
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsotope-dilution LC-MS/MS analysis of the elastin crosslinkers desmosine and isodesmosine in acute cerebral stroke patients†
Ayame Mikagi a,
Ryosuke Tashirob,
Tomoo Inoue*abc,
Riki Anzawaa,
Akiho Imuraa,
Takahiro Tanigawaa,
Tomohisa Ishidac,
Takashi Inouec,
Kuniyasu Niizuma*de,
Teiji Tominagab and
Toyonobu Usuki*a
a,
Ryosuke Tashirob,
Tomoo Inoue*abc,
Riki Anzawaa,
Akiho Imuraa,
Takahiro Tanigawaa,
Tomohisa Ishidac,
Takashi Inouec,
Kuniyasu Niizuma*de,
Teiji Tominagab and
Toyonobu Usuki*a
aDepartment of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioicho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: t-usuki@sophia.ac.jp
bDepartment of Neurosurgery, Tohoku University Graduate School of Medicine, 1-1 Seiryo-machi, Aoba-ku, Sendai 980-8574, Japan. E-mail: tomoo49@gmail.com
cDepartment of Neurosurgery, Sendai Medical Center, 2-11-12 Miyagino, Miyagino-ku, Sendai 983-8520, Japan
dDepartment of Neurosurgical Engineering and Translational Neuroscience, Graduate School of Biomedical Engineering, Tohoku University, 2-1 Seiryo-machi, Aoba-ku, Sendai 980-8575, Japan. E-mail: niizuma@nsg.med.tohoku.ac.jp
eDepartment of Neurosurgical Engineering and Translational Neuroscience, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Aoba-ku, Sendai 980-8575, Japan
First published on 7th November 2022
Abstract
Utilizing chemically synthesized an isotopically labeled internal standard, isodesmosine-13C3,15N1, an isotope-dilution LC-MS/MS method was established. Concentrations of desmosine and isodesmosine in plasma of acute cerebral stroke patients and healthy controls were determined. The concentration of desmosines was markedly higher in plasma from acute stroke patients compared with healthy controls. Desmosines are thus novel biomarkers for evaluating the extent of vascular injury after acute cerebral stroke.
Introduction
Homeostasis of the extracellular matrix is critical for maintaining vascular integrity. The extracellular matrix proteins elastin and collagen are particularly important for the maintenance of vascular walls. Elastin is a major component of the fibers that provide elasticity to the alveoli, skin, ligaments, blood vessels, and other components of the body.1,2 Elastin fibers contribute to the elastic characteristic of the arterial wall, whereas collagen is a principal load-bearing component of vascular walls. Along with collagen accumulation, the elastic fibers augment stiffness and resist degradation and fragmentation induced by various stimuli. However, disruption of the elaborate mechanism regulating elastin homeostasis due to pathological conditions such as chronic obstructive pulmonary disease (COPD) or vascular injury results in irreversible degradation of elastin-containing tissues. These observations suggest elastin has potential value as a biomarker of tissue and vascular damage.Quantitative measurement of the products of extracellular matrix degradation could enhance our understanding of tissue and vascular injuries. Desmosine and isodesmosine (Fig. 1) are crosslinked pyridinium-based amino acids that exist only in the extracellular matrix protein elastin.3,4 However, the precise measurement of desmosine and isodesmosine is technically challenging. Recent analyses of desmosine and isodesmosine have been developed using liquid chromatography-mass spectrometry (LC-MS) or liquid chromatography-tandem mass spectrometry (LC-MS/MS: see Fig. S0†).5–13 Notably, patients with exacerbated COPD exhibit higher urinary and blood desmosine levels than healthy controls.14 Desmosine and isodesmosine can thus be used as biomarkers to gauge the severity of COPD.
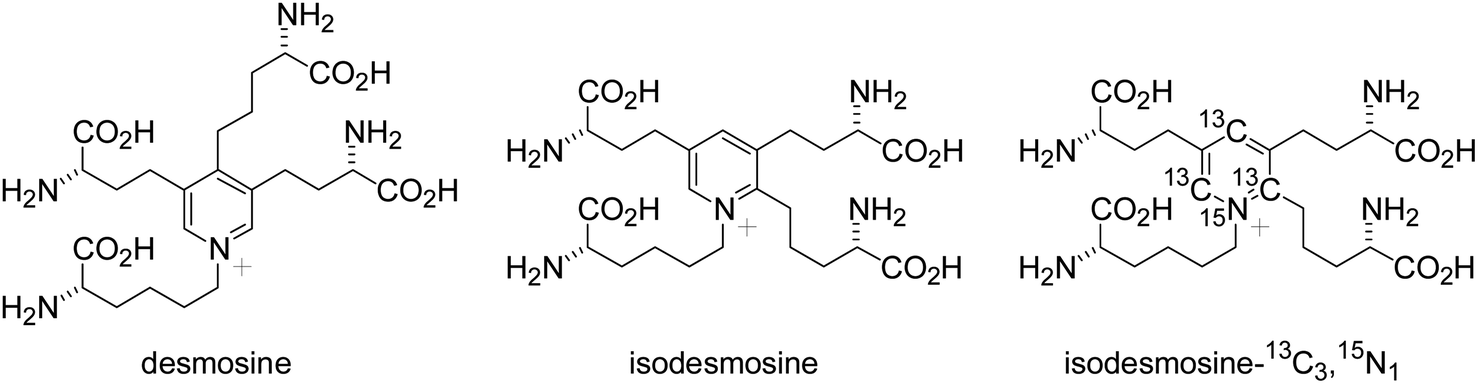 | ||
| Fig. 1 Structures of desmosine, isodesmosine, and isodesmosine-13C3,15N1. | ||
Although deuterated desmosine internal standards derived from natural products are commercially available, these standard compounds are not stable during the acid hydrolysis process required to dissociate desmosine and isodesmosine in tissues. As an isotopically labeled internal standard for LC-MS/MS analyses, a previous study utilized synthetic desmosine-d4, in which four deuterium atoms were incorporated into desmosine.13 Even though isotopically labeled internal standards are among the most favorable compounds for sensitive and accurate quantitative measurements,15 the synthetic desmosine-d4 isotopic standard contained impurities such as desmosine-d2, desmosine-d3,16,17 which led to inaccurate quantitation. Furthermore, heptafluorobutyric acid (HFBA), which is commonly used as an ion pair reagent in separations of compounds with similar polarity, inhibits MS ionization and also carries a risk of contamination.18 Therefore, the development of highly pure isotopically labeled internal standards would enable more-precise quantitation of desmosine and isodesmosine.
In the present study, we chemically synthesized isotopically labeled isodesmosine-13C3,15N1 (Fig. 1)19 via praseodymium-promoted Chichibabin pyridinium synthesis, as we reported the total synthesis of desmosine and isodesmosine.20,21 Using isotopically labeled isodesmosine-13C3,15N1 as an internal control, we established an isotope-dilution LC-MS/MS method. Additionally, the method developed in this work should be free from any ion-pair reagents for routine analysis of desmosines.
