
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare prenylated isoflavonoids from the young twigs of Millettia extensa and their cytotoxic activities†
Sarot Cheenpracha *a,
Ratchanaporn Chokchaisiria,
Surat Laphookhieobc,
Thunwadee Limtharakuld and
Chutamas Thepmaleee
*a,
Ratchanaporn Chokchaisiria,
Surat Laphookhieobc,
Thunwadee Limtharakuld and
Chutamas Thepmaleee
aDivision of Chemistry, School of Science, University of Phayao, Phayao 56000, Thailand. E-mail: cheenpracha@gmail.com; sarot.ch@up.ac.th
bCenter of Chemical Innovation for Sustainability (CIS), School of Science, Mae Fah Luang University, Chiang Rai 57100, Thailand
cMedicinal Plants Innovation Center of Mae Fah Luang University, Chiang Rai 57100, Thailand
dDepartment of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, The Graduate School and Research Center on Chemistry for Development of Health Promoting Products from Northern Resources, Chiang Mai University, Chiang Mai 50200, Thailand
eDivision of Biochemistry, School of Medical Sciences, University of Phayao, Phayao 56000, Thailand
First published on 24th October 2022
Abstract
Three new isoflavonoids, millexatins N–P (1–3), along with seven known compounds (6–10), were isolated from the acetone extract of the young twigs of Millettia extensa. The structures were characterized by NMR spectroscopic and mass spectrometric analyses. Millexatin N (1) is an unusual geminal diisoprenylated isoflavone with a modified ring A. Millexatin P (3) is an unusual isoflavone with a cyclohexyl substituent on ring B, which is extremely rare in nature. The isolated metabolites (1, 2, and 6–10) were evaluated for cytotoxicities against MDA-MB231, Huh-7, KKU-100 and normal human dermal fibroblast (NHDF) cell lines. Only compounds 1, 6 and 8 showed cytotoxicities against all cell lines with IC50 values ranging from 13.9 to 30.9 μM.
Introduction
Isoflavonoids possess a 3-phenyl-chromen-4-one scaffold found abundantly in the family Leguminosae, and many of them are considered for promising pharmacological properties and health benefits on multitarget tissues.1–3 Especially the prenylated forms have more potential to be developed and utilized for medicinal purposes and as lead compounds due to lipophilicity, as compared to nonprenylated forms, leading to high affinity with cell membranes and significant pharmacological activity.1The genus Millettia, belonging to the family Fabaceae, is widespread predominantly in the subtropical and tropical regions of Africa, Asia, and Australasia.4 Many species of this genus such as M. extensa, M. conraui, M. brandisiana, and M. auriculata have been used for the treatment of infected wound, tonic, skin infection, cough, boils, sores, insecticide, haematonic, and fish poison.5–8 Various types of compounds have been isolated from this genus, including isoflavonoids, chalcones, benzofuran-chalcones, rotenoids, and flavonoids,9–15 some of which showed valuable anti-estrogenic,16 anti-inflammatory,12 antibacterial,11 antiplasmodial,17,18 NAD(P)H quinine oxidoreductase 1-inducing,14 inhibitory effects on NLRP3 inflammasome activation,19 and cytotoxic20 activities.
Millettia extensa (Benth.) Baker, named “Kao Khruea” in Thai, is widely distributed in most tropical areas and it is also mostly found in the northern part of Thailand. As a traditional medicinal plant, the barks and roots have been used for the treatment of sprains, scabies, contraceptive, and protective medicine for women after childbirth.21 Previous phytochemical studies into M. extensa have revealed prenylated isoflavonoids as one of the major classes of bioactive compounds with antibacterial and anti-inflammatory activities.11,12 With the aim for searching for bioactive constituents from the medicinal plants growing in northern region of Thailand,22 the acetone extract of the stem barks of M. extensa was investigated. Three new isoflavonoids (1–3) together with seven known compounds (4–10) (Fig. 1) and cytotoxic activities against human cancer cell lines of some isolated compounds are described in this paper.
