
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic supramolecular polymerization of water-soluble porphyrins: balancing ionic attraction and steric repulsion to govern stacking†
Chisako Kanzaki,
Hiroshi Yoneda,
Shota Nomura,
Takato Maeda and
Munenori Numata *
*
Department of Biomolecular Chemistry, Graduate School of Life and Environmental Sciences, Kyoto Prefectural University, Shimogamo, Sakyo-ku, Kyoto 606-8522, Japan. E-mail: numata@kpu.ac.jp; Fax: +81-75-703-5132
First published on 26th October 2022
Abstract
We have synthesized novel water-soluble anionic porphyrin monomers that undergo pH-regulated ionic supramolecular polymerization in aqueous media. By tuning the total charge of the monomer, we selectively produced two different supramolecular polymers: J- and H-stacked. The main driving force toward the J-aggregated supramolecular polymers was the ionic interactions between the sulfonate and protonated pyrrole groups, ultimately affording neutral supramolecular polymers. In these J-aggregated supramolecular polymers, amide groups were aligned regularly along polymer wedges, which further assembled in an edge-to-edge manner to afford nanosheets. In contrast, the H-aggregated supramolecular polymers remained anionic, with their amide NH moieties acting as anion receptors along the polymer chains, thereby minimizing repulsion. For both polymers, varying the steric bulk of the peripheral ethylene glycol (EG) units controlled the rates of self-assembly as well as the degrees of polymerization. This steric effect was further tunable, depending on the solvation state of the EG chains. Accordingly, this new family of supramolecular polymers was created by taking advantage of unique driving forces that depended on both the pH and solvent.
Introduction
Supramolecular polymerization of π-conjugated molecules is an attractive way to create functional materials by amplifying the effects of the inherent molecular functionality. Among the various π-conjugated molecules, porphyrin derivatives are attractive because they have a characteristic absorption band in the visible region, making their supramolecular polymers fascinating candidates for practical electro-optical materials.1–7 Over the years, many porphyrin-based supramolecular polymers have been created by tuning the structures of their monomers, especially by varying the peripheral groups attached to the porphyrin cores.8–11 Similar to the general supramolecular polymerization strategies of π-conjugated monomers, self-assembly of porphyrin monomers is commonly driven primarily by strong π-stacking, with the assistance of other noncovalent interactions (e.g., hydrogen bonding). Thus, changing of polarity or composition of the solvent, or varying the temperature, can trigger the self-assembly of porphyrins and allow control over polymerization processes.12–15 Despite an abundance of porphyrin structures, relatively little attention has been paid to their use in other categories of polymerization—for example, by exploiting their ionic interactions, as in the widespread practical preparation of covalent polymers. Any comprehensive means of applying such approaches would be highly attractive for the production of new families of supramolecular polymers through simple acid/base reactions. To this end, our attention has turned to finding ways to exploit new classes of water-soluble porphyrin monomers featuring cationic and anionic reactive moieties, as well as using them to achieve entire polymerization processes through tuning of their π-stacking in aqueous media. Supramolecular polymerization in aqueous media requires different strategies to control the noncovalent interactions relative to those performed in organic solvents.16–18 In this sense, there remains much room to design novel water-soluble porphyrins capable of serving as ionic monomers.Among the many potential candidate porphyrin derivatives,19–23 [5,10,15,20-tetrakis(4-sulfonatophenyl)]porphyrin (TPPS) is a fascinating example of a rare water-soluble self-assembling porphyrin. Under acidic conditions, H2TPPS4 is converted to the protonated species H4TPPS4, which undergoes spontaneous slipped-stack self-assembly (forming J-aggregates), mediated by intermolecular electrostatic interactions between its sulfonate and protonated pyrrole groups. The J-aggregates of H4TPPS4 produced in aqueous media have been regarded as an artificial model of the natural photosynthesis antenna composed of bacteriochlorophylls.24 Although H2TPPS4 derivatives have potential to serve as ionic monomers for fine supramolecular polymerization, it has remained difficult to isolate single pieces of their J-aggregates—because they tend to assemble in a face-to-face manner to form crystalline fibers.25 Therefore, it has not been possible to categorize this type of self-assembly as an example of forming one-dimensional supramolecular polymers without bundling. Exploiting new porphyrin monomers based on H2TPPS4, without being susceptible to bundling of J-aggregates, would lead to the production of new families of supramolecular polymers capable of assembling through ionic interactions.
Several research groups have synthesized H2TPPS4 derivatives in which some of the sulfonic acid moieties have been replaced by nonionic groups (e.g., H2TPPS2 and H2TPPS3).26 Studies of these modified porphyrins have revealed that they are still capable of self-assembly through ionic interactions, even with a low number of sulfonate groups, thereby leading to the creation of tape- and sheet-like27 and as tubular25,28,29 structures. Although these porphyrin derivatives have potential to produce a variety of porphyrin-based supramolecular structures, there have been no previous attempts to synthesize H2TPPS4 derivatives in which two sulfonic acid moieties at opposite meso (trans) positions have been replaced by nonionic groups. We suspected that trans-H2TPPS2 derivatives with various peripheral nonionic units would maintain their ability to undergo one-dimensional self-assembly through interactions of alternating sulfonate (anion) and protonated pyrrole (cation) units, thereby functioning as monomers for ionic supramolecular polymerization in aqueous media, with peripheral nonionic units placed at the wedges potentially suppressing the inherently strong tendency of the resultant supramolecular polymers to undergo bundling.16,30,31
Accordingly, in this study we synthesized novel H2TPPS4 derivatives by positioning two nonionic oligo(ethylene glycol) (EG) units at trans positions. As a standard structure, we introduced EG units through amide groups (H2TPPS2-NHCO-EG), in which two sulfonic acid moieties could potentially interact with the central pyrrole groups under acidic conditions, thereby leading to the creation of J-aggregates. To investigate whether the steric bulk of the EG units influenced the degree of polymerization, we synthesized five H2TPPS2-NHCO-EG derivatives (Fig. 1 and Scheme S1†) having different numbers of EG units [H2TPPS2-NHCO-EGx; x = 2, 4, 6, 8, and 18 (where x represents the number of EG units in a single molecule)]. Each EG unit exerted a different steric effect when it existed in a different solvation state when changing the solvent (e.g., protic or aprotic co-solvents). To highlight the influence of the amide spacer on the self-assembly process, we designed another series of monomers (H2TPPS2-O-EGx; x = 2, 4) in which EG units were introduced at the meso-phenyl position through ether bonds (Fig. 1, and Scheme S2†).
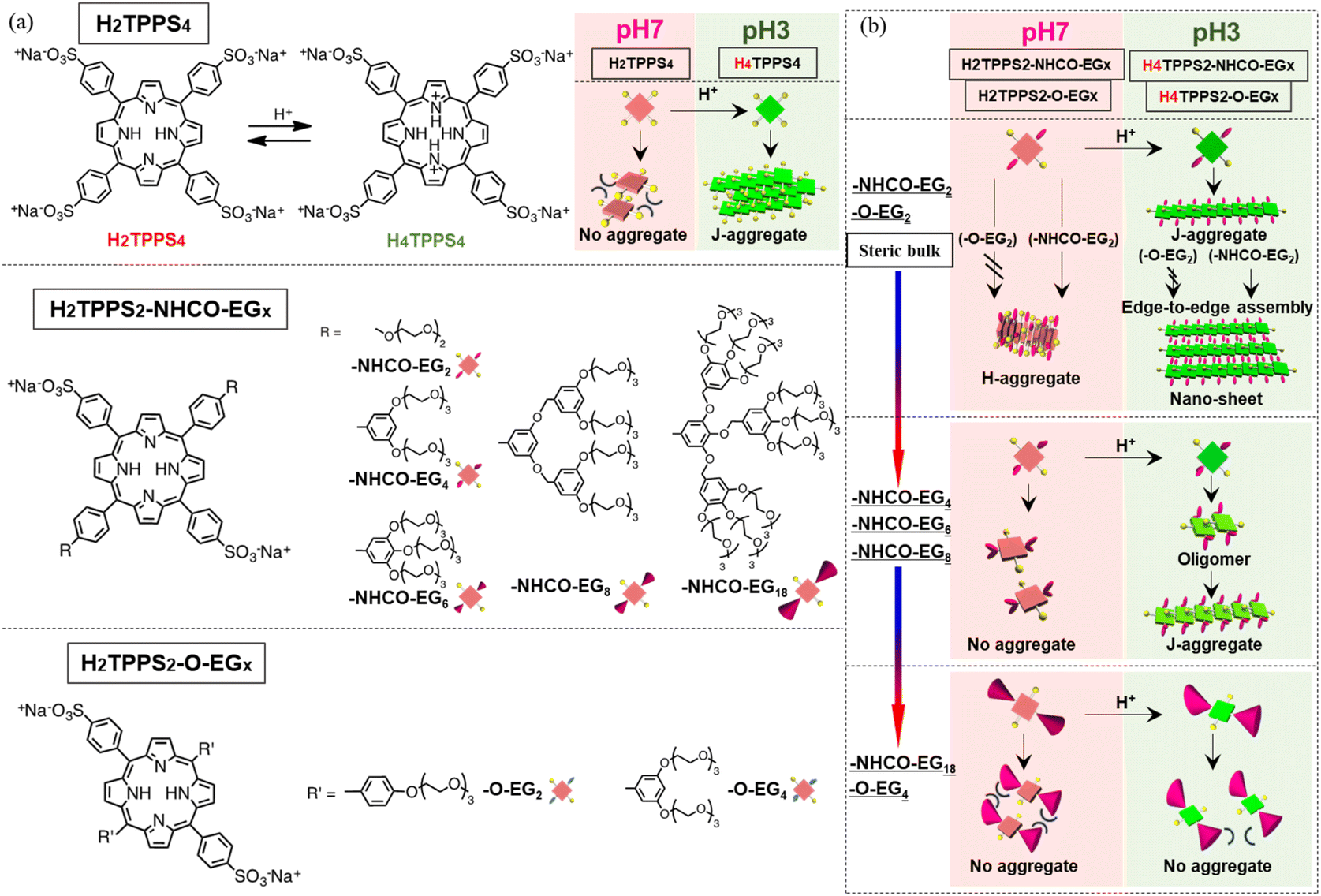 | ||
| Fig. 1 (a) Molecular structures of H2TPPS4, H2TPPS2-NHCO-EGx (x = 2, 4, 6, 8, 18), and H2TPPS2-O-EGx (x = 2, 4); the DFT-calculated structures of the H2TPPS2-NHCO-EGx (x = 2, 4, 6, 8, 18) and H2TPPS2-O-EGx (x = 2, 4) derivatives are provided in the ESI (Fig. S18†). (b) Schematic representation of EG unit dependent supramolecular polymerization toward H- and J-aggregates. Synthetic procedures and spectral data for these porphyrins are available in the ESI.† | ||
We observed several interesting effects that were dependent on the steric bulk. For example, for the H2TPPS2-NHCO-EGx (x = 2, 4) and H2TPPS2-O-EGx (x = 2) derivatives presenting smaller EG units, supramolecular polymerization could be realized with suppression of bundling, with high degrees of polymerization, with the pH directing two independent pathways—one toward slipped stacking (J-aggregate) and the other toward a novel type of face-to-face stacking (H-aggregate). Furthermore, the J-aggregate polymers created from H2TPPS2-NHCO-EGx (x = 2) underwent additional two-dimensional assembly in a unique edge-to-edge manner, due to the regular alignment of amide groups along the polymer edges—in stark contrast to the bundling, driven by face-to-face π-stacking, observed for the parent H2TPPS4 (Fig. S20†). Moreover, for both the J- and H-stacking modes, the degrees of polymerization were tunable by varying the EG units' steric bulk, which was affected not only by the number of EG chains but also by solvent effects (using both protic and aprotic co-solvents). H2TPPS2-NHCO-EGx (x = 6, 8) with larger EG units underwent self-assembly only under acidic conditions, affording J-aggregates, whereas H2TPPS2-NHCO-EGx (x = 18) did not possess any self-assembly ability. Remarkably, these H2TPPS2-NHCO-EGx (x = 6, 8) derivatives with larger EG units could be detected; in some cases, we could isolate the reactive oligomer intermediates before they reached their final supramolecular polymers, due to very slow elongation processes.