Cerebral stroke is a devastating cerebrovascular disorder composed of ischemic stroke (cerebral infarction) and hemorrhagic stroke (intracerebral hemorrhage and subarachnoid hemorrhage).22–24 Regardless of stroke subtype, cerebral stroke can result in death or disability, resulting in significant socio-economic burden worldwide.25 Despite the remarkable advances in treatment modalities for acute ischemic stroke, such as intravenous thrombolysis and endovascular thrombectomy, the indications of these treatments are limited to acute ischemic stroke due to their relatively small therapeutic windows and the specialized expertise required for endovascular thrombectomy.22,26 With regard to hemorrhagic stroke, surgical interventions are currently performed to reduce mass effects of hemorrhaging and prevent re-bleeding.27,28 However, no effective treatment is available to reduce secondary brain injuries resulting from cell death, inflammation, oxidative stress, vascular injuries, and brain edema.22,29,30 In particular, vascular injury caused by inflammation and oxidative stress is a primary mechanism of brain injury, as vascular injuries worsen brain edema and enhance inflammation.22,29,30 Although biomarkers of oxidative stress and inflammation have been developed, there are no useful biomarkers available to detect vascular injuries after acute cerebral stroke.31,32
In the present study, we hypothesized that products of extracellular matrix degradation would be useful biomarkers of vascular injury after acute cerebral stroke. Specifically, we hypothesized that quantification of desmosine and isodesmosine levels could be used to detect vascular injuries after acute cerebral stroke. The concentrations of desmosine and isodesmosine were measured in acute cerebral stroke patients and healthy controls using an established isotope-dilution LC-MS/MS method.
Results and discussion
Establishment of the isotopically labeled LC-MS/MS method
A Shimadzu LCMS-8030 plus system, which exhibits high sensitivity and good reproducibility, was used for LC-MS/MS analyses. The initial attempt to select an appropriate high-performance liquid chromatography (HPLC) column focused on waters symmetry C18 and YMC-Pack ODS-AM, which are typical octadecylsilane (ODS) columns. A gradient system of water-acetonitrile (MeCN) was utilized for the elution solvent. As trifluoroacetic acid is less and might have some effects on the MS instruments, 0.1% (v/v) formic acid (FA) was used to enhance both column retention and ionization during MS. However, the reproducibility of the desmosine and isodesmosine peaks was not acceptable. Alternatively, we used a Supelco Discovery HS F5-3 pentafluorophenyl (F5)-type column, which exhibits unique selectivity characteristics due to π–π stacking interactions with aromatic compounds, delocalized electron density induced by fluorine, and weaker hydrogen bonds.33 Using this column, as shown in Table S1,† the reproducibility of desmosine and isodesmosine peaks was improved using a water-acetonitrile gradient system with the addition of 0.1% (v/v) FA without an ion pairing reagent such as HFBA. However, chromatographic separation of desmosine and isodesmosine was not accomplished under any LC conditions.We then revised our strategy to optimize the tandem MS/MS method and establish appropriate equations for calculations. Major factors that can affect analytical sensitivity include selection of ions and the voltage of each MS compartment.34 Optimizations were performed for general parameters, including the voltage of MS compartments with flow injection without a column. The program began with determination of the precursor ion followed by product ion search, which enumerates desirable product ions. The precursor ion was set according to the molecular weight of desmosine. A protonated double-charged ion (m/z 263.25) was selected as the precursor ion, and product ions exhibiting high intensity were selected in the second step. Voltage optimization was performed for these precursor and product ions automatically.35
Based on observed m/z values and the structures of desmosine and isodesmosine, the structures of some fragment ions were estimated (Fig. S1†) using multi-reaction monitoring (MRM) mode. It should be noted that isodesmosine formed the same fragments, but the relative intensity of the ions enabled them to be distinguished from desmosine.36 The m/z 232.10 and 397.25 ions were clearly different; the m/z 232.10 ion was easier to detect than the m/z 397.25 ion from desmosine. However, in the case of isodesmosine, the area of the m/z 397.25 ion peak was larger than that of the m/z 232.10 ion peak. Although the 84.15 m/z ion peak exhibited the greatest area among the peaks of both desmosine and isodesmosine, it was not a favorable ion because peaks with a low m/z value are often associated with noise resulting from impurities such as peptides or plasticizers. Therefore, the product ions m/z 232.10 and 397.25 exhibiting high intensity formed from precursor ion m/z 263.65 were determined to be the best targets for detecting desmosine and isodesmosine. The optimization was also applied to isodesmosine-13C3,15N1 (Fig. S2†). Detected ions at m/z 265.65 and 401.25 corresponded to m/z 263.65 and 397.25 peaks of isodesmosine. Optimized MS/MS conditions for desmosine, isodesmosine, and isodesmosine-13C3,15N1 are summarized in Table S2.†
In order to analyze human plasma samples, calibration curves were drawn for fragment ions m/z 232.10 and 397.25 with isotopic internal standard isodesmosine-13C3,15N1. An example calibration sample (0.005 ppm) is shown in Fig. S3.† As shown in the MS chromatogram, two fragments (m/z 232.10 and 397.25) were clearly observed. In all samples, the retention times of the internal standard and isodesmosine exhibited good reproducibility at approximately 12 min. The peak area ratios between the isotopic standard and isodesmosine were calculated to draw calibration curves (Fig. S4 and S5†). In order to obtain greater accuracy, calibration curves were drawn for each sample group. Satisfactory Rr1 (correlation coefficient value) and Rr2 (coefficient of determination value) indicated that all calibration points were successfully analyzed. Compared with the curve for fragment ion m/z 232.10, fragment ion m/z 397.25 exhibited better accuracy in both stroke and control samples, as the function of fragment ion m/z 397.20 was closer to the zero point, and Rr2 was >0.999. Therefore, product ion m/z 397.25 was selected for the calibration of isodesmosine.
The reproducibility of calibration samples was confirmed using the m/z 397.25 ion (Tables 1 and S3†). The mean concentration indicates the concentration of a sample as predicated from the calibration curve. Standard deviation, relative standard deviation, and signal-to-noise (S/N) ratio were also calculated. The limit of quantitation (LOQ) was determined at S/N = 10 unless otherwise stated in Table S3.† According to the analytical results, the LOQ of isodesmosine based on the m/z 397.25 fragment was 0.005 ppm for stroke samples and 0.01 ppm for healthy control samples. This difference derived from instrument or column conditions, because the two calibration curves were drawn before the respective analyses, which were carried out on different days.
