
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNano-optical method for transforming a single yeast cell using exogenous genes
Yao-Xiong Huang *,
Ji-Wang Yang and
Zhuo Wang
*,
Ji-Wang Yang and
Zhuo Wang
Department of Biomedical Engineering, Ji Nan University, Guangzhou, China 510632. E-mail: tyxhuang@jnu.edu.cn; Fax: +86-20-85223742; Tel: +86-20-85223742
First published on 7th November 2022
Abstract
We report a highly efficient nano-optical method for transforming a single yeast cell using exogenous genes. It used laser tweezers or micromanipulators to immobilize the cell immersed in a DNA solution and created a transient nano-sized hole on its cell wall concurrently with laser scissors to deliver nano moles of DNA into the cell. With this method, one can directly transfer the naked DNA of exogenous genes into yeast cells for transformation. We successfully transformed S. cerevisiae yeasts respectively with GFP (Green Fluorescent Protein) plasmid and the nucleic acid extraction of a bacteria GF1 from the gut of Coptotermes formosanus termites. The experimental results demonstrated that the recombinants had high survival rate and transformation efficiency (28%). The recombinant GFP–yeast system showed green fluorescence for generations. GF1 DNA sequences were incorporated into the yeast genome as a heritable component with stable expression for multi-generations so that the recombinant GF1–yeast had a strong capability of digesting biomass as GF1. Our method would apply to different cells with cell walls for various gene transformations.
Introduction
Yeast is an ideal versatile model organism for gene transformation because it has a high endogenous rate of homologous recombination. With a sophisticated molecular system, it can clone exogenous genes much easier than more complex eukaryotes to result in efficient expression of the desired gene.1 So the DNA transformation of yeast cells has attracted much attention since 1928.2,3 However, as yeast cells have lignified plant cell walls, delivering DNA into yeast cells has been an enormous technical challenge.The conventional methods3 used to remove the cell wall obstacle for delivering DNA into yeast cells include spheroplasting,4 Lic/ssDNA/PEG,5 electroporation,6 glass beads,7 and biolistic transformation.8 Though they can achieve yeast cell transformation by chemical or electrical treatments, the mechanisms by which the plasmid DNA traverses the cell membrane are still unknown for most of them.3 They also shared some limitations: only offered population/multiple-cell transformation but not targeted at a specific single cell present in a cluster of other cells and unable to generate pores on precise spatial positions on the cell wall. It is difficult to determine which cells and how many of them had been transformed, so the transformation efficiency is uncertain and of limited reliability. Although single-cell electroporation can use patch electrodes for single-cell transformation,9,10 it was mainly used for mammalian cell transformation but not yet applied to yeast cells. There was a report about the single-cell injection of Schizosaccharomyces pombe cells. The method used a piezo-impact micromanipulator to inject the cells constrained by microfabricated channels.11 However, they only injected fluorescently labeled phalloidin into the cells to observe actin cytoskeletal structures and the disruption of the cytokinetic ring, but not exogenous genes for transformation. It is not sure if such an approach that would induce intense deformation for 10 s on the cell during the mechanical injection is suitable for gene transformation.
Later, a method of DNA transfection by laser microbeam cell surgery was also proposed.12 It used a nanosecond or a femtosecond laser to puncture a self-healing submicrometer hole in the cell membrane to inject nano-molar concentrations of membrane-impermeable molecules, such as DNA plasmids, into the targeted living cells. The technique has several advantages over the conventional ones. It is sterile intrinsically and requires no mechanical contact during the transfection. It can perform single-cell transfection and apply to all types of cells. Since then, there have been numerous researches on optical transfections of various mammalian cells. They used different laser technologies such as Bessel beam or fiber delivery system for the target transfections13–16 or cooperating the technique of optical tweezers to insert the plasmid-coated micro-particles into the cell.17,18 However, up to now, it has been mainly applied to mammalian cell transfection except one for rice cell transformation by the laser puncture in combination with the pretreatment of the cell in a hypertonic buffer to generate a gradient of negative osmotic potential.19 Though a study had reported the combined use of the optical microsurgery and trapping for intracellular organelle manipulation and extraction of yeast cells,17 their laser microsurgery was used to break the cell, and the laser tweezer was employed to take the intracellular organelle out from the broken cell. No one had applied both the optical technologies for yeast transformation.
Transformation refers to introducing foreign DNA into bacterial, yeast, or plant cells, while transfection means to transfer the foreign DNA into mammalian cells.20 The cell membranes of mammalian cells are thin, soft, and with good viscoelasticity. So it is easier to create a self-healing hole of submicrometer aperture in the cell membrane to transfer foreign DNA into the cell by the laser beam. However, yeast cells have cell walls that are stiff, brittle, and thicker (about 100–200 nm thick) relative to mammalian cell membranes.21,22 They are also quite different from the plant cell walls like rice cell walls, for the yeast cell walls are composed mainly of mannoprotein fibrous β1,3 glucan and chitins, while plant cell walls are cellulose and pectin.23 So yeast cell walls are more brittle than rice cell walls, and it is much more difficult to create a penetrated hole on the wall without hurting the cell. Less laser energy could not ablate the yeast cell wall, while excess irradiation would disrupt it and lead to cell damage. For a successful transformation, laser parameters need to be optimized to create a hole with suitable nanosize on the cell to allow nano-mole DNA plasmids to enter. But the holes should be able to self-reseal after the optoporation to leave the cells intact and healthy. Moreover, after the transformation, the cell should survive, have an efficient expression of the exogenous genes, then divide and proliferate with multigenerational inheritances of the integrated genes. So the vital point for yeast transformation by optoporation is how to create a nano-sized hole that penetrates the cell wall of 100–200 nm thick only but does not induce other disturbances in the cell.
Another difference between the optoporations on yeast and mammalian cells is that, the mammalian cells usually adhere to the surface of the culturing dishes but do not freely move away during the short period of optoporation. So it is easier to target at a specific point for perforation, and it is unnecessary to use manipulators to hold the cells. Therefore, the previously reported studies of laser transfection on mammalian cells only need to use one laser for optoporation except for Waleed et al.24 They also employed the laser tweezers besides the femtosecond laser for optoporation. But they only used them to trap and insert the plasmid-coated micro-particles into the cell instead of holding, or manipulating the cells. Unlike mammalian cells, yeasts are suspension cells. The impact of the laser irradiation would push the cell away from the laser focus so that the irradiation is hard to achieve at the same point in the entire optoporation process to create the hole. For this reason, it is quite an immense technical challenge for the optical method on yeast cells for gene transformation. It may be one of the reasons why no yeast transformation was performed by optoporation up to the present.
