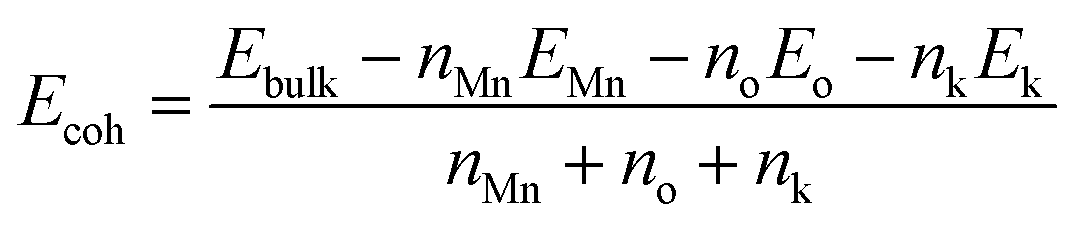DOI:
10.1039/D2RA05493K
(Paper)
RSC Adv., 2022,
12, 30549-30556
Promoting the mechanism of OMS-2 for gas adsorption in different K+ concentrations
Received
1st September 2022
, Accepted 8th October 2022
First published on 26th October 2022
Abstract
Catalytic combustion technology is an efficient and green method to deal with low concentration methane. Gas adsorption over the catalyst surface is a key step in the catalytic combustion process, which has attracted much interest. In this work, the first-principles density functional theory calculation method has been applied to explore the adsorption processes of CH4 and O2 molecules on the surface of cryptomelane type manganese oxide octahedral molecular sieves (OMS-2). In addition, the effect of K+ concentration in the OMS-2 tunnel on the adsorption of the two gaseous molecules has also been investigated. The results of adsorption energy and structural characteristics show that the adsorption energies of CH4 and O2 molecules over the catalyst surface are favorable. Adsorption sites of CH4 are the K+ and O sites, among which the K+ site is the most stable adsorption site. In addition, Mn sites are favorable for adsorbing O2 molecules. The interactions between the catalyst and the adsorbed CH4 and O2 are enhanced with the increasing tunnel potassium ions. It should be noted that with the increasing strength of the adsorption energies, equilibrium distances from the two gaseous molecules to the active sites become shorter and the bond lengths of C–H and O–O bonds become longer. Moreover, the adsorption sites of CH4 on the catalyst surface increase with the increasing K+ concentration. Bader charge and cohesive energy calculations reveal that the tunnel K+ can balance charges and help strengthen the structural stability of OMS-2. Interestingly, the electronegativity of the catalyst has been altered after introducing K+, which leads to better adsorption of gaseous CH4 and O2. The microscopic mechanism of the effect of K+ concentration on the adsorption of CH4 and O2 over the catalyst surface paves the way for further deciphering the mechanism underlying the catalytic oxidation process and helps design more efficient catalysts for methane utilization.
1. Introduction
Ventilation air methane emissions from coal mines have drawn worldwide attention because of the high global warming potential and the loss of methane resources.1 Therefore, exploration and the development of ventilation air methane capture and utilization is technically challenging and has important environmental and safety concerns. Among the alternatives, catalytic combustion technology has been considered as the most effective way to solve this problem because of its efficient and green purification means.2,3 Catalysts used in methane catalytic combustion are mainly divided into non-noble metal catalysts4–6 (such as perovskite and hexaaluminate-type catalysts) and noble metal catalysts7,8 (such as supported Pd and Pt catalysts), wherein non-noble metal catalysts have a wide application prospect due to their low price, easy availability and strong thermal stability.
Among these non-noble metal catalysts, the special properties of manganese oxide molecular, such as high catalytic activity and thermal stability, low cost and easy to obtain, make it a new star material.9–11 OMS-2 is a type of manganese oxide sieve with edge- and corner-sharing MnO6 octahedra forming a 2 × 2 tunnel structure.12 The key features for the high catalytic activities are mainly on account of the mixed valency of Mn (2+, 3+ and 4+), porous structure, easy release of lattice oxygen and acidic sites.13 In addition, for the 0.46 nm tunnel cavity, some cations (such as Li+, Na+ and K+) can be introduced into the tunnel to alter its chemical and physical properties. Many efforts have been invested to explore the effect of introduced metal cations on its catalytic activity. Ni et al.14 found that the introduction of alkali earth metal ions (Mg2+, Ca2+ and Sr2+) into the tunnel of OMS-2 can obviously change the catalytic activity for benzene oxidation. Among them, the Sr2+-doped catalyst exhibited the best catalytic activity and was even superior to the commercial 0.5% Pt/Al2O3 catalyst. In addition, Hao et al.15 revealed that introducing Co ions into the OMS-2 tunnel can effectively improve the catalytic activity of OMS-2. Further evidence of this remarkable effect of introducing metal cations has been provided by Hong et al.,16 who demonstrated that ozone decomposition was promoted after introducing Na+ into OMS-2 tunnels by facilitating lattice defect formation. Recently, Ming et al.17 found that doping metal cations (Li+, Na+, Mg2+, Ca2+, Fe3+, Zn2+ and Ag+) in OMS-2 catalysts promoted the selective catalytic reduction. It is widely known that the K+ ion is close to the dimensions of the 2 × 2 tunnels of OMS materials and has been used as an ideal cationic template in OMS-2 tunnels.18–20 Luo et al.21 found that K+ concentration significantly affected the magnetism of OMS-2. Hou et al. enhanced the oxidation of benzene18 and arsenite22 by tuning the K+ concentration in the OMS-2 tunnel. Moreover, Hamaguchi et al.23 investigated the effect of K+ content on the NO-adsorption properties and suggested that the adsorption sites of NOx were the K sites.
In light of the above developments, it can be seen that lots of efforts have been directed toward the development of the OMS-2. Nevertheless, insufficient work has been reported on methane adsorption over the OMS-2 surface. Recently, our group has reported that the positions of oxygen vacancies have influence on the adsorption capacity of the OMS-2 (110) surface for methane.24 Do any other factors exist? Further, how does K+ concentration affect methane adsorption? In addition, most of the studies focus on the experimental aspect. The detailed information and the mechanism of the adsorption process still remain unanswered.