| Concentration of isodesmosine (ppm) | Area ratio | Mean concentration (ppm) | Area ratio SD | Area ratio RSD |
|---|---|---|---|---|
| a SD: standard deviation; RSD: relative standard deviation. | ||||
| 0.005 | 0.0631258 | 0.00917 | 0.0179 | 0.4218 |
| 0.0333533 | 0.00473 | |||
| 0.030962 | 0.00437 | |||
| 0.01 | 0.0705102 | 0.01027 | 0.0074 | 0.1094 |
| 0.0589324 | 0.00854 | |||
| 0.0726177 | 0.01059 | |||
| 0.02 | 0.139143 | 0.02052 | 0.0062 | 0.0456 |
| 0.128068 | 0.01886 | |||
| 0.138298 | 0.02039 | |||
| 0.05 | 0.31623 | 0.04695 | 0.0094 | 0.0290 |
| 0.32687 | 0.04853 | |||
| 0.335067 | 0.04976 | |||
| 0.1 | 0.696939 | 0.10377 | 0.0193 | 0.0285 |
| 0.675204 | 0.10052 | |||
| 0.658539 | 0.09804 | |||
Measurement of the plasma concentrations of desmosine and isodesmosine in acute stroke patients using isotope-dilution LC-MS/MS
A total of 17 plasma samples obtained from stroke patients (samples S1–S9) and healthy control subjects (samples C1–C8) were hydrolyzed and purified as previously reported.11–13 A comparison of the peak area ratios of desmosine and isodesmosine with the internal standard is shown in Table 2. Raw values for fragment ions m/z 232.10 and 397.25 are shown in the ESI (Tables S4 and S5†). Quantitative analysis of desmosine and isodesmosine was possible for all samples in which the peak area could be determined for both the m/z 232.10 and 397.25 channels. Therefore, peak areas for desmosine and isodesmosine were obtained for seven samples (S1–S3, S6, S8, S9, and C3). Four samples (S4, S5, S7, and C7) were detected in the m/z 397.25 channel, but they were below the LOQ due to lack of detection of the m/z 232.10 channel. Hence, the peak area ratio of desmosine and isodesmosine in these samples (S4, S5, S7, and C7) was above the limit of detection (LOD) but under the LOQ. In summary, the areas of desmosine and isodesmosine in all plasma samples from stroke patients were above the LOD. Quantitative analysis of desmosine and isodesmosine was thus possible for six of nine samples from stroke patients (S1, S2, S3, S6, S8 and S9). Among the eight healthy control group samples, the peak area ratio of desmosine and isodesmosine was above the LOD in two samples (C3 and C7). Quantitative analysis of desmosine and isodesmosine was only possible for one control sample (C3); the other five samples (C1, C2, C4, C5, and C8) were below the LOD.| Desmosine | Isodesmosine | Desmosine + isodesmosine | Comment | |
|---|---|---|---|---|
| a All values refer to mean area ratio obtained from calibration curves. S: stroke; C: control; LOQ: limit of quantitation; LOD: limit of detection; ND: not detected. | ||||
| S1 | 0.03701812 | 0.031877437 | 0.068895558 | |
| S2 | 0.014672097 | 0.047289901 | 0.061961998 | |
| S3 | 0.024437845 | 0.03809611 | 0.062533955 | |
| S4 | — | — | <LOQ | Fragment 232.15 was ND |
| S5 | — | — | <LOQ | Fragment 232.15 was ND |
