
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSERS combined with PCR as a potent tool for detecting mutations: a case study of tomato plants†
Samyabrata Sen‡
 ab,
Divya Chalapathi‡ab,
Jayaprakash Targollic and
Chandrabhas Narayana*ab
ab,
Divya Chalapathi‡ab,
Jayaprakash Targollic and
Chandrabhas Narayana*ab
aChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bengaluru, India. E-mail: cbhas@rgcb.res.in
bSchool of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research, Bengaluru, India
cBiodecipher, Bengaluru, India
First published on 15th December 2022
Abstract
Conventional methods of detecting economically essential mutations have several disadvantages. Even though fluorescence-based methods are still the best option, Surface-Enhanced Raman Spectroscopy (SERS) may soon emerge as an alternative to the current techniques for detecting these mutations, because of its ability to detect molecular vibrational signatures. We were able to identify and develop a PCR-based SERS assay that can relentlessly differentiate between different types of indels, and SNPs as demonstrated in the case of tomato genome related to tomato yellow leaf curl virus and root-knot nematodes, diseases that are economically significant to the global agriculture industry and where the selection of resistant crops is the best solution. This tri-primer assay utilizes mutation-specific forward primers and SERS probes tagged with FAM and Cy3 dyes, specific for each allele of a particular gene (Ty-3 and Mi-1). The unique Raman spectral features of these dyes enabled to perform of multiplexing, which made it possible to detect not only the indel type but also the zygosity in a single experiment. Moreover, this technique successfully differentiated between two different SNP-based alleles. Therefore, due to its efficient multiplexing capability and lack of the need for quenchers, it has the potential to become a powerful onsite and offsite screening tool in the not-too-distant future.
Introduction
Although it has been demonstrated extensively that mutations can cause various types of cancer in humans and animals,1–5 occasionally, mutations like indels and Single Nucleotide Polymorphisms (SNPs) allow some varieties of crops to exhibit resistance against specific forms of pathogens due to their unique genes, and plants lacking these genes, therefore become susceptible to infection. Thus, screening resistant crops are one of the most effective methods to counter the threats posed by plant pathogens, the primary culprits against the agricultural industry which cause extensive damage to various economically significant crops.Fluorescence-based techniques have proven to be superior to many gel-based methods that were employed previously.6 FRET (Förster resonance energy transfer) techniques and molecular beacons have emerged as dependable assays for SNP and indel detection. Even though these are the best techniques available today, they too are not devoid of drawbacks, especially when it comes to multiplexing, which implements multiple probes for the detection of multiple types of mutations at the same time. The limited availability of these reporter–quencher combinations causes a hindrance in their multiplexing capability along with the costs for tagging both dyes. Moreover, the detection system is often monochromatic, which results in non-uniform fluorescence of different dyes, thus restricting the number of probes that can be utilized.7
Raman spectroscopy can identify molecules based on their vibrational fingerprints, which gets around most problems with fluorescent approaches. It relies primarily upon the inelastic scattering of photons. Surface-enhanced Raman spectroscopy (SERS) is a technique in which the Raman signal is multiplied when a Raman reporter is near a nanostructured metal surface. There is no need for a quencher in this method, which adds to its cost-effectiveness compared to the dual-labeled probes used in fluorescence-based PCR methods. We have successfully used this to create a dependable detection system in which mutations can be identified and differentiated in similar time scales. To demonstrate the assay, we chose tomato alleles in the Ty-3 and Mi-1 genes resistant to or susceptible to tomato yellow leaf curl virus (TYLCV) and root-knot nematodes, respectively, owing to their infamy in agribusiness.
Tomato yellow leaf curl virus (TYLCV) is considered one of the most destructive types of viruses8 that can cause complete yield loss in the tomato (Solanum lycopersicum) crop.9 This disease is transmitted among the most important culinary vegetables in the world, by the Silverleaf whitefly (Bemisia tabaci), a major pest for the Solanaceae family. Characteristic manifestation of TYLCV disease includes extreme hindering of plant height, a decrease in leaf size, curling of leaves, chlorosis on leaves and flowers, and reduction of the production of fruits.10 It has been assessed that around 7 million hectares can encounter TYLCV contamination or mixed viral infections yearly.11 Plants, in turn, use a variety of defense mechanisms to shield themselves from viral intrusion like RNA silencing.12 Besides TYLCV, infection of root nematodes is a severe problem faced by tomato cultivators worldwide.13 They cause decreased yield in adult crops but are lethal to young plants, being responsible for approximately 5% of global crop loss.14 The Mi-1 gene comes into play during this nematode infection, as it confers resistance against three common root-knot nematodes, i.e., Meloidogyne incognita, M. javanica, and M. arenaria.15
SERS as a technique has recently become an extremely dependent detection assay16 for its ability to identify analytes down to the single molecular level17,18 not only in the field of biology but also for the screening of toxic industrial chemicals and explosives19,20 and to study electrochemistry at a molecular level.21 SERS has recently made a splash on the biology stage as well. It has been used in imaging live cells and tissues because of its non-destructive nature. SERS has also been demonstrated to be valuable for recognizing proteins, antigens, and infections like Salmonella, Giardia, and Cryptosporidium.22 SERS biosensors are also being utilized in the successful and accurate detection of a variety of diseases including different types of cancers,23,24 Alzheimer's disease,25 and Parkinson's disease.26 Recently SERS and PCR have been used in conjunction to detect SAR Cov2 (ref. 27) and Influenza A virus.28 In plants, SERS has been wielded for the detection of plant hormones29 and pathogen detection in the early stages of the disease.30 SERS assays have also been invented which can detect specific bacterial DNA sequences.31–34 Mandrile et al. have designed a non-destructive Raman spectroscopic assay that is capable of detecting the DNA of the tomato yellow leaf curl sardinia virus (TYLCSV) and tomato spotted wilt virus (TSWV) in the very early stages of infection, before the symptoms are visible, in artificially inoculated tomato plants.30 The benefits of exploring SERS for DNA detection include the requirement for low DNA quantities, quick scan times, and the ability to multiplex with a variety of dyes due to the low full width at half maximum (FWHM) of the characteristic peaks.