In recent years, with the rapid development of computing technology and hardware, first-principles computation becomes an important tool for material design, synthesis and property prediction. Many experimental phenomena and microscopic reaction mechanisms have been well explained by theoretical research. Adsorption of reactant molecules on the catalyst surface is the first and key step in the catalytic reaction process. To this end, the reactants CH4 and O2 molecules were selected to explore the adsorption mechanism over the OMS-2 catalyst surface by a theoretical calculation method. In addition, the effects of tunnel K+ concentrations (OMS-2 without K+, with 1 atom and 2 atom% K+) on the adsorption process were also analyzed. From the investigation of the geometric and energetic features of the complexes formed by the catalyst and the gaseous molecules, we suggest that the tunnel K+ concentrations are essential for gas adsorption.
2. Computational details
All calculations were carried out using the Vienna Ab initio Simulation Package (VASP).25,26 The Perdew–Burke–Ernzerhof (PBE) functional and the projector augmented wave (PAW) method were used for density functional theory (DFT) calculations.27 To accurately describe the highly localized Mn 3d-orbitals, DFT + U calculations were performed with a value of U = 4 eV applied to the Mn 3d states. A cut-off energy of 500 eV and 2 × 2 × 1 Monkhorst–Pack k-point mesh were used throughout the calculations. All calculations were converged until the maximum force on all atoms was smaller than 0.02 eV Å−1. Meanwhile, the electronic optimization steps were fewer than 10−4 eV. Adsorption energy (Eads) of the gas molecules on the OMS-2 catalyst surface can be calculated by eqn (1):| | |
Eads = Ecom − ECH4 − EOMS-2
| (1) |
where Ecom, ECH4 and EOMS-2 represent the total energy of the adsorbed configuration, the isolated methane molecule and the optimized OMS-2 slab, respectively.
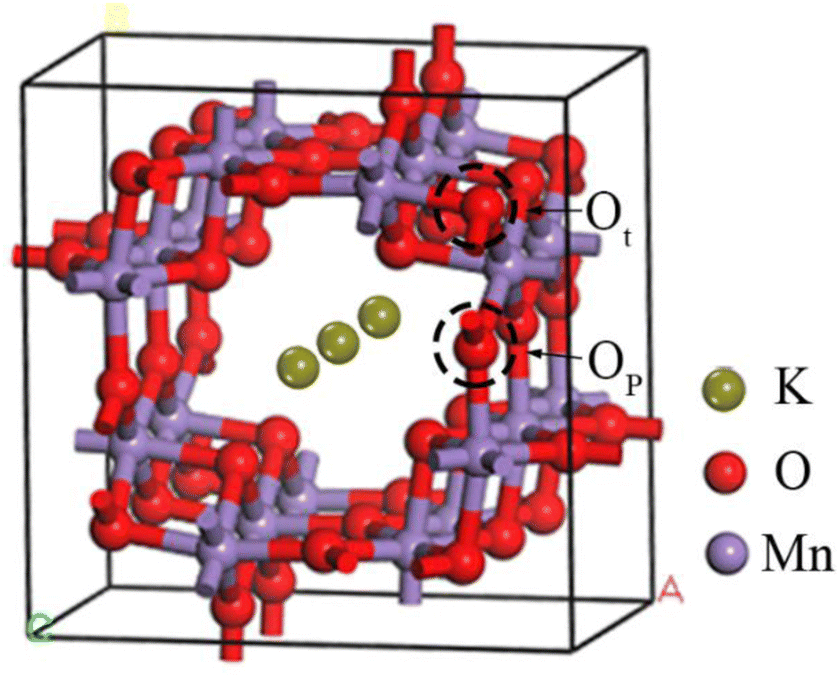
The unit cell model has been established based on the crystal structure information of OMS-2 in the Materials Project28 database. As shown in Fig. 1, the crystal parameters after optimization are a = b = 9.74563 Å, c = 5.81139 Å, and α = β = γ = 90°. In addition, it has been reported that the OMS-2 (110) surfaces have the lowest surface energy and dominate the largest surface area.29 Therefore, the (110) surfaces were selected to explore the gas adsorption in different K+ concentrations. The slab (Fig. 2), with a (2 × 2) supercell and a vacuum layer of 15 Å, was constructed to model the OMS-2 (110) surface.30 In addition, gas molecules (CH4 and O2) placed at all possible adsorption sites over the catalyst surface were optimized. Furthermore, it is worth noting that two types of oxygen atoms30 are found in the structure of OMS-2, which are distinguished by Ot and Op respectively.
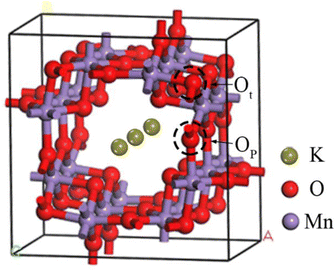 |
| | Fig. 1 Unit cell structure of OMS-2. | |
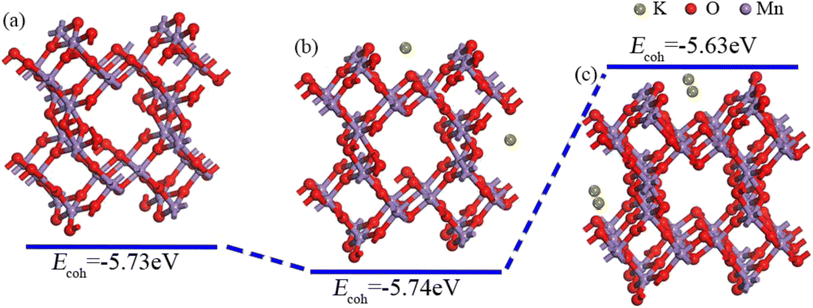 |
| | Fig. 2 OMS-2 (110) surface under different K+ concentrations. (a) Without K+, (b) OMS-2 with 1 atom% K+, and (c) OMS-2 with 2 atom% K+. | |
3. Results and discussion
3.1 Cohesion energy calculations
Cohesion energy (Ecoh) is representative of the energy needed to decompose a crystal into atoms, which is an important parameter to evaluate the stability of a crystal model.31 A lower cohesion energy shows a higher crystal stability. Herein, Ecoh has been calculated from the following equation:| |
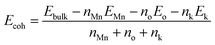 | (2) |
wherein Ebulk, EMn, Eo and Ek are the energies of OMS-2, Mn atom, O atom and K+, respectively. nMn, no and nk represent the number of Mn, O and K+ atoms, respectively.
Fig. 2 shows the OMS-2 (110) surface and the corresponding Ecoh under different K+ concentrations. It is apparent that the cohesion energy of OMS-2 is −5.73 eV. Upon introducing 1 atom% K+ into the tunnel of OMS-2 (K1-OMS-2), the cohesion energy becomes −5.74 eV, suggesting a significant enhancement of the catalyst stability. Interestingly enough, the cohesion energy decreases to −5.63 eV after increasing K+ to a certain extent (2 atom% K+, K2-OMS-2), which indicates that an appropriate K+ concentration can stabilize the Mn–O skeleton.
