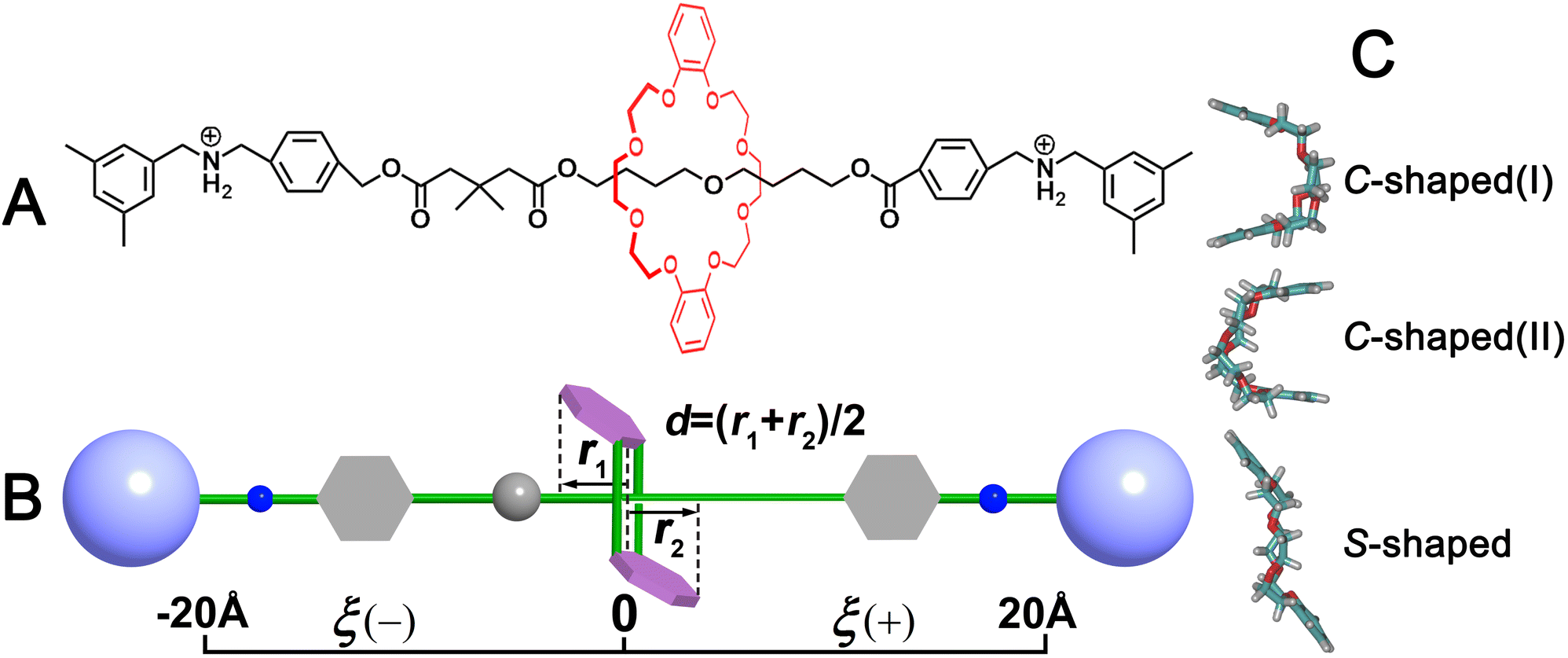 | ||
| Fig. 1 (A) Chemical structure of the rotaxane formed by C[8] and an axle molecule with two amino recognition sites. (B) Transition coordinates (ξ, d) for free-energy calculations are utilized to explore the putative transition pathways. ξ denotes the projection along the z-direction of the Cartesian space of the vector connecting the centroid of C[8] and that of the axle. r1 and r2 are defined as the projection onto the z-axle of the orientation of the benzo groups of C[8]. d was defined as d = (r1 + r2)/2 to describe the conformational change of C[8]. (C) Two C-shaped conformations were derived from d = ± 3.0 Å and one S-shaped conformation was derived from d = 0 Å. | ||
2 Simulation details
2.1 Molecular models
The molecular model of a rotaxane formed by a C[8] host and a dumbbell-shaped axle was constructed, as depicted in Fig. 1. The available three-dimensional crystal structure39 was used for extracting the initial coordinate of C[8]. The molecule assembly was energy-minimized using a conjugate-gradient algorithm and then immersed respectively in a pre-equilibrated box of chloroform and water using the solvate module of the visualization program VMD.40 For adapting to periodic boundary conditions, any atom of the complex must distance each edge of the boxes from at least 15 Å. Two chloride ions were placed 10 Å away from the rotaxane to ensure electric neutrality in each solvent box. The detail of the molecular assemblies examined in this work is given in Table S1.