Our purpose was to approach the problems of yeast transformation by developing a novel nano-optical method. The method would use laser tweezers to immobilize the yeast cell and simultaneously employ laser scissors to ablate its cell wall for a penetrative nano-sized hole to transform the Saccharomyces cerevisiae yeast. The technique is highly sophisticated as it needs to operate the two lasers in a finely coordinated operation. Although Berns group and our lab had applied these techniques to chromosome cutting25,26 or welding,26 chromosomes are smaller, lighter, and easier to be handled than the yeast cells suspended in solution. So we should explore the optimal way and laser parameters for yeast cell immobilization and optoporation. Accordingly, besides using laser tweezers, we would also use micromanipulators to hold the yeast cells for optoporation. Thereby we can learn which means of yeast cell immobilization is better relative to conducting optoporation on the yeast cells without immobilization. We would use two exogenous genes to transform yeast cells: the Green Fluorescent Protein (GFP) plasmids and the nucleic acid of a bacterium GF1 found in termites' gut and could produce efficient cellulase. The reason for choosing the two genes was that, after the transformation, the visualization of the GFP fluorescence in the recombinant cells could provide direct evidence for the achievement and efficiency of each transformation way to help us obtain the optimal parameters for our nano-optical method. Similarly, the transformed GF1 cellulase in the recombinant yeast cells would digest the biomass in Congo red plates to appear transparent zones, thus providing visible evidence for the successful transformation. We would also evaluate the transformation efficiency and reliability by culturing the transformed yeast cells for passages of generations to see if the bacterial cellulase/GFP DNA had become incorporated as a heritable component of the yeast genome and remained for multi-generations.
It would be a new technical challenge for laser ablation and trapping on living cell nano-scaled manipulation. The resolution to the problem would let us achieve a new way of efficient yeast cell transformation. It will also provide a powerful means for transforming any suspending cells with glycan or lignified cell walls.
Experimental
Exogenous genes and reagents
Green fluorescent protein (GFP) shuttle plasmid was purchased from Kang Wei Reagent Company, Beijing, China.The termite-associated symbiotic cellulase nucleic acid was extracted from the bacterial colony GF1 (a Novosphingobium panipatense) in the gut of Coptotermes Formosanus termites (provided by the Guang Dong Institute of Entomology) that screened by the Congo red dye method27 and showed strong capability of digesting biomass. Firstly, take 1 ml of 4 ml cultured bacteria that had been cultured for two days to centrifuge at 1.6 × 104 g for 2 min. After discarding its supernatant, it was mixed with the left 3 ml cultured bacteria for centrifugation again. Following the centrifugation and discarding of the supernatant, added 600 ml bacteria lysate into the tube. Next, the mixture was incubated at 80 °C for 5 minutes. When they cooled to room temperature, added 3 ml RNase buffer to perform enzymolysis at 37 °C for 30 minutes and then cooled to room temperature again. Later, the solution was mixed with 200 ml of protein sediment reagent and put in an ice bath for 5 minutes, then centrifuged at 1.78 × 104 g for 2 minutes. The supernatant was then transferred to another 1.5 ml centrifugal tube and mixed evenly with 600 ml isopropanol, centrifuged at 1.78 × 104 g for 10 minutes. Once removing the supernatant and adding 600 ml 10% ethanol into the tube, the mixture was centrifuged again at 1.78 × 104 g for 5 minutes. After discarding the supernatant, the pellet was put on a clean bench for 15 minutes to evaporate the ethanol, and then added 100 ml of DNA lysates into the tube to re-dissolve the DNA at 65 °C. Finally, the solution was stored at 4 °C and taken as the nucleic acid extraction of the termite-associated symbiotic cellulase for gene transformation.