Results and discussion
We have synthesized novel H2TPPS4 derivatives, H2TPPS2-NHCO-EGx; x = 2, 4, 6, 8, and 18, according to Scheme S1 (for details see the ESI†). Briefly, we started all syntheses using compound 1, having two amino groups at trans meso positions, as a common intermediate.32 After treated it with conc. H2SO4, sulfonated porphyrin intermediate (compound 3) was obtained as tetrabutylammonium (TBA) salt.28 Different EG unites (compound 5–8) were also synthesized according to the reported methods.33,34 Finally, after condensation reaction, the resultant solution was passed through an ion exchange column to yield a purple-colored solid.35First, we investigated the proton-triggered self-assembly of the H2TPPS2-NHCO-EGx derivatives in MeOH/water as the mixed solvent. All of the H2TPPS2-NHCO-EGx and H2TPPS2-O-EGx derivatives were soluble in MeOH, functioning as a common good solvent. Therefore, we could use a common procedure for preparation of all of the H2TPPS2-NHCO-EGx (H2TPPS2-O-EGx) samples, independent of the nature of the EG units. Briefly, after solubilizing the H2TPPS2-NHCO-EGx (H2TPPS2-O-EGx) derivatives in MeOH, we added aqueous HCl and examined their self-assembly in the MeOH/aqueous HCl mixed solvents (for details, see the ESI†). We adjusted a final concentration to 25 μM, with a MeOH/water solvent composition of 25/75 (v/v) and a final acidity of pH 3. Fig. 2 presents the resultant UV-vis spectra of all of the H2TPPS2-NHCO-EGx derivatives (for UV-vis spectra recorded with other MeOH/water compositions, see Fig. S19†). These spectra revealed that the monomeric states of these H2TPPS2-NHCO-EGx derivatives in MeOH featured their Soret bands near 416 nm (red line); upon addition of aqueous HCl, however, these peaks commonly shifted to near 431 nm, consistent with the protonated parent H4TPPS4.18,23 Accordingly, we suspected that these H2TPPS2-NHCO-EGx derivatives would have potential to serve as ionic monomers in their cationic H4TPPS2-NHCO-EGx forms. Although all of the H2TPPS2-NHCO-EGx derivatives had a common proton-accepting porphyrin core, the self-assembly of their H4TPPS2-NHCO-EGx structures following protonation depended on the number of EG units. In particular, we observed significant differences in their rates of self-assembly and degrees of polymerization. Therefore, we investigated the self-assembly of each H4TPPS2-NHCO-EGx derivative in detail to determine the effects of the EG units.
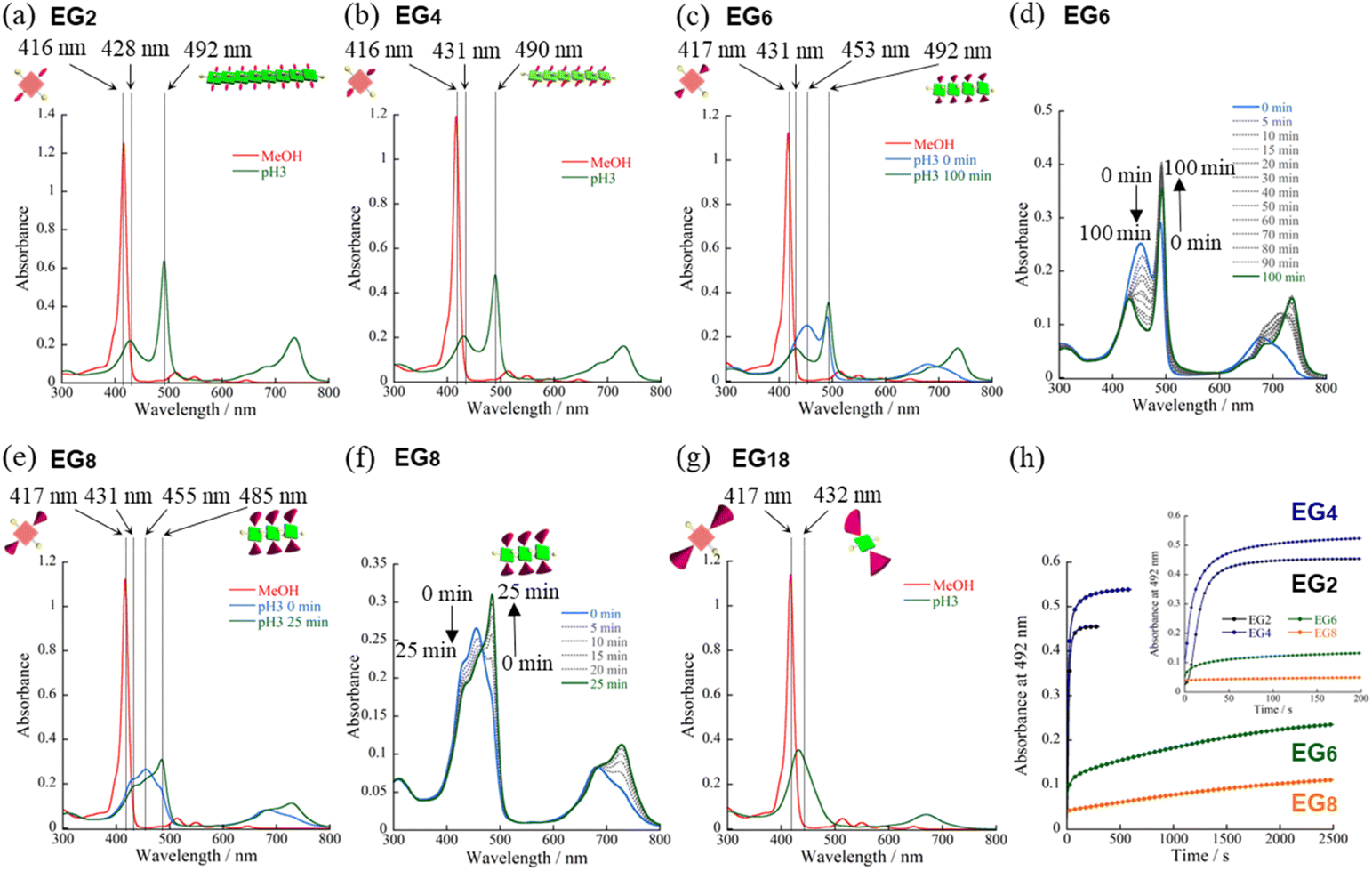 | ||
| Fig. 2 (a–g) UV-vis spectral changes of H2TPPS2-NHCO-EGx derivatives in MeOH/water solvent upon addition of aqueous HCl. (a) H2TPPS2-NHCO-EG2, (b) -EG4, (c and d) -EG6, (e and f) -EG8, (g) -EG18; (d and f) time-dependent spectral changes for (d) -EG6 and (f) -EG8. Red lines in (a), (b), (c), (e), and (g) are the spectra of the monomeric H2TPPS2-NHCO-EGx derivatives in MeOH; green lines are the spectra in MeOH/aqueous HCl (25/75; pH 3) (final concentration, 25 μM; optical path length, 1 mm; r.t.). (h) Plots of time-dependent absorption changes, recorded in MeOH/aqueous HCl (25/75, pH 3) (5.0 μM; optical path length, 3 mm; r.t.). | ||
Proton-triggered self-assembly of H2TPPS2-NHCO-EGx (x = 2, 4) in MeOH/water
In the cases of H2TPPS2-NHCO-EG2 and -EG4, their Soret bands at 416 nm, assignable to non-protonated monomers, were almost completely absent at pH 3, with these peaks red-shifted to 492 and 490 nm, respectively. These spectral changes suggested that J-aggregates had formed predominantly for both H2TPPS2-NHCO-EG2 and -EG4 (Fig. 2a and b). As discussed below, the rates of self-assembly of these two monomers were faster than those for the other derivatives presenting larger EG units (Fig. 2h). Although the nature of their self-assembly appeared to be similar to that of the parent H2TPPS4, to compare their rates of J-aggregate formation quantitatively with respect to the other H2TPPS2-NHCO-EGx derivatives, we considered their degrees of polymerization (aagg).36here, AJ-aggregate and Aprotonated monomer represents the absorption maximum of J-aggregate and protonated monomer, respectively.