† Two coarse variables, ξ and d, describing translocation and conformational change of C[8], respectively (defined in Fig. 1B), were chosen to form the transition coordinate. The transition pathways ξ and d were extending from −20 to +20 Å and −3.9 to +3.9 Å, and instantaneous values of both the forces were accrued in bins 0.1 Å wide. The more simulation details can be seen in ESI.†3 Results and discussion
3.1 Free-energy landscapes underlying the shuttling movement
The two-dimensional free-energy landscapes, characterizing the shuttling motion and conformational change of C[8] along the axle of the rotaxane in CHCl3 and water, are depicted in Fig. 2A and B. With a cursory glance, these profiles reveal that (i) overall, the barriers of the free-energy landscape in CHCl3 are significantly higher than that in water; (ii) each map features two basins separated by one wide ridge, and the ridge in the free-energy landscape in water is wider than that in CHCl3; and (iii) symmetric basin areas are around ξ > +10.0 Å and ξ < −10.0 Å for CHCl3 and around ξ > +12.0 Å and ξ < −12.0 Å for water. The minima of the low-energy areas in the two solvents can be found at ξ = −16.0 Å, d = −2.4 Å and ξ = +16.0 Å, d = +2.4 Å, namely, the positively charged amino sites. In the corresponding structures shown in Fig. 3a and c, C[8] adopts