The cellulase activities of the GF1 bacterial colonies and the recombinant GF1–yeast cells were measured following the method of Miller.28 The DNA sequence analysis and identification of GF1 were performed by BGI Genomics Co., Ltd (Shenzhen, China) using the bacterial colonies. Its growth curve, optimum catalytic conditions, and cellulase production and activity were also determined using methods similar to that described previously.29,30
Experimental setup
The experimental setup is shown in Fig. 1. It was a P.A.L.M Microlaser Combisystem (P.A.L.M. AG, Germany) that was based on a Zeiss inverted microscope and equipped with a pulsed nitrogen laser (with pulse duration of 3 ns) as the laser scissors and a CW YAG laser as the laser tweezers (see Fig. 5). The wavelengths of the pulsed nitrogen laser and the YAG laser were 337 nm and 1064 nm, respectively. The diameter of the nitrogen laser micro beam was focused to 0.6 μm. Its laser pulse (duration: ∼3 nanosecond, pulse repeat frequency: 10 Hz) was of 160 μJ to 230 μJ energy per pulse. The power densities of the laser tweezers for trapping the cells were 100 × 109 W m−2 to 200 × 109 W m−2. The system was adapted to equip with a two-arm multi-joystick robotic micromanipulator for the micromanipulation of cells. It controlled the capillary tubes to either hold the yeast cells during the optoporation or transport the cells. The screw syringes connected to the capillary tubes provided stable negative pressure for holding the cells.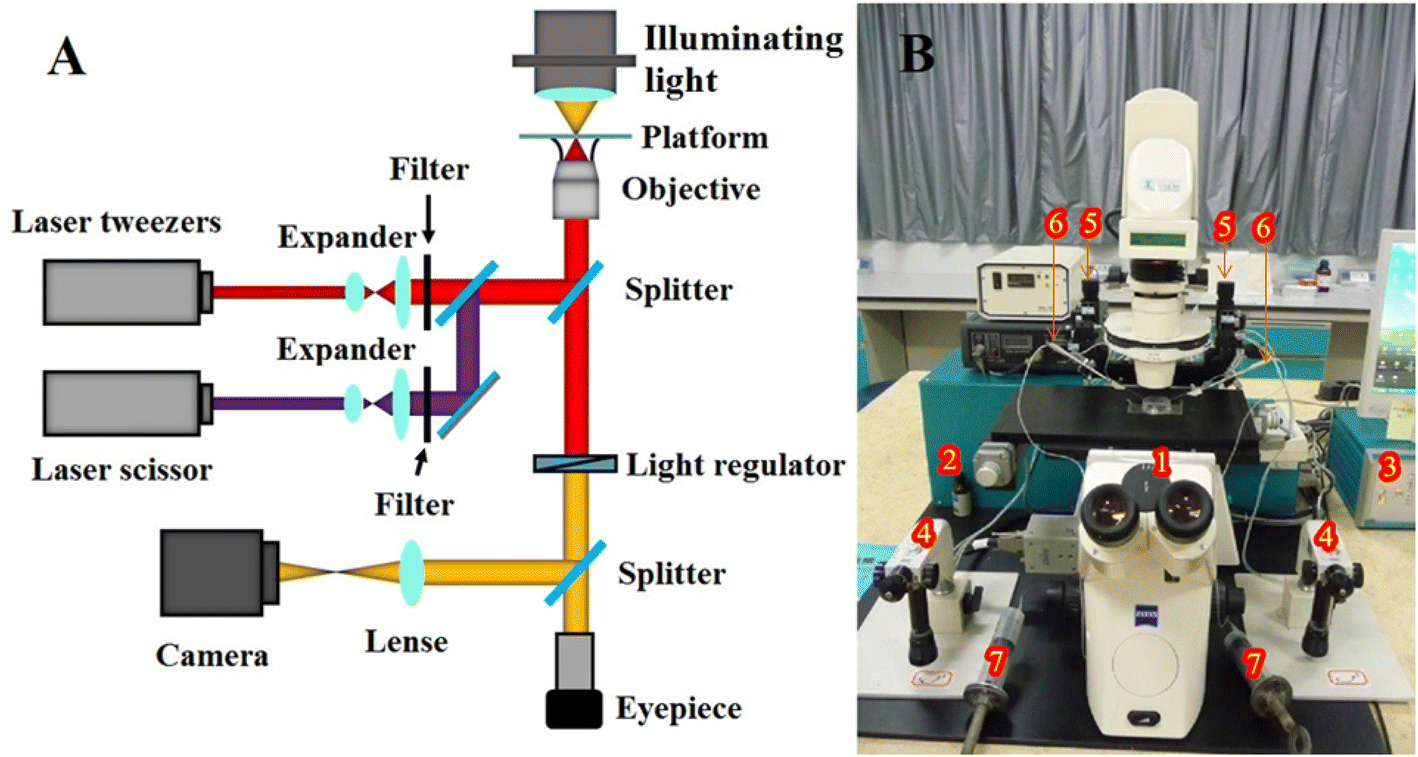 | ||
| Fig. 1 The structure of the microlaser combisystem (A) and the whole experimental set up (B). (1) The microscope; (2) laser interface and lasers (laser scissor and laser tweezers); (3) control unit; (4) joystick of the micromanipulation system; (5) micromanipulation system; (6) capillary tube; (7) screw syringe for providing negative pressure; (8) sample vessel. | ||
Transformation method
The selected yeast cells were cultured 10 h or overnight in Yeast Extract Peptone Dextrose Medium (YPD) and washed twice with PBS solution at 2.7 × 103 g. After that, 50 ml of them was placed in a hypertonic solution with either the GFP plasmid (0.2 μg μL−1 in concentration) or the DNA extraction of the termite bacterial cellulase (10-fold diluted GF1 gDNA). Then optoporation was performed on the cells with the nitrogen laser beam to ablate their cell walls and plasma membranes for a transient nano-sized hole. The optoporation was conducted in three ways, not only on the yeast cells immobilized by the laser tweezers but also those held by a negative pressure capillary tube or suspended freely in the DNA solution. During the experiment, the irradiation energy of the nitrogen laser varied from 160 μJ per pulse to 230 μJ per pulse, and the irradiation time changed from 1 to 5 seconds. After the optoporation, the transformed yeast cells were immediately transferred to a new YPD culture medium by a negative pressure capillary tube. The cell viability and transform efficiency for the different parameters and means were compared to determine the optimal way and parameters for optoporation.Evaluation of the transforming efficiency and stability