However, few studies on the complex DNA of higher organisms have been conducted. The development of a SERS-based strategy for identifying the DNA of eukaryotes like plants and animals could pave the way for early disease detection in the world of diagnostics. SERS has already been shown to be an effective technique for identifying significant mutations,35 particularly mutations linked to colorectal cancer36 in human cell lines. Strategies for animals and humans must go through extensive trial and testing, as well as numerous ethical clearances before they can be converted into diagnostic tools. Given the high demand for agricultural products, the technology transfer of a strategy for DNA detection in plants is comparatively easier and very important.
Our SERS-based PCR assay aims to screen resistant plants and weed out susceptible plants to help the plant breeding program long before an infection occurs, allowing us to stop breeding that set of plants and provide resistant seeds to farmers. Our method uses the plant's innate genetic ability to prevent virus attack and distinguishes between homozygous and heterozygous, resistant, and susceptible tomato alleles. Multiplexing has also been accomplished, which makes it easier to detect mutations in unknown DNA sequences. This technique shows promise and, with further refinement, could be used to detect mutations in animal DNA as well.
Materials and methods
Materials
Spermine tetrahydrochloride (N,N′-bis(3-aminopropyl)-1,4-butanediamine tetrahydrochloride) (ST) and silver nitrate (AgNO3, 99.9999% trace metals basis) from Sigma Aldrich; molecular biology grade water from Himedia and TaqMan™ Genotyping Master Mix from Thermo Fisher Scientific.Oligonucleotide design
The genome of Solanum lycopersicum was downloaded from NCBI and the locations of the mutations were obtained with the help of previously identified markers from Kim et al. in the case of Ty-3 indels37 and the case of Mi-1 indels15 and Jung et al. in the case of Ty-3 SNP.38 The sequences of the probes and primers were then designed manually (Tables S1–S3†). They were ordered primarily from integrated DNA technologies.DNA samples
The tomato seeds were germinated, and the leaves were harvested for genomic DNA isolation. When the plants reached the 3rd or 4th leaf stage, the DNA samples were extracted via the CTAB method. The concentration of the DNA was adjusted to 50 ng μL−1. The zygosity and the resistance/susceptibility of the samples were analyzed via a gel-based method. Fig. S3a and S3b† show the gel data, indicating the corresponding alleles to the indels in the Ty-3 and Mi-1 plants respectively. Escherichia coli plasmid DNA (XL-10 Gold), used as a negative control, was obtained via alkaline lysis protocol.Polymerase chain reactions
Each reaction tube contained 12.5 μL TaqMan™ Genotyping Master Mix and 2.5 μL of DNA (50 ng μL−1). For single detection, 2 μL (2.5 μM) forward primers (FP) and reverse primers (RP) each, and 1 μL (2.5 μM) designed SERS probes were added. For multiplex reactions, 2 μL of both the FPs as well as 1 μL of both the SERS probes were added. Molecular biology grade water was used to make up the total volume of 25 μL. The reactions were carried out in T100 Thermal Cycler (Bio-Rad Laboratories, Inc). The following was the cycling protocol: 15 minutes at 94 °C for hot start activation, then, 30 repeats of 45 s at 94 °C, 60 s at 62 °C and 60 s at 72 °C, followed by a 60 s final extension at 72 °C. After the PCR completion, the samples were gradually cooled to 25 °C.SERS
After the PCR was complete, 2.5 μL from each reaction tube was added to 125 μL of 1× phosphate buffer saline (PBS). To it, 10 μL of 400 μM or 0.01 M ST was added and incubated for 6 min.The silver nanoparticles (AgNPs) for the SERS experiments were synthesized using the Lee Meisel method39 and characterized using UV-visible absorption spectroscopy and TEM. The synthesized AgNPs were concentrated to 20×, in a micro-centrifuge (MiniSpin, Eppendorf) at 7200 rpm for 10 min 8 μL of the concentrated AgNPs was added to 2 μL of the incubated PCR product. The Raman spectra of this mixture were collected in liquid mode using a 532 nm (2.3 mW) incident laser, through an 1800 grooves per mm diffraction grating with an acquisition time of 10 s per window, on the LabRam HR Evolution (Horiba, Ltd). The obtained Raman spectra were baseline corrected using a user-defined baseline. The required Raman peaks were fit using a Lorentzian curve and the statistical distribution of intensities was mapped using box plots.