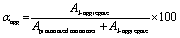
From the UV-vis spectra, we estimated the values of aagg of H2TPPS2-NHCO-EG2 and -EG4 to be 75 and 71%, respectively. Although we could not compare these values directly with that of the parent H2TPPS4, due to the latter's poor solubility in MeOH/water, the value of aagg of the parent H2TPPS4 in water (pH 3) was 11% (Fig. S20a†). Taking this number into consideration, it is obvious that the values of aagg of H2TPPS2-NHCO-EG2 and -EG4 were both much higher (Table 1). This behavior can be explained by considering the solvophobic effect caused by replacement of the ionic groups with non-ionic counterparts. This conclusion was supported by experiments in which we used DMF instead of MeOH/water (Fig. 3). In addition to the solvophobic effect, replacing the sulfonate groups with nonionic EG2 and EG4 units would also decrease the degree of electrostatic repulsion between adjacent porphyrin units, thereby facilitating intermolecular interactions.
| EG2 | EG4 | EG6 | EG8 | EG18 | |
|---|---|---|---|---|---|
| AJ-aggregate | 0.62 | 0.48 | 0.33 | 0.26 | 0.04 |
| Aprotonated monomer | 0.20 | 0.20 | 0.15 | 0.19 | 0.35 |
| αagg | 75.2 | 70.5 | 69.3 | 57.8 | 10.1 |
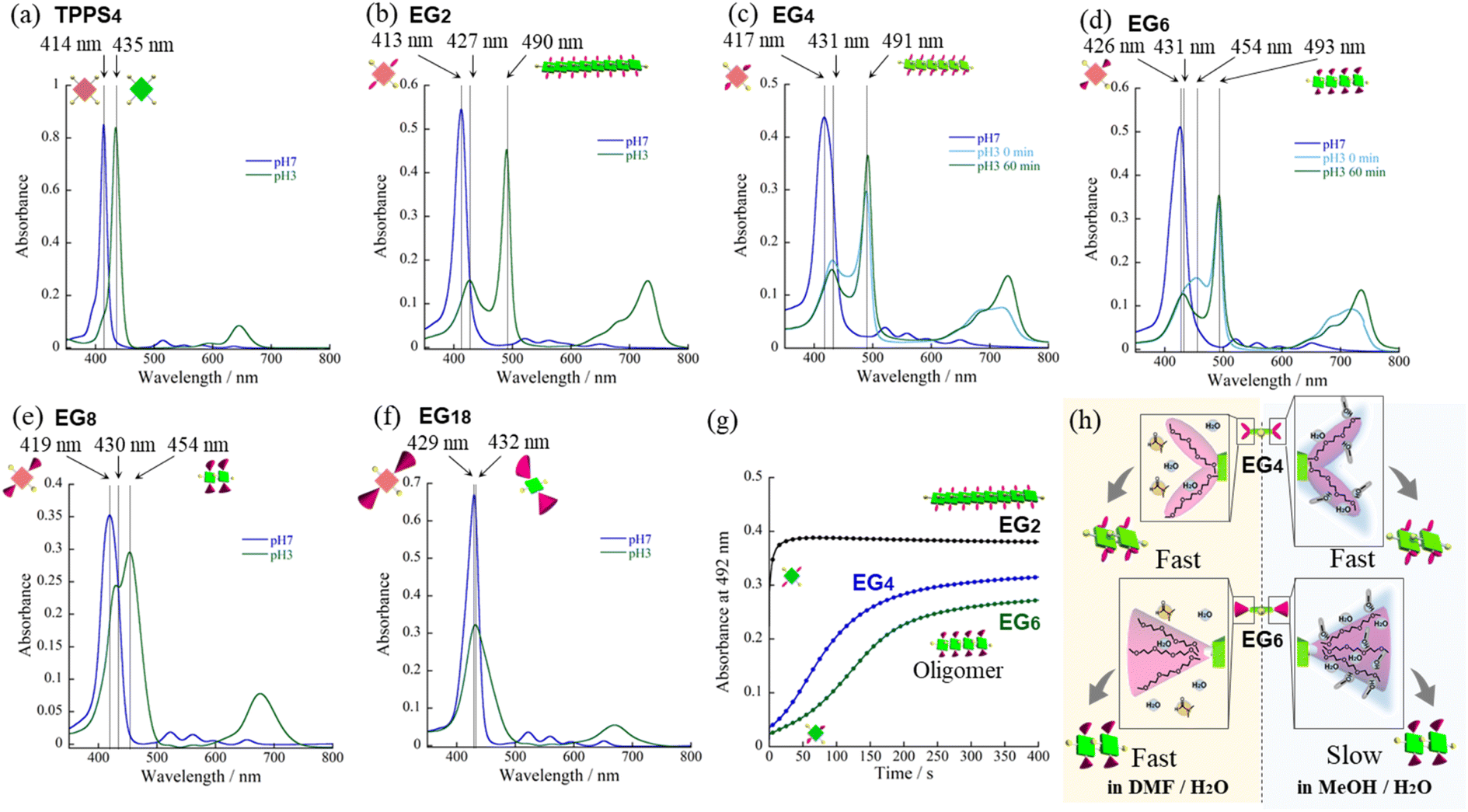 | ||
| Fig. 3 UV-vis spectra of (a) H2TPPS4, (b) H2TPPS2-NHCO-EG2, (c) -EG4, (d) -EG6, (e) -EG8, and (f) -EG18 in DMF/water = 2/98 mixed solvent (25 μM; optical path length, 1 mm; r.t.). Blue lines are the spectra recorded in DMF/water = 2/98 (pH 7). Sky-blue and green lines are the spectra recorded in DMF/aqueous HCl, 2/98 (pH 3). Sky-blue lines in (c) and (d) are the spectra recorded immediately after addition of aqueous HCl (0 min); the green lines are the spectra obtained after 60 min. (g) Plots of time-dependent absorption changes against time for H2TPPS2-NHCO-EG2, -EG4 and -EG6 in DMF/water (5.0 μM, optical path length, 3 mm, r.t.). For the spectra recorded in DMF/aqueous HCl, 2/98 (pH 2); see Fig. S27.† (h) Schematic representation of solvation around EG chains with protic (MeOH) and aprotic (DMF) solvent. | ||
Fig. 4a and b present atomic force microscopy (AFM) images of the self-assembled structures of H2TPPS2-NHCO-EG2 and -EG4. Consistent with their high values of aagg, H2TPPS2-NHCO-EG2 and -EG4 self-assembled to form extended J-aggregate nanofibers in MeOH/water = 25/75. We estimated the average lengths of the H2TPPS2-NHCO-EG2 and -EG4 nanofibers to be 290 and 180 nm, respectively. These values are comparable with that of the crystalline fibers of the parent H2TPPS4 (350 nm) (Fig. S20†). Note that the crystalline fibers are extended through aggregation of many short nanofibers. Notably, the H2TPPS2-NHCO-EG2 and -EG4 nanofibers were much thinner than the H2TPPS4 fibers. From height profiles of the AFM images, we found that the heights of the parent nanofibers ranged from 1.5 to 15 nm (Fig. S20†), whereas the H2TPPS2-NHCO-EG2 nanofibers were of almost uniform thickness (1.5–3.0 nm), while the nanofibers of H2TPPS2-NHCO-EG4 were of uniform height (1.5 nm). This height of 1.5 nm implies that the π-surface of a single piece of the supramolecular polymer had been positioned on the mica surface with an inclination of approximately 12°; this angle was supported by the density functional theory (DFT)-calculated structures of the dimer (Fig. 4c). Taking the thickness of the parent H2TPPS4 fibers with multi-dispersity (Fig. S20b†), these findings imply that the inherently strong stacking of the J-aggregate fibers was suppressed by the peripheral EG units, thereby enabling us to isolate single strands of the J-aggregate supramolecular polymers. X-ray scattering patterns of H2TPPS2-NHCO-EG2 and -EG4, as shown in Fig. 5a, revealed a scattering peak at a value of q of 19.7, implying similar packing distances (0.32 nm). The somewhat broadened peak for H2TPPS2-NHCO-EG4 might have arisen from the steric bulkiness of its EG4 units.