| S6 | 0.036334632 | 0.017706312 | 0.054040945 | |
| S7 | — | — | <LOQ | Fragment 232.15 was ND |
| S8 | 0.02580029 | 0.030987129 | 0.056787419 | |
| S9 | 0.02564125 | 0.02081456 | 0.04645581 | |
| C1 | — | — | <LOD | Fragments 232.15/397.25 were ND |
| C2 | — | — | <LOD | Fragments 232.15/397.25 were ND |
| C3 | 0.015242721 | 0.031290451 | 0.046533172 | |
| C4 | — | — | <LOD | Fragments 232.15/397.25 were ND |
| C5 | — | — | <LOD | Fragments 232.15/397.25 were ND |
| C6 | — | — | <LOD | Fragments 232.15/397.25 were ND |
| C7 | — | — | <LOQ | Fragment 397.25 was ND |
| C8 | — | — | <LOD | Fragments 232.15/397.25 were ND |
Based on the peak area ratio, the concentration of desmosine, isodesmosine, and their total amount in plasma were calculated (Fig. 2 and 3, Table S6†). The relationship between the amount of desmosine and the patient's pathology was also confirmed. Plasma concentrations of desmosine and isodesmosine were elevated in stroke patients compared with healthy volunteers (0.05810 vs. 0.005817, unpaired t-test, P < 0.05. Fig. 3). In contrast, there were no obvious differences in desmosine concentration between ischemic versus hemorrhage stroke patients (representative cases are shown in Fig. 4).
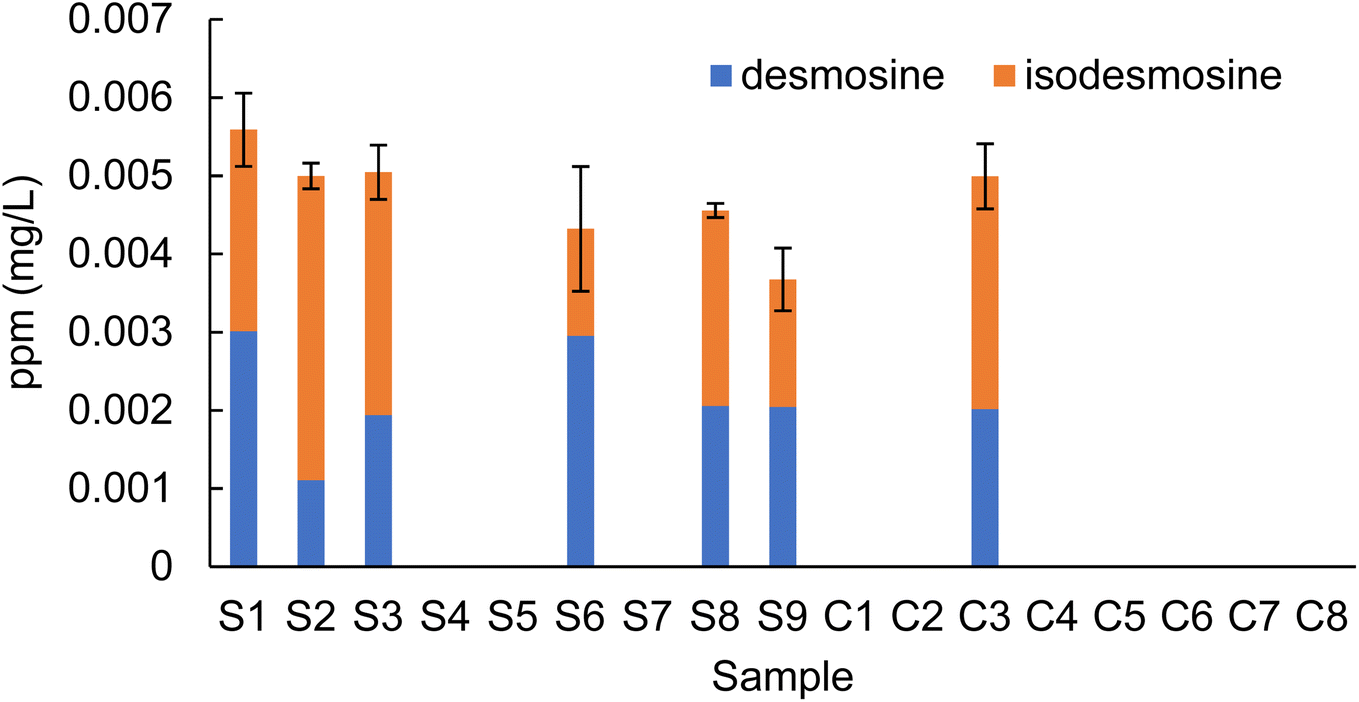 | ||
| Fig. 2 Concentrations of desmosine and isodesmosine in plasma (n = 3). S: stroke; C: control. | ||
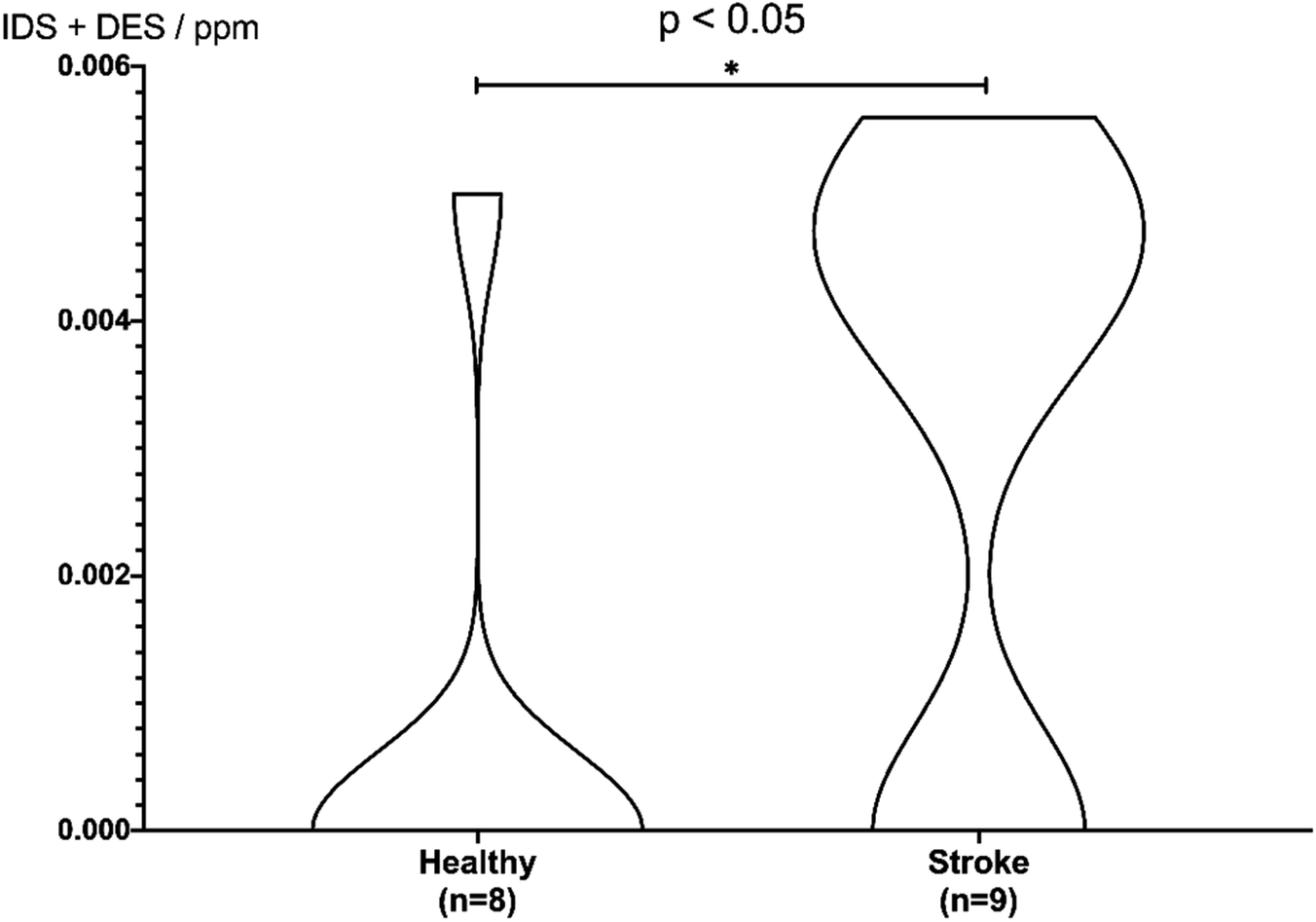 | ||
| Fig. 3 Violin plot for discrimination of stroke patients. Differences in concentrations of desmosine and isodesmosine were analyzed using a two-sided unpaired t-test. | ||
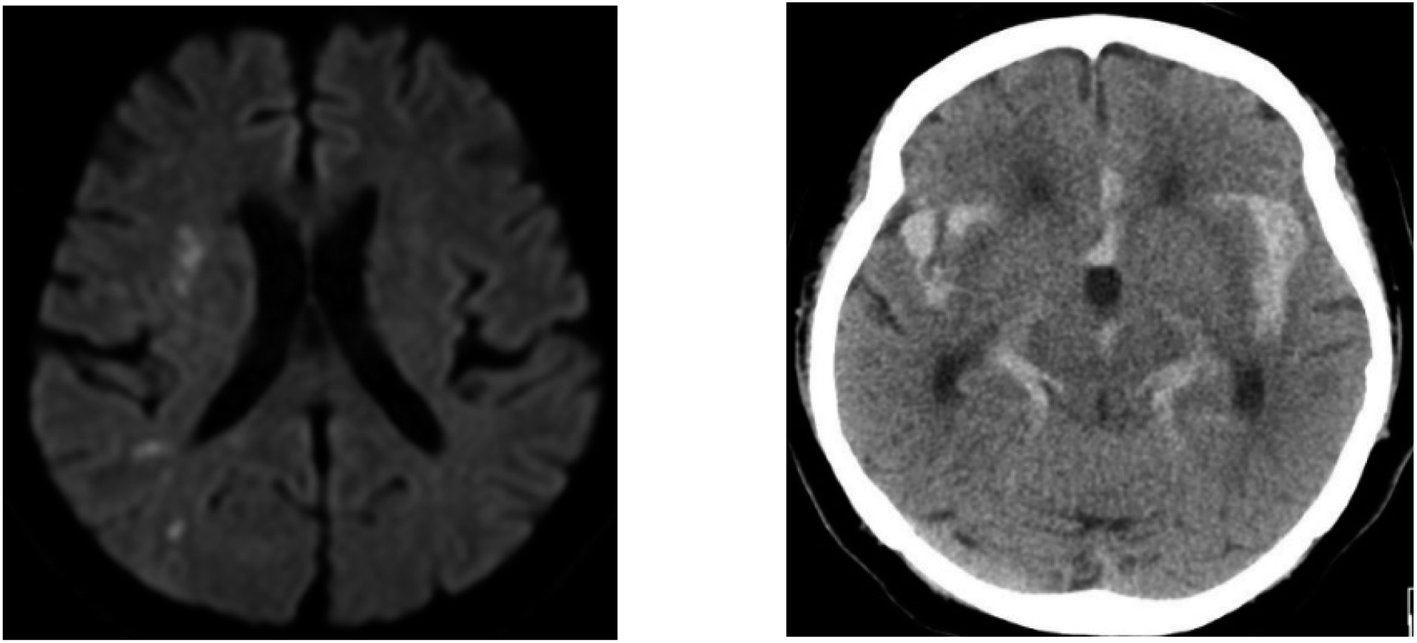 | ||
| Fig. 4 Representative cases of stroke. Left: a 55 year-old man (S6) with cerebral infarction containing 0.00432 ppm of desmosine. Diffusion-weighted magnetic resonance imaging demonstrated multiple high-intensity spots indicating small infarctions. Right: a 77 year-old woman (S9) with subarachnoid hemorrhage containing 0.00367 ppm of desmosine. Computed tomography revealed diffuse hemorrhaging in the basal cisterns. | ||
Several issues must be addressed to interpret desmosine and isodesmosine levels in plasma (Fig. 1 and 2). First, the number of plasma samples analyzed was small. Second, elevated plasma elastin levels are not solely derived from vascular injuries caused by acute cerebral stroke. A history of COPD and smoking habit can affect elastin dynamics. Plasma concentrations of desmosine and isodesmosine should therefore be carefully interpreted in patients who have a history of COPD or smoking habit. Finally, the optimal time point for measuring plasma elastin levels remains unclear. The progression of a brain injury depends on a variety of factors, such as stroke subtype and location of the stroke. Future studies including larger cohorts should be conducted to validate the findings of the present study.