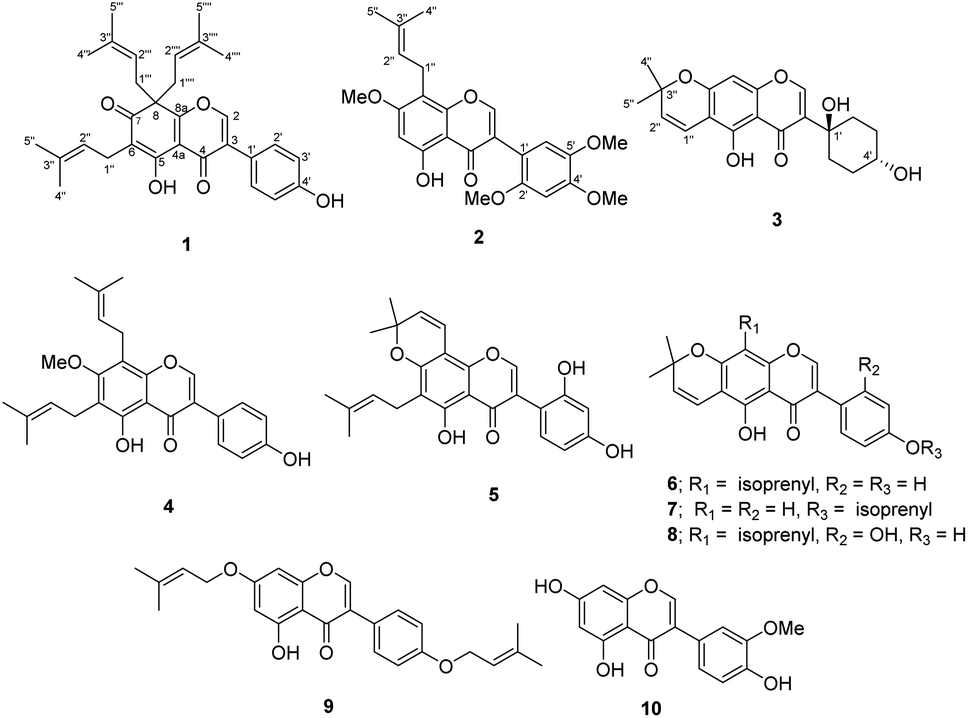 | ||
| Fig. 1 Structures of compounds 1–10. | ||
Experimental
General experimental procedures
Optical rotation was obtained on a JASCO P-2000 polarimeter in MeOH. The UV spectra were recorded with a PerkinElmer UV-vis spectrophotometer, whereas the IR spectra were obtained using a PerkinElmer FTS FT-IR spectrophotometer. The Bruker Ultrashield 500 MHz NMR spectrometer was used to measure the 1D and 2D NMR spectra of the isolated compounds. Chemical shifts (δ) are expressed in ppm with CDCl3 (δH 7.24 and δC 77.0) using TMS as an internal reference, reference to the solvent signals. A MicroTOF, Bruker Daltonics mass spectrometer was employed to acquire HRESIMS spectra. Column chromatography (CC) was performed on silica gel 60H (5–40 μm), silica gel 100 (63–200 μm) and Sephadex LH-20 (Pharmacia Fine Chemical Co., Ltd., Sweden). Semipreparative HPLC separations were performed on a Waters 626 liquid chromatography system equipped with a Grace C18 column (Econosil C18, 10 μm, 10.0 i.d. × 250 mm) and a Waters 486 Tunable Absorbance detector (Waters, USA). Fractions obtained from CC were monitored by TLC using precoated plates of silica gel 60 F254 (Merck).Plant material
The young twigs of Millettia extensa (Benth.) Baker, were collected in August 2021 at Maeka, Phayao Province, Thailand (GPS: 19°01′32.7′′N 99°53′24.2′′E) and were authenticated by Mr Martin van de Bult, Doi Tung Development Project. A voucher specimen (UP-CNP002) was deposited at the Chemistry of Natural Products for Sustainability Laboratory, School of Science, University of Phayao.Extraction and isolation
The air-dried and ground young twigs of M. extensa (1.8 kg) were extracted by percolation with acetone (4 L) at room temperature (3 days × 2), and the solvent was evaporated under reduced pressure to give acetone extract (236.4 g). The crude extract was subjected to silica gel column chromatography (CC), eluting with 100% hexanes to 100% acetone, to provide seven fractions (A–G). Fraction A (2.5 g) was further purified through Sephadex LH-20 (MeOH/CH2Cl2, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to afford five subfractions (A1–A5). Subfraction A4 (840.4 mg) was subjected on silica gel CC, eluting with acetone–hexanes (1:19, v/v) to yield compounds 7 (8.2 mg) and 9 (7.7 mg). The separation of fraction C (17.4 g) by silica gel CC, eluting with 100% CH2Cl2 and an increasing polarity with MeOH, afforded five subfractions (C1–C5). Subfraction C5 (5.3 g) was purified by Sephadex LH-20 (100% MeOH), following purified by silica gel CC (acetone–hexanes, 1:4, v/v) to obtain five subfractions (C5a–C5e). Subfraction C5b (150.5 mg) was chromatographed on a silica gel CC with MeOH–CH2Cl2 (1:99, v/v) to yield compounds 1 (9.1 mg), 2 (5.9 mg), 4 (2.6 mg) and 6 (8.0 mg). Fraction E (11.5 g) was loaded to silica gel CC eluting with a MeOH–CH2Cl2 gradient solvent system (0:1 to 1:9, v/v) and nine subfractions were obtained (E1–E9). Subfraction E2 (1.7 g) was separated by Sephadex LH-20 with 100% MeOH as eluent to afford five subfractions (E2a–E2e). Recrystallization of subfraction E2b with acetone–hexanes (1:4, v/v) gave compound 10 (3.1 mg). Subfraction E2c (87.4 mg) was achieved by silica gel CC eluting with acetone–hexanes (1:4, v/v) to yield compounds 5 (2.8 mg) and 8 (6.7 mg). Purification of fraction F (1.1 g) by Sephadex LH-20 eluting with MeOH and subjected to silica gel CC eluting with MeOH–CH2Cl2 (1:49, v/v) yielded five subfractions (F1–F5). Subfraction F4 (15.2 mg) was further purified by semipreparative C18 HPLC with MeOH–H2O (75:25, v/v) to yield compound 3 (2.1 mg, tR 13.2).