Results and discussion
The main principle of our strategy lies in the fact that the DNA binds well to a complementary region in the other strand as compared to a random sequence. We created a customized forward primer that is very specific to the target and has a tail sequence that is distinct and not complementary to the tomato genome. Only in the presence of the target will this tail sequence and its complement become amplified. The technique also required the creation of a unimolecular probe with a bottom oligo whose sequence is similar to the tail of the forward primer and an upper oligo that is smaller and bound to a Raman active dye. In the absence of a target and subsequent complementary forward primer tail sequence, these two components, which are joined by a HEG spacer (hexaethylene glycol), hybridize, shutting the probe and leaving the dye buried, resulting in a weaker Raman signal. By generating a Tm difference of 15 °C, the bottom oligo portion has been tailored to have a greater affinity for the tail complementary sequence than the smaller oligo. When the target DNA is present, increased selective interaction with the probe frees the dye molecule to allow for an enhanced Raman signal (Fig. S1†).As seen in Fig. 1, the steps involved in the PCR followed by SERS are represented schematically. After the denaturation step of PCR, the allele-specific FP and the RP hybridize with their specific targets during the annealing phase. The tail of the FP fails to bind to the DNA, but the other part of it gets extended in the next step. In the second round of PCR, the complementary region of the tail is created by the extension of the RP. In the third round of the PCR, the SERS probe binds with the tail complementary region allowing the dye-tagged part to become free. So, once the probe binds to the target, the dye becomes accessible to the AgNPs during SERS detection of the dye. However, since both the DNA backbone and the AgNPs are negatively charged, detection of the dye signal becomes difficult due to their mutual repulsion. ST neutralizes the charge of the DNA backbone, allowing the nanoparticles to approach the dye and generate a stronger SERS signal.40,41 ST also aggregates nanoparticles, thus adding to our advantage of increased SERS hotspots, thus showing an improved SERS signal.42,43 Our study was carried out using two different concentrations of ST, 400 μM and 0.01 M. These two concentrations were the extreme values provided by Graham et al. that could be used to properly differentiate the presence and absence of a sequence through SERS.31
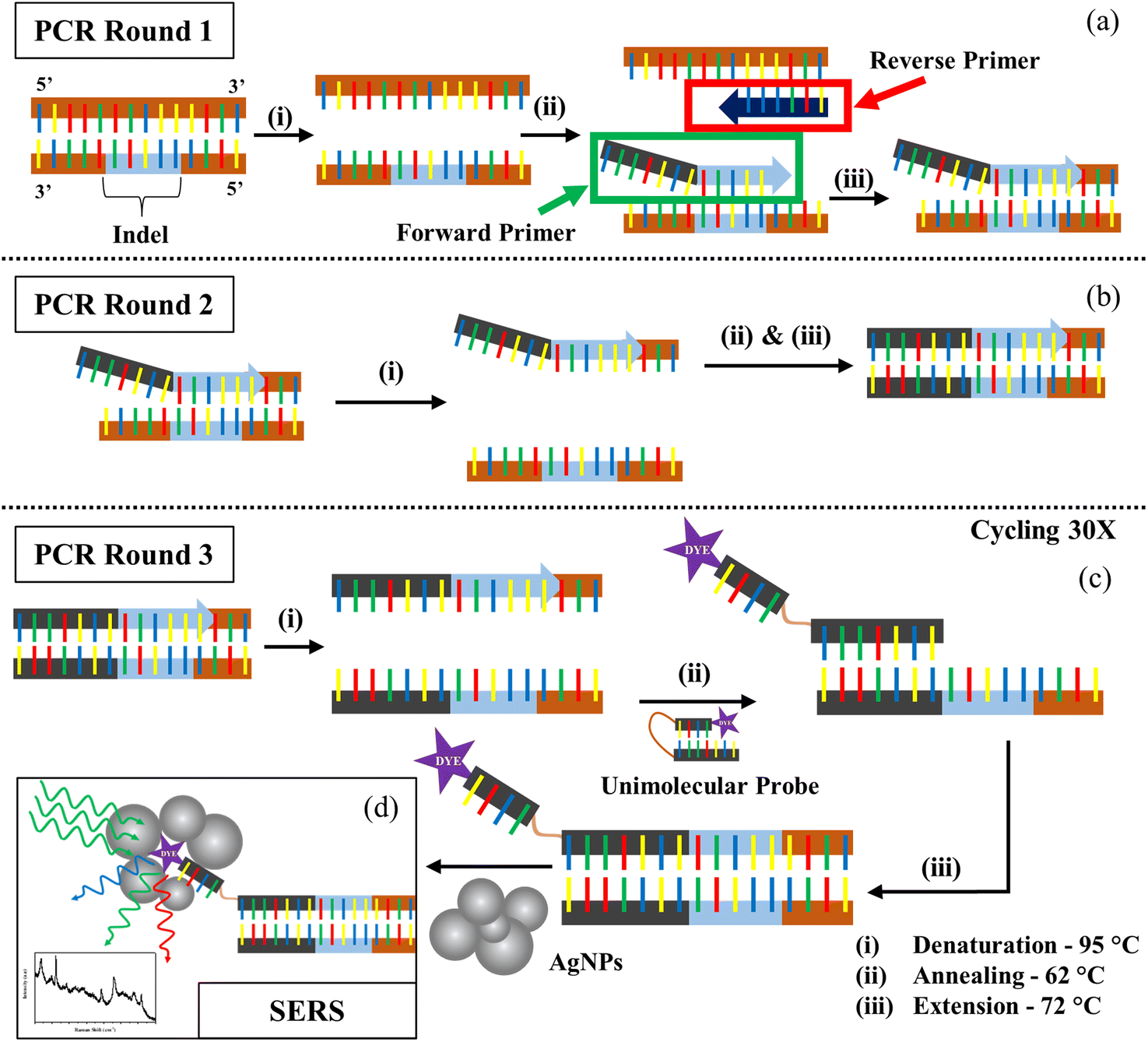 | ||
| Fig. 1 Schematic representation of the PCR-based SERS strategy. (a) Annealing of mutation-specific FP and RP, with the non-complementary tail of the FP failing to anneal. (b) Formation of the unique, complementary region of the hanging tail. (c) SERS probe binds to its target, the complementary region of the tail. (d) Increased Raman signal from the exposed dye-tagged region of the SERS probe. | ||