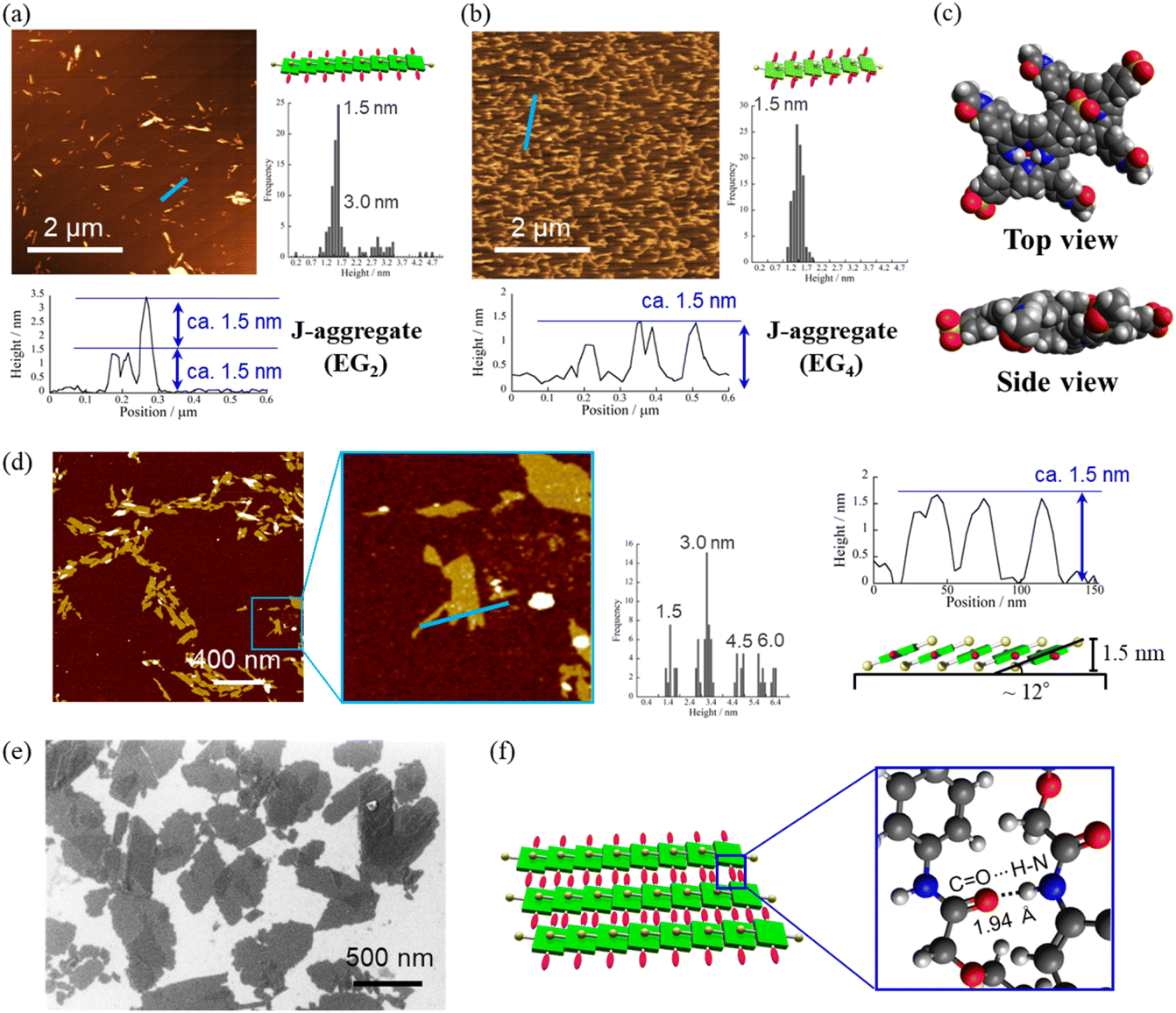 | ||
Fig. 4 AFM images, height profiles and height distributions of J-aggregated supramolecular polymers created from (a) H2TPPS2-NHCO-EG2 and (b) -EG4 [25 μM, MeOH/aqueous HCl (pH 3); mica substrate]. (c) DFT-calculated structures of the J-aggregate dimer of H2TPPS2-NHCO-EG2 and an illustration of the polymer on a surface. DFT calculations were performed at the B3LYP/6-31G+** level with empirical dispersion correction. For simplicity, EG groups have been replaced by CH3 units. (d) AFM and (e) SEM images of sheet structures formed from the supramolecular polymers [these images were obtained after leaving the solution in (a) for 9 h]. Height distributions were measured from AFM images of at least 100 fibers or sheets. (f) Schematic representation of the sheet structures and DFT-calculated structure for EG2 units forming CONH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds (B3LYP/6-31G**). For other AFM and SEM images, see Fig. S21 and S22,† respectively. C hydrogen bonds (B3LYP/6-31G**). For other AFM and SEM images, see Fig. S21 and S22,† respectively. | ||
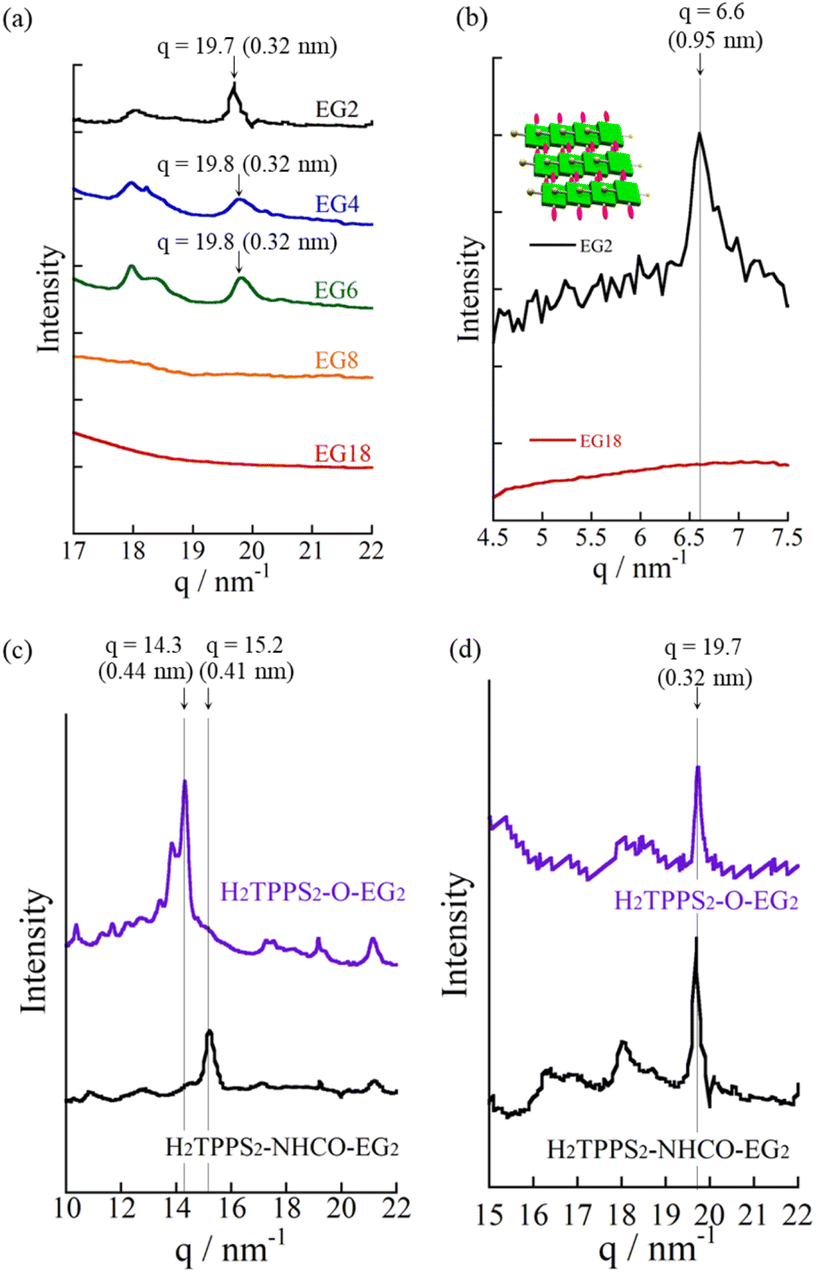 | ||
| Fig. 5 (a) XRD patterns of (a) J-aggregated supramolecular polymers formed from H4TPPS2-NHCO-EG2 (black line), -EG4 (blue line), -EG6 (green line), -EG8 (orange line), and -EG18 (red line) [MeOH/aqueous HCl (pH 2), 25/75] and (b) J-aggregate sheets formed from H4TPPS2-NHCO-EG2 (black line) and -EG18 (red line). (c and d) XRD patterns of (c) H- and (d) J-aggregated supramolecular polymers formed from H4TPPS2-NHCO-EG2 (black line) and H4TPPS2-O-EG2 (purple line). | ||
To identify the mechanism of self-assembly of these monomers, we fitted the time-dependent UV-vis spectral changes of H2TPPS2-NHCO-EG2 and -EG4 to the autocatalytic mode.37 The plots revealed that, similar to the parent H2TPPS4, the self-assembly processes of both H2TPPS2-NHCO-EG2 and -EG4 obeyed a cooperative mechanism (Fig. S23†). Indeed the elongation mechanism of H2TPPS2-NHCO-EG2 and -EG4 was similar to that of the parent H2TPPS4.37 Notably, the J-aggregated supramolecular polymers formed from these monomers were all overall non-ionic (except at both termini), in contrast to the J-aggregate of the parent H2TPPS4, which featured anionic wedges along the fibers, making its fibers overall anionic.
Reflecting their non-ionic properties, the created J-aggregated supramolecular polymers of H2TPPS2-NHCO-EG2 underwent unique hierarchical self-assembly. Upon standing the resultant solution of pH 3.0–4.0 for 6 h, the J-aggregate polymers assembled to create sheet-like structures. AFM and SEM images revealed (Fig. 4d and e) that the sheets extended to micrometer size, with the surfaces being almost flat all over the entire range. Remarkably, the height profiles of these sheets revealed the sheets tended to pile-up and the interval of the thicknesses comparable with heights of the J-aggregated polymers (1.5 nm), implying that edge-to-edge assembly had occurred, rather than the face-to-face stacking observed for the parent J-aggregate. Changes in the time-dependent infrared (IR) spectra support such an edge-to-edge assembly mode (Fig. S24†). The free amide I and II bands of H2TPPS2-NHCO-EG2 appeared at 1695 and 1596 cm−1, respectively. Upon adding aqueous HCl to the monomers, with accompanying formation of the supramolecular polymers, these peaks shifted to lower wavelengths (1690 and 1581 cm−1, respectively), suggesting that amide N–H⋯OC hydrogen bonding occurred in an interdigitated manner along the supramolecular polymers. The possibility of such hydrogen-bonding occurring was supported by DFT-calculated structures of the dimer of EG units (Fig. 4f). The preference for edge-to-edge assembly was further supported by the appearance of a new diffraction peak at a value of q of 6.6 (0.95 nm) in the X-ray scattering pattern (Fig. 5b). This peak corresponded to the edge-to-edge distance of the porphyrin core.
To gain insight into the kinetics of the self-assembly of H2TPPS2-NHCO-EG2 and -EG4, we plotted their values of Δabs at 492 nm with respect to time. Fig. 2h reveals that J-aggregate formation for both H2TPPS2-NHCO-EG2 and -EG4 was almost complete within 60 s. At least under the present conditions, there were no significant differences in the rates observed for H2TPPS2-NHCO-EG2 and -EG4, suggesting no steric effects for the bulkier EG units. As described below, the steric effect of the EG4 units became conspicuous, however, when using DMF/water mixed solvents.
Self-assembly of H2TPPS2-NHCO-EGx (x = 2, 4) in DMF/water
For an aqueous HCl solution (pH 3) of the parent H2TPPS4, we estimated the value of aagg to be 11%. In the present MeOH/water = 25/75 solvent, we could not directly compare the values of aagg of H2TPPS2-NHCO-EG2 and -EG4 with that of H2TPPS4. We could, however, compare their aggregation when employing DMF in place of MeOH, because the parent H2TPPS4 was soluble in water even in the presence of 2% of DMF. In the same solvent of DMF/water = 2/98, H2TPPS2-NHCO-EG2 also displayed self-assembly ability (Fig. 3b). The estimated value of aagg (77%) was comparable with that in MeOH/water = 25/75. In contrast to the high aggregation ratio of H2TPPS2-NHCO-EG2, the parent H2TPPS4 underwent almost no self-assembly in the same solvent (Fig. 3a and Table 2), even at pH 2. UV-vis spectra revealed the effective conversion of H2TPPS4 to protonated H4TPPS4 in DMF/water = 2/98 at pH 2. It is likely that DMF interacted with H4TPPS4 tightly through CODMF⋯NH2+ (protonated pyrrole) or (Me)2NDMF⋯NH2+ interactions, which would hamper π-stacking of the porphyrin units. Alternatively, the non-protic nature of DMF decreased the local polarity of the solvent water molecules surrounding the porphyrin cores. Regardless, the different nature of the self-assembly of the parent H2TPPS2, even in DMF/water = 2/98, highlighted the stronger aggregation ability of H2TPPS2-NHCO-EG2. Along this line of discussion, as described above, we did not identify any significant discrepancies in the values of aagg or the rates of aggregation for H2TPPS2-NHCO-EG2 and -EG4 in MeOH/water, due to their inherently strong aggregation. Even in DMF/water, the value of aagg of H2TPPS2-NHCO-EG4 was comparable with that of H2TPPS2-NHCO-EG2 (Fig. 3c). We did, however, detect a clear discrepancy in the rates of aggregation of these monomers. As revealed in the time-dependent spectra in Fig. 3g, the absorption peak corresponding to the J-aggregate of H2TPPS2-NHCO-EG4 required over 60 min to become saturated, in contrast to the spontaneous J-aggregate formation of H2TPPS2-NHCO-EG2. These results imply that steric effects caused the self-assembly of H2TPPS2-NHCO-EG4 to become slower than that of H2TPPS2-NHCO-EG2. Thus, DMF/water as the mixed solvent revealed a slight, but significant, discrepancy in the steric effects of the EG2 and EG4 units.