C-shaped conformations. The least free-energy pathways connecting these two minima represent the most reasonable transition path for the shuttling of C[8] between two amino sites. It can be seen that C[8] almost adopts S-shaped conformations (see Fig. 3b) during shuttling unless it is located in amino sites.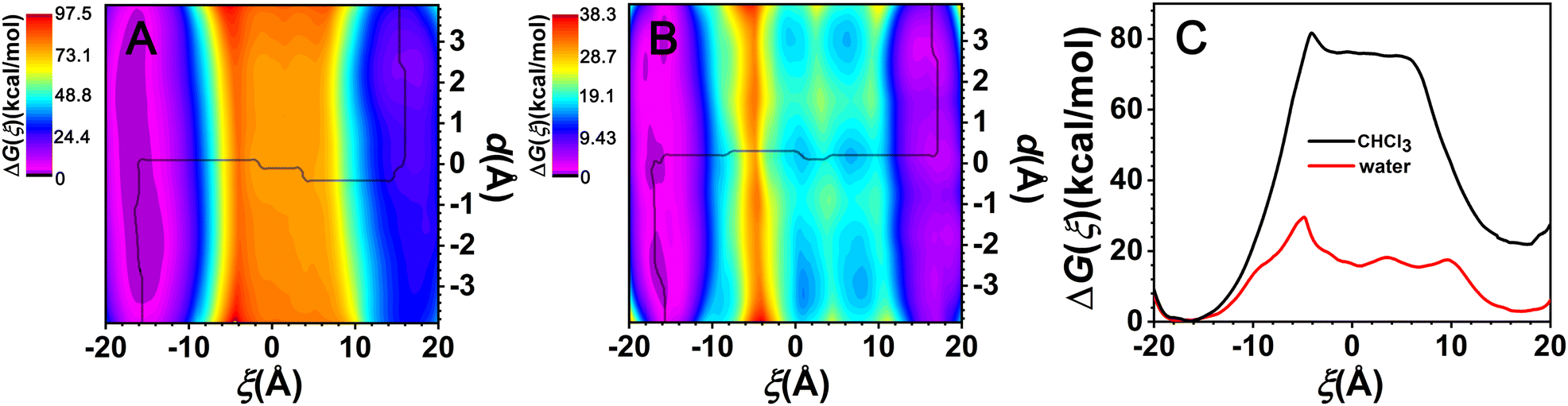 | ||
| Fig. 2 Free-energy landscapes characterizing the shuttling and conformational change of the rotaxane in CHCl3 (A) and water (B). The black lines show the least free-energy pathways. (C) Free-energy changes for the shuttling movements in two different solvents along ξ according to the least free-energy pathways. | ||
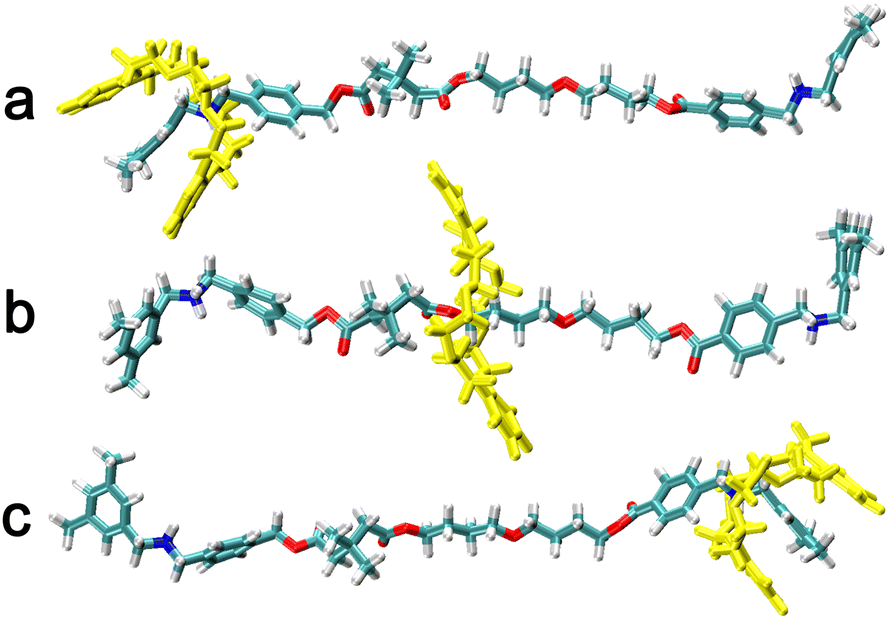 | ||
| Fig. 3 Representative structures during shuttling. The C[8] is located in (a) and (c) the amino sites and (b) the middle of the axle. | ||
Fig. 2C shows the one-dimensional free-energy profiles (ODFEs) for shuttling along ξ according to the least free-energy pathways. From the results in CHCl3, the energy barrier to be overcome for shuttling was inferred to be equal to about 81.6 kcal mol−1, while the barrier in water was estimated to be only 29.5 kcal mol−1, thereby indicating that the shuttling rate of the latter is significantly faster than that of the former. Moreover, the two maxima all appear at about ξ = −4.5 Å, wherein C[8] overlaps with the neopentane moieties between the two ester groups.