ε): 201 (4.10), 260 (3.64), 355 (3.10) nm; IR (neat) νmax 3304, 2955, 1661, 1548, 1426, 1401 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, Table 1; HRESIMS m/z 497.2299 [M + Na]+ (calcd for [C30H34O5Na]+, 497.2298).
:1, v/v) to afford five subfractions (A1–A5). Subfraction A4 (840.4 mg) was subjected on silica gel CC, eluting with acetone–hexanes (1:19, v/v) to yield compounds 7 (8.2 mg) and 9 (7.7 mg). The separation of fraction C (17.4 g) by silica gel CC, eluting with 100% CH2Cl2 and an increasing polarity with MeOH, afforded five subfractions (C1–C5). Subfraction C5 (5.3 g) was purified by Sephadex LH-20 (100% MeOH), following purified by silica gel CC (acetone–hexanes, 1:4, v/v) to obtain five subfractions (C5a–C5e). Subfraction C5b (150.5 mg) was chromatographed on a silica gel CC with MeOH–CH2Cl2 (1:99, v/v) to yield compounds 1 (9.1 mg), 2 (5.9 mg), 4 (2.6 mg) and 6 (8.0 mg). Fraction E (11.5 g) was loaded to silica gel CC eluting with a MeOH–CH2Cl2 gradient solvent system (0:1 to 1:9, v/v) and nine subfractions were obtained (E1–E9). Subfraction E2 (1.7 g) was separated by Sephadex LH-20 with 100% MeOH as eluent to afford five subfractions (E2a–E2e). Recrystallization of subfraction E2b with acetone–hexanes (1:4, v/v) gave compound 10 (3.1 mg). Subfraction E2c (87.4 mg) was achieved by silica gel CC eluting with acetone–hexanes (1:4, v/v) to yield compounds 5 (2.8 mg) and 8 (6.7 mg). Purification of fraction F (1.1 g) by Sephadex LH-20 eluting with MeOH and subjected to silica gel CC eluting with MeOH–CH2Cl2 (1:49, v/v) yielded five subfractions (F1–F5). Subfraction F4 (15.2 mg) was further purified by semipreparative C18 HPLC with MeOH–H2O (75:25, v/v) to yield compound 3 (2.1 mg, tR 13.2).
ε): 201 (4.10), 260 (3.64), 355 (3.10) nm; IR (neat) νmax 3304, 2955, 1661, 1548, 1426, 1401 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, Table 1; HRESIMS m/z 497.2299 [M + Na]+ (calcd for [C30H34O5Na]+, 497.2298).
| No. | Millexatin N (1)a | Millexatin O (2)a | Millexatin P (3)a | |||
|---|---|---|---|---|---|---|
| δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | δC, type | δH, mult. (J in Hz) | |
| a NMR data were recorded in chloroform-d. | ||||||
| 2 | 152.6, CH | 8.00, s | 154.9, CH | 7.95, s | 151.7, CH | 7.86, s |
| 3 | 128.8, C | 119.7, C | 127.4, C | |||
| 4 | 178.9, C | 179.1, C | 183.0, C | |||
| 4a | 114.6, C | 106.5, C | 106.0, C | |||
| 5 | 164.1, C | 161.0, C | 152.1, C | |||
| 6 | 115.4, C | 95.2, CH | 6.44, s | 101.1, C | ||
| 7 | 195.3, C | 162.4, C | 159.9, C | |||
| 8 | 57.0, C | 107.9, C | 100.4, CH | 6.29, s | ||
| 8a | 174.2, C | 154.6, C | 162.0, C | |||
| 1′ | 121.4, C | 111.0, C | 71.0, C | |||
| 2′ | 130.1, CH | 7.40, d (8.7) | 150.1, C | 29.8, CH2 | 2.15, m | |
| 1.91, m | ||||||
| 3′ | 116.0, CH | 6.94, d (8.7) | 98.2, CH | 6.66, s | 28.2, CH2 | 2.12, m |
| 1.64, m | ||||||
| 4′ | 157.2, C | 152.1, C | 65.6, CH | 4.16, brs | ||
| 5′ | 116.0, CH | 6.94, d (8.7) | 143.1, C | 28.2, CH2 | 2.12, m | |
| 1.64, m | ||||||
| 6′ | 130.4, CH | 7.40, d (8.7) | 115.1, CH | 6.91, s | 29.8, CH2 | 2.15, m |
| 1.91, m | ||||||
| 1′′ | 21.0, CH2 | 3.09, d (6.8) | 21.5, CH2 | 3.44, d (7.2) | 114.4, CH | 6.66, d (10.1) |
| 2′′ | 121.9, CH | 5.07, brt (6.8) | 122.0, CH | 5.20, brt (7.2) | 127.5, CH | 5.59, d (10.1) |
| 3′′ | 131.9, C | 132.6, C | 78.3, C | |||
| 4′′ | 18.0, CH3 | 1.73, s | 17.8, CH3 | 1.82, s | 28.3, CH3 | 1.49, s |
| 5′′ | 26.0, CH3 | 1.65, s | 25.8, CH3 | 1.70, s | 28.3, CH3 | 1.49, s |
| 1′′′/1′′′′ | 38.7, CH2 | 2.85, dd (13.9, 7.1) | ||||
| 2.62, dd (13.9, 7.1) | ||||||
| 2′′′/2′′′′ | 117.1, CH | 4.69, t (7.1) | ||||
| 3′′′/3′′′′ | 136.0, C | |||||
| 4′′′/4′′′′ | 17.9, CH3 | 1.54, s | ||||