3.2 Effects of K+ concentration on CH4 adsorption over the OMS-2 (110) surface
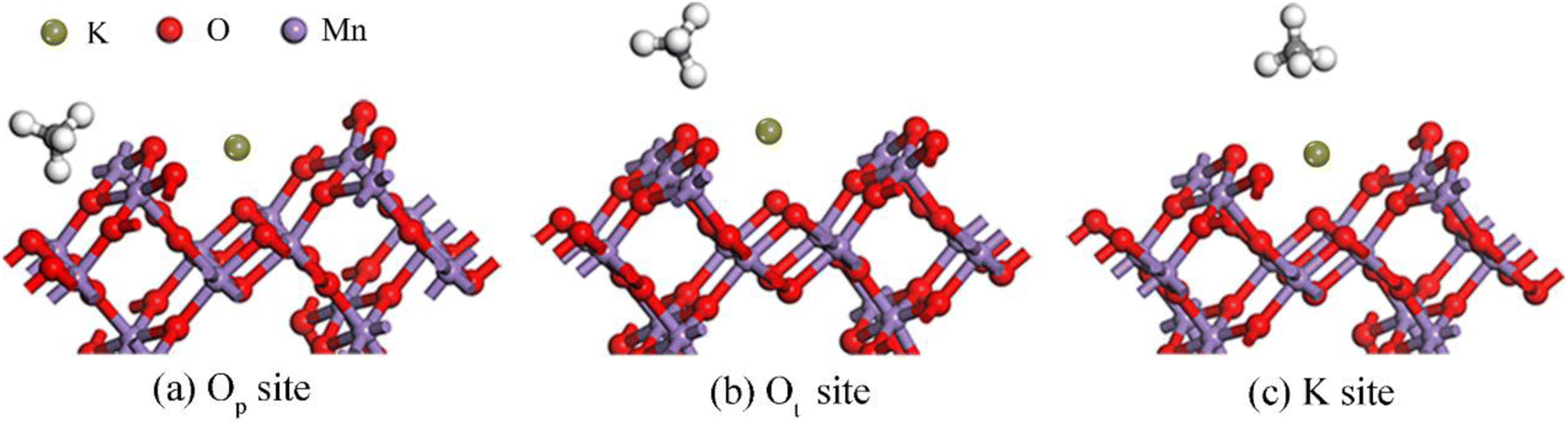
3.2.1 Adsorption properties of CH4 on the OMS-2 (110) surface. In order to explore the adsorption properties of CH4 on the OMS-2 (110) surface, all possible adsorption configurations were optimized. As shown in Fig. 3, the most stable adsorption site of CH4 is the Ot-top site with two H atoms of CH4 interacting with the Ot atom. In addition, the C–H bond lengths, interacting with the catalyst surface, are slightly elongated from its isolated state (shown in Table 1), implying that the electronic structure of CH4 has minimal perturbation after adsorption over the OMS-2 (110) surface. The adsorption energy is −1.13 eV, exhibiting a weak chemical adsorption, which is in line with the change of structural parameters.
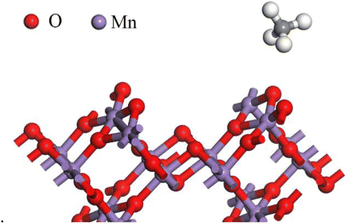 |
| | Fig. 3 Stable structure of CH4 on the OMS-2 (110) surface. | |
Table 1 Adsorption energy of CH4 molecules on the OMS-2 (110) surface (Eads, eV), bond length of C–H (d, Å) and the average bond length of C–H (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) , Å)
, Å)
| Adsorption site |
Eads |
Chemical bond |
d |
|
| Ot |
−1.13 |
C–H1 |
1.095 |
1.0955 |
| C–H2 |
1.096 |
|
3.2.2 Adsorption properties of CH4 on the K1-OMS-2 (110) surface. To further test the influence of K+ on the adsorption of CH4 over the catalyst surface, we have tested all the possible adsorption configurations. The adsorption energies and the corresponding structural parameters are summarized in Fig. 4 and Table 2. Apparently, a significant difference exists after introducing K+ into the tunnel of OMS-2. Three different stable adsorption states (Op, Ot and K+ sites) are found. Upon CH4 adsorption at Op and Ot sites, only one H atom of CH4 interacts with the adsorption site. The C–H bond lengths are also increased, suggesting a weakened C–H bond energy of CH4. In addition, the adsorption energies of O sites are −1.15 eV and −1.92 eV, stronger than that over the OMS-2 surface (−1.13 eV). Interestingly, the distance from the C atom of CH4 to the adsorption site decreases with the increase of adsorption energy. For the adsorption of CH4 on the K+ site, three H atoms of CH4 interact with K+. The C–H bond lengths are further increased compared with that on the O site. The adsorption energy is −2.07 eV, with the closest distance from CH4 to the adsorption site. In light of the above discussion, we can conclude that the most stable adsorption site of CH4 on the K1-OMS-2 (110) surface is the K+ top site. All the adsorption energies of CH4 are enhanced compared with that over the OMS-2 (110) surface, indicating that the tunnel K+ of OMS-2 can affect methane adsorption. In addition, the C–H bond lengths of CH4 are also increased, which demonstrates that the changes of the C–H bond lengths of CH4 are in line with that of the adsorption energies.