| EG2 | EG4 | EG6 | EG8 | EG18 | TPPS4 | |
|---|---|---|---|---|---|---|
| AJ-aggregate | 0.45 | 0.36 | 0.37 | 0.04 | 0.03 | 0.00 |
| Aprotonated monomer | 0.14 | 0.14 | 0.16 | 0.24 | 0.32 | 0.84 |
| αagg | 76.7 | 71.3 | 70.4 | 14.3 | 9.2 | 0.0 |
Self-assembly of H2TPPS2-NHCO-EGx (x = 6, 8, and 18) in MeOH/water
On the basis of the findings for H2TPPS2-NHCO-EG2 and -EG4 in MeOH/water, we investigated the self-assembly of H2TPPS2-NHCO-EGx derivatives with larger EG units (i.e., H2TPPS2-NHCO-EG6, -EG8 and -EG18) in the same solvent (MeOH/water = 25/75) at the same value of pH. First, we focused on the self-assembly of H2TPPS2-NHCO-EG6. In contrast to the spontaneous polymerizations of H2TPPS2-NHCO-EG2 and -EG4, H2TPPS2-NHCO-EG6 underwent very slow polymerization in MeOH/water = 25/75. As displayed in Fig. 2c, upon protonation, a broad and predominant absorption band appeared initially at 453 nm—we did not observe this signal for the parent H2TPPS4 or for H2TPPS2-NHCO-EG2 or -EG4. Fig. 2d reveals that this new peak gradually red-shifted to 492 nm (corresponding to the J-aggregate) over a period of 100 min. From the UV spectrum recorded after 100 min we obtained a value of aagg of 69%, similar to that for H2TPPS2-NHCO-EG4. We assign the temporary peak at 453 nm to the oligomeric intermediates formed as precursors to the J-aggregates. DLS data further supported this view; that is, the hydrodynamic radius was gradually increase over a period of 100 min. The resultant hydrodynamic radius was smaller than that of J-aggregate of H2TPPS2-NHCO-EG2, suggesting the formation of oligomeric species (Fig. S33†). Because the absorption peak at 453 nm appeared immediately after addition of the acid, protonated H4TPPS2-NHCO-EG6 monomers presumably afforded such oligomers spontaneously. Furthermore, no time-dependent changes occurred to the peak at 431 nm, corresponding to H4TPPS2-NHCO-EG6 monomers, suggesting that almost quantitative conversion of the H4TPPS2-NHCO-EG6 monomers to the oligomers occurred spontaneously after protonation.To estimate the length of the oligomers, we used an exciton coupling model to evaluate the absorption maxima of the oligomeric intermediates (Fig. S25†).38 From the estimated wavelengths of the oligomeric intermediates with different chain lengths, we deconvoluted the time-dependent UV-vis spectra using Gaussian functions (Fig. S26†). The deconvoluted spectra revealed that the oligomeric intermediates were initially composed of some two to nine H4TPPS2-NHCO-EG6 monomers. In general, during one-dimensional self-assembly of the parent H2TPPS4, oligomeric intermediates are not detectable because of very rapid elongation processes. Indeed, in a previous paper, we estimated the rate of elongation of H2TPPS4 to be on the order of milliseconds.39 The steric effects of the EG6 units rendered its oligomers to be quite long-lived, with the π-surfaces of the porphyrin cores presumably being covered with EG chains. Here, we note that in the EG6 unit, addition of a EG chain to the 4′-position of the peripheral phenyl ring in the EG4 units would have induced significant steric bulk. Accordingly, solvation of each EG chain in the EG6 unit in MeOH/water would cause significant changes in the steric bulk, as evidenced by the distinct self-assembly behavior of H2TPPS2-NHCO-EG4 and -EG6 (Fig. 3h). The effective solvation of EG units in protic MeOH/water co-solvents enhanced the steric effects of the EG units. This effect was further confirmed for H2TPPS2-NHCO-EG8. Fig. 2e and f reveal that the absorption band assignable to the J-aggregates of H2TPPS2-NHCO-EG8 gradually increased over a period of 25 min, whereas the broad peak near 455 nm, corresponding to the oligomers, remained even after the system had reached thermodynamic equilibrium. Similar to the case of H4TPPS2-NHCO-EG6, DLS data is evident the slow formation of oligomers. The rates of self-assembly became slower for H2TPPS2-NHCO-EG6 and -EG8 than those for H2TPPS2-NHCO-EG2 and -EG4, even when using MeOH/water as the solvent. As displayed in Table 1, although the value of aagg for H2TPPS2-NHCO-EG6 (69%) was comparable with those of H2TPPS2-NHCO-EG2 and -EG4, its rate of self-assembly was much slower than that those of H2TPPS2-NHCO-EG2 and -EG4, highlighting the effect of the steric bulk of the EG units over the porphyrin core. In other words, larger EG units stabilized the H4TPPS2 core kinetically, even when it favored strong aggregation through ionic interactions. This steric effect was more conspicuous for H2TPPS2-NHCO-EG18. As revealed from Fig. 2g, only the broad peak at 432 nm, assignable to protonated H4TPPS2-NHCO-EG18, was present even after the system had reached equilibrium, implying that not even oligomer formation had occurred. DLS data also confirmed that H4TPPS2-NHCO-EG18 remained as a monomer even after protonation (Fig. S33†). Furthermore, in contrast to the case of H4TPPS2-NHCO-EG6 and H4TPPS2-NHCO-EG8, we observed no time-dependent changes in the hydrodynamic radius. Here, we emphasize that the protonated H4TPPS2-NHCO-EG18 was a zwitterionic monomer that we might have expected to have high aggregation ability in the absence of its EG units. The steric bulk of those EG18 units made the highly reactive protonated core species isolatable.
Consistent with the UV-vis spectra, AFM images revealed that no fiber structures formed when the EG units were larger than EG8 (Fig. S21†). For H2TPPS2-NHCO-EG6, despite the value of aagg being 69%, we could not observe extended nanofibers through AFM. The corresponding X-ray diffraction (XRD) pattern, however, did provide evidence for the formation of J-aggregates. In Fig. 5a, the diffraction peak at a value of q of 19.8 is consistent with a π-stacking distance. As stated above, we observed similar diffraction peaks in the patterns of the J-aggregates prepared from H2TPPS2-NHCO-EG2 and -EG4. Such common diffraction patterns imply that even large EG units did not affect the π-stacking distance in the final J-aggregates. Because the diffraction peak broadened slightly upon increasing the steric bulk, larger EG units (e.g., EG4 and EG6) presumably disturbed the π-stacking interactions slightly, correlated with the slower rates of self-assembly of these monomers relative to that of H2TPPS2-NHCO-EG2. The common diffraction patterns indicated that larger EG units affected the rates of self-assembly more significantly than they did the packing mode in the J-aggregates.