| 5′′′/5′′′′ | 25.9, CH3 | 1.50, s | ||||
| OH-5 | 13.13, s | 12.97, s | 13.59, s | |||
| OMe-7 | 56.1, CH3 | 3.93, s | ||||
| OMe-2′ | 56.2, CH3 | 3.97, s | ||||
| OMe-4′ | 56.9, CH3 | 3.82, s | ||||
| OMe-5′ | 56.6, CH3 | 3.88, s | ||||
ε): 205 (4.15), 269 (4.11), 330 (3.12) nm; IR (neat) νmax 3332, 2958, 1706, 1635, 1589, 1435, 1252 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, Table 1; HRESIMS m/z 449.1578 [M + Na]+ (calcd for [C24H26O7Na]+, 449.1576).ε): 202 (4.11), 268 (3.65), 310 (3.22) nm; IR (neat) νmax 3430, 2970, 1665, 1589, 1430, 1254, cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, Table 1; HRESIMS m/z 359.1493 [M + H]+ (calcd for [C20H23O6]+, 359.1489).Cell lines and culture
Human CCA cell lines, namely KKU-100 (Japanese Collection of Research Bioresources Cell Bank (JCRB), National Institute of Biomedical Innovation, Japan), MDA-MB-231 and Huh-7 (ATCC, American Type Culture Collection), and normal human dermal fibroblast (PromoCell, Germany) were cultured in Dulbecco's modified Eagle's medium and Ham's F-12 nutrient mixture (DMEM/F12; Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific) and penicillin (100 U mL−1)–streptomycin (100 μg mL−1) at 37 °C in a humidified atmosphere of 5% CO2. The cells were sub-cultured twice per week by following the standard trypsinization protocol.Cytotoxicity assay
The procedure for the cytotoxic assay was assessed against KKU-100, MDA-MB-231, Huh-7, and normal human dermal fibroblast (NHDF) cell lines by the MTT method. Cell lines (5 × 103 cells per well) were seeded in a 96-well plate for 24 hours. Various tested compound concentrations were added to the 96-well plate and incubated for 24 h. Ten microliters of MTT solution (5 mg mL−1) were added and incubated for 2 h. Then, cell supernatants were removed, and DMSO was added to dissolve the formazan product. A microplate spectrophotometer measured light absorbent at 540 nm. The IC50 was calculated by using GraphPad Prism version 8. Doxorubicin (Sigma) and 0.75% DMSO (RCI Labscan) were used as a positive and negative control.Results and discussion
The air-dried and ground young twigs of M. extensa were extracted with acetone to afford a dark green crude extract. Purification of this extract by repeated chromatographic separation gave three undescribed isoflavones (1–3), along with seven known compounds that were identified as millewanin F (4),23 millexatin F (5),11 scandenone (6),11 2′-deoxyisoauriculatin (7),24 auriculatin (8),25 7,4′-di-O-prenylgenistein (9),26 and 3′-methylorobol (10)27 upon comparison of their physical and spectroscopic data with published values.Compound 1 was obtained as yellow viscous oil. Its molecular formula was deduced as C30H34O5 on the basis of the HRESIMS ion peak at m/z 497.2299 [M + H]+ (calcd for [C30H34O5Na]+, 497.2298), which accounted for 14 degrees of unsaturation. The IR spectrum displayed absorptions due to hydroxy group (3304 cm−1) and conjugated carbonyl (1661 cm−1) functionalities. Its UV spectrum at λmax 260 and 355 nm were consistent with an isoflavone chromophore that was corroborated with a singlet at δH 8.00 (H-2) in the 1H NMR spectrum, and the 13C NMR signals at δC 152.6 (C-2), 128.8 (C-3), and 178.9 (C-4). Analysis of the 13C NMR data (Table 1) of 1 displayed 30 carbon resonances, which were classified by their chemical shifts and HSQC spectrum as six methyls [δC 25.7, 25.9 (2), 17.7 and 17.9 (2)], eight sp2 methines [δC 152.6, 130.4 (2), 121.9, 117.1 (2) and 116.0 (2)], three sp3 methylenes [δC 38.7 (2) and 21.0], 11 non-hydrogenated carbons [δC 174.2, 164.1, 157.2, 136.0 (2), 131.9, 128.8, 121.4, 115.4, 114.6, and 57.0], and two conjugated