3.2 Free-energy decomposition
According to the above-mentioned analysis, the rotaxane can shuttle between two sites and the shuttling rate is apparently distinct in two solvents. Then, what are the driving forces of the shuttling motion? For this purpose, the free-energy contributions along ξ extracted from the potentials of means force by dividing the total free energy into physically meaningful individual components and binning, averaging, and integrating the force. The results are shown in Fig. 4.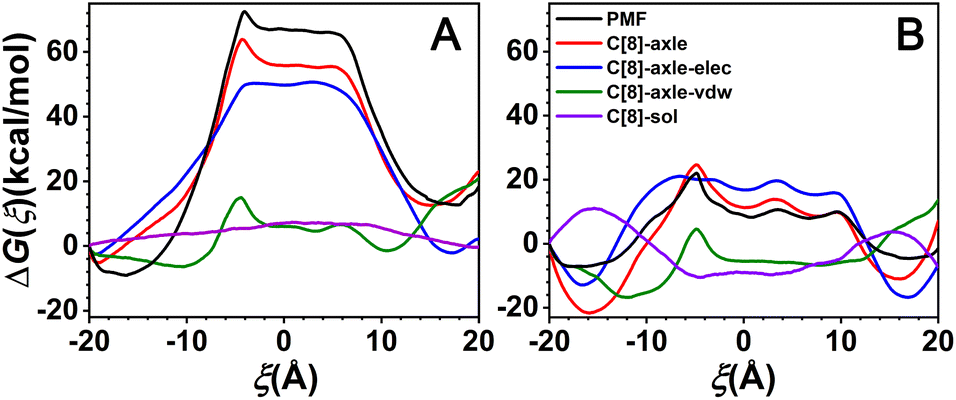 | ||
| Fig. 4 Breakdown of the total free-energy profiles into C[8]-axle and C[8]-sol contributions, which were further decomposed into electrostatic (C[8]-axle-elec) and van der Waals (C[8]-axle-vdw) contributions, for the shuttling processes of the rotaxane in CHCl3 (A) and water (B). ODFEs are the one-dimensional free-energy profiles for shuttling along ξ. | ||
As shown in Fig. 4, the C[8]-axle interactions feature two basins separated by one wide barrier and show similar tendencies as the ODFEs, which indicates that the C[8]-axle interactions constitute the main contributions to the barriers of the ODFEs. The C[8]-axle interactions were further decomposed into van der Waals (C[8]-axle-vdw) and electrostatic (C[8]-axle-elec) contributions. The C[8]-axle-vdw contributions for all solvent conditions generally feature a wide valley, which is because of the suitable size of the C[8] cavity to include the linear molecule. It also illustrates that the solvents have little effects on the C[8]-axle-vdw. It is noteworthy that there is a little bump at about ξ = −4.5 Å, which is in agreement with the maxima of the ODFEs. The contribution to the bump is the steric hindrance caused by the neopentane moieties. What is more, the right tail of the C[8]-axle-vdw is higher than the left tail, which could be attributed to the slight asymmetry of the structure, thereby resulting in the asymmetry of the free-energy profiles. The C[8]-axle-elec contributions feature two basins separated by one wide barrier and show similar tendencies as the ODFEs, which illustrates that the electrostatic interactions determine the stable binding sites of C[8] and constitute the main driving force responsible for shuttling. Furthermore, the C[8]-axle-elec in CHCl3 is markedly stronger than that in water, suggesting that the polarity of the solvents has a significant impact on it; the stronger the solvent polarity, the weaker the C[8]-axle-elec. It can be further observed in Fig. 4 that the C[8]-sol contribution in CHCl3 presents a low mound, indicating that it has little effect on the ODFE. By contrast, the C[8]-sol contribution in water possesses two bumps, spanning ξ ≤ −12 Å and ξ ≥ 12 Å, separated by a wide and flat domain, implying that while C[8] is located in the amino sites, the C[8]–sol interactions are unfavorable for the shuttling process. Accordingly, the C[8]–sol interactions in the water partly contribute to ODFE.
To appreciate the effect of the solvents on C[8]–sol interactions, the distributions of the solvent molecules around the left amino moiety were monitored. This was achieved by computing the bidimensional radial distribution functions, g(r; ξ), of the C atom of CHCl3 or the O atom of water with respect to the centroid of the selected moieties. The results are gathered in Fig. 5.