Based on these results, we conclude that the plasma of stroke patients contains increased levels of desmosine and isodesmosine due to vascular injuries. The present research suggests that desmosine and isodesmosine could be useful as novel biomarkers for vascular injuries after acute cerebral stroke. Further studies should be conducted to validate the diagnostic value of desmosine and isodesmosine measurements for evaluating vascular injuries caused by acute cerebral stroke. This analytical method provided reliable quantitative measurement of desmosines in human plasma samples with good reproducibility.37
Conclusion
We developed a novel quantitative LC-MS/MS method to analyze desmosine and isodesmosine levels using isotopically labeled internal standard isodesmosine-13C3,15N1. We measured the concentrations of desmosine and isodesmosine in plasma collected from nine acute cerebral stroke patients and eight healthy controls. The results showed that plasma from acute cerebral stroke patients contains higher desmosine levels than plasma from healthy controls, suggesting the potential value of desmosine and isodesmosine as biomarkers for evaluating the extent of vascular injury after acute cerebral stroke.Materials and methods
Study population and clinical data collection
Stroke patients were prospectively recruited for this study between December 2018 and February 2019. Patients with a first-ever acute cerebral stroke whose onset was within 24 h of admission were considered for eligibility in the present study. The diagnosis of stroke was based on brain computed tomography or magnetic resonance imaging. The exclusion criteria were recent history of conditions involving elastic fiber defects such as COPD, asthma, bronchiectasis, interstitial lung disease, cardiovascular diseases, hypertension, diabetes, renal failure, and obesity.38 Patients with craniotomy, transient ischemic attack, subarachnoid hemorrhage, or functional disability before the index stroke were also excluded.Nine acute stroke patients were included in the present study. Eight healthy volunteers who had no previous stroke episodes and no remarkable lesions on magnetic resonance imaging were registered. Table 2 shows baseline demographic and other characteristics of acute stroke patients and healthy volunteers. This research was conducted in accordance with the Helsinki declaration. Research procedures were approved by the Institutional Review Board of Sendai Medical Center (approval number: 29-6). Written informed consent was obtained from each subject's family or relatives before inclusion in the study.
Plasma isolation
A total of 5 mL of peripheral blood was collected into a heparin-containing tube from each participant enrolled in this study. Plasma was isolated by centrifugation of peripheral blood at 1500×g at 4 °C for 10 min. Plasma samples were immediately frozen and preserved in a −80 °C freezer until further analysis.General experimental procedure
All LC-MS/MS analyses were conducted using a Shimadzu LC-20AD, DGU-20 A5R, SPD-20A, SIL-20AC, CBM-20A, CTO-20AC, and LCMS-8030 plus system. Nebulizer gas for MS/MS was provided using a N2 supplier model 24F SLP-07E-S53C (System Instruments, Tokyo, Japan). HPLC for separation was conducted on Shimadzu LC-6AD, DGU-20 A3R, SPD-20A, FCV-12AH, and FRC-10A instruments. The results were collected and analyzed using Shimadzu Labsolution software. Nebulizing gas was set at 2.0 L min−1; DL temperature was set at 250 °C; heat block temperature was set at 400 °C; drying gas flow was 15 L min−1; CID gas was set at 230 kPa; and ion gauge vacuum was stable at 1.72 × 10−3 Pa. Solvents or additives such as MeCN and FA were HPLC grade and purchased from commercial suppliers. Isodesmosine-13C3,15N1 was chemically synthesized in our previous research.10 All columns for HPLC separation were purchased from commercial suppliers, including the symmetry C18 (250 mm × 4.6 mm, Waters, MA, USA), YMC-Pack ODS-AM (150 mm × 4.6 mm, YMC, Kyoto, Japan), and Supelco Discovery HS F5-3 (3 μm, 150 mm × 2.1 mm, Sigma Aldrich, St. Louis, MO, USA). A Supelguard guard cartridge (3 μm, 2.1 mm, Sigma Aldrich) was used alongside the Supelco Discovery HS F5-3 column. Terumo syringes (1 mL) (Terumo, Tokyo, Japan) and Millex LH filters (0.45 μL) (Merck Millipore, MO, USA) were also used.Hydrolysis of plasma samples11–13
Analytical plasma sample (volume of each sample was shown in Table S7†) and 1 μL of isodesmosine-13C3,15N1 (100 ppm) were distributed into a small-scale vial with deionized water and an equal amount of 6 N HCl aq was added. The vial was tightly capped, then heated to 110 °C for 24 h without stirring. The color of plasma samples changed from clear pale yellow to brown with precipitation. These samples were then cooled to 60 °C, and HCl was completely evaporated by an air pump.Cellulose column preparation11–13
A column to purify hydrolyzed sample was prepared using 130 mg of 5% CF1 cellulose powder. A 3 mL mixture of 1-butanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetic acid:H2O (4:1:1 [v/v/v]) was prepared and then poured into the cellulose. The mixture was stirred for 24 h at room temperature to obtain a slurry. The slurry was charged into a syringe with a mesh cap and pressure was applied by pushing a pipette rubber to compress.
:acetic acid:H2O (4:1:1 [v/v/v]) was prepared and then poured into the cellulose. The mixture was stirred for 24 h at room temperature to obtain a slurry. The slurry was charged into a syringe with a mesh cap and pressure was applied by pushing a pipette rubber to compress.
Purification of an analytical plasma sample11–13
According to previous studies, 1 mL of premixed solution (1-butanol:acetic acid:deionized water; 4:1:1 [v/v/v]) was added in order to dissolve the sample and apply it to the prepared cellulose column. This procedure was repeated twice. After draining, the column was washed three times with 3 mL of pre-mixture followed by elution of desmosines using deionized water (3 mL). The water phase was collected and removed using an air pump at 60 °C for a few hours. Following this procedure, the sample was brought to 201 μL in an HPLC vial using deionized water and a Millex filter. Each sample was stored at 4 °C, and 20 μL of each sample was injected into the LC-MS/MS system using an auto-sampler and analyzed by the optimized method (Tables S1 and S2†).