carbonyl carbons (δC 195.3 and 178.9). In the 1H NMR spectrum (Table 1) of 1, a downfield signal at δH 13.13 (1H, s) was assigned to a hydrogen-bonded hydroxy group at C-5. The 1H and 13C NMR signals showed resonances for an isoprenyl unit [δH/δC 5.07 (1H, brt, J = 6.8 Hz, H-2′′)/121.9, 3.09 (2H, d, J = 6.8 Hz, H2-1′′)/21.0, 1.73 (3H, s, H3-4′′)/18.0, 1.65 (3H, s, H3-5′′)/26.0, and δC 131.9 (C-3′′)] and a geminal diisoprenyl group [δH/δC 4.69 (2H, t, J = 7.1 Hz, H-2′′′, H-2′′′′)/117.1, 2.85 (2H, dd, J = 13.9, 7.1 Hz, H2-1′′′a, H2-1′′′′a)/38.7, 2.62 (2H, dd, J = 13.9, 7.1 Hz, H2-1′′′b, H2-1′′′′b), 1.54 (6H, s, H3-4′′′, H3-4′′′′)/17.9, 1.50 (6H, s, H3-5′′′, H3-5′′′′)/25.9, and δC 136.0 (C-3′′′/C-3′′′′)]. These spectroscopic data revealed that it is structurally closely related to millexatin A, an isoflavone that was isolated from the stems of M. extensa.11 The major difference found was that a set of ABC spin-coupled aromatic protons of millexatin A was replaced with the two doublet resonances for the para-aromatic protons on the B ring [δH 7.40 (2H, d, J = 8.7 Hz, H-2′, H-6′), and 6.94 (2H, d, J = 8.7 Hz, H-3′, H-5′)]. The HMBC correlation (Fig. 2) of the methine proton H-2′/H-6′ with C-3, C-1′, C-3′ and C-4′ allowed the assignments of H-2′/H-6′, and H-3′/H-5′ of ring B. The long-range 1H–13C correlations from OH-5 (δH 13.13) to C-4a, C-5, C-6, and from H2-1′′ (δH 3.09) to C-5, C-6 and C-7 suggested the location of an isoprenyl unit at C-6 and a conjugated ketone carbonyl group at C-7. The HMBC correlations observed between the methylene protons H2-1′′′/H2-1′′′′ (δH 2.85 and 2.62) and C-7, C-8 and C-8a confirmed the attachment of a geminal diisoprenyl group at C-8 (Fig. 2). On the basis of these data, the structure of compound 1, which features an unusual isoflavone with a geminal diisoprenyl unit on a modified ring A, was characterized as millexatin N.
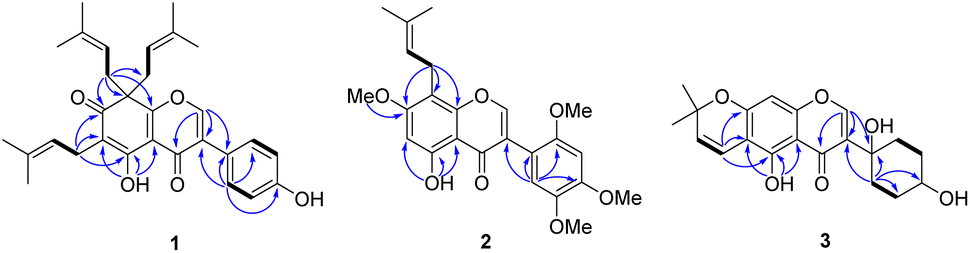 | ||
| Fig. 2 Selected 1H → 13C HMBC (blue arrows) and COSY (bold line) correlations of 1–3. | ||
Compound 2 was isolated as a yellow viscous oil and exhibited a [M + Na]+ ion peak at m/z 449.1578 (calcd. for [C24H26O7Na]+, 449.1576) in the HRESIMS analysis, consistent with a molecular formula of C24H26O7. The IR and UV spectra were similar to those of 1, suggesting the presence of an isoflavone skeleton. The NMR spectroscopic data (Table 1) indicated resonances typical of an isoprenyl moiety [δH/δC 5.20 (1H, brt, J = 7.2 Hz, H-2′′)/122.0, 3.44 (2H, d, J = 7.2 Hz, H2-1′′)/21.5, 1.82 (3H, s, H3-4′′)/17.8, 1.70 (3H, s, H3-5′′)/25.8, and δC 132.6(C-3′′)], three aromatic protons [δH/δC 6.91 (1H, s, H-6′)/115.1, 6.66 (1H, s, H-3′)/98.2, and 6.44 (1H, s, H-6)/95.2], and four methoxy groups [δH/δC 3.97 (3H, s, OMe-2′)/56.2, 3.93 (3H, s, OMe-7)/56.1, 3.88 (3H, s, OMe-5′)/56.6, and 3.82 (3H, s, OMe-4′)/56.9], along with a hydrogen-bonded hydroxy group [δH 12.97 (1H, s, 5-OH)]. In addition, a characteristic resonance for H-2 of an isoflavone at δH 7.95 (1H, s)/δC 154.9 was observed in the 1H NMR spectrum, which was further confirmed by the key HMBC correlations (Fig. 2) from H-2 (δH 7.95) to C-3, C-4, C-8a and C-1′. The HMBC correlations of H-6 (δH 6.44) with C-4a, C-5, C-7 and C-8, of H2-1′′ (δH 3.44) with C-7, C-8 and C-8a, and of 7-OMe (δH 3.93) with C-7 showed that an isoprenyl moiety and a methoxy group were attached to C-8 and C-7, respectively. Another methoxy groups at δH 3.97, 3.88, and 3.82 were placed at C-2′, C-5′ and C-4′, respectively, based on its HMBC correlations shown in Fig. 2 and the NOESY correlations of H-6′/OMe-5′ and OMe-2′/H-3′/OMe-4′. Therefore, the structure of compound 2 was established as shown and named millexatin O.