Many standard chromophore dyes can be used to label the oligos for SERS detection, including HEX, TAMRA, R6G, ROX, Cy3, Cy5, FAM, TET, etc.44–46 These molecules have good Raman scattering cross-sections and hence can produce good Raman signals in short scan intervals. During SERS, as the fluorophore is close to a metallic surface, the fluorescence gets quenched, resulting in a good-quality Raman spectrum with no fluorescent background. These dyes have a distinct number of well-separated peaks that can be assigned to various vibrational modes of the molecule. The narrow FWHM of the peaks in their Raman spectra easily facilitates exploring multiplexing options using a single excitation laser. We chose the two dyes with minimum peak overlaps in their spectra for our studies: FAM(6-carboxyfluorescein) and Cy3(cyanine-3). We designed the unimolecular probes to have a FAM tag for detecting the DNA having the insertion indel, and the Cy3-tag for the deletion indel. As seen in Fig. S2,† the FAM and Cy3 molecules have distinct Raman spectral signatures. The 1636 cm−1 peak originating from the Xanthene ring's C–C stretching47 is a unique spectral feature, selected for the analysis of FAM-tagged oligos. Similarly, for the Cy3 tagged oligos, the 1589 cm−1 peak corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch48 was used.
N stretch48 was used.
The synthesized AgNPs had a plasmon peak around 434 nm (Fig. S3a†) predicting an average size range of 50–70 nm and zeta potential of −13.3 mV (Fig. S3b†), confirming a citrate cap on them. The TEM images indicate the general size and shape distribution of the AgNPs (Fig. S3c and S3d†). The oligo sequences corresponding to the FP and RP, insertion, and deletion-specific unimolecular probes that were designed and used to conduct the PCR experiments are given in Table S1.†
Before conducting the SERS experiments, the Raman spectra of the controls were obtained to be assured of no background peaks overlapping the dye's unique spectral peaks (Fig. S5†). The many SERS spectra collected over 3 experiments prove consistency and their averages provided a consolidated picture. For the FAM-tag probe mixtures, we obtained the intensity of the 1636 cm−1 peak through a Lorentzian fit after a user-defined baseline correction, and similarly, the intensities of the 1589 cm−1 peak were obtained for the Cy3-tag probe mixtures. The no-template control (NTC) sample was prepared by replacing the DNA with water in the PCR tube to act as a control for the SERS experiments.
We initially used 400 μM ST for the SERS experiments using the obtained PCR product. As seen in Fig. 2a, for the FAM-tag probes, the box plot of the intensities of the 1636 cm−1 peak shows an average higher intensity for the insertion-specific DNA, compared to the NTC and deletion-specific DNA. As the SERS experiments are conducted at room temperature, a few of the unimolecular probes can remain unfolded. This results in the small but finite intensity of the peak in the cases of NTC and deletion-specific DNA and also causes a distribution of intensities for each case. The average Raman spectra (Fig. 2b) clearly distinguish the peak intensities for the samples. In presence of the insertion sequence (homozygous resistant allele) in the Ty-3 gene, the tail corresponding to the insertion-specific FP had its complement synthesized in a larger amount compared to the deletion-specific FP, during the PCR. As a result, the FAM-tagged probe hybridized to the complementary region, freeing the dye and thus generating an increased SERS signal (blue). In the case of NTC and negative samples, the absence of the target sequence (deletion) forced the FAM-tagged probe to self-hybridize thus yielding a lower Raman intensity.