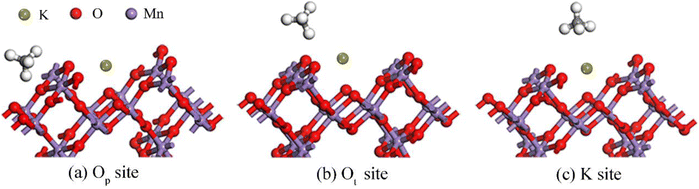 |
| | Fig. 4 Stable structures of CH4 on the K1-OMS-2 (110) surface. | |
Table 2 Adsorption energy of CH4 molecules on the K1-OMS-2 (110) surface (Eads, eV), bond length of C–H (d, Å), average bond length of C–H (, Å) and the distance from the C atom of CH4 to the adsorption site (r, Å)
| Adsorption site |
Eads |
Chemical bond |
d |
|
r |
| Op |
−1.15 |
C–H1 |
1.097 |
1.097 |
3.507 |
| Ot |
−1.92 |
C–H1 |
1.097 |
1.097 |
3.483 |
| K+ |
−2.07 |
C–H1 |
1.099 |
1.0977 |
3.457 |
| C–H2 |
1.097 |
| C–H3 |
1.097 |
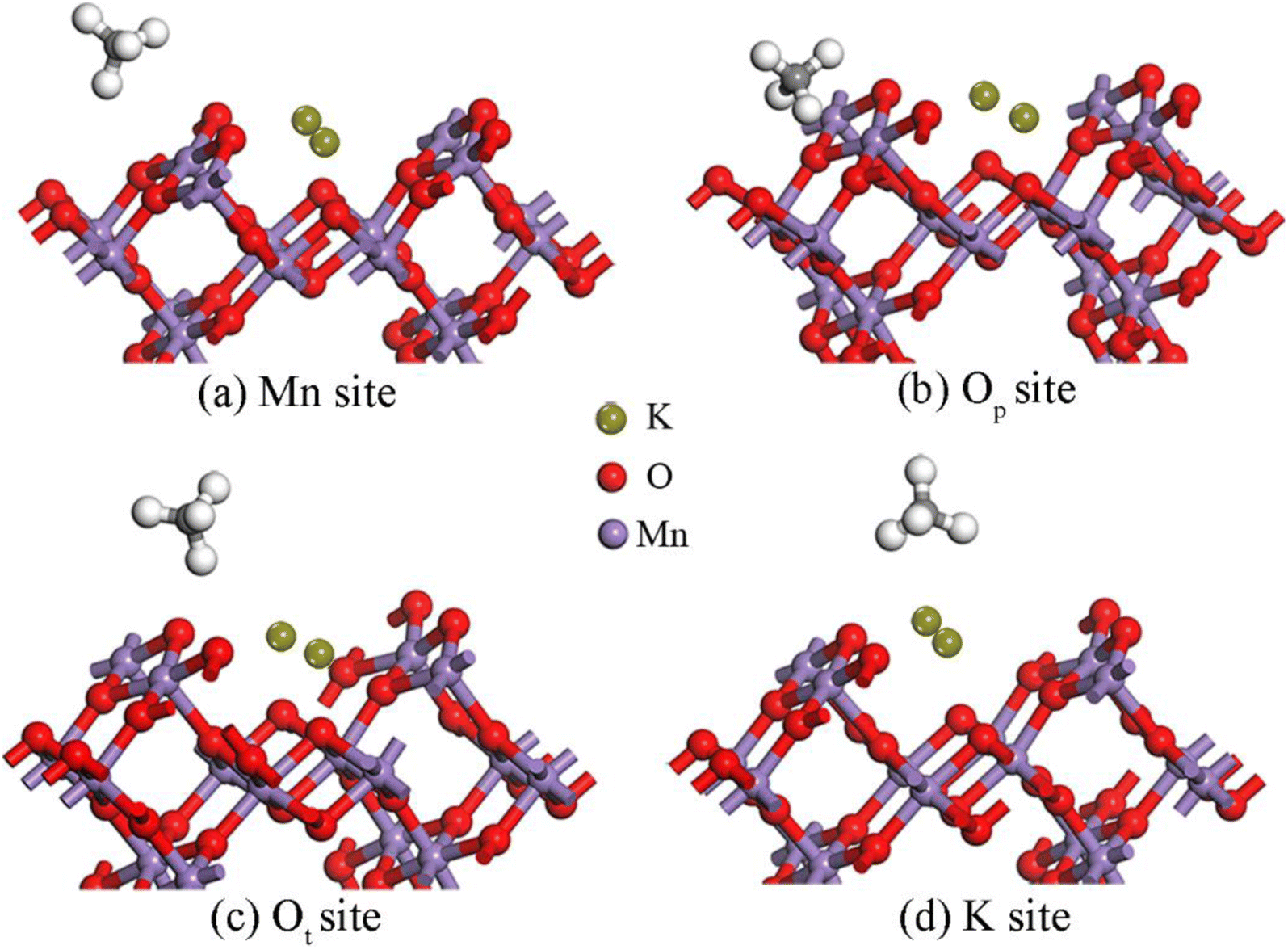
3.2.3 Adsorption properties of CH4 on the K2-OMS-2 (110) surface. To delve further into the distinct adsorption of CH4 over the OMS-2 (110) surface under different K+ concentrations, the adsorption properties of CH4 on the K2-OMS-2 (110) surface were investigated. All stable adsorption configurations are shown in Fig. 5. Apparently, four different stable adsorption states (Mn, Op, Ot and K+ sites) are obtained. Upon CH4 adsorption at the Mn site, two H atoms of CH4 interact with the Mn atom, with the 3.632 Å distance from the C atom to the Mn site, yielding an adsorption energy of −0.53 eV. In addition, the C–H bond lengths of methane are elongated to a certain degree. Upon CH4 locating at Op and Ot sites, only one H atom interacts with the adsorption site, which is similar to that over the K1-OMS-2 surface. In addition, with the increasing K+ concentration, both the adsorption energies (−3.81 eV and −4.21 eV) at the two sites are enhanced. Meanwhile, methane becomes closer to the adsorption site accompanied by a longer C–H bond length. The most favorable configuration is the CH4 adsorbing on the K+-top site with three H atoms interacting with K+, with the Eads equal to −6.10 eV. As expected, the C atom is closest to the adsorption site (3.424 Å) accompanied by the longest C–H bond length. Moreover, it can be clearly found from analysis of the data in Table 3 that the longer the C–H bond length, the closer the C atom is to the adsorption site, and the stronger the interaction between methane and the surface. On the basis of these findings, we can conclude that the adsorption energy of CH4 on the OMS-2 (110) surface is enhanced with the increase of K+ concentration.