Preparation of calibration samples
Synthetic isodesmosine samples (0.005, 0.01, 0.02, 0.05, 0.1 ppm) were prepared in 200 μL total volume, and 1 μL of isodesmosine-13C3,15N1 (100 ppm) was added to each sample and mixed. Samples were stored at 4 °C, and 20 μL of each sample was injected into the LC-MS/MS system using an auto-sampler and analyzed by the optimized method summarized in Tables S1 and S2.† The calibration curve was drawn twice: before analysis of the stroke samples (samples S1–S9) and before analysis of the control samples (samples C1–C8) in order to reflect instrument conditions.Optimization of LC-MS/MS conditions
An LCMS-8030 plus system was used for all LC-MS/MS analyses using a Supelco Discovery HS F5-3 (3 μm, 150 mm × 2.1 mm) column unless otherwise noted. HPLC conditions were set as mobile phase A (0.1% FA in MeCN) and B (0.1% FA in water) with a programmed linear flow rate to obtain sharp and reproducible peaks (Table S1†).MS/MS conditions were determined by precursor ion search followed by product ion search, which enumerates desirable product ions (Table S2†). The precursor ion was set according to the desmosine molecular mass (m/z = 526.3). Product ions were searched between m/z 80 and m/z 500 under conditions in which the minimum intensity was >1000 and ion tolerance was 0.5. After these processes, the precursor ion selected was m/z 263.65. For the m/z 263.65 ion, some product ions exhibiting high intensity were selected (m/z 232.10 and 397.25). As the MS/MS system uses a quadrupole analyzer, the voltages of Q1, Q3, and collision energy were also optimized. Conditions for the analysis of isodesmosine-13C3,15N1 (m/z = 530.3) were also determined as done for desmosine.
Quantitation was performed in MRM mode for desmosines using isodesmosine-13C3,15N1 as the internal standard. The optimized HPLC and MS/MS conditions were utilized for all analyses. Dwell time (400 ms for internal standard, 50 ms for desmosines) and pause time (1.0 ms) were newly optimized for LC-MS/MS analyses.
Calculations and data processing
In this study, a calculation step was required to determine the concentrations of desmosine, isodesmosine. This calculation would help discriminate inseparable desmosine, isodesmosine and obtain the isodesmosine peak area.Parameters given by analysis of clinical samples:
| A = [area ratio of 232.10 (in clinical sample)] |
| B = [area ratio of 397.25 (in clinical sample)] |
Constants (determined by analysis of a synthetic sample):
| Cda = [desmosine's area of 232.10] |
| Cdb = [desmosine's area of 397.25] |
| Cia = [isodesmosine's area of 232.10] |
| Cib = [isodesmosine's area of 397.25] |
Unknowns:
| Da = [area ratio of 232.10 (amount of desmosine in clinical sample)] |
| Db = [area ratio of 397.25 (amount of desmosine in clinical sample)] |
| Ia = [area ratio of 232.10 (amount of isodesmosine in clinical sample)] |
| Ib = [area ratio of 397.25 (amount of isodesmosine in clinical sample)] |
Eqn (1)–(6) below were established for each sample group (stroke or control) and referred the same calibration curves and corresponding instrument conditions. The mixed raw peak contained desmosine and isodesmosine, as indicated in eqn (1) and (2).
| A = Da + Ia | (1) |
| B = Db + Ib | (2) |
Based on the theory and reproducibility of the LC-MS/MS instrument, for each compound, the area ratio of the product ions should be stable under the same instrument conditions, as shown in eqn (3) and (4).
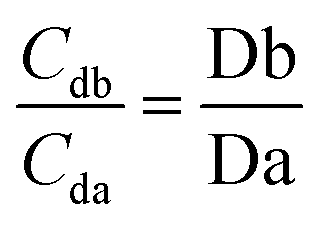 | (3) |
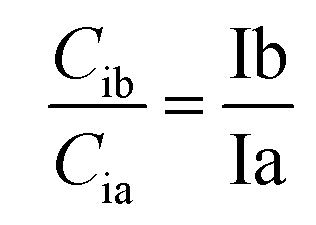 | (4) |
When the concentrations of desmosine and isodesmosine are the same, eqn (5) and (6) can be obtained.
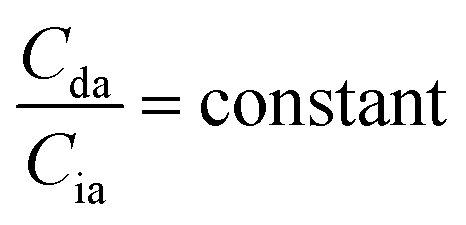 | (5) |
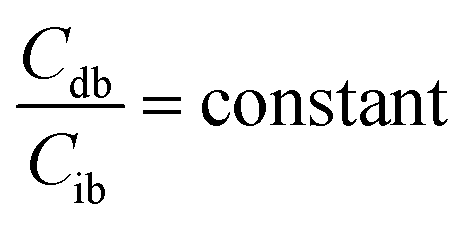 | (6) |
The constants (Cda, Cdb, Cia, and Cib) were determined by analysis of synthetic desmosine and isodesmosine (Table S8†). Each sample was prepared to 0.01 ppm, and the resulting peak area was obtained for the m/z 232.10 and 397.25 ions. The constants were obtained separately for stroke and control samples, similar to the calibration curves.
Based on these equations, Ib and Db were calculated as shown below. From eqn (3), eqn (1) was assigned as follows.