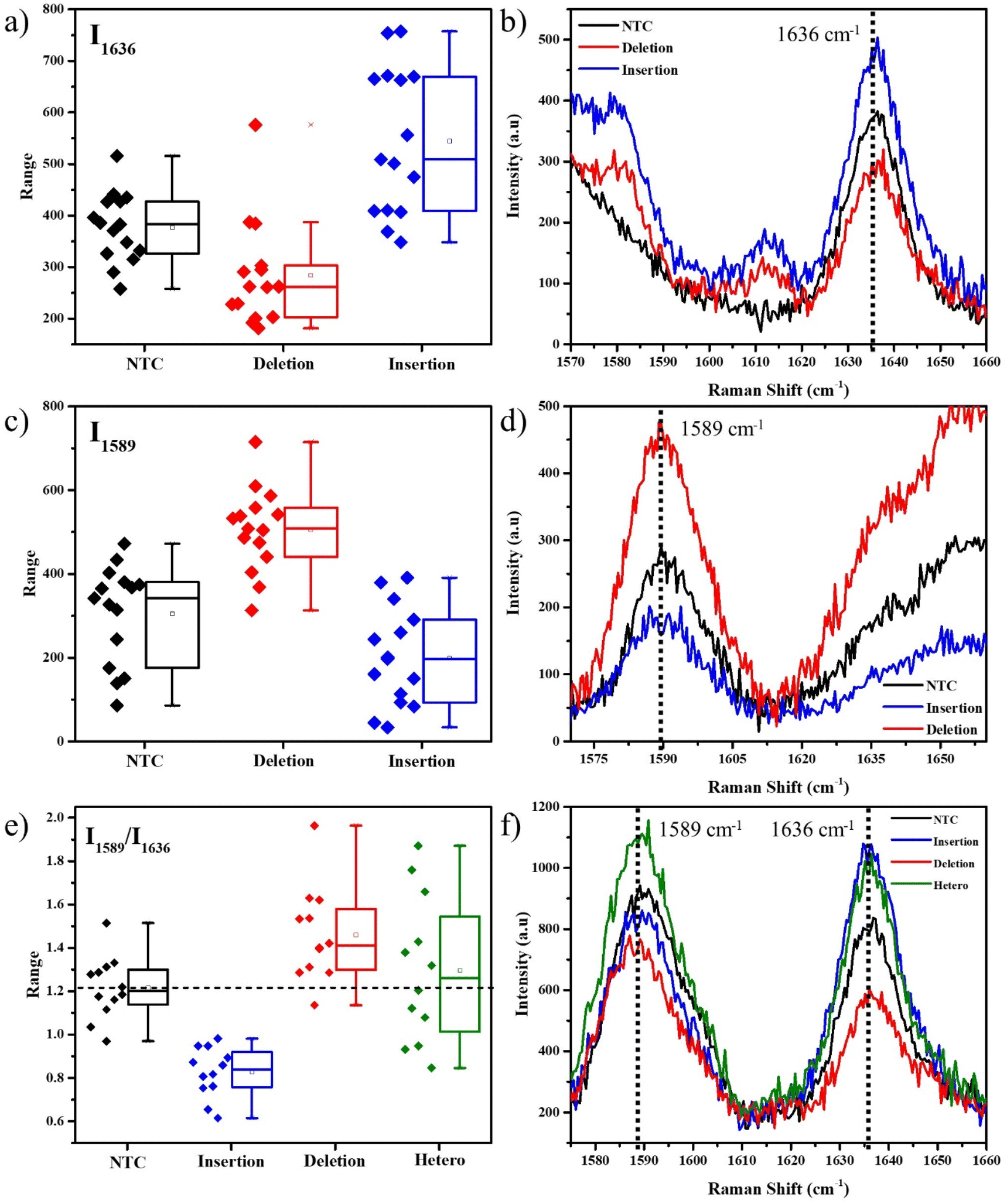 | ||
| Fig. 2 1636 cm−1 peak analysis for the FAM-labelled SERS probe containing PCR products, (a) box plot representing the 1636 cm−1 peak intensities, (b) SERS spectra in the unique regions. 1589 cm−1 peak analysis for the Cy3-labelled SERS probe containing PCR products, (c) box plot representing the 1589 cm−1 peak intensities, (d) SERS spectra in the unique regions. Multiplexing SERS spectral analysis of the PCR products containing both FAM-tagged and Cy3-tagged probes – intensity ratio of 1589 cm−1 and 1636 cm−1 peaks, (e) box plot representation, and (f) SERS spectra of the unique spectral region. Black – no DNA-template control, blue – DNA of resistant allele (insertion indel), red – DNA of susceptible allele (deletion indel), green – heterozygous DNA (both deletion and insertion indel). | ||
Similarly, for the Cy3-tag probes, the box plot of intensities of the 1589 cm−1 peaks (Fig. 2c) and the average Raman spectra (Fig. 2d) represented a higher average intensity for the deletion-specific DNA as compared to the NTC and the insertion-specific DNA. The complementary region of the tail of the deletion-specific FP is the target of the Cy3-tagged probes. This region was amplified when the DNA template was that of a susceptible tomato allele. From Fig. 2, the differences in the box plots clearly show that our strategy can be used to differentiate between alleles corresponding to susceptible and resistant species of tomato based on their Ty-3 gene composition.