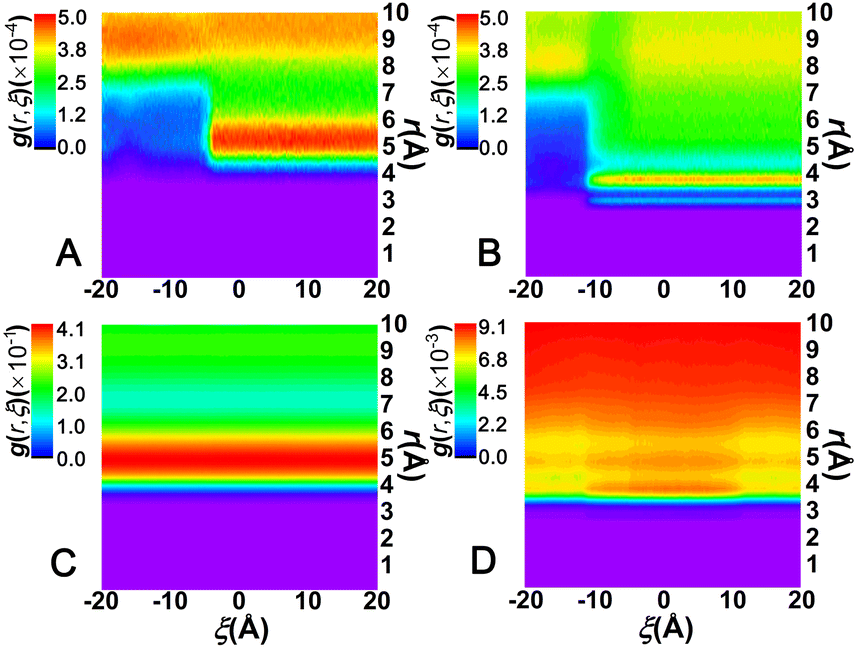 | ||
| Fig. 5 Variation in the radial distribution function of the center of mass (left amino moiety)-C (CHCl3)/O (water) pair as a function of r, the distance separating the pair of atoms, for the shuttling process (A) in CHCl3, (B) in water. Variation in the radial distribution function of the center of mass (C[8])-C (CHCl3)/O (water) pair as a function of r for the shuttling process (C) in CHCl3 and (D) in water. | ||
As shown in Fig. 5A and B, the maximum density found in the region spanning 5 ≤ r ≤ 6 Å in CHCl3 and 3 ≤ r ≤ 4 Å in water corresponds to the first solvation shell of the left amino moiety, illustrating that the amino moieties are closer to water molecules than to chloroform molecules. The solvation shell of the left amino moiety is discontinuous along ξ, for ξ ≤ −6 Å in CHCl3 and ξ ≤ −12 Å in water, which means that the solvation shell is disrupted as C[8] locates on the amino moiety. With the former moving away from the latter, the solvation shell in water forms faster than that in CHCl3, indicating that water could promote the motion of C[8], namely, that water could act as a lubricant and accelerate the shuttling rate of the rotaxane. According to Fig. 5C, the first solvation shell of C[8], found in the region spanning 4 ≤ r ≤ 6 Å, is practically unchanged during shuttling and the density is the highest. However, as shown in Fig. 5D, the density of the solvation shell in water is up to 9.10 × 10−3 and evidently weaker than that in CHCl3, which is up to 0.41. What is more, the density of the first solvation shell in water is the lowest and is slightly weakened as C[8] is located in the amino sites in water. These features illustrate that C[8] has a clear affinity toward CHCl3 and the solvation shell is not affected during shuttling, and instead, the solvation shell can be disrupted because of the weak affinity of C[8] and water.
On balance, both the C[8]-axle and C[8]–sol interactions act on the ODFEs together. Thereinto, the C[8]-axle-elec contributions constitute the main factor for the emergence of the free-energy barriers. The solvent with higher polarity could have a stronger binding ability with the amino moieties, and then weaken the electrostatic interactions between C[8] and the axle, finally accelerating the shuttling rate of the rotaxane. Besides the polarity of the solvents, the hydrogen-bonding (H-bonding) interactions could be one of the main reasons for contributing to the C[8]-axle-elec interactions.