The fluorescent images of the recombinant GFP–yeast cells were observed and analyzed with the Zeiss inverted microscope for transforming efficiency evaluation. While the PCR amplification and gel electrophoresis were used to determine if the transformed yeast cell had expressed the termite bacterial cellulase genes. The cellulase activity of the recombinant yeast cells was tested by the Congo red dye method and the DNS assay.30 The cells were also re-plated several times and grown for spread cultivation. The transformation efficiency was defined as the percentage of the cells that not only remained viable but also retained termite bacterial gene expression for generations within all the transformed cells. After transferring for generations, the production and fermentation abilities and the cellulase activity of the 5th, 10th, and 15th generations were tested in comparison with that of the first generation to evaluate the stabilities of the termite cellulase gene expression and heredity of the recombinant yeast cells.Statistical analysis
The experiments were repeated at least three times and in triplicate parallels when possible. Results are presented as mean ± standard deviation. Statistical significance was evaluated by Student's t-test. Values of P < 0.05 were considered statistically significant.Results
Transformation of S. cerevisiae cells with GFP
To get the optimal conditions of laser irradiation for gene transformation by optoporation, we tried the energies of the nanosecond ultraviolet pulsed laser ranging from 160 μJ to 230 μJ per pulse, and the irradiation time for each of them from 1 to 5 seconds. After tests on thousands of yeast cells, we found that when the irradiation energy per pulse was higher than 196 μJ, it would induce an ejection of the cellular contents so that the cells were lethally damaged (see Fig. 2(A)). However, if the energy per pulse was less than 165 μJ, it was hard to ablate the cell wall to produce a transient hole in the cell (see Fig. 2(B)). The optimal parameters of the irradiation were 182 μJ per pulse with a pulse frequency of 10 Hz for 2–3 s (so the total number of the laser pulse was 20–30, and the total energy for the optoporation was in the range of 3.64 × 10−3 to 5.46 × 10−3 J). Such irradiation would not induce perceptible deformation but create a penetrative hole of about 500–600 nm in diameter on the cell suspended in the solution of GFP plasmid and immobilized by the laser tweezers, as shown in Fig. 2(C). The hole size was almost the same as the laser beam size at the focal point, it allowed sufficient DNA plasmids to enter it (see the enlarged phase contrast image of the irradiated yeast). While after the optoporation, the hole could immediately self-reseal in a couple of seconds, and the cell kept on surviving. The budding cells were found on the fifth-day post optoporation. Fig. 2 also shows the yeast cells after the optoporation (Fig. 2(D)). The fluorescence displayed by the cells (Fig. 2(E)) indicated that the transformation was successful and the cell could express the GFP gene.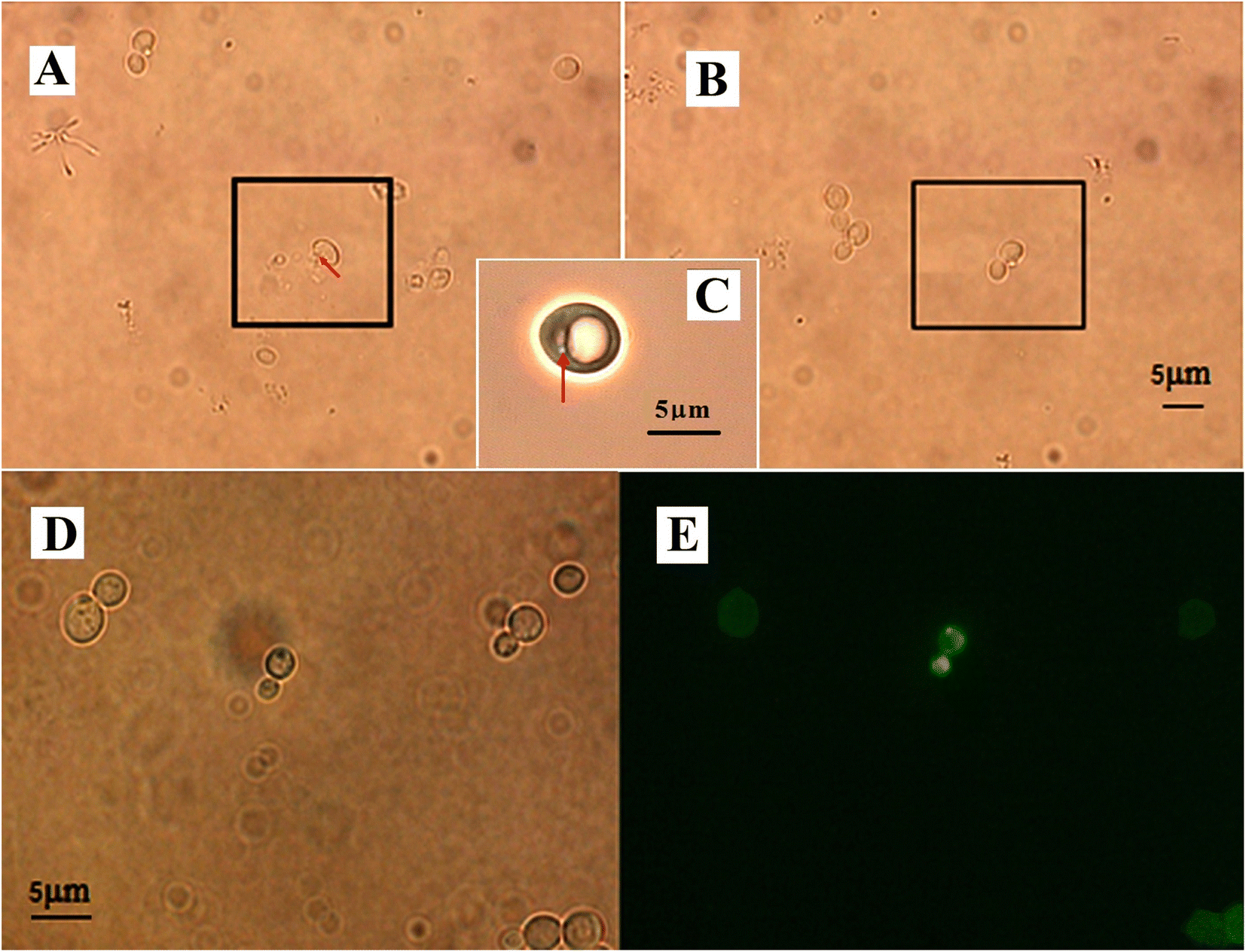 | ||
| Fig. 2 The yeast cells during and after the optoporation. (A) The cell was damaged by the irradiation energy higher than 196 μJ, the red arrow indicates an ejection of the cellular contents from the damaged cell. (B) The irradiation of energy less than 165 μJ couldn't produce a transient pore in the cell. (C) The 182 μJ irradiation created a transient nano-sized hole on the cell as indicated by the red arrow. (D) The cells after the optoporation and (E) exhibited GFP fluorescence. | ||
Table 1 illustrates the transformation efficiencies obtained by the three means mentioned above. The transformation efficiency = (cells remaining viable after optoporation and retained the ability of exogenous gene expression for generations/all the cells under transformation) × 100%. From the table, we can see that the transformation efficiency was increased from 5% to 28% for using laser tweezers to immobilize the cells and to 23% using a negative pressure capillary tube, indicating that holding the yeast cells for optoporation is necessary for efficient transformation.