The unique isolated peaks of FAM and Cy3 make SERS multiplexing a possibility, thus helping determine the nature of an unknown tomato DNA sample. For the multiplexing studies, both the probes along with their corresponding FPs were added into the tubes for conducting the PCR. The peak intensity ratio I1589/I1636, corresponding to the signature peaks of Cy3 and FAM was obtained for all the spectra. A box plot for these intensity ratios and the corresponding SERS spectral peaks are represented in Fig. 2e and f. In the absence of any DNA, both the dye molecules would be equally but limitedly accessible to the AgNPs. As their Raman scattering cross-sections are different, slight differences in the intensities corresponding to their peaks are observed. The average intensity ratio of the NTC sample was obtained from the box plot as 1.2. This was used as a reference for further analysis. A higher ratio compared to NTC, indicated a larger number of Cy3 dye molecules accessible to the AgNPs as compared to the FAM, thus confirming that the probes were in the presence of DNA from the susceptible allele of tomato, i.e., the deletion indel. A higher intensity ratio than NTC indicates the DNA is from a susceptible tomato allele, while a lower ratio indicates the DNA is from a resistant tomato allele.
This multiplexing experiment was conducted for a random DNA where we used DNA extracted from E. coli. In the absence of either of the two probe-specific sequences, a few non-specific bindings with both probes resulted in an intensity ratio equal to NTC (Fig. S6†), confirming the absence of either of the two target DNA sequences. This method was also tested in the case of heterozygous DNA samples, which contain both the alleles for insertion and deletion. As seen in Fig. 2e, it appeared as a widely scattered box plot, due to signals from both alleles. However, the mean value of 1.3 was found to be close to that of NTC since both the FAM and Cy3 signals were almost identical in intensity. This is due to the nature of the heterozygous DNA, which tends to have equal quantities of both deletion and insertion indels, thus overall resulting in an average equivalent to that of NTC, or E. coli, where either no DNA is present or the DNA doesn't match.