3.3 Analysis of the H-bonding and π–π stacking interactions
The variation in the average number of intermolecular H-bonds formed between the solvents and the axle and between the solvents and the C[8] by analyzing the trajectories is shown in Fig. 6. The H-bonds could be formed between the O atoms of C[8] as acceptors and N–H of the axle as donors. The largest number of the H-bonds between the axle and C[8] is about 1.8 in both solvents, illustrating that the axle will form H-bonds with C[8] with preference to the solvents, while C[8] is located at the amino sites. The difference is that the H-bonds between the axle and C[8] can be maintained when −18 < ξ < −6 Å in CHCl3, and only when −18 < ξ < −12 Å in water. It indicates that as C[8] moves away from the amino sites, and the H-bonds between the axle and C[8] in water will be released more easily than those in CHCl3. Meanwhile, it can also be found that the number of H-bonds between the axle and solvents increases from 0.6 to 1.2 in CHCl3 and from 1.8 to 3.6 in water, illustrating that the axle can form more H-bonds with water molecules than with CHCl3 molecules. More stable H-bonds between water and the axle impel the H-bonds between C[8] and the axle to be broken earlier and promote the motion of C[8]. Furthermore, the turning points of the curves are consistent with those of the C[8]-axle-elec, illustrating that the H-bonding interactions are one of the main reasons for contributing to the C[8]-axle-elec.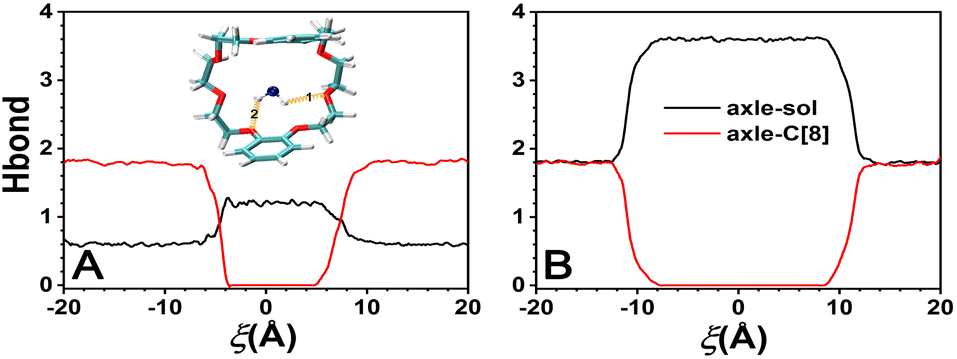 | ||
| Fig. 6 Variation of the average number of intermolecular H-bonds formed between the axle and the solvents (axle–sol, colored black) and between the axle and the C[8] (axle-C[8], colored red) in CHCl3 (A) and water (B). The inset shows hydrogen bonds formed between the N–H donors of the axle and the acceptor O atoms of the C[8] (N–H⋯O). The hydrogen-bonding criteria are (i) the angle N–H⋯O > 135° and (ii) the distance N⋯O < 3.5 Å. The hydrogen bonds are highlighted in gold. | ||
However, according to Fig. 3a and c, C[8] usually adopts C-shaped conformations, while it is near the benzene rings at both ends of the axle. Why? To this end, the π–π stacking interaction, which is a direct attractive non-covalent interaction between aromatic moieties,41 is considered. The variation in π–π interactions between the axle and C[8] by analyzing the trajectories is shown in Fig. 7. It can be seen that only while C[8] locates at both ends of the axle, π–π stacking interactions are formed between C[8] and the axle. That is to say that C[8] adopts suitable conformations, namely, C-shaped ones, for matching the axle to form the intermolecular forces with the latter, thereby stabilizing the system. The interactions in CHCl3 are stronger than these in water, which illustrates that the stronger the polarity of the solvent is, the stronger the interactions are. However, relative to the H-bonds, π–π stacking interactions are weak and thus make no difference to the ODFE, i.e., the shuttling rate of the rotaxane.