| a 1 – Just using the laser beam to create holes on the yeast cells free in suspension. 2 – Holding the cells with laser tweezers for the optoporation. 3 – Holding the cells with a negative pressure capillary tube for the optoporation. | |||
|---|---|---|---|
| Ways | 1 | 2 | 3 |
| Efficiency (%) | 5.0 ± 0.8 | 28.1 ± 0.3 | 23.0 ± 0.4 |
Transformation of S. cerevisiae cells with the nucleic acid of GF1
The transformation of S. cerevisiae cells with the nucleic acid of GF1 was performed using the optimal laser parameters for optoporation combined with laser tweezers for holding the cells. Fig. 3 illustrates the process of the transformation. From Fig. 3(A), we can see that the yeast cell was firstly immobilized by the laser trapping with a power density of 150 × 109 W m−2. After selecting the irradiating point by turning over the cell with the laser tweezers (as indicated by the vertex angle of the green triangle), the 182 μJ ultraviolet pulsed laser beam (pulse duration ∼3 ns at 10 Hz) shot at the spot for 2–3 s. The micropipette placed beside the cell was for transferring the cell after the optoporation. It sucked the cell in immediately after the optoporation (Fig. 3(B)) and put the cell into a new incubation medium solution of YPD (Fig. 3(C)). Fig. 4(A) shows a budding recombinant cell found on the fifth day after the optoporation. The clear zone in the Congo red dye plate produced by the supernatant of the recombinant yeast cells shown in Fig. 4(B) indicated that the recombinant GF1–yeast cells had the hydrolysis cellulose capability comparable to that of GF1 (see Fig. 4(D)). The transformation efficiency of yeast cells with GF1 was similar to that of transforming GFP into the cells. | ||
| Fig. 3 The process of the transformation on a yeast cell with GF1. | ||
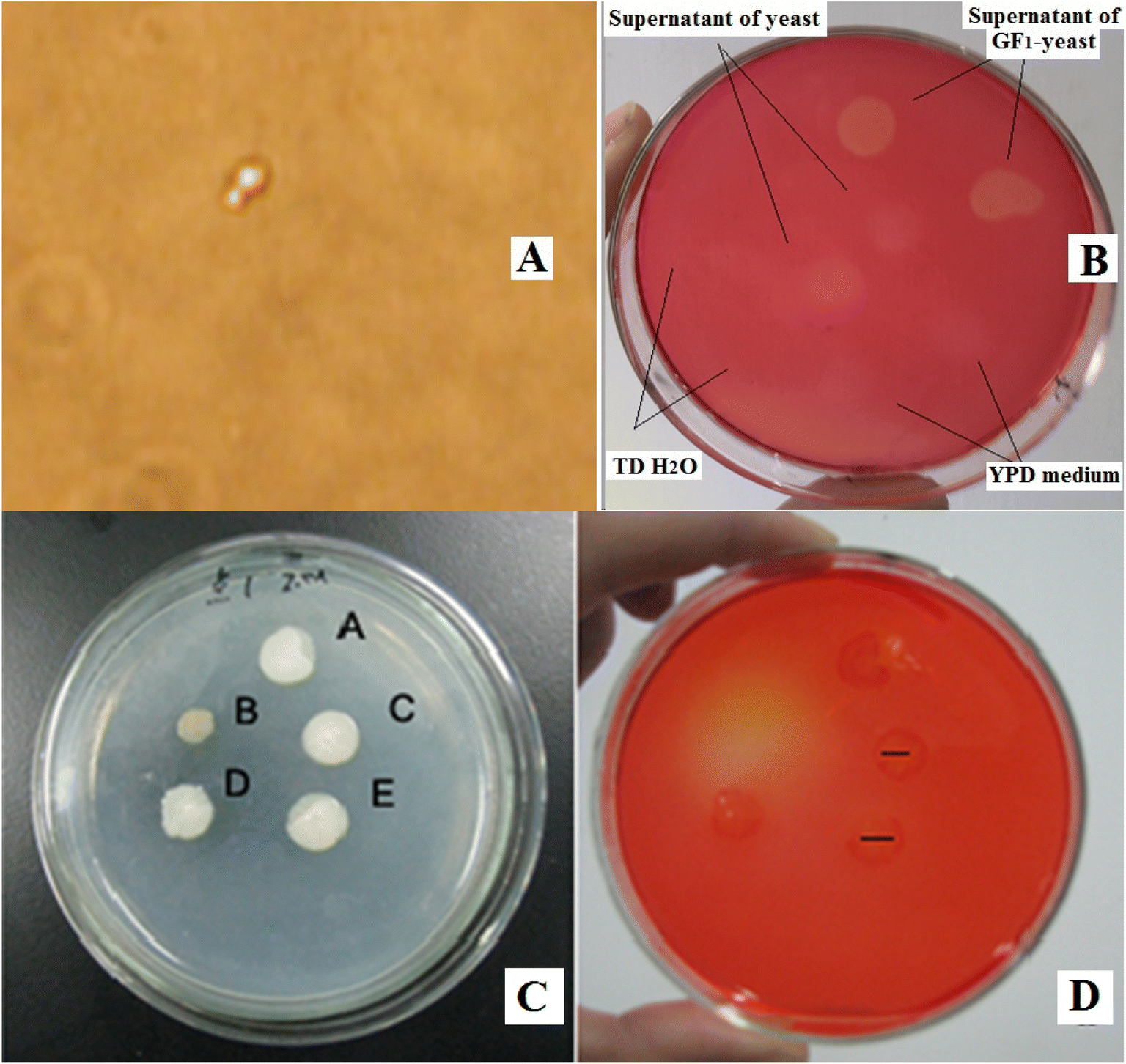 | ||
| Fig. 4 (A) A budding recombinant cell on the fifth day after the transformation, and (B) the clear zone in the Congo red dye plate produced by the supernatant of the recombinant yeast cells. (C) The GF1 bacterial colonies and (D) the clear zones produced by them in the Congo red dye plate on the sixth day. | ||
The cellulase gene expression and heredity stabilities of the recombinant cells
Fig. 5(A) shows the PCR gel electrophoresis of the transformed yeast (rY), GF1, and the wild-type S. cerevisiae yeast (Y). We can see that the recombinant yeast rY also had a single band at ∼1600 bp as GF1 (also see the inset of Fig. 5(A)) while there was none in wild-type yeast (Y), indicating that the transformed yeast expressed the genes of GF1. Table 2 lists the data about the production and fermentation abilities and the cellulase activities of the first, 5th, 10th, and 15th generations of the recombinant yeast cells. They suggested that the termite-associated cellulase gene expression was stable and with heredity stability in the recombinant yeast cells. Fig. 5(B) shows the phase-contrast images of the recombinant yeast cells (the 5th generation). We can see that the cells grew well with ordinary shape and size. Fig. 5(C) and (E) respectively illustrate the enzyme activity of GF1 at different temperatures and pH values.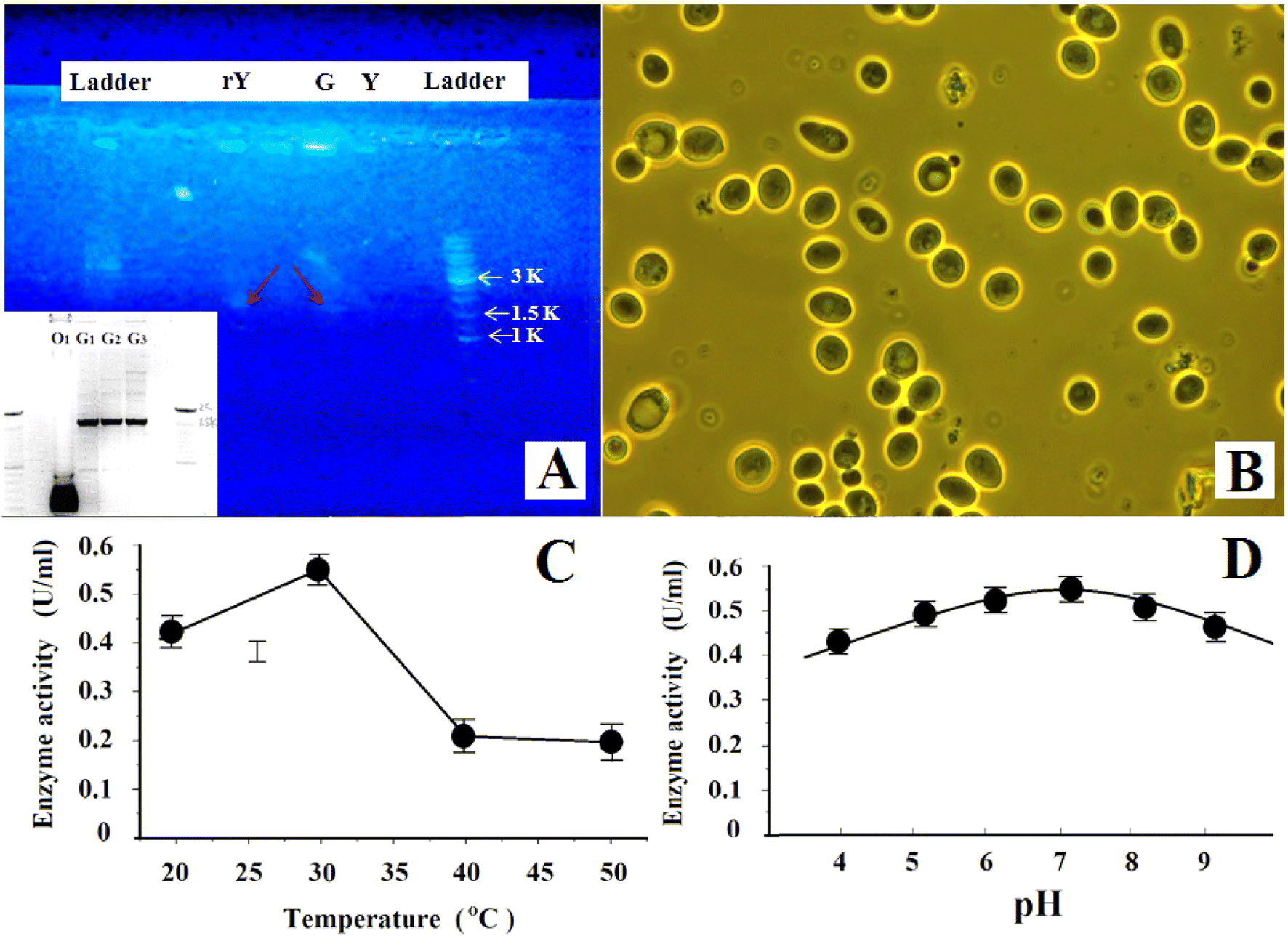 | ||
| Fig. 5 (A) The PCR gel electrophoresis of GF1 (G) and the transformed yeast (rY), in comparison with that of wild type yeast (Y). (B) The phase contrast image of the recombinant yeast cells (the 5th generation). (C). The enzyme activity of GF1 at different temperature. (D) The enzyme activity of GF1 vs. pH. | ||