Self-assembly of H2TPPS2-NHCO-EGx (x = 6, 8, and 18) in DMF/water
H2TPPS2-NHCO-EG4 and -EG6 appeared to be an ideal pair for investigating the influence of the solvation of EG units on the self-assembly behavior in the presence of different co-solvents (i.e., MeOH and DMF), because they have the same number of peripheral phenyl rings but different numbers of EG units.40–42 In DMF/water = 2/98, the self-assembly of H2TPPS2-NHCO-EG6 was similar to that of H2TPPS2-NHCO-EG4 (Fig. 3c and d)—in contrast to the case when using MeOH/water as the mixed solvent (Fig. 2b and c). The plot of absorbance at 492 nm with respect to time (Fig. 3g) revealed that these two monomers had similar rates of self-assembly, indicating that the EG6 units exerted no effective steric effects on the self-assembly process. DMF is a typical aprotic polar solvent that would not solvate EG chains as effectively as would MeOH. We suspected, however, that the number of EG units would directly influence the self-assembly of H2TPPS2-NHCO-EGx in DMF/water. As displayed in Fig. 3e, in contrast to the effective J-aggregate formation from H2TPPS2-NHCO-EG4 and -EG6, elongation of the protonated H4TPPS2-NHCO-EG8 was terminated at the oligomer stage, as confirmed by the presence of an absorption peak at 454 nm even after thermodynamic equilibrium had been reached. As described above for Fig. 2e, the shift in the absorption peak of H4TPPS2-NHCO-EG8 in MeOH/water from 455 to 485 nm was indicative of the further extension of oligomers. The UV-vis spectra in DMF/water revealed that the self-assembly of H4TPPS2-NHCO-EG8 became weaker in this solvent. DLS data also confirmed that the hydrodynamic radius of H4TPPS2-NHCO-EG8 in DMF/water was smaller than that in MeOH/water, supporting our view (Fig. S34†). This behavior can be explained by considering that DMF solvated the central pyrrole unit, thereby suppressing its ionic interactions with the sulfonic acid groups, as well as its π-stacking. Similar to the situation in MeOH/water, H4TPPS2-NHCO-EG18 displayed no self-assembly ability in DMF/water, due to the steric bulk (Fig. 3f).Attempted face-to-face stacking of anionic H2TPPS2-NHCO-EGx (x = 2, 4, 6, 8, and 18) in MeOH/water
Having identified the self-assembly behavior of the series of H2TPPS2-NHCO-EGx derivatives in acidic media, we investigated whether the examined porphyrin monomers could undergo supramolecular polymerization through direct π-stacking under neutral media to afford H-aggregated supramolecular polymers. We emphasize that no direct π-stacking interactions have been observed for the parent H2TPPS4 unless the assistance of polymers or surfactants, due to strong electrostatic repulsion among its sulfonic acid groups.43 We expected the present H2TPPS2-NHCO-EGx derivatives might encounter similar problems, but that electrostatic repulsion would probably be weaker in their H-aggregates because the number of sulfonic acid groups had halved. Expecting strong π-stacking interactions, we prepared MeOH solutions (100 μM) of the H2TPPS2-NHCO-EGx derivatives and mixed each one with pure water to adjust the final concentration to 25 μM. For H2TPPS2-NHCO-EG2, the Soret band at 416 nm, corresponding to non-protonated monomer, blue-shifted to approximately 414 nm upon addition of water. To clarify the effect of water on the aggregation behavior of H2TPPS2-NHCO-EG2, we changed the water content in increments from MeOH/water = 100/0 to 5/95. Fig. 6a reveals that the absorbance maxima blue-shifted upon increasing the water content, accompanied by a hypsochromic effect, indicative of strong π-stacking interactions. To clarify the cause of the peak shift, we deconvoluted the spectra using Gaussian functions. In the deconvoluted spectra (Fig. S28†), we assigned the blue-shifted peak at 405 nm to the H-aggregate.43 Fig. 6b presents the spectra for the other H2TPPS2-NHCO-EGx derivatives in MeOH/water = 25/75. In contrast to the clear blue-shift for H2TPPS2-NHCO-EG2, the corresponding peaks of H2TPPS2-NHCO-EG6, -EG8, and -EG18 were red-shifted slightly (by 15–20 nm) upon increasing the water content, suggesting a weak tendency to form slipped stacks, even in neutral media. As for H2TPPS2-NHCO-EG4, we observed no shift in the absorption band, presumably because the inherent blue-shift originating from H-aggregate formation was cancelled by the red-shift caused by steric repulsion. These spectral changes suggested that tight face-to-face π-stacking had been realized for H2TPPS2-NHCO-EG2. When the EG units were larger than EG4, the porphyrin core still had a strong tendency to undergo tight face-to-face stacking, but it was inhibited by the steric bulk of the EG units. Combined with the fact that H2TPPS2-NHCO-EG4 and -EG6 displayed strong abilities to form J-aggregates in this same solvent at pH 3, the effect of steric repulsion of the EG units when larger than EG4 became more conspicuous when assembling the H-aggregates than it was in the case of the J-aggregates. AFM images (Fig. 7a) revealed that, reflecting its strong π-stacking capability, H2TPPS2-NHCO-EG2 self-assembled to form extended fibrous structures. To the best of our knowledge, this example is the first of H-aggregated supramolecular polymer formed from an H2TPPS4 derivative. The formation of well-extended polymers implies that H2TPPS2-NHCO-EG2 stacks favorably while avoiding electrostatic repulsion. From height profiles of the H-aggregate, we estimated the average diameter of the supramolecular polymers to be 2 nm, consistent with the calculated dimensions of H2TPPS2-NHCO-EG2. Furthermore, consistent with the UV-vis spectra, the average length of the H-aggregated supramolecular polymers tended to increase upon increasing the water content. In contrast to the well-extended supramolecular polymers of H2TPPS2-NHCO-EG2, we observed no fibrous structures for the other H2TPPS2-NHCO-EGx derivatives. These results confirmed that even increasing the steric bulk slightly inhibited direct π-stacking interactions.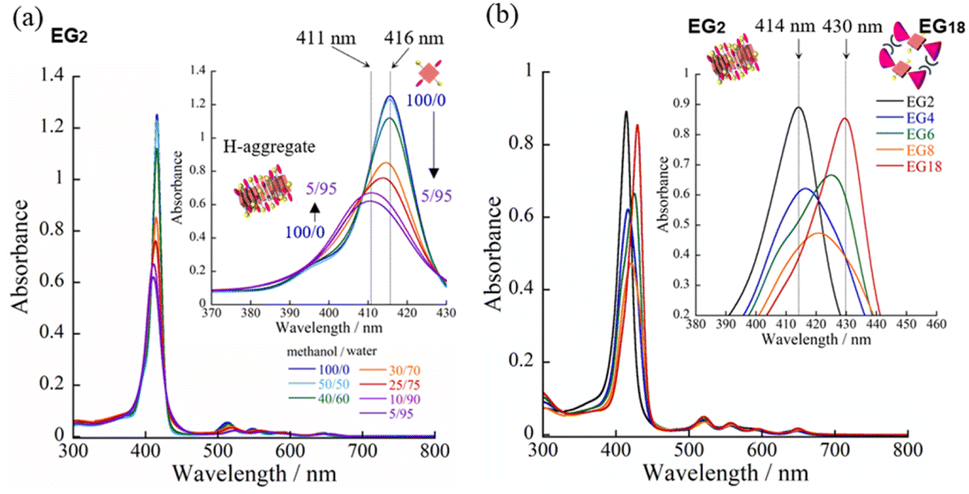 | ||
| Fig. 6 (a) UV-vis spectra of H2TPPS2-NHCO-EG2 recorded at various MeOH/water ratios (from 100/0 to 5/95); inset: expanded view of the Soret band. (b) UV-vis spectra of H2TPPS2-NHCO-EGx derivatives upon mixing with pure water (MeOH/water = 25/75); inset: expanded view of the Soret band (25 μM; optical path length, 1 mm; r.t.). | ||
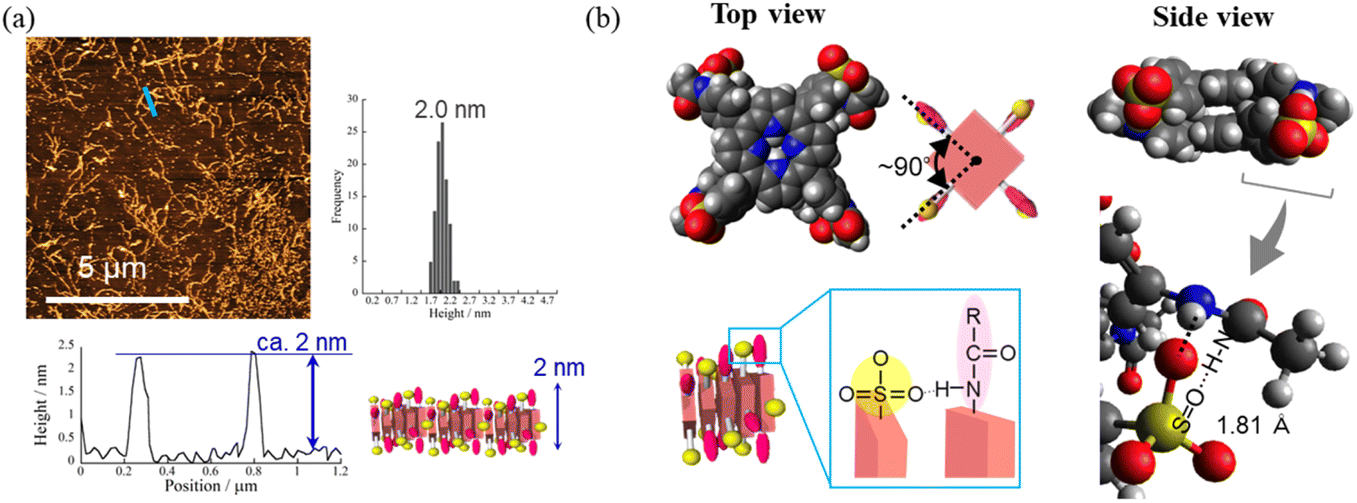 | ||
| Fig. 7 (a) AFM image, height profile and height distribution of the H-aggregated supramolecular polymer formed from H2TPPS2-NHCO-EG2 (25 μM; MeOH/water, 25/75; mica substrate). Illustration of face-to-face stacking assisted by [N–H⋯OS] hydrogen bonding. Height distribution was measured from AFM images of at least 100 fibers. (b) DFT-calculated structures of the H-aggregate dimer of H2TPPS2-NHCO-EG2, performed at the B3LYP/6-31G+** level with empirical dispersion correction. Arrow: [N–H⋯OS] hydrogen bonding. For simplicity, EG groups have been replaced by CH3 units. | ||
To gain insight into how the H2TPPS2-NHCO-EG2 stacks overcame electrostatic repulsion among the sulfonic acid groups, we performed DFT calculations of its dimer. The DFT calculations revealed that hydrogen bonding between the amide NH and sulfonic acid groups (N–H⋯OS) played an essential role in the formation of the H-aggregate (Fig. 7b). In this structure, the porphyrin units were offset by approximately 90° to avoid direct electrostatic repulsion between the sulfonate groups. The offset stacking minimized such repulsion and enabled the N–H⋯OS hydrogen bonds to form along the polymer chain, with amide NH unit acting as a sort of anion binder to contribute to the stabilization of the polymers. The XRD patterns of the H-aggregated supramolecular polymers supported this notion. Fig. 5c reveals a diffraction peak at a value of q of 15.2 (0.41 nm), corresponding to the π-stacking distance. This value for the H-aggregates was slightly greater than that for the corresponding J-aggregates (0.32 nm; Fig. 5d). Furthermore, as we describe in the following section, the supramolecular polymer of H2TPPS2-O-EG2, for which there was no possibility of assistance from hydrogen bonding, provided a diffraction peak at a value of q of 14.3 (0.44 nm; purple line in Fig. 5c). The relatively shorter π-stacking distance for H2TPPS2-NHCO-EG2 is consistent with the existence of the assisting hydrogen bonding interactions. Finally, to gain insight into the mechanism of polymerization of H2TPPS2-NHCO-EG2, we plotted the absorbance at 405 nm, arising from the H-aggregate, with respect to temperature. This absorption data fitted well to an isodesmic model of supramolecular polymerization (Fig. S29†).44 The competing supramolecular polymerization pathway, such as cooperative and isodesmic, can be widely seen in other monomer systems.31 The presence of different pathway can be employed to control the function of the supramolecular polymers.