Compound 3, isolated as a yellow viscous oil, was deduced to have a molecular formula of C20H22O6 from its HRESIMS ion peak at m/z 359.1493 [M + H]+ (calcd. for [C20H23O6]+, 359.1489), indicating 10 degrees of unsaturation. The IR absorptions implied the presence of hydroxy (3430 cm−1) and carbonyl (1665 cm−1) functionalities. The 13C NMR and HMQC spectra (Table 1) revealed 20 carbon resonances for one carbonyl carbon (δC 183.0), eight non-hydrogenated carbons, of which one was a sp3 oxygenated carbon (δC 71.0), four sp2 methines, including one oxygenated carbon (δC 151.7), four methylenes, and two methyls. Comparison of its 1D NMR data of 3 (Table 1) with compound 7 (ref. 24) suggested that these two compounds shared the same isoflavone skeleton with a hydrogen-bonded hydroxy group [δH 13.59 (1H, s, OH-5)], an olefinic proton [δH 7.86 (1H, s, H-2)/δC 151.7], an aromatic proton [δH 6.29 (1H, s, H-8)/δC 100.4], and a 1,1-dimethylallyl group [δH/δC 6.66 (1H, d, J = 10.1 Hz, H-1′′)/114.4, 6.59 (1H, d, J = 10.1 Hz, H-2′′)/127.5, 1.49 (6H, s, H3-4′′ and H3-5′′)/28.3, and δC 78.3 (C-3′′)]. The difference between them was the absence of an isoprenyl group and four aromatic protons of ring B. Compound 3 displayed signals for 4-hydroxycyclohexyl moiety [δH/δC 4.16 (1H, brs, H-4′)/65.6, 2.15 (2H, m, H2-2′a, H-6′a)/29.8, 2.12 (2H, m, H2-3′a, H-5′a)/28.2, 1.91 (2H, m, H2-2′b, H-6′b) and 1.64 (2H, m, H2-3′b, H-5′b), and δC 71.0 (C-1′)] instead of the coupled aromatic protons in 7. In the HMBC spectrum of 3 (Fig. 2), the correlations from H2-2′/H2-6′ to C-3, C-1′ and C-4′, from H-2 to C-4, C-8a, C-3 and C-1′ and the low field chemical shift of C-1′ at δC 71.0 indicated that hydroxy group was attached at C-1′ and 4-hydroxycyclohexyl moiety at C-3. These spectroscopic data were similar to those of 2-(trans-1,4-dihydroxy-2-cyclohexenyl)-5-hydroxy7-methoxychromone previously isolated from the fern of Phegopteris connectilis.28 Additionally, the HMBC correlations of OH-5 (δH 13.59) and H-1′′ (δH 6.66) with C-5 confirmed a 1,1-dimethylallyl group at C-6 and C-7. The relative configuration of 3 was established by analysis of NOESY data (Fig. 3) and 1H–1H couplings. The small J value (J = <1 Hz) between H-3′/H-4′ and H-4′/H-5′ and the cross-peaks of 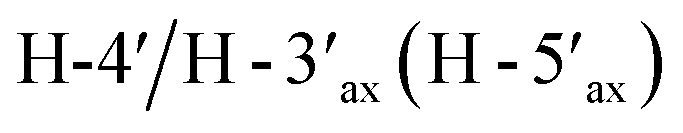 and
and 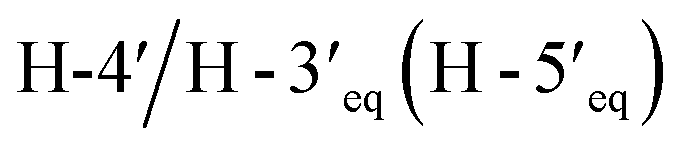 in the NOESY spectrum indicated that the hydroxy group at C-4′ was α-axially oriented. In addition, the NOESY correlations of H-2 with
in the NOESY spectrum indicated that the hydroxy group at C-4′ was α-axially oriented. In addition, the NOESY correlations of H-2 with 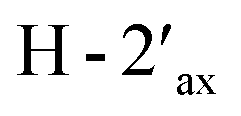 and
and 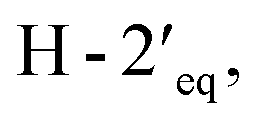 and of
and of 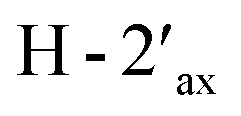 with
with 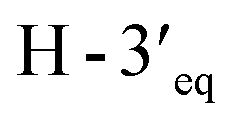 and
and 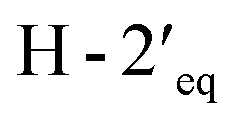 implied that the hydroxy group at C-1′ was in an β-axial direction. These were in good agreement with the relative configurations at C-1′ and C-4′ to that of related compound in the literature.28 The observation of a specific rotation value ([α]27D + 0.20 (c 0.05, MeOH)) and the lack of a Cotton effect was observed, suggesting 3 to be a racemate. Thus, the structure of compound 3 was named millexatin P.
implied that the hydroxy group at C-1′ was in an β-axial direction. These were in good agreement with the relative configurations at C-1′ and C-4′ to that of related compound in the literature.28 The observation of a specific rotation value ([α]27D + 0.20 (c 0.05, MeOH)) and the lack of a Cotton effect was observed, suggesting 3 to be a racemate. Thus, the structure of compound 3 was named millexatin P.