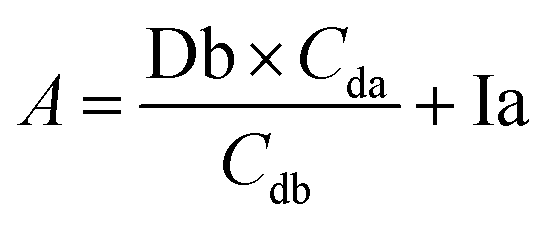
Based on (4),
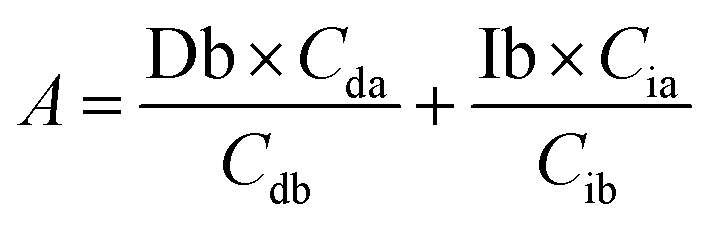
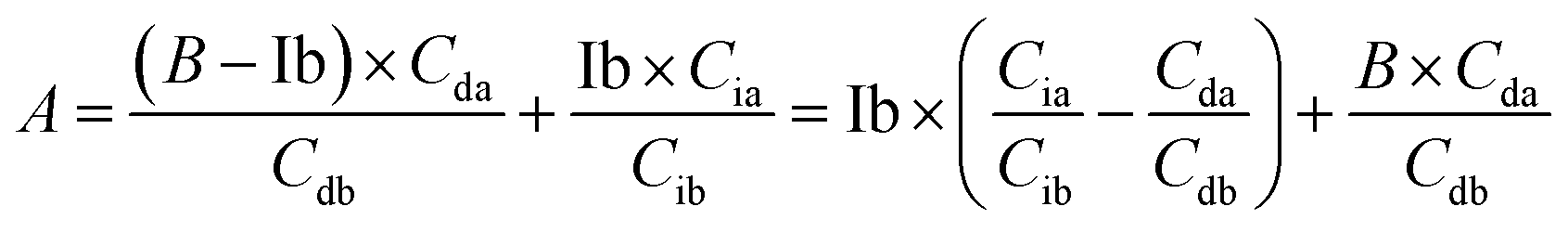
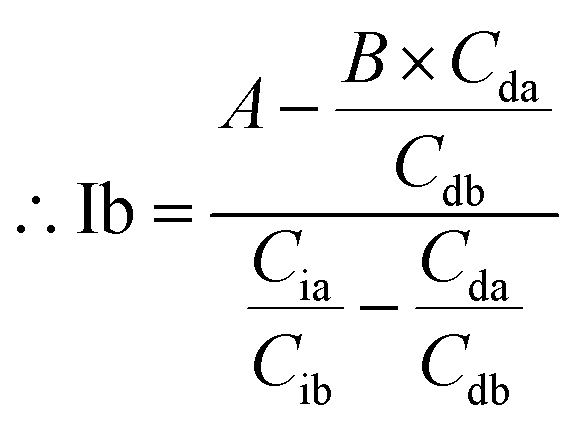
The area of desmosine should be modified to apply the calibration curve drawn for isodesmosine. Therefore, the area ratio of desmosine was obtained from Ib and converted into an area ratio of the same concentration of isodesmosine using eqn (6).
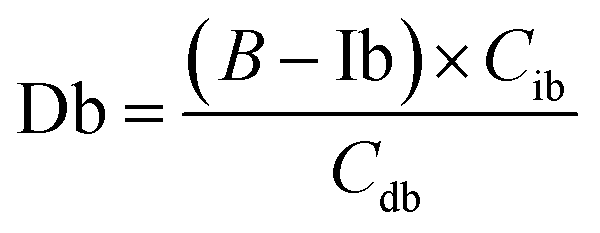
The equations for desmosine and isodesmosine are summarized in eqn (7) and (8). The concentrations of isodesmosine (Ib), desmosine (Db), and their total concentration (Ib + Db) were determined from the calibration curve showing the relationship between isodesmosine's peak area ratio and its concentration compared with the internal standard. The calculated total level, which was determined based on the peak area of the corresponding sample, was above the LOQ for each sample.
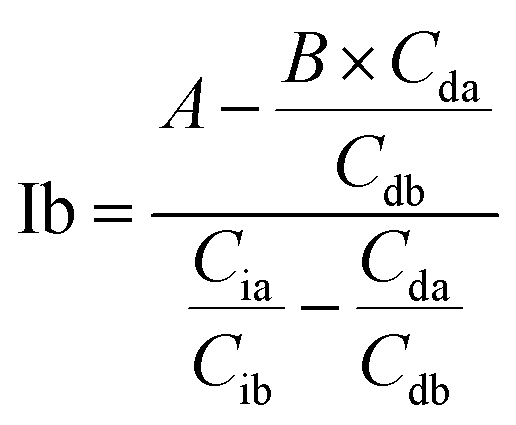 | (7) |
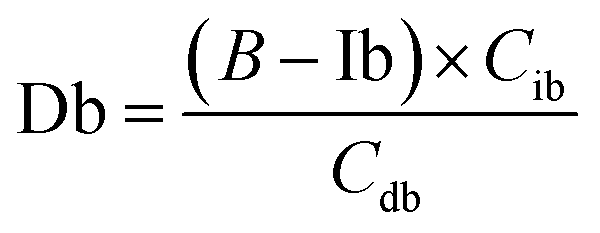 | (8) |
The concentration obtained by the calibration curve was converted into the concentration in plasma. The original volume of plasma samples is noted in Table S7.† Because each synthetic isodesmosine sample for calibration (200 μL) was diluted to 201 μL, precise concentration should be considered. Also, all plasma samples were diluted by the addition of internal standard (1 μL) and then concentrated to 201 μL. Therefore, the original concentration in plasma can be described by eqn (9). It should be noted that according to the isotope-dilution method, the concentration of internal standard was set as the same (1 μL of 100 ppm internal standard was contained in the 201 μL sample) between the calibration and plasma samples.
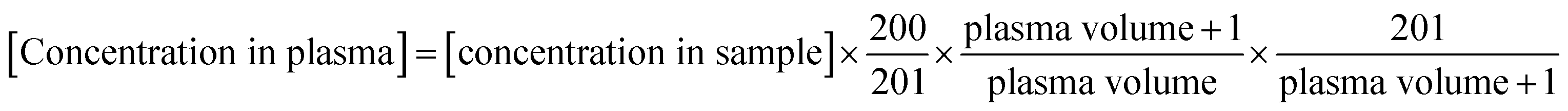 | (9) |
To confirm the discrimination of total concentration, stroke and control sample data were analyzed using an unpaired t-test (Fig. 3). Differences were evaluated using a two-sided test with an alpha level of 0.05. Samples S4–S5, S7, C1–C2, and C4–C8, which could not be quantified, were regarded as 0 ppm. Consequently, as the difference between plasma and healthy samples met the significance level of 5% (P = 0.027), stroke patients could be discriminated from healthy subjects based on the total concentration of desmosine and isodesmosine.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA, USA). A two-sided unpaired t-test with an alpha level of 0.05 was conducted to evaluate the difference in plasma desmosine and isodesmosine levels between stroke patients and healthy controls. Samples S4–S5, S7, C1–C2, and C4–C8, which could not be quantified, were regarded as 0 ppm.Author contributions
A.M. established the LC-MS/MS methods and analyzed clinical samples; A.M., R.T., Tomoo I., K.N., and T.U. contributed to manuscript preparation; R.A. analyzed control samples by LC-MS/MS; A.I. conducted preliminary LC-MS/MS experiments; T. Tanigawa prepared the isotopically labeled compound; Tomoo I., Tomohisa I., Takashi I. collected clinical samples of stroke patients; Tomoo I., K.N., T. Tominaga, and T.U. supervised the work; T.U. handled the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) through a KAKENHI grant (JP19K09498 to T. U.). Fellowships (to A. M.) from Yoshida Scholarship Foundation and JSPS are also acknowledged. We thank Ms. Manami Kobayashi (Shimadzu Co., Ltd) for valuable suggestions. Tomoo I. and T. U. thank the Kogyokusha School (Shinagawa, Tokyo, Japan).Notes and references
- J. Rosenbloom, W. R. Abrams and R. Mecham, FASEB J., 1993, 7, 1208–1218 CrossRef CAS.