Self-assembly of H2TPPS2-O-EGx (x = 2, 4) in MeOH/water
Next, to highlight the influence of the spacer length on the self-assembly process, another series of modified H2TPPS2 derivatives, H2TPPS2-O-EGx (x = 2, 4) derivatives, were synthesized according to Scheme S2.† Firstly, we synthesized 5,15-dibromo-10,20-disulfophenyl porphyrin (compound 18) as a common intermediate.45 EG units were then introduced at the meso positions of the sulfonated porphyrin through the Suzuki coupling reactions. After ion exchange, H2TPPS2-O-EGx was obtained as a purple solid.46Our series of tested H2TPPS2-NHCO-EGx derivatives possessed self-assembly abilities in both acidic and neutral media, forming supramolecular polymers through different pathways, where the strong desire for aggregation of the porphyrin cores being balanced by the steric bulk of the EG units as well as their solvation state. Furthermore, their hydrogen bonding ability, originating from the amide moieties, played an essential role in realizing offset H-stacking under neutral conditions. On the basis of these findings, we investigated the necessity of the amide groups in our molecular design. To obtain another series of modified H2TPPS2 derivatives, but without amide linkers, we synthesized H2TPPS2-O-EGx (x = 2, 4) derivatives (Fig. 1a) in which the peripheral EG units were connected directly to the meso positions of the porphyrin core. With this design, the spacer between the porphyrin core and the peripheral EG units became shorter than that in the H2TPPS2-NHCO-EGx series. Considering that the steric bulk of the EG units affected the self-assembly of the H2TPPS2-NHCO-EGx derivatives in an ON/OFF manner, we suspected that the shorter spacers would further this tendency. Following the same procedure used for the H2TPPS2-NHCO-EGx series, we prepared MeOH solutions (100 μM) of the H2TPPS2-O-EGx (x = 2, 4) derivatives and mixed each one with aqueous HCl to adjust its final concentration to 25 μM, with a MeOH/water composition of 25/75 (v/v) and a final acidity of pH 3. In the case of H2TPPS2-O-EG2, Fig. 8a reveals that the absorption peak at 416 nm, corresponding to monomeric H2TPPS2-O-EG2, disappeared and a red-shifted peak appeared at 488 nm, assignable to the J-aggregate. These spectral changes were effectively identical to those of the corresponding H2TPPS2-NHCO-EG2 derivative; that is, the value of aagg (71%) and the rate of aggregation for H2TPPS2-O-EG2 were comparable with those for H2TPPS2-NHCO-EG2. XRD patterns supported these findings; Fig. 5d reveals a diffraction peak at a value of q of 19.7 (0.32 nm), in accordance with that observed for H2TPPS2-NHCO-EG2. Furthermore, AFM confirmed the presence of well-extended nanofibers (Fig. 8c). From the height profiles of the AFM images, we estimated an average diameter of 1.5 nm, indicative of a single piece of the supramolecular polymer. In this molecular design, the peripheral EG units once again effectively suppressed further bundling. The formation of supramolecular polymers similar to those the case of H2TPPS2-NHCO-EG2 implies that the amide linkages in the latter had no significant effect on the J-aggregate stacking. The DFT-calculated structures of the H2TPPS2-O-EG2 dimer (Fig. 8d) supported this notion. Most importantly, in contrast to the additional hierarchical assembly of the H2TPPS2-NHCO-EG2 polymers, the resultant supramolecular polymers of H2TPPS2-O-EG2 underwent no further assembly. Thus, the CO⋯H–N (amide) hydrogen bonding interactions at the wedges were essential for sheet formation from the H2TPPS2-NHCO-EG2 polymers.
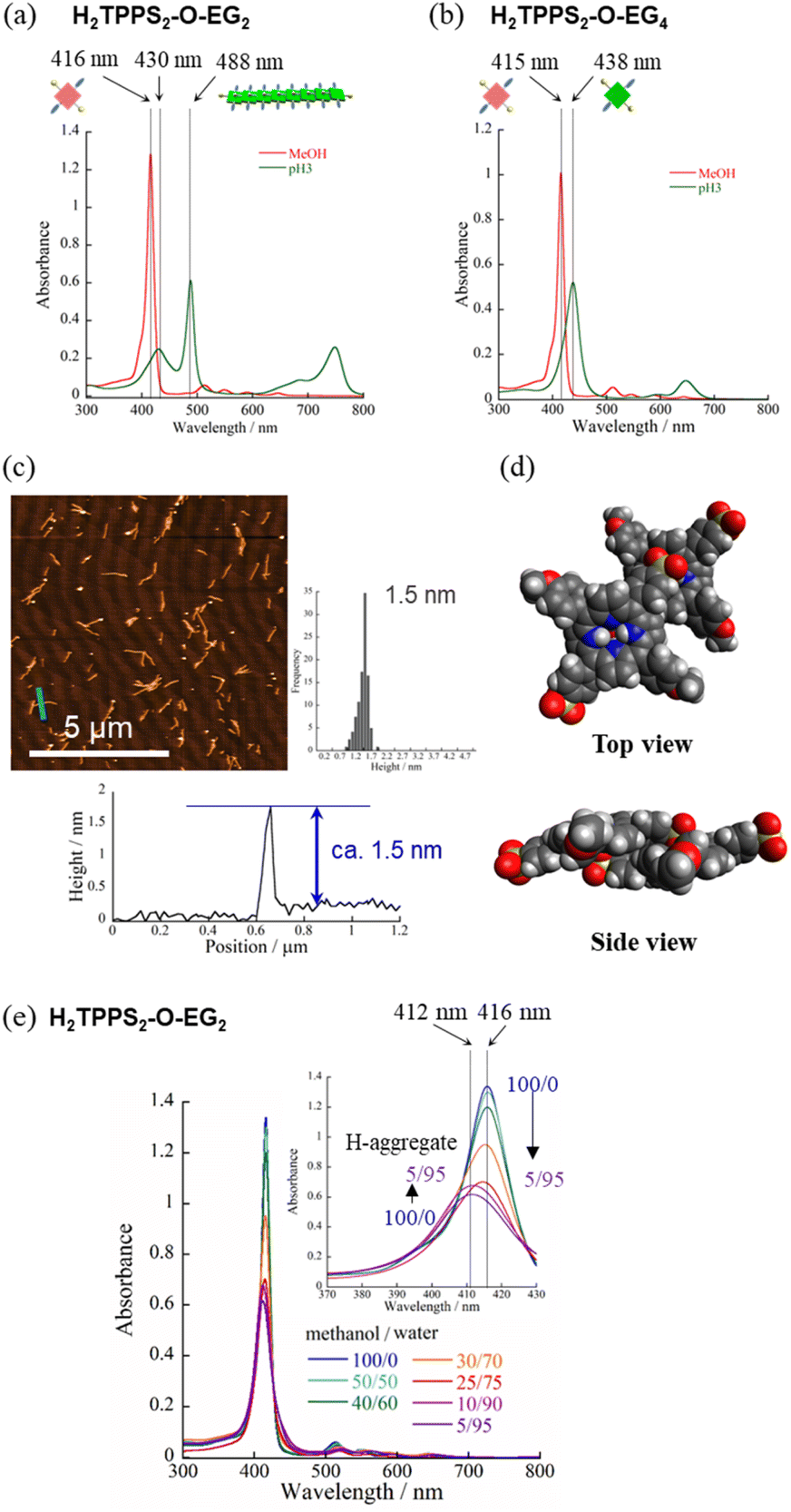 | ||
| Fig. 8 UV-vis spectra of (a) H2TPPS2-O-EG2 and (b) H2TPPS2-O-EG4 in MeOH/aqueous HCl (25/75; pH 3). (c) AFM image, height profile and height distribution of J-aggregated supramolecular polymers of H2TPPS2-O-EG2. Height distribution was measured from AFM images of at least 100 fibers. (d) DFT-calculated structure of the J-aggregated dimer of H2TPPS2-O-EG2 (B3LYP/6-31G+** level with empirical dispersion correction). (e) UV-vis spectra of H2TPPS2-O-EG2 recorded at various MeOH/water ratios (from 100/0 to 5/95); inset: expanded view of the Soret band (final concentration, 25 μM; optical path length, 1 mm; r.t.). | ||
Unlike the effective J-aggregate formation that occurred for H2TPPS2-O-EG2, for H2TPPS2-O-EG4 we observed an absorption peak at 438 nm, corresponding to the protonated species (i.e., H4TPPS2-O-EG2), but no J-aggregation peak, even after reaching thermodynamic equilibrium (Fig. 8b). This result is in sharp contrast to the successful J-aggregate formation from H2TPPS2-NHCO-EG4. As described in the previous section, we observed no significant discrepancies in the behavior of H2TPPS2-NHCO-EG2 and -EG4 in MeOH/water. Here, the self-assembly behavior of H2TPPS2-O-EG4 resembled that of H2TPPS2-NHCO-EG18, suggesting that even EG4 units could now cover the porphyrin surface. In this case, we suspect that the shorter spacer length enhanced the steric effect of the peripheral EG4 units.
When we mixed MeOH solutions (100 μM) of the H2TPPS2-O-EG2 with pure water to adjust the final concentration to 25 μM, clear blue-shifting of the absorption maximum occurred upon increasing the water content. Fig. 8e summarizes the solvent-dependent UV-vis spectral changes. Contrary to the prediction that unless the N–H⋯OS hydrogen bond, electrostatic repulsion between the sulfonate groups would suppress face-to-face stacking, these spectral changes were, in essence, identical to those of H2TPPS2-NHCO-EG2, suggesting the formation of a similar H-aggregate. Fig. S30 and S31† present the spectra deconvoluted using Gaussian functions and an isodesmic fitting. AFM images, however, did not reveal the formation of any fibrous structures. Therefore, we conclude that no effective supramolecular polymerization occurred. Together with the observed spectral changes, is seems that H2TPPS2-O-EG2 has a strong tendency to undergo π-stacking, but its long-range interactions were suppressed, presumably because of electrostatic repulsion. From DLS data shown in Fig. S35,† we confirmed that the hydrodynamic radius was smaller than that of H-aggregate prepared from H2TPPS2-O-EG2, suggesting the formation of oligomers. The XRD pattern of the resultant aggregate was consistent with this view. From Fig. 5c we estimate a π-stacking distance of 0.44 nm (q = 14.3), slightly longer than that of the H-aggregate of H2TPPS2-NHCO-EG2 (q = 15.2; 0.41 nm). In the absence of any assisting hydrogen bonding interactions, the π-stacking interactions alone were not sufficiently strong to ensure H-aggregate fibers. Here, it should be noted that we could confirm the formation of H-aggregate fibers of H2TPPS2-O-EG2 by AFM when increasing the concentration of sodium salts (Fig. S32†). This finding would suggest the view that the π-stacking interactions between H2TPPS2-O-EG2 could be assisted by EG units-Na+ interactions. Unless the assistance of hydrogen bonds or EG units-Na+ interactions, effective supramolecular polymerization could not be occurred only through the π-stacking interactions. Regular alignment of the porphyrin units, offset by 90°, could be realized with the assistance of hydrogen bonds, making them essential interactions to ensure supramolecular polymerization of the anionic monomers while overcoming neighboring electrostatic repulsion.