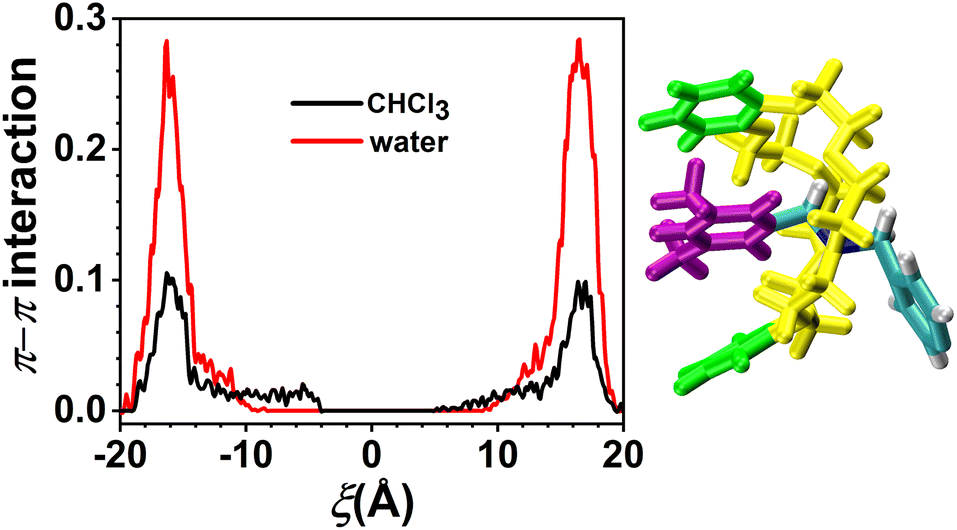 | ||
| Fig. 7 Variation in the average number of π–π stacking interactions in CHCl3 (colored black) and water (colored red). The π–π stacking criterion is that the distance between the benzene rings of C[8] (colored green) and these of the axle (colored purple) are below 4. | ||
4. Conclusions
Two-dimensional free-energy calculations were utilized for investigating the mechanisms of the crown ether-based rotaxane. The results clearly indicate that the barriers in CHCl3 are significantly higher than those in water and the shuttling of the flexible C[8] along the axle is highly coupled with the conformational transition. The C[8] would adopt an S-shaped conformation during shuttling, unless it could be located in the amino sites with a C-shaped conformation adopted. The main driving force for shuttling is derived from electrostatic interactions between C[8] and the axles by partitioning the free-energy profiles into different components. Both the polarity of the solvents and the capability of H-bonds formation in the solvents have an effect on the barriers and then help to modulate the shuttling of the rotaxane. In detail, the strong solvent polarity can spur C[8] to move away from both the amino moieties and the solvation shell of the latter forms and then activate this motion. Moreover, the H-bonds can show a faster shuttling rate in the solvents due to greater interactions between solvent molecules and the axle due to its release, as C[8] moves away the amino sites. In general, water can act as a lubricant and accelerate the shuttling rate of the rotaxane.The results in this article introduce a comprehensible solvent modulation ability for shuttling in the crown ether-based rotaxanes and disentangle the effects of the structural modifications on the motions. In the future work, the rotaxanes with crown ether and deprotonated amino sites in several distinguished other solvents will be compared to explore the response mechanisms of the crown ether/amino rotaxanes to environmental change.
Author contributions
Conceptualization, supervision, writing – review and editing, funding acquisition, Shuang-Shuang Wang; data curation, writing – original draft preparation, Zhen Wu and Shuang-Shuang Wang; formal analysis, resources, Yan-Zhen Yin; formal analysis and discussion, Zi-Lin Zhang, and Yan-Jun Zhang; investigation, Hai-Xin Shi and Shu-Fei Jiao. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study is supported by Natural Science Foundation of Guangxi Province (No. 2018JJB160116), Marine Science Guangxi First-Class Subject, Beibu Gulf University (No. DTB003).References
- A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- Q. Zhang, S. J. Rao, T. Xie, X. Li, T. Y. Xu, D. W. Li, D. H. Qu, Y. T. Long and H. Tian, Chem, 2018, 4, 2670–2684 CAS.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584–588 CrossRef CAS PubMed.
- M. W. Ambrogio, C. R. Thomas, Y. L. Zhao, J. I. Zink and J. F. Stoddart, Acc. Chem. Res., 2011, 44, 903–913 CrossRef CAS.
- J. J. Yu, Z. Q. Cao, Q. Zhang, S. Yang, D. H. Qu and H. Tian, Chem. Commun., 2016, 52, 12056–12059 RSC.
- M. Xue, Y. Yang, X. D. Chi, X. Z. Yan and F. H. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed.
- J. C. Chang, S. H. Tseng, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, Nat. Chem., 2017, 9, 128–134 CrossRef CAS.
- H. Li and D. H. Qu, Sci. China Chem., 2015, 58, 916–921 CrossRef CAS.