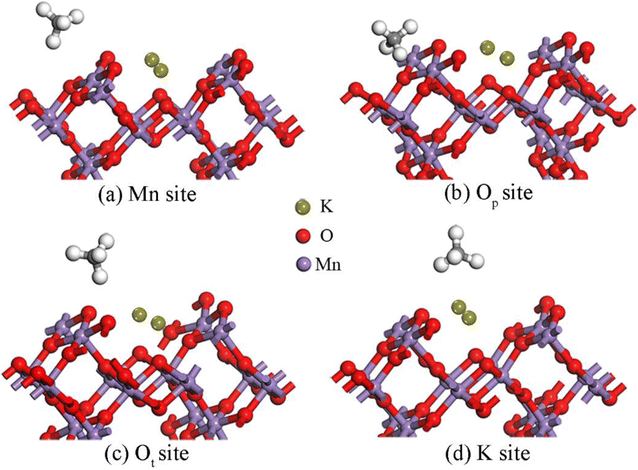 |
| | Fig. 5 Stable structures of CH4 on the K2-OMS-2 (110) surface. | |
Table 3 Adsorption energy of CH4 molecules on the K2-OMS-2 (110) surface (Eads, eV), bond length of C–H (d, Å), average bond length of C–H (, Å) and the distance from the C atom of CH4 to the adsorption site (r, Å)
| Adsorption site |
Eads |
Chemical bond |
d |
|
r |
| Mn |
−0.53 |
C–H1 |
1.098 |
1.097 |
3.632 |
| C–H2 |
1.096 |
| Op |
−3.81 |
C–H1 |
1.097 |
1.097 |
3.424 |
| Ot |
−4.21 |
C–H1 |
1.098 |
1.098 |
3.472 |
| K |
−6.10 |
C–H1 |
1.100 |
1.098 |
3.424 |
| C–H2 |
1.096 |
| C–H3 |
1.098 |
3.3 Adsorption properties of O2 on the OMS-2 (110) surface under different K+ concentrations
The adsorption properties of O2 over the catalyst surface are very important to reveal the catalytic combustion mechanism. All possible adsorption configurations under different K+ concentrations were investigated systematically. As depicted in Fig. 6, only one adsorption site (Mn site) can be found after O2 adsorption over the OMS-2 catalyst surface, in which an O atom of the O2 molecule tilts downward to interact with the Mn site. For the adsorption performances under low K+ concentration (K1-OMS-2), the distance from the Mn-site to the closer O atom of O2 is 3.239 Å, accompanied by an elongated O–O bond of the O2 molecule. The adsorption energy is −0.90 eV. In addition, it can be seen from Table 4 that the O–O bond length is further elongated to 1.276 Å after increasing K+ concentration. The corresponding adsorption energy is −5.67 eV, and the distance from the closer O atom of O2 to the Mn site is shortened to 2.27 Å. Similar to the result of CH4 adsorption, the adsorption energy between O2 and the catalyst surface is enhanced with the increase of K+ concentrations, and the distance from the O2 molecule to the active site becomes closer with an elongated O–O bond.
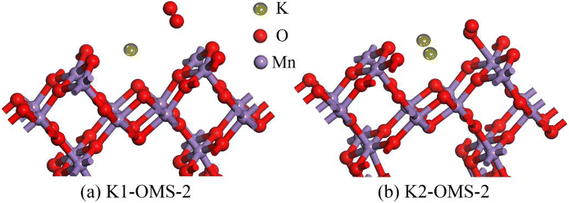 |
| | Fig. 6 Stable structures of O2 on the catalyst surface. | |
Table 4 Adsorption energy of O2 molecules on the catalyst surface (Eads, eV), bond length of O2 molecules (d, Å) and the distance from the closer O atom of O2 to the adsorption site (r, Å)
| |
Eads |
Chemical bond |
d |
r |
| K1-OMS-2 |
−0.90 |
O–O |
1.236 |
3.239 |
| K2-OMS-2 |
−5.67 |
O–O |
1.276 |
2.27 |
3.4 Bader charge analysis
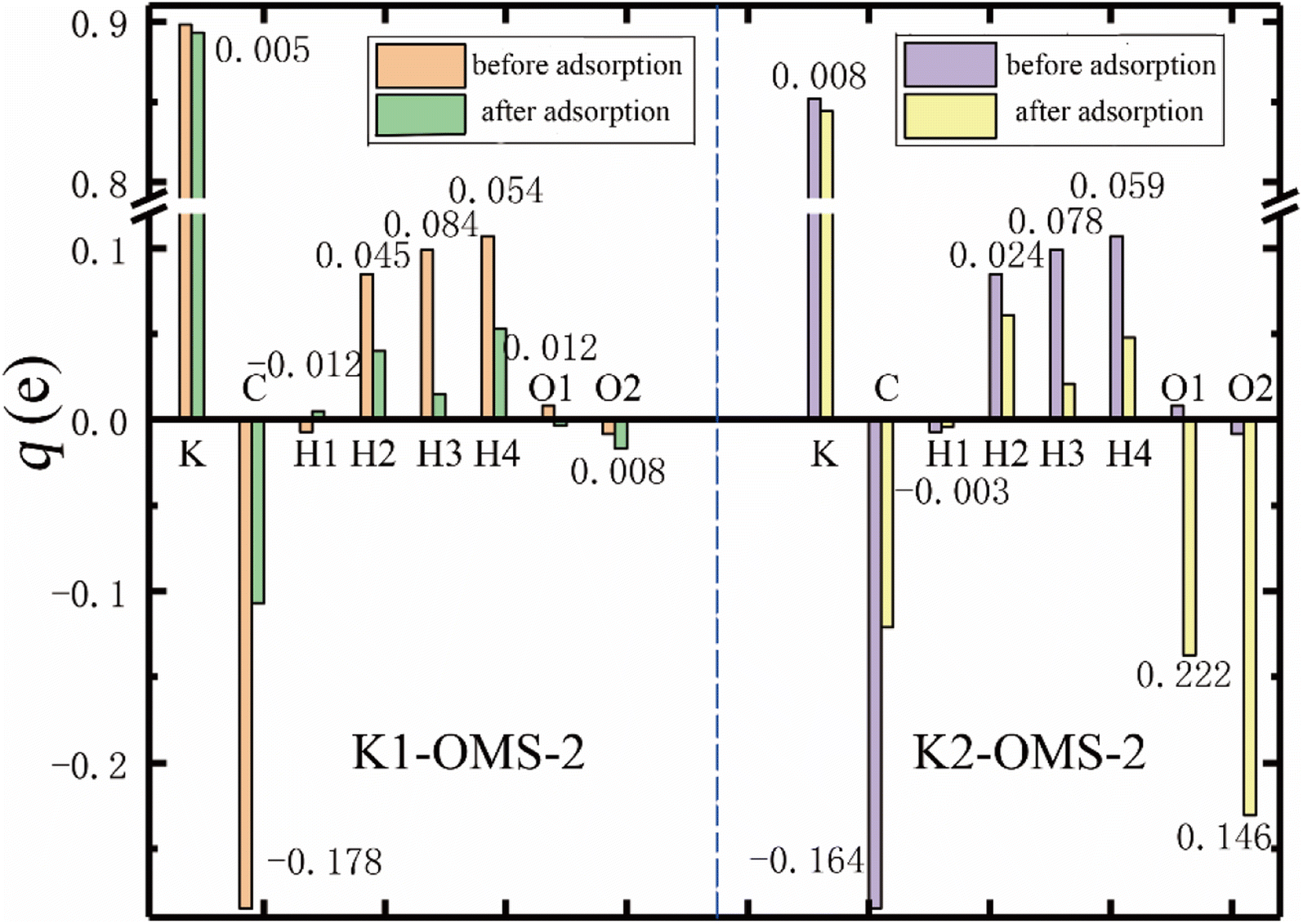
Charge transfer provides very useful information on the adsorption and diffusion of molecules on the catalyst surface.32 To get a deeper understanding of the effect of K+ concentrations on the adsorption process, charge changes of gas molecules and catalyst surface have been calculated by Bader charge analysis.33 Fig. 7 shows the charge distribution of the most stable adsorption configuration of CH4 and O2 molecules on the catalyst surface at different K+ concentrations. As can be seen, CH4 loses electrons and K+ gains electrons, which indicates charge transfer from CH4 to K+ in all adsorption processes. In addition, the charges obtained by K+ after CH4 adsorption over K1-OMS-2 and K2-OMS-2 surfaces are 0.005e and 0.008e, indicating that the charge transfer increases with the increasing K+ concentration. This trend of the charge transfer is consistent with that for the stability of CH4 adsorbing over the catalyst surface. Furthermore, the two O atoms of the O2 molecule obtain electrons after adsorption over the catalyst surface. The numbers of electrons obtained by the O2 molecule increase with the increasing K+ concentration, which is related to a stronger interaction between the O2 molecule and the catalyst surface.