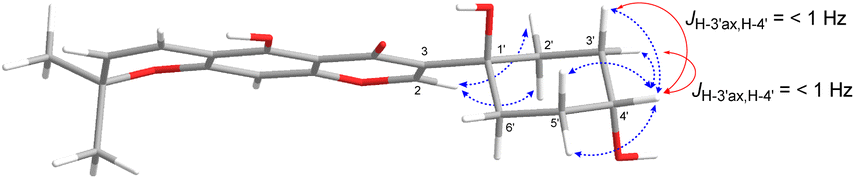 | ||
| Fig. 3 Key NOESY cross-peaks (blue) and coupling constants (red) of 3. | ||
Most of the isolated isoflavones (1, 2, 6–10) were tested for their cytotoxic activities against breast (MDA-MB231), hepatocellular carcinoma (Huh-7), cholangiocarcinoma (KKU-100) and normal human dermal fibroblasts (NHDF) using the MTT assay, with doxorubicin as the positive control (Table 2). Compounds 1, 6 and 8 exhibited cytotoxic activity against all the cell lines with IC50 values ranging from 13.9 to 30.9 μM. Compound 1 was found to be the best cytotoxic effect against Huh-7 cell line. However, this compound was relatively cytotoxic. Compounds 2, 7, 9 and 10 were inactive toward all (IC50 > 100 μM). It is interesting to note that compound 6 with an isoprenyl moiety at C-8 had better activity than 7, which lacked this substituent.
Conclusions
In conclusion, two undescribed modified isoflavones (1 and 3) and one undescribed methylated isoflavone (2) together with seven known compounds (4–10) were isolated from the young twigs of M. extensa. Compounds 1 is a rare isoflavone with a geminal diisoprenyl unit on a modified ring A, which compound 3 is unusual isoflavone with saturated ring B. Several modified isoflavones were also isolated from the genus Millettia.15,29,30 Moreover, compounds 1, 6 and 8 were found to have cytotoxic effect against MDA-MB231, Huh-7, KKU-100 and normal human dermal fibroblasts cell lines.Author contributions
S. Cheenpracha, contributed to developing the concept, experimental work, formal analysis and writing–original draft; R. Chokchaisiri, S. Laphookhieo, T. Limtharakul, contributed to experimental design and writing–review & editing; C. Thepmalee, contributed to experimental design, writing–review & editing. All authors have read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was supported by the Thailand Science Research and Innovation Fund and the University of Phayao (Grant No. FF65-RIM050). TL thanks Chiang Mai University for partial support. The funders had no other role during the experiment and preparation of the manuscript. We thank Mr Martin van de Bult, Doi Tung Development Project, for the plant identification.Notes and references
- S. B. Ateba, M. A. Mvondo, S. Djiogue, S. Zingué, L. Krenn and D. Njamen, Front. Pharmacol., 2019, 10, 952 CrossRef CAS PubMed.
- L. Hanski, N. Genina, H. Uvell, K. Malinovskaja, Å. Gylfe, T. Laaksonen, R. Kolakovic, E. Mäkilä, J. Salonen, J. Hirvonen, M. Elofsson, N. Sandler and P. M. Vuorela, PLoS One, 2014, 9, e115115 CrossRef PubMed.
- M. Mahmoud, M. R. A. Abdollah, M. E. Elsesy, D. A. Abou El Ella, S. K. Zada and M. F. Tolba, Phytother. Res., 2022, 36, 1310–1325 CrossRef CAS PubMed.
- R. Jena, D. Rath, S. S. Rout and D. M. Kar, Saudi Pharm. J., 2020, 28, 1686–1703 CrossRef CAS PubMed.
- S. Das and S. Ganapaty, Asian Pac. J. Trop. Dis., 2014, 4, S870–S873 CrossRef.
- V. Fuendjiep, A. E. Nkengfack, Z. T. Fomum, B. L. Sondengam and B. Bodo, Phytochemistry, 1998, 47, 113–115 CrossRef CAS.
- P. Pailee, C. Mahidol, S. Ruchirawat and V. Prachyawarakorn, Phytochemistry, 2019, 162, 157–164 CrossRef CAS PubMed.
- J. Sharma, S. Gairola, Y. P. Sharma and R. D. Gaur, J. Ethnopharmacol., 2014, 158, 140–206 CrossRef PubMed.
- F. Ngandeu, M. Bezabih, D. Ngamga, A. T. Tchinda, B. T. Ngadjui, B. M. Abegaz, H. Dufat and F. Tillequin, Phytochemistry, 2008, 69, 258–263 CrossRef CAS PubMed.