- S. Y. Dong, J. Y. Yuan and F. H. Huang, Chem. Sci., 2014, 5, 247–252 RSC.
- X. Song, X. F. Zhu, S. Qiu, W. Tian and M. H. Liu, Angew. Chem., Int. Ed., 2022, e202208574 CAS.
- V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., 2012, 124, 5256–5259 CrossRef.
- P. J. Altmann and A. Pöthig, Angew. Chem., Int. Ed., 2017, 56, 15733–15736 CrossRef CAS PubMed.
- Z. B. Zhang, C. Y. Han, G. C. Yu and F. H. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC.
- H. H. Fu, X. G. Shao, C. Chipot and W. S. Cai, Chem. Sci., 2017, 8, 5087–5094 RSC.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed.
- S. J. Rao, Q. Zhang, J. Mei, X. H. Ye, C. Gao, Q. C. Wang, D. H. Qu and H. Tian, Chem. Sci., 2017, 8, 6777–6783 RSC.
- J. S. Cui, Q. K. Ba, H. Ke, A. Valkonen, K. Rissanen and W. Jiang, Angew. Chem., Int. Ed., 2018, 130, 7935–7940 CrossRef.
- K. Zhu, V. N. Vukotic, N. Noujeim and S. J. Loeb, Chem. Sci., 2012, 3, 3265–3271 RSC.
- C. J. Zhang, S. J. Li, J. Q. Zhang, K. L. Zhu, N. Li and F. H. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. D. Chi, Z. Zhang and F. H. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- C. Casati, P. Franchi, R. Pievo, E. Mezzina and M. Lucarini, J. Am. Chem. Soc., 2012, 134, 19108–19117 CrossRef CAS PubMed.
- M. Bazzoni, L. Andreoni, S. Silvi, A. Credi, G. Cera, A. Secchi and A. Arduini, Chem. Sci., 2021, 12, 6419–6428 RSC.
- Y. Han and C. F. Chen, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef PubMed.
- S. S. Wang, Y. Z. Yin, J. Gao, X. T Liang and H. X. Shi, J. Braz. Chem. Soc., 2021, 32, 1963–1971 CAS.
- P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932–11942 CrossRef CAS.
- Z. Meng, J. F. Xiang and C. F. Chen, J. Am. Chem. Soc., 2016, 138, 5652–5658 CrossRef CAS PubMed.
- R. R. Tao, Q. Zhang, S. J. Rao, X. L. Zheng, M. M. Li and D. H. Qu, Sci. China: Chem., 2019, 62, 245–250 CrossRef CAS.
- H. Zhang, X. G. Shao, C. Chipot and W. S. Cai, J. Phys. Chem. C, 2019, 123, 11304–11309 CrossRef CAS.
- Y. Abe, H. Okamura, K. Nakazono, Y. Koyama, S. Uchida and T. Takata, Org. Lett., 2012, 14, 4122–4125 CrossRef CAS.
- S. Corra, C. D. Vet, J. Groppi, M. L. Rosa, S. Silvi, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2019, 141, 9129–9133 CrossRef CAS PubMed.
- S. J. Cantrill, S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1363–1366 CrossRef CAS.
- T. Takata, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS.
- M. Mao, X. K. Zhang, T. Y. Xu, X. D. Wang, S. J. Rao, Y. Liu, D. H. Qu and H. Tian, Chem. Commun., 2019, 55, 3525–3528 RSC.
- K. L. Zhu, V. N. Vukotic and S. J. Loeb, Chem.–Asian J., 2016, 11, 3258–3266 CrossRef CAS.
- S. l. Du, H. H. Fu, X. G. Shao, C. Chipot and W. S. Cai, J. Phys. Chem. C, 2018, 122, 9229–9234 CrossRef CAS.
- M. Howard, S. M. D. Colquhoun, J. F. Stoddart, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1986, 253–257 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- L. J. Riwar, N. Trapp, B. Kuhn and F. Diederich, Angew. Chem., Int. Ed., 2017, 56, 11252–11257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05453a |
| This journal is © The Royal Society of Chemistry 2022 |