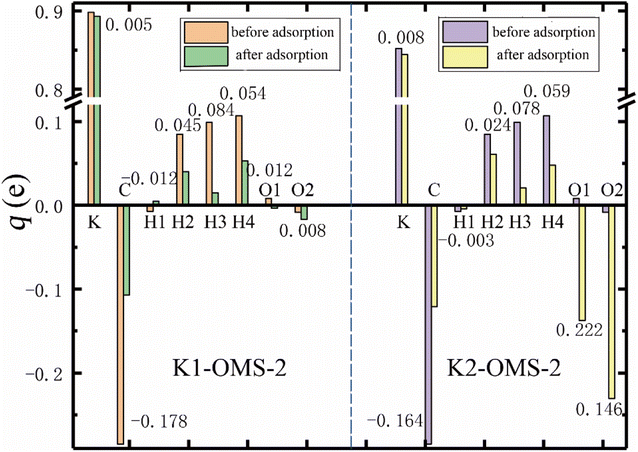 |
| | Fig. 7 Bader charge distribution (q, e) of CH4 and O2 molecules before and after adsorption on the catalyst surface. The numbers in the figure indicate the charge changes after adsorption (Δq, e). | |
Bader charges of different active sites on the catalyst surface were also calculated (as shown in Fig. 8a). Clearly, the presence of K+ changes the electronegativity of Mn and O atoms on the catalyst surface. There is a negative linear relationship between the concentration of K+ and the electronegativity of Mn and O atoms. The higher the concentration of K+, the more obvious the changes of Mn and O electronegativity. It has been reported that the lower average oxidation state (AOS) of the Mn atom is related to the decreased charges on Mn atoms.34 Hence, it can be inferred that the AOS of the Mn atom is decreased with the increasing K+ concentration. In addition, as depicted in Fig. 8a, the introduction of tunnel K+ changes the electronegativity of the OMS-2 skeleton, resulting in a strengthened interaction between the catalyst surface and the gas molecules.
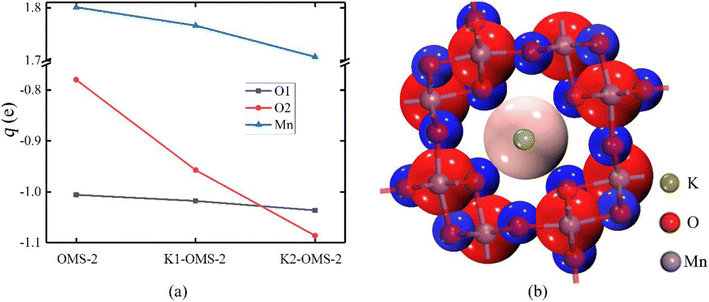 |
| | Fig. 8 (a) Charge changes (q, e) of adsorption sites on the catalyst surface with the change of K+ concentration. (b) The charge colorings on the K2-OMS-2. Red and bule represent positive and negative charges. | |
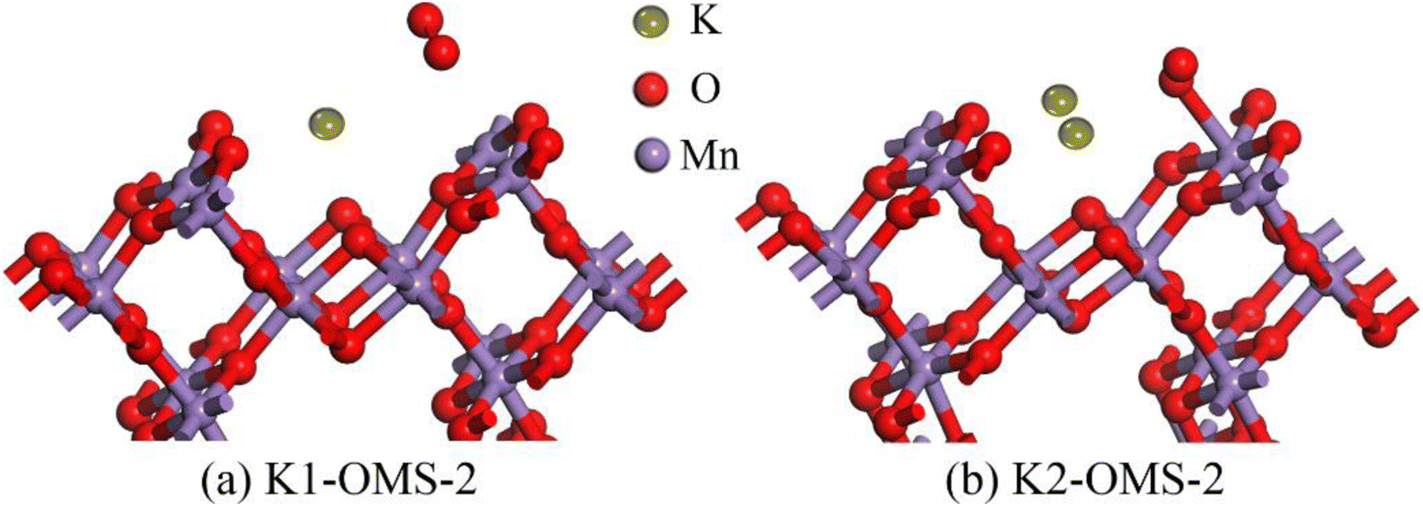
Fig. 8b shows the charge colorings on the K2-OMS-2. It can be clearly seen that the positively charged K+ is surrounded by negatively charged O atoms in the tunnel. Moreover, the radius of K+ is well consistent with the radius of the OMS-2 tunnel. It can, therefore, be concluded that the introduction of K+ can balance the charge and stabilize the tunnel structure, in line with previous results of cohesion energy.