- G. Palazzino, P. Rasoanaivo, E. Federici, M. Nicoletti and C. Galeffi, Phytochemistry, 2003, 63, 471–474 CrossRef CAS.
- A. Raksat, W. Maneerat, R. J. Andersen, S. G. Pyne and S. Laphookhieo, J. Nat. Prod., 2018, 81, 1835–1840 CrossRef CAS PubMed.
- A. Raksat, W. Maneerat, N. Rujanapun, R. J. Andersen, S. G. Pyne and S. Laphookhieo, J. Nat. Prod., 2019, 82, 2343–2348 CrossRef CAS PubMed.
- B. Sritularak, K. Likhitwitayawuid, J. Conrad, B. Vogler, S. Reeb, I. Klaiber and W. Kraus, J. Nat. Prod., 2002, 65, 589–591 CrossRef CAS PubMed.
- W. Wang, N. Li, J. Wang, G. Chen, R. Huang, W. Zhao, J. Li and Y. Si, Phytochemistry, 2016, 131, 107–114 CrossRef CAS PubMed.
- E. Yankep, J. T. Mbafor, Z. T. Fomum, C. Steinbeck, B. B. Messanga, B. Nyasse, H. Budzikiewicz, C. Lenz and H. Schmickler, Phytochemistry, 2001, 56, 363–368 CrossRef CAS PubMed.
- C. Ito, M. Itoigawa, M. Kumagaya, Y. Okamoto, K. Ueda, T. Nishihara, N. Kojima and H. Furukawa, J. Nat. Prod., 2006, 69, 138–141 CrossRef CAS PubMed.
- M. Marco, T. Deyou, A. Gruhonjic, J. Holleran, S. Duffy, M. Heydenreich, P. A. Firtzpatrick, G. Landberg, A. Koch, S. Derese, J. Pelletier, V. M. Avery, M. Erdélyi and A. Yenesew, Phytochem. Lett., 2017, 21, 216–220 CrossRef CAS.
- A. Yenesew, S. Derese, J. O. Midiwo, H. A. Oketch-Rabah, J. Lisgarten, R. Palmer, M. Heydenreich, M. G. Peter, H. Akala, J. Wangui and P. Liyala, Phytochemistry, 2003, 64, 773–779 CrossRef CAS PubMed.
- X. Ma, M. Zhao, M. H. Tang, L. L. Xue, R. J. Zhang, L. Liu, H. F. Ni, X. Y. Cai, S. Kuang, F. Hong and L. Wang, J. Nat. Prod., 2020, 83, 2950–2959 CrossRef CAS PubMed.
- T. Deyou, M. Marco, M. Heydenreich, F. Pan, A. Gruhonjic, P. A. Fitzpatrick, A. Koch, S. Derese, J. Pelletier, K. Rissanen, A. Yenesew and M. Erdélyi, J. Nat. Prod., 2017, 80, 2060–2066 CrossRef CAS PubMed.
- U. Quattrocchi, CRC World Dictionary of Medicinal and Poisonous Plants: Common Names, Scientific Names, Eponyms, Synonyms, and Etymology, CRC Press, Taylor & Francis, 2012, p. 2510 Search PubMed.
- S. Cheenpracha, R. Chokchaisiri, L. Ganranoo, T. Maneerat, N. Rujanapun, R. Charoensup, S. Laphookhieo, N. Injan and S. Nokbin, Phytochem. Lett., 2022, 49, 192–196 CrossRef CAS.
- C. Ito, T. Murata, M. Itoigawa, K. Nakao, M. Kumagai, N. Kaneda and H. Furukawa, Planta Med., 2006, 72, 424–429 CrossRef CAS PubMed.
- N. MinHaj, H. Khan, S. K. Kapoor and A. Zaman, Tetrahedron, 1976, 32, 749–751 CrossRef CAS.
- S. Ganapaty, V. Nair, D. R. Devi, S. T. Pannakal, H. Laatsch and B. Dittrich, Nat. Prod. Commun., 2014, 9, 937–940 CrossRef CAS.
- O. Pancharoen, A. Athipornchai, A. Panthong and W. C. Taylor, Chem. Pharm. Bull., 2008, 56, 835–838 CrossRef CAS PubMed.
- K. Asres, P. Mascagni, M. J. O'Neill and D. Phillipson, Z. Naturforsch. C, 1985, 40, 617–620 CrossRef.
- K. P. Adam, Phytochemistry, 1999, 52, 929–934 CrossRef CAS.
- C. Ito, M. Itoigawa, H. T. Tan, H. Tokuda, X. Y. Mou, T. Mukainaka, T. Ishikawa, H. Nishino and H. Furukawa, Cancer Lett., 2000, 152, 187–192 CrossRef CAS PubMed.
- A. K. Singhal, N. C. Barua, R. P. Sharma and J. N. Baruah, Phytochemistry, 1983, 22, 1005–1006 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS, NMR spectra of compounds 1–3. See DOI: https://doi.org/10.1039/d2ra05950a |
| This journal is © The Royal Society of Chemistry 2022 |