| Function | First generation | 5th | 10th | 15th | Wild-type yeast |
|---|---|---|---|---|---|
| Cell count after 24 h incubation | 3.6 × 107 | 3.7 × 107 | 3.6 × 107 | 3.5 × 107 | 3.8 × 107 |
| Fermentation rate (g d−1) | 1.51 ± 0.05 | 1.49 ± 0.04 | 1.50 ± 0.05 | 1.48 ± 0.03 | 1.51 ± 0.05 |
| Cellulase activity (U mL−1) | 0.48 ± 0.03 | 0.46 ± 0.04 | 0.44 ± 0.03 | 0.45 ± 0.03 | 0.01 ± 0.01 |
Discussion
We have developed an efficient nano-optical method that transferred exogenous genes (GFP and termite-associated cellulase DNA) into Saccharomyces cerevisiae cells by the combined operation of laser optoporation, laser trapping, and micromanipulation. With the nano-optical method, we have not only performed the first yeast transformation but also established an efficient GFP–yeast recombinant system and a novel termite cellulase DNA recombinant yeast system. Our technique differed from the conventional ones for yeast transformation such as spheroplasting, LiAc/ssDNA/PEG, electroporation, glass bead method, and biolistic method and showed advantages in several aspects.Our method was distinct from the conventional ones in using laser light to create a transient nano-sized hole on the yeast cell wall to deliver the exogenous DNA into the specific targeting single cell. Unlike the chemical and electrical methods, the mechanisms by which how did the plasmid DNA traverses the cell wall are still unknown.3 Ours for transferring plasmid DNA into yeast cells is clear enough. The hole created by the laser beam provided a clear nano-sized entering path for the exogenous DNA. The impacting force of the pulsed laser helped to compel more DNAs to enter the cell and increase their uptake into the cytoplasm. The achievement of yeast transformation with our method depended on three factors. The first was the laser energy for the optoporation. As aforementioned, the proper laser energy should create a transient hole with appropriate nano sizes in the cell wall that would self-reseal after the optoporation to leave the cells intact and healthy. By employing different laser pulse energies for the optoporation on thousands of cells, we found that the suitable one for a pulsed nitrogen laser was between 165 μJ per pulse and 196 μJ per pulse. Since the effect of the nanosecond ultraviolet pulse laser on the cell is photochemical,17 the irradiation of the optimal pulse energy 182 μJ only created a penetrative hole of about 500–600 nm in diameter on the cell wall precisely at the laser-focused point, but without other disturbances even perceptible deformation on the cell. According to the studies reported previously,11,31,32 the hole of similar sizes could self-reseal immediately after the perforation due to the combined effects of cell wall regulation and remodeling, spontaneous deplasmolysis, osmotic pressure, membrane surface tension, line tension, etc. The quick resealing of the hole lets the cell keep intact and alive.
The second factor was the irradiation time. Based on our observation about the cell survival and transform efficiency, the laser optoporation should last for 2–3 s (corresponding to 20–30 laser pulses). With such a length of time, the laser could pierce the cell wall of 100–200 nm thick, and the created hole could keep open for some time to allow sufficient exogenous DNA to get in but self-reseal soon afterward. Too short a time (less than 2–3 s) could not produce a penetrative hole, and too long would induce cell damage.