3.5 Effects of K+ concentration on CO2 adsorption over the OMS-2 (110) surface
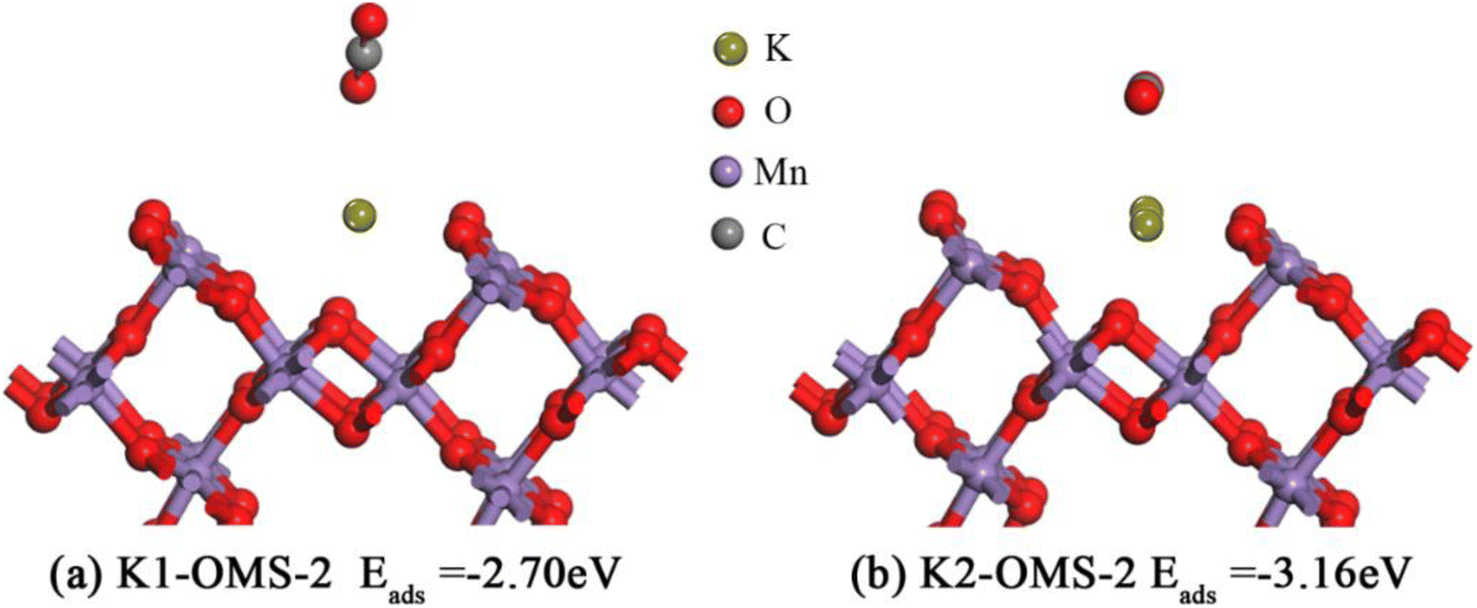
We also extend our investigation to CO2 adsorption over the OMS-2 (110) surface. It is widely known that CO2, one of the products of methane catalytic combustion, can adsorb on the catalyst surface, hindering the reaction. Therefore, it is very important to study the adsorption properties of CO2 on the catalyst surface. As depicted in Fig. 9, CO2 molecules are found to have the most stable states when they are adsorbed on the K+ site, with O atoms of CO2 interacting with K+. In addition, it should be noted that the CO2 molecule is almost parallel to the K2-OMS-2 catalyst plane with two O atoms of CO2 interacting with K+. It is, therefore, reasonable to deduce that the adsorption energy of CO2 molecules on the catalyst surface becomes stronger with the increasing K+ concentration. The adsorption energies (Eads, eV) are also calculated. As displayed in Fig. 9, the calculation results (−2.70 eV to −3.16 eV) are consistent with the above deduction.
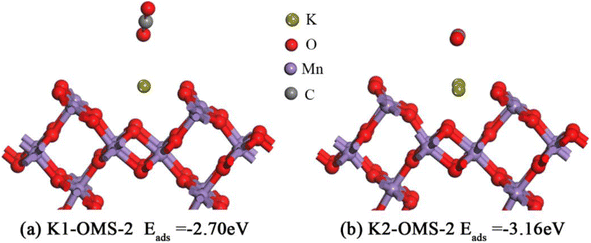 |
| | Fig. 9 Stable structures of CO2 on the catalyst surface. | |
4. Conclusions
K+ is the ideal cationic template in the OMS-2 tunnel and the K+ concentrations significantly affect gas adsorption. The first-principles density functional theory calculation method has been carried out to investigate the inner mechanism for the CH4 and O2 molecule adsorption under different K+ concentrations. Bader charge analysis indicates that the positive charge of the tunnel potassium ions can neutralize the negative charge of the O atom in the skeleton of OMS-2. Interestingly, the van der Waals radius of K+ is in good agreement with the radius of the OMS-2 tunnel. It can, therefore, be concluded that K+ can balance the charge and stabilize the tunnel structure. Adsorption energy and structural parameters show that CH4 and O2 molecules are favorably adsorbed over the catalyst surface. CH4 is preferentially adsorbed on the K+ and O sites, wherein the K+ site is the most stable adsorption site. O2 molecules preferentially reside at the Mn site to form the most favorable configurations. In addition, two different types of oxygen adsorption sites have been found. CH4 molecules tend to react with the Ot species. Moreover, with the increasing strength of the adsorption energies, equilibrium distances from the two gaseous molecules to the active sites become shorter and the bond lengths (C–H and O–O bond) become longer; especially, the adsorption performances between the catalyst and the gas molecules are enhanced with the increase of K+ concentration. Meanwhile, the adsorption sites of CH4 over the catalyst surface are also increased. Furthermore, we also extend our investigation to CO2 adsorption over the OMS-2 (110) surface. As expected, the adsorption energy becomes stronger with the increasing K+ concentration. In summary, the tunnel K+ can change the electronegativity of OMS-2, strengthening the interaction between the catalyst surface and gas molecules. The results presented here offer a broadened view of the subtle effects of the tunnel K+ on the gas adsorption of OMS-2, while providing the theoretical basis for the exploration and development of ventilation air methane capture and utilization.