- L. Debelle and A. M. Tamburro, Int. J. Biochem. Cell Biol., 1999, 31, 261–272 CrossRef CAS.
- S. M. Partridge, D. F. Elsden and J. Thomas, Nature, 1963, 197, 1297–1298 CrossRef CAS.
- J. Thomas, D. F. Elsden and S. M. Partridge, Nature, 1963, 200, 651–652 CrossRef CAS PubMed.
- M. Boutin, C. Berthelette, F. G. Gervais, M.-B. Scholand, J. Hoidal, M. F. Leppert, K. P. Bateman and P. Thibault, Anal. Chem., 2009, 81, 1881–1887 CrossRef CAS PubMed.
- T. Miliotis, C. Lindberg, K. F. Semb, M. van Geest and S. Kjellstrom, J. Chromatogr. A, 2013, 1308, 73–78 CrossRef CAS PubMed.
- O. Albarbarawi, A. Barton, D. Miller, C. McSharry, R. Chaudhuri, N. C. Thomson, C. N. A. Palmer, G. Devereux and J. T.-J. Huang, Bioanalytical, 2013, 5, 1991–2001 CAS.
- J. Lamerz, A. Friedlein, N. Soder, P. Cutler and H. Dobeli, Anal. Biochem., 2013, 436, 127–136 CrossRef CAS PubMed.
- S. Ongay, G. Hendriks, J. Hermans, M. van den Berge, N. H. T. ten Hacken, N. C. van de Merbel and R. Bischoff, J. Chromatogr. A, 2014, 1326, 13–19 CrossRef CAS PubMed.
- S. Ma, S. Lieberman, G. M. Turino and Y. Y. Lin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12941–12943 CrossRef CAS PubMed.
- S. Ma, Y. Y. Lin and G. M. Turino, Chest, 2007, 131, 1363–1371 CrossRef CAS PubMed.
- S. Ma, G. M. Turino and Y. Y. Lin, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1893–1898 CrossRef CAS PubMed.
- S. Ma, G. M. Turino, T. Hayashi, H. Yanuma, T. Usuki and Y. Y. Lin, Anal. Biochem., 2013, 440, 158–165 CrossRef CAS PubMed.
- J. T.-J. Huang, R. Chaudhuri, O. Albarbarawi, A. Barton, C. Grierson, P. Rauchhaus, C. J. Weir, M. Messow, N. Stevens, C. McSharry, G. Feuerstein, S. Mukhopadhyay, J. Brady, C. N. A. Palmer, D. Miller and N. C. Thomson, Thorax, 2012, 67, 502–508 CrossRef PubMed.
- J. Wieling, Chromatographia, 2002, 55, S107–S113 CrossRef CAS.
- R. Suzuki, H. Yanuma, T. Hayashi, H. Yamada and T. Usuki, Tetrahedron, 2015, 71, 1851–1862 CrossRef CAS.
- D. Watanabe, R. Suzuki and T. Usuki, Tetrahedron Lett., 2017, 58, 1194–1197 CrossRef CAS.
- F. Gosetti, E. Mazzucco, D. Zampieri and M. C. Gennaro, J. Chromatogr. A, 2010, 1217, 3929–3937 CrossRef CAS PubMed.
- T. Tanigawa, A. Komatsu and T. Usuki, Bioorg. Med. Chem. Lett., 2015, 25, 2046–2049 CrossRef CAS PubMed.
- T. Usuki, T. Sugimura, A. Komatsu and Y. Koseki, Org. Lett., 2014, 16, 1672–1675 CrossRef CAS PubMed.
- N. Tanaka, M. Kurita, Y. Murakami and T. Usuki, Eur. J. Org. Chem., 2018, 21, 6002–6009 CrossRef.
- B. C. V. Campbell, D. A. De Silva, M. R. Macleod, S. B. Coutts, L. H. Schwamm, S. M. Davis and G. A. Donnan, Nat. Rev. Dis. Primers, 2019, 5, 70 CrossRef PubMed.
- R. L. Macdonald and T. A. Schweizer, Lancet, 2017, 389, 655–666 CrossRef.
- C. Cordonnier, A. Demchuk, W. Ziai and C. S. Anderson, Lancet, 2018, 392, 1257–1268 CrossRef.
- G. J. Hankey, Lancet, 2017, 389, 641–654 CrossRef.
- W. H. Powers, A. A. Rabinstein, T. Ackerson, O. M. Adeoye, N. C. Bambakidis, K. Becker, J. Biller, M. Brown, B. M. Demaerschalk, B. Hoh, E. C. Jauch, C. S. Kidwell, T. M. Leslie-Mazwi, B. Ovbiagele, P. A. Scott, K. N. Sheth, A. M. Southerland, D. V. Summers and D. L. Tirschwell, Stroke, 2019, 50, e344–e418 CrossRef PubMed.
- R. L. Macdonald and T. A. Schweizer, Lancet, 2017, 389, 655–666 CrossRef.
- C. Cordonnier, A. Demchuk, W. Zhai and C. S. Anderson, Lancet, 2018, 392, 1257–1268 CrossRef.
- R. F. Keep, Y. Hua and G. Xi, Lancet Neurol., 2012, 11, 720–731 CrossRef CAS PubMed.
- A. A. Rabinstein, Lancet Neurol., 2011, 10, 593–595 CrossRef.
- G. J. L. Ng, A. M. L. Quek, C. Cheung, T. V. Arumuga and R. C. S. Seet, Neurochem. Int., 2017, 107, 11–22 CrossRef CAS PubMed.
- A. Simpkins, M. Janowski, H. S. Oz, J. Roberts, G. Bix, S. Dor and A. M. Stowe, Transl. Stroke Res., 2020, 11, 615–627 CrossRef PubMed.
- Sigma Aldrich (Millipore Sigma) HPLC Discovery HS F5 (Nov/2/2021), https://www.sigmaaldrich.com/analytical-chromatography/hplc/columns/discovery-hplc/hs-f5.html.
- H. J. Gross, Mass Spectrometry: A Textbook, Springer 2017 Search PubMed.
- Shimadzu Application Data Sheet No. 98 Automatic Optimization of Transitions and Collision Energies (Nov/2/2021), https://solutions.shimadzu.co.jp/an/n/en/gcms/jpo214019.pdf.
- C. U. Schräder, A. Heinz, P. Majovsky and C. E. H. Schmelzer, J. Am. Soc. Mass Spectrom., 2015, 26, 762–773 CrossRef CAS PubMed.
- M. Hirose, T. Kobayashi, N. Tanaka, A. Mikagi, H. Wachi, Y. Mizutani and T. Usuki, Bioorg. Med. Chem., 2021, 52, 116519 CrossRef CAS PubMed.
- A. J. Cocciolone, J. Z. Hawes, M. C. Staiculescu, E. O. Johnson, M. Murshed and J. E. Wagenseil, Am. J. Physiol. Heart Circ. Physiol., 2019, 315, H189–H205 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and data. See DOI: https://doi.org/10.1039/d2ra06009d |
| This journal is © The Royal Society of Chemistry 2022 |