The third one was the way to perform the optoporation. Though the laser pulse energy was a pivotal point for determining whether an optoporation could create a penetrative hole, our experiments on the three means of optoporation suggested that holding the cells or not would strongly affect the success rate of the transformation and its efficiency. Retaining precise focalization on the cells in suspension during the optoporation was difficult if the cells were not immobilized. So it was necessary to hold the cells for optoporation to ensure the laser irradiation was at the same spot in the entire 2–3 s process. Compared with using a negative pressure micropipette for holding, trapping the cells with a laser tweezer for optoporation was easier to handle and more convenient to turn them around to select target points. Although during the trapping process, the cells might absorb some energies of the infrared laser light. There is no need to worry about the possible thermal effect of the laser tweezers on the cell. The previous studies demonstrated that even after manipulating the yeast cells for several minutes with a similar laser power density as that used in our method, the absorbed laser energy by the cell walls was not sufficient to cause visible disruption or other observable side effects on the cell.17 Our experiment also proved that the 2–3 seconds of trapping the yeast cell for optoporation with the laser tweezers did not disturb the yeast transformation. On the contrary, it led to higher transformation efficiency than manipulating the cell with a micropipette.
Our method was also different from the previous works using laser optoporation for mammalian cell transfection12–15 and rice cell transformation.19 As aforementioned, yeasts differed from mammalian cells or rice cells in cellular structures and components. Their stiff, brittle, and thick cell wall makes the optoporation more difficult than on the mammalian cell membranes and rice cells. So we had to explore a new set of laser parameters (including the wavelength, pulse duration, repetition frequency, pulse number, and lasted time) for the optoporation to ensure that it would only create a penetrative nano-size hole but did not induce cell wall cracking. Moreover, mammalian cells are attached to the cultured plate. They would not move away during the optoporation process, so the previous works of laser transfections on mammalian cells only used one laser for optoporation. But yeasts are suspension cells. Therefore, besides the laser for optoporation, we used one more laser for laser trapping (or micromanipulators) to immobilize and manipulate the cells. So the combined use of laser puncturing and trapping was a new approach to yeast transformation. Our nano-optical method for yeast cell transformation has opened up a new way for the optical transformation of cells with stiff, brittle thick cell walls.
The transformation efficiency of the conventional ones, such as LiAc/ssDNA/PEG, was only several transformants per 104 yeast cells to several percent.3,33,34 So the transformation efficiency of our method (see Table 1) was very high compared with theirs. The high transformation efficiency might be due to our unique way of transformation. The laser beam created a clear passage for the exogenous genes to get into the yeast cell. The passing hole was kept open for 2–3 s so that enough exogenous genes could get in to increase their efficient uptake and recombination into the target locus. The laser light did not induce perceptible deformation or other damage to the cell but only created a transient nano-sized hole. So the cell could have high survival rate and transformation efficiency. Although how the exogenous genes moved to the yeast nucleus and became established was unclear and invites further investigation. There was a possibility that the laser beam pierced both the cell wall and nuclear membranes and directly sent the exogenous genes into the nucleus. The S. cerevisiae cells themselves also have host nuclear import machinery and DNA replication machinery for the nucleus uptake of exogenous DNA and DNA replication inside the nucleus.35 At any rate, the fluorescence appearing in the GFP–yeast recombinant system indicated the incorporation of GFP genes into the yeast. The PCR amplification and the gel electrophoresis shown in Fig. 4(C) suggested that the recombinant GF1–yeast cells also had a single band at ∼1600 bp as GF1, thus providing direct evidence for the GF1 DNA sequences presented in the genome of the transformed yeast strain and the transformation had occurred in the yeast system. Besides, the clear zone produced by the supernatant of the recombinant yeast (rY) cells shown in the Congo red dye plate (Fig. 4(B)) indicated that the GF1–yeast system could degrade CMC. Although the supernatant of the intact yeast cells also induced a faint zone in the plate (see Fig. 4(B)) implied that the wild-type yeast cells might secrete a small amount of cellulase themselves, it was not comparable to that produced by the transformed yeast. As shown in Table 2, the cellulase activity of the recombinant yeast was similar to that of the donor GF1. Such a strong capacity for degrading cellulose should be due to the effect of the incorporated termite bacterial cellulase genes. It further proved that the termite bacterial cellulase DNA should have been integrated into the yeast cell and let the cell have and remain the biomass digesting capability for multi-generations with high gene expression stability and hereditary stability. The comparison between the enzyme activities of GF1 and that of the recombinant GF1–yeast cells, on the other hand, indicated that the recombinant yeast had a high endogenous rate of termite bacterial gene recombination and could secrete cellulase with high secretory capacity. It also suggested that the expression host had little effect on the functionality of the GF1. Inversely, the recombination only slightly affected the fermentation rate of the host yeast by reducing it from ∼1.53 g d−1 to about 1.50 g d−1. All these results indicated that using our method, it is possible to directly use the termite nucleic acid to transform S. cerevisiae cells without the help of any vectors or carriers. Based on our knowledge, this is the first report of transferring bacteria gDNA into yeast without using any vectors or carriers.
In summary, we have demonstrated the validity of our nano-optical method for yeast cell transformation. The method showed several advantages over preexisting transformation methods: (1) it can perform precise nano-scale manipulation on living yeast cells for transformation. (2) It has a definite mechanism of transporting exogenous genes into yeast cells through the transient nano-sized hole created by the laser optoporation. (3) It can perform single-cell transformation and target any point of the cell for optoporation. (4) Although the simultaneous joint operation of the laser scissors and laser tweezers for the optoporation is a sophisticated work, its process is simple, just holding the cell and shooting it with the laser beam, and easy to perform under visual control. (5) It can directly transfer naked DNA into yeast cells for transformation. (6) The survival rate and transformation efficiency of the yeasts transformed using our method were much higher than the conventional methods. Its limitations included needing expensive equipment equipped with laser scissors and laser tweezers together and well-trained technical personnel to handle dosimetric and optical alignment issues that occur with complex bio-photonic systems frequently. We believe that our method would also work with other eukaryotic cells of cell walls for gene transformations and will broadly apply to different aspects of gene engineering.