Conclusions
The porphyrin derivatives H2TPPS2-NHCO-EGx (x = 2, 4, 6, 8, 18) and H2TPPS2-O-EG2 function as monomers for ionic supramolecular polymerization in aqueous solvents, with the driving force for assembly being switchable depending on the pH as well as the composition of the solvent. In particular, the polymerizations of H2TPPS2-NHCO-EG2 and -EG4 resulted in the isolation of single strands of J- and H-aggregated supramolecular polymers. In the case of J-aggregate formation, as for the H2TPPS2-NHCO-EGx (x = 2, 4) and H2TPPS2-O-EGx (x = 2) derivatives presenting smaller EG units, supramolecular polymerization could be realized with suppression of bundling. The J-aggregate polymers created from H2TPPS2-NHCO-EGx (x = 2) underwent two-dimensional assembly through the edge-to-edge (N–H⋯OC) hydrogen-bonding interactions. The degree of polymerization (aagg) was dramatically higher than that of the parent H2TPPS4. Because the steric bulk of the EG units weakened adjacent monomer–monomer interactions in the J-aggregate, the rate of self-assembly of H2TPPS2-NHCO-EG4 was slower than that of H2TPPS2-NHCO-EG2, and the chain growth process was even more strongly suppressed for H2TPPS2-NHCO-EG18. Controlling the monomer reactivity is essential for the supramolecular co-polymerizations. The present concept, therefore, will lead to create various supramolecular polymers having desired inner complexity with a combination of various monomers under acidic conditions. By reducing the number of sulfonate anion of the parent H2TPPS4, the creation of face-to-face H-aggregates was first achieved. In the H-aggregates, the steric bulk of the EG units affected the assembly processes in an ON/OFF manner; that is, only H2TPPS2-NHCO-EG2 formed a stable H-aggregate, with a combination of amide–anion (N–H⋯OS) and π-stacking interactions. Unlike the parent H2TPPS4, upon protonation of the pyrrole, the local charge at the center and the periphery of the porphyrin ring can be balanced. Therefore, the created J-stacked supramolecular polymers were neutral; the H-stacked ones were anionic. Both occurred through supramolecular polymerizations of ionic monomers by tuning the balance of charge distribution in the porphyrin. The new family of supramolecular polymers was created by taking advantage of unique driving forces that depended on both the charge distribution and steric effects. We believe that the present ionic porphyrins function as common monomers for supramolecular polymerization in aqueous media, thereby extending the frontiers of this field.
Author contributions
The manuscript was written through contribution of all authors, led by MN. Most experimental works were performed by CK. HY, SN, and TM were contributed to the syntheses of a series of porphyrins. Experiments were planned by CK and MN in discussion with HY, SN, and TM.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The synchrotron radiation experiments were performed at the BL40B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal no. 2020A0845, 2021A1590, 2021A1591, 2021B1267, 2021B1778, 2022A1728, and 2022A1725). DFT calculations were performed at the Research Center for Computational Science, Okazaki, Japan. This study was supported by the ZE Research Program, IAE (ZE2022B-10); the Asahi Glass Foundation and Iketani Science and Technology Foundation; and JSPS KAKENHI (grant no. 22K19070 and 22H02065). C. K. thanks the JSPS Grant-in-Aid for JSPS Fellows Grant (no. 21J11175). C. K. and M. N. thank Peter Glink for polishing the language of our manuscript.Notes and references
- M. O. S. Penelope and J. Brothers, Fundamentals of Porphyrin Chemistry: A 21st Century Approach, 2022 Search PubMed.
- R. Takahashi and Y. Kobuke, J. Am. Chem. Soc., 2003, 125, 2372–2373 CrossRef CAS PubMed.
- J. M. Park, K. I. Hong, H. Lee and W. D. Jang, Acc. Chem. Res., 2021, 54, 2249–2260 CrossRef CAS PubMed.
- M. S. Choi, T. Yamazaki, I. Yamazaki and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 150–158 CrossRef CAS PubMed.
- K. Yamasumi and H. Maeda, Bull. Chem. Soc. Jpn., 2021, 94, 2252–2262 CrossRef CAS.
- H. Tanaka, Y. Haketa, Y. Bando, R. Yamakado, N. Yasuda and H. Maeda, Chem.–Asian J., 2020, 15, 494–498 CrossRef CAS PubMed.
- C. C. You and F. Würthner, Org. Lett., 2004, 6, 2401–2404 CrossRef CAS PubMed.
- S. P. Wang, W. Lin, X. Wang, T. Y. Cen, H. Xie, J. Huang, B. Y. Zhu, Z. Zhang, A. Song, J. Hao, J. Wu and S. Li, Nat. Commun., 2019, 10, 1399 CrossRef PubMed.
- E. Weyandt, I. A. W. Filot, G. Vantomme and E. W. Meijer, Chem.–Eur. J., 2021, 27, 9700–9707 CrossRef CAS PubMed.
- E. Weyandt, L. Leanza, R. Capelli, G. M. Pavan, G. Vantomme and E. W. Meijer, Nat. Commun., 2022, 13, 248 CrossRef CAS PubMed.
- A. Tsuda, M. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198–8202 CrossRef CAS PubMed.
- M. Sugimoto, Y. Kuramochi and A. Satake, ACS Omega, 2020, 5, 6045–6050 CrossRef CAS PubMed.
- A. Satake, Y. Suzuki, M. Sugimoto and Y. Kuramochi, Chem.–Eur. J., 2020, 26, 669–684 CrossRef CAS PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. I. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- K. Venkata Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133–1139 CrossRef CAS PubMed.
- E. Krieg, M. M. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- Z. Li, C. J. t. Zeman, S. R. Valandro, J. P. O. Bantang and K. S. Schanze, J. Am. Chem. Soc., 2019, 141, 12610–12618 CrossRef CAS PubMed.
- R. Rubires, J. Crusats, Z. El-Hachemi, T. Jaramillo, M. López, E. Valls, J.-A. Farrera and J. M. Ribó, New J. Chem., 1999, 23, 189–198 RSC.
- R. Rotomskis, R. Augulis, V. Snitka, R. Valiokas and B. Liedberg, J. Phys. Chem. B, 2004, 108, 2833–2838 CrossRef CAS.
- A. D. Schwab, D. E. Smith, C. S. Rich, E. R. Young, W. F. Smith and J. C. de Paula, J. Phys. Chem. B, 2003, 107, 11339–11345 CrossRef CAS.
- K. Kano, K. Fukuda, H. Wakami, R. Nishiyabu and R. F. Pasternack, J. Am. Chem. Soc., 2000, 122, 7494–7502 CrossRef CAS.
- P. J. Collings, E. J. Gibbs, T. E. Starr, O. Vafek, C. Yee, L. A. Pomerance and R. F. Pasternack, J. Phys. Chem. B, 1999, 103, 8474–8481 CrossRef CAS.
- J. M. Ribó, J. Crusats, J.-A. Farrera and M. L. Valero, J. Chem. Soc. Chem. Commun., 1994, 681–682 RSC.
- J. V. Hollingsworth, A. J. Richard, M. G. Vicente and P. S. Russo, Biomacromolecules, 2012, 13, 60–72 CrossRef CAS PubMed.
- Z. El-Hachemi, T. S. Balaban, J. L. Campos, S. Cespedes, J. Crusats, C. Escudero, C. S. Kamma-Lorger, J. Llorens, M. Malfois, G. R. Mitchell, A. P. Tojeira and J. M. Ribo, Chem.–Eur. J., 2016, 22, 9740–9749 CrossRef CAS PubMed.
- Z. El-Hachemi, C. Escudero, F. Acosta-Reyes, M. T. Casas, V. Altoe, S. Aloni, G. Oncins, A. Sorrenti, J. Crusats, J. L. Campos and J. M. Ribó, J. Mater. Chem. C, 2013, 1, 3337–3346 RSC.
- K. Hosomizu, M. Oodoi, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, M. Isosomppi, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, J. Phys. Chem. B, 2008, 112, 16517–16524 CrossRef CAS PubMed.
- A. Arlegui, Z. El-Hachemi, J. Crusats and A. Moyano, Molecules, 2018, 23, 3363 CrossRef PubMed.
- R. Rotomskis, R. Augulis, V. Snitka, R. Valiokas and B. Liedberg, J. Phys. Chem. B, 2004, 108, 2833–2838 CrossRef CAS.
- R. van der Weegen, A. J. Teunissen and E. W. Meijer, Chem.–Eur. J., 2017, 23, 3773–3783 CrossRef CAS PubMed.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7810–7819 CrossRef CAS PubMed.
- T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 135, 3576–3582 CrossRef CAS PubMed.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS PubMed.
- A. Bertin, J. Steibel, A. I. Michou-Gallani, J. L. Gallani and D. Felder-Flesch, Bioconjugate Chem., 2009, 20, 760–767 CrossRef CAS PubMed.
- H. Kitagishi, H. Kawasaki and K. Kano, Chem.–Asian J., 2015, 10, 1768–1775 CrossRef CAS PubMed.
- For the pH-dependent aggregation of these various types of monomers, it was difficult to define common values of aagg using each absorption maximum, assuming 0 and 100% aggregation states, according to the widely employed definition. Accordingly, here we define relative values of aagg by simply taking the absorption ratio between the monomer (protonated species) and J-aggregate. We could apply this approach to all of the monomers in this study..
- R. F. Pasternack, C. Fleming, S. Herring, P. J. Collings, J. dePaula, G. DeCastro and E. J. Gibbs, Biophys. J., 2000, 79, 550–560 CrossRef CAS PubMed.
- A. Sorrenti, Z. El-Hachemi, J. Crusats and J. M. Ribo, Chem. Commun., 2011, 47, 8551–8553 RSC.
- C. Kanzaki, A. Inagawa, G. Fukuhara, T. Okada and M. Numata, ChemSystemsChem, 2020, 2, e2000006 CrossRef CAS.
- F. Würthner, J. Org. Chem., 2022, 87, 1602–1615 CrossRef PubMed.
- M. F. J. Mabesoone, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 19781–19798 CrossRef CAS PubMed.
- Y. Lin, M. Penna, M. R. Thomas, J. P. Wojciechowski, V. Leonardo, Y. Wang, E. T. Pashuck, I. Yarovsky and M. M. Stevens, ACS Nano, 2019, 13, 1900–1909 CAS.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- M. M. Smulders, M. M. Nieuwenhuizen, T. F. de Greef, P. van der Schoot, A. P. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- X. Jiang, F. Gou and H. Jing, J. Catal., 2014, 313, 159–167 CrossRef CAS.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05542b |
| This journal is © The Royal Society of Chemistry 2022 |