Author contributions
Shuangli Du: investigation and writing-original draft, Huan Zhang: formal analysis and review, Cunbao Deng and Xuefeng Wang: supervision and validation, Ruicong Zhai: data curation, and Zhijie Wen: supervision, review and editing. All authors analyzed data and contributed to writing the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (U1810206, 52004170), the Open Fund for Key Laboratory of Mining Disaster Prevention and Control (MDPC202009) and the Foundation of Taiyuan University of Technology (2022QN131).
References
- X. Wang, F. Zhou, Y. Ling, Y. Xiao, B. Ma, X. Ma, S. Yu, H. Liu, K. Wei and J. Kang, Energy Fuels, 2021, 35, 15398–15423 CrossRef CAS.
- D. Tian, K. Li, Y. Wei, X. Zhu, C. Zeng, X. Cheng, Y. Zheng and H. Wang, Phys. Chem. Chem. Phys., 2018, 20, 11912–11929 RSC.
- S. Wang, C. Zhao, S. Li and Y. Sun, Phys. Chem. Chem. Phys., 2017, 19, 30874–30882 RSC.
- F. Miao, D. Mao, X. Guo, J. Yu and H. Huang, J. Technol., 2019, 19, 242–248 Search PubMed.
- H. Zhang, S. Du and X. Wang, Nat. Gas Chem. Ind., 2021, 46, 10–14, 127 CAS.
- W. Hu, J. Lan, Y. Guo, X. Cao and P. Hu, ACS Catal., 2016, 6, 5508–5519 CrossRef CAS.
- M. Hou, X. Zhang, C. Fu, W. Cen and J. Chen, Phys. Chem. Chem. Phys., 2020, 22, 4692–4698 RSC.
- X. Wang, X. Zhang, M. Cao, X. Wang, Q. Gao, C. Deng and X. Huang, Energy Fuels, 2022, 36, 6999–7005 CrossRef CAS.
- K. Wantala, T. Suwannaruang, J. Palalerd, P. Chirawatkul and R. Khunphonoil, Surf. Interfaces, 2021, 25, 101286 CrossRef.
- S. GUO, G. Zhang, Z. K. Han, S. Zhang and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 622–630 CrossRef CAS PubMed.
- D. S. Pisal and G. D. Yadav, Mol. Catal., 2021, 491, 110991 CrossRef.
- T. Gao, G. Marianne, K. Frank, N. Reinhard and F. Helmer, J. Phys. Chem. C, 2008, 112, 13134–13140 CrossRef CAS.
- E. Cockayne and L. Li, Chem. Phys. Lett., 2012, 544, 53–58 CrossRef CAS.
- C. Ni, J. Hou, L. Li, Y. Li, M. Wang, H. Yin and W. Tan, Chemosphere, 2020, 250, 126211 CrossRef CAS PubMed.
- Y. Hao, L. Li, Z. Lu, X. YU and X. Yang, Appl. Catal., B, 2020, 279, 119373 CrossRef.
- W. Hong, T. Zhu, Y. Sun, H. Wang and F. Shen, Environ. Sci. Technol., 2019, 53, 13332–13343 CrossRef PubMed.
- S. Ming, P. Wang, P. Liu, J. Duan and T. Li, Chem. Eng. J., 2020, 379, 122287 CrossRef CAS.
- J. Hou, L. Liu, Y. Li, M. Mao, H. Lv and X. Zhao, Environ. Sci. Technol., 2013, 47, 13730–13736 CrossRef CAS PubMed.
- J. Liu, V. Makwana, J. Cai, L. S. Steven and M. Aindow, J. Phys. Chem. B, 2003, 107, 9185–9194 CrossRef CAS.
- Q. Feng, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 319–333 RSC.
- J. Luo, H. T. Zhu, J. K. Liang, G. H. Rao, J. B. Li and Z. M. Du, J. Phys. Chem. C, 2010, 114, 8782–8786 CrossRef CAS.
- J. Hou, Z. Sha, W. Hartely, W. Tan and M. Wang, Environ. Pollut., 2018, 238, 524–531 CrossRef CAS PubMed.
- T. Hamaguchi, T. Tanaka, N. Takahashi, Y. Tsukamoto, N. Takagi and H. Shinjoh, Appl. Catal., B, 2016, 19, 234–239 CrossRef.
- R. Zhai, C. Deng, S. Du and L. Li, Chem. Phys., 2023, 564, 111708 CrossRef CAS.
- G. Kresse and J. Furthmuller, Appl. Catal., B, 1996, 54, 11169–11186 CAS.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 77, 3865–3868 CrossRef PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen and A. K. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- D. A. Tompsett, S. C. Parker and M. S. Islam, J. Mater. Chem. A, 2014, 2, 15509–15518 RSC.
- G. Zhu, J. Zhu, W. Jiang, Z. Zhang, J. Wang, Y. Zhu and Q. Zhang, Appl. Catal., B, 2017, 209, 729–737 CrossRef CAS.
- Z. Chen, G. Li, H. Zheng, X. Shu, J. Zou and P. Peng, Appl. Surf. Sci., 2017, 420, 205–213 CrossRef CAS.
- D. Meng, W. Luo, G. Li and H. C. Chen, Acta Phys. Sin., 2009, 58, 8224–8229 CrossRef CAS.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- Z. Song, Z. Yan, X. Yang, H. Bai, Y. Duan, B. Yang and L. Leng, Chem. Phys. Lett., 2018, 695, 216–221 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Huan Zhanga,
Cunbao Denga,
Xuefeng Wanga,
Ruicong Zhaia and
Zhijie Wen*b
a,
Huan Zhanga,
Cunbao Denga,
Xuefeng Wanga,
Ruicong Zhaia and
Zhijie Wen*b