Author contributions
YXH conceived and designed the experiments. JWY and ZHW performed the experiments and data/evidence collection. YXH, JWY and ZHW analyzed the data. YXH wrote the paper.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported partly by the Chinese National Natural Science Foundation (Nos. 30940019 and 60377043), Guang Dong Provincial Science and Technology Foundation (No. 2015B010105006 and 2013B060100011), and Guang Zhou Science and Technology Foundation (No. 2014Y2-00508). We would also like to gratefully acknowledge support from the GeneForge Ltd and J Schroeder.References
- J. C. Mell and S. M. Burgess, Encyclopedia of Life Sciences, 2003, DOI:10.1038/npg.els.0000821.
- F. Griffith, J. Hyg., 1928, 27, 113–159 CrossRef CAS PubMed.
- R. D. Gietz and R. A. Woods, BioTechniques, 2001, 30, 816–831 CrossRef CAS PubMed.
- A. A. Eddy and D. H. Williamson, Nature, 1957, 179, 252–1253 CrossRef.
- H. Ito, K. Murata and A. Kimura, Agric. Biol. Chem., 1983, 47, 1691–1692 CAS.
- E. Neumann, S. Kakorin, I. Tsoneva, B. Nikolova and T. Tomov, Biophys. J., 1996, 71, 868–877 CrossRef CAS PubMed.
- M. C. Costanzo and T. D. Fox, Genetics, 1988, 120, 667–670 CrossRef CAS.
- S. A. Johnston, P. Q. Anziano, K. Shark, J. C. Sanford and R. A. Butow, Science, 1988, 240, 1538–1541 CrossRef CAS PubMed.
- K. Haas, W.-C. Sin, A. Javaherian, Z. Li and H. T. Cline, Neuron, 2001, 29, 583–591 CrossRef CAS.
- S. Kar, L. Mohan, K. Dey, P. Shinde, H.-Y. Chang, M. Nagai and T. S. Santra, J. Micromech. Microeng, 2018, 28, 123002 CrossRef CAS.
- D. Riveline and P. Nurse, Nat. Methods, 2009, 6, 513–515 CrossRef CAS PubMed.
- M. Tsukakoshi, S. Kurata, Y. Nomiya, Y. Ikawa and T. Kasuya, Appl. Phys. B: Photophys. Laser Chem., 1984, 35, 135–140 CrossRef.
- W. Tao, J. Wilkinson, E. J. Stanbridge and M. W. Berns, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4180–4184 CrossRef CAS PubMed.
- U. K. Tirlapur and K. König, Nature, 2002, 418, 290–291 CrossRef CAS PubMed.
- X. Tsampoula, V. Garcés-Chávez, M. Comrie, D. J. Stevenson, B. Agate, C. T. A. Brown, F. Gunn-Moore and K. Dholakia, Appl. Phys. Lett., 2007, 91, 053902–053904 CrossRef.
- X. Tsampoula, K. Taguchi, T. Čižmár, V. Garces-Chavez, N. Ma, S. Mohanty, K. Mohanty, F. Gunn-Moore and K. Dholakia, Opt. Express, 2008, 16, 17007–17013 CrossRef CAS PubMed.
- J. Ando, G. Bautista, N. Smith, K. Fujita and V. R. Daria, Rev. Sci. Instrum., 2008, 79, 103705 CrossRef PubMed.
- M. Waleed, S.-U. Hwang, J.-D. Kim, I. Shabbir, S.-M. Shin and Y.-G. Lee, Biomed. Opt. Express, 2013, 4, 1533–1547 CrossRef PubMed.
- Y. Guo, H. Liang and M. W. Berns, Physiol. Plant., 1995, 93, 19–24 CrossRef CAS.
- Z. X. Chong, S. K. Yeap and W. Y. Ho, Peer J, 2021, 9, e11165, DOI:10.7717/peerj.11165.
- V. Dupres, Y. F. Dufre and J. r. J. Heinisch, ACS Nano, 2010, 4, 5498–5504 CrossRef CAS.
- N. Maclean, J. Bacteriol., 1964, 88, 1459–1466 CrossRef CAS PubMed.
- P. Tanger, M. E. Vega-Sánchez, M. Fleming, K. Tran, S. Singh, J. B. Abrahamson, C. E. Jahn, N. Santoro, E. B. Naredo, M. Baraoidan, J. M. C. Danku, D. E. Salt, K. L. McNally, B. A. Simmons, P. C. Ronald, H. Leung, D. R. Bush, J. K. McKay and J. E. Leach, Bioenergy Res., 2015, 8, 1165–1182 CrossRef CAS.
- M. Waleed, S.-U. Hwang, J.-D. Kim, I. Shabbir, S.-M. Shin and Y.-G. Lee, Biomed. Opt. Express, 2013, 4, 1533–1547 CrossRef PubMed.
- W. He, Y. Liu, M. Smith and M. W. Berns, Microsc. Microanal., 1997, 3, 47–52 CrossRef CAS.
- Y.-X. Huang, L. Li, L. Yang and Y. Zhang, Biomed. Opt. Express, 2018, 9, 1783–1794 CrossRef CAS PubMed.
- P. Dheeran, N. Nandhagopal, S. Kumar, Y. K. Jaiswal and D. K. Adhikari, J. Ind. Microbiol. Biotechnol., 2012, 39, 851–860 CrossRef CAS PubMed.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- C. A. Uchima, G. Tokuda, H. Watanabe, K. Kitamoto and M. Ariokaa, Appl. Environ. Microbiol., 2012, 78, 4288–4293 CrossRef CAS PubMed.
- D.-s. Liu, Q. Shen, Z.-x. Wang and Q. Zhuang, J. Cellul. Sci. Technol., 2004, 12, 17–19 Search PubMed.
- C. S. Buer, K. T. Gahagan, A. Grover, J. Swartzlander and P. J. Weathers, Biotechnol. Bioeng., 1998, 60, 348–355 CrossRef CAS.
- Y. Zhen, M. Radulovic, M. Vietri and H. Stenmark, EMBO J., 2021, 40, e106922 CrossRef CAS PubMed.
- C. Bruschi, A. Comer and G. Howe, Yest, 1987, 3, 131–137 CAS.
- R. H. Schiestl and R. D. Gietz, Curr. Genet., 1989, 16, 339–346 CrossRef CAS PubMed.
- B. Lacroix and V. Citovsky, mBio, 2016, 7(4), 008633, DOI:10.1128/mBio.00863-16.
| This journal is © The Royal Society of Chemistry 2022 |