To evaluate the assay's dependability in another situation, we used the same protocol but to detect the indels in a separate gene involved with a different disease. The mi-1 gene present in tomatoes is involved in the conferring of resistance against root-knot nematodes. Deletion of a specific sequence causes the resistance of the plant against the pathogen, while insertion makes it vulnerable to the nematode's attack. Markers for these mutations were used to develop the primers and probes accordingly (Table S2†).15 kept the same system of detecting insertions with FAM-tagged probes and deletions with Cy3-tagged probes. The results were similar to the previous experiments, with distinct differences in dye intensities in the presence of the two different mutations (Fig. 3). This experiment demonstrates that this strategy can be used to detect indels in any DNA.
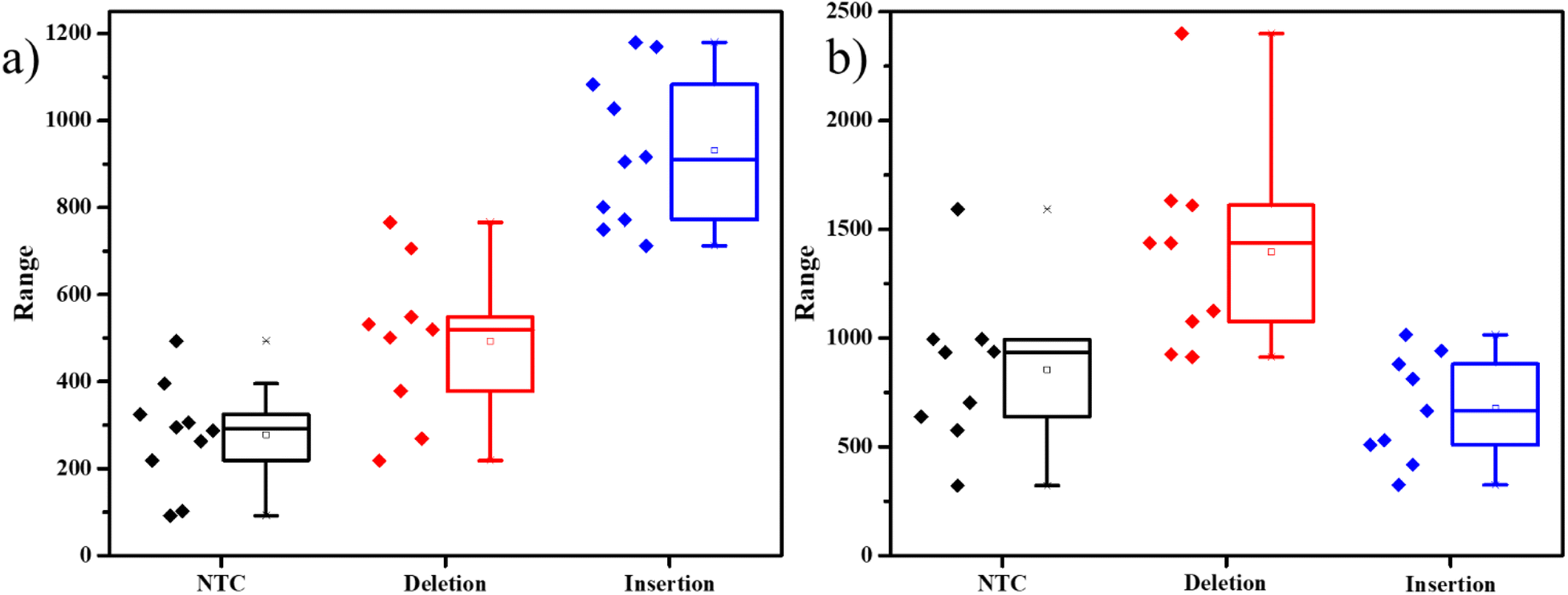 | ||
| Fig. 3 Box plots of SERS spectral analysis of the PCR products for Mi-1 gene containing (a) intensity of 1636 cm−1 peak corresponding to FAM-tagged probe and (b) intensity of 1589 cm−1 peak corresponding to Cy3-tagged probes. Black – no DNA-template control, red – DNA of resistant allele (deletion indel), blue – DNA of susceptible allele (insertion indel). | ||
The SERS experiments with 400 μM ST, were able to differentiate the insertion indels from the deletion indels clearly on average but had a few data points in the box plots that were overlapping. This indicates that there are comparable free unimolecular probes to the opened unimolecular probes. To increase the access of the dye molecules in the opened probes to the AgNPs, we further increased the ST concentration to a higher value of 0.01 M. As seen in the TEM figures, represented in Fig. S7,† with an increase in ST concentration, larger clusters of AgNPs are formed. The inset in Fig. S7a† shows a slight grey tinge to the AgNPs, and the TEM image in Fig. S7a† shows agglomerated AgNPs in the size range of 500–800 nm, for 400 μM ST. When the ST concentration was increased to 0.01 M, the inset of Fig. S7b† shows a dark grey color to the AgNPs indicating the formation of larger particles, which is very clearly seen in the TEM image, indicating agglomerates in the size range of 2–3 μm. The formation of such large aggregates, allowed for a greater number of SERS hotspots for the molecules to sit in. SERS studies on the PCR products with 0.01 M ST indicated a larger separation in the intensities represented by the box plots. As seen in Fig. 4a, the average SERS intensity of the FAM peak corresponding to the insertion-specific DNA was very large as compared to the NTC and deletion-specific DNA. Similarly, in Fig. 4b, the SERS intensity of the Cy3 peak was considerably higher for the deletion-specific indel compared to NTC or the insertion-specific indel. In, Fig. 4, the box plots of the heterozygous samples, marked their corresponding average values midway to the two homozygous samples. This was as expected, as the heterozygous sample is a mix of both. Similarly, the average intensities of the E. coli DNA were matching the NTC and deletion (for FAM-tagged probe) or insertion (for Cy3-tagged probe), as there was no amplification of this DNA. Therefore, a higher concentration of ST allowed for more SERS hotspots, hence easy differentiation of the indels under study.
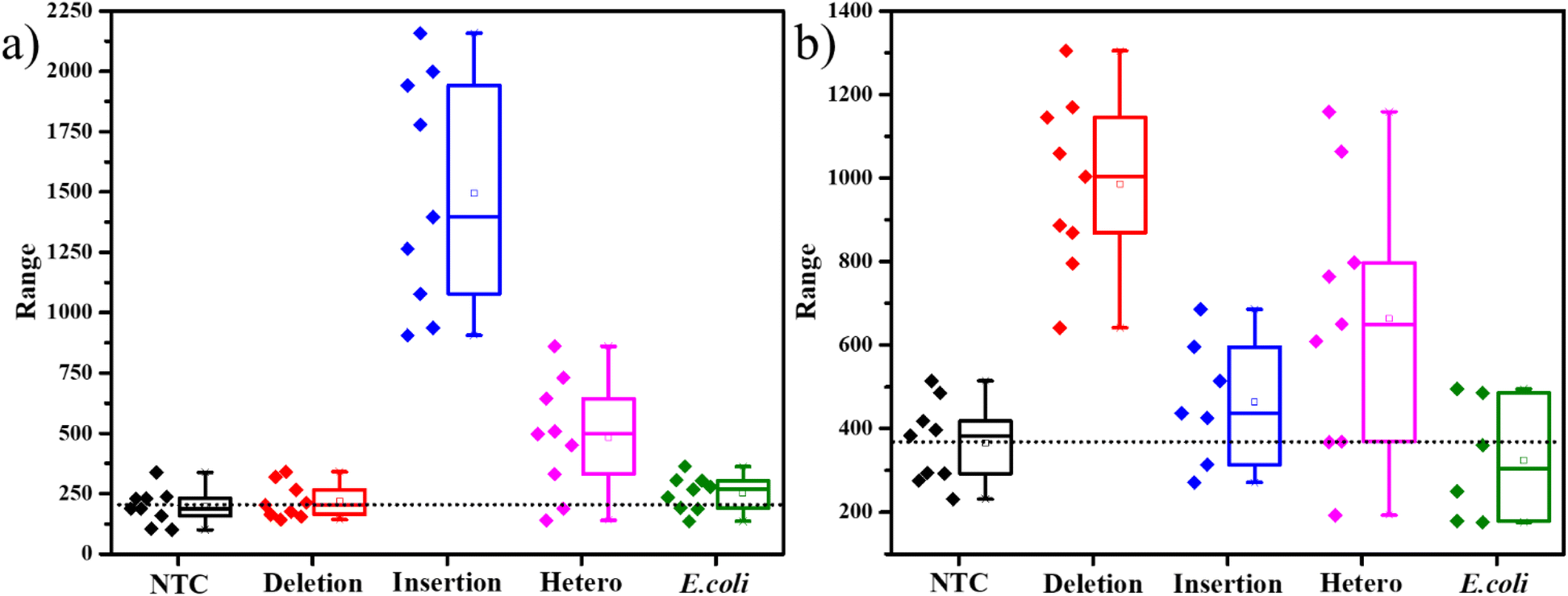 | ||
| Fig. 4 Box plot representations of intensities of unique peaks from SERS analysis, (a) 1636 cm−1 peak for the FAM-tagged SERS probe containing PCR products, (b) 1589 cm−1 peak for the Cy3-tagged SERS probe containing PCR products. Black – no DNA-template control (NTC), red – DNA of susceptible allele (deletion indel), blue – resistant DNA of resistant allele (insertion indel), magenta – heterozygous DNA (both deletion and insertion indels), green – E. coli (negative control – bacterial DNA). | ||
The Ty-3 gene also exhibits the phenomenon of SNP, in which the C-allele confers the resistance against TYLCV, but the T-allele makes it susceptible.38 We developed probes based on the available markers, where the FAM-tagged probe was designed to detect the C-allele (Table S3†). As expected, the mean intensity of the 1636 cm−1 peak was the highest in the case of the C-allele while T-allele showed an average lower intensity, even lesser than NTC itself (Fig. 5). Thus, it is evident that this assay can also be used to detect SNP in plant genomes. As this strategy involves the detection of the tail of the FP and not the original DNA strand itself, throughout the PCR cycles, the probability of non-specific binding reduces. Also, as a part of the design of the FP for SNP detection, we have created a mismatch in the 3rd base pair, thus if the required allele is absent, there is higher confidence of reduction in non-specific hybridization.49
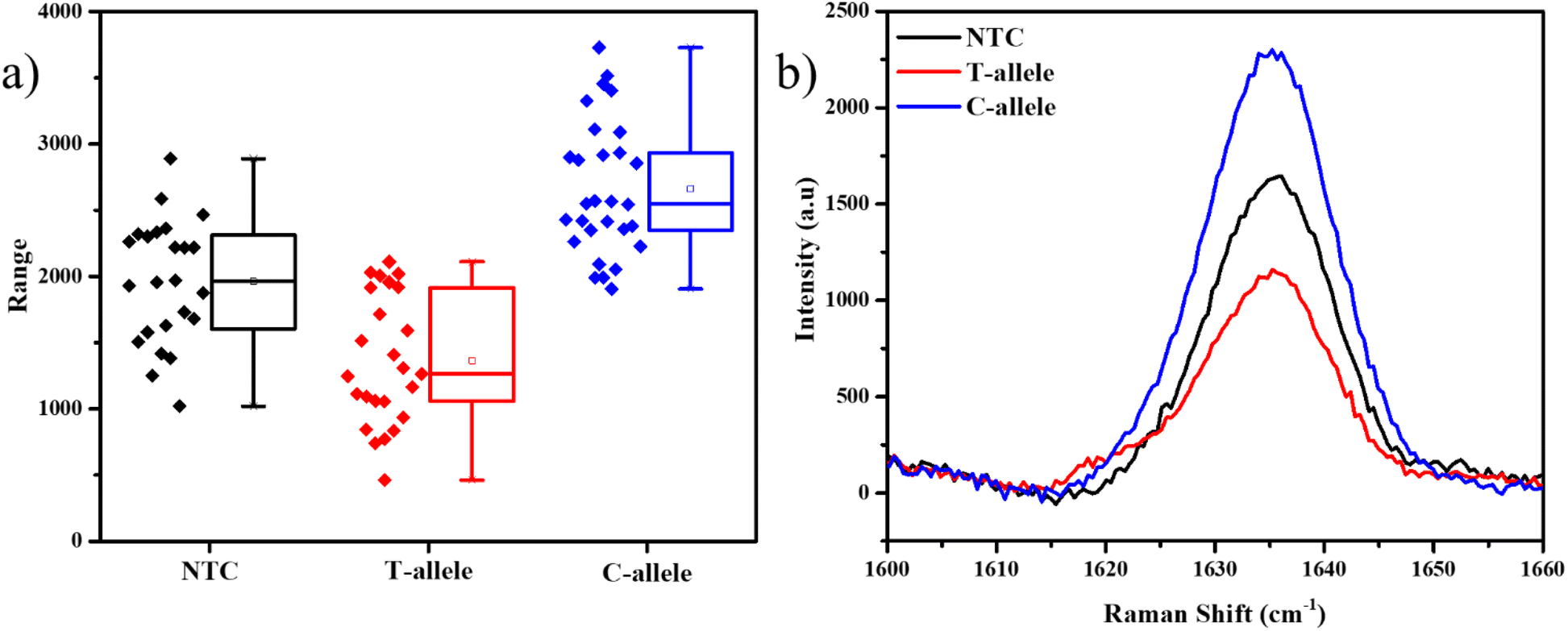 | ||
| Fig. 5 1636 cm−1 peak analysis for the FAM-labelled SERS probe containing PCR products, (a) box plot representing the 1636 cm−1 peak intensities, (b) SERS spectra in the unique regions. Black – no DNA-template control (NTC), red – DNA of susceptible allele (T-allele), and blue – DNA of resistant allele (C-allele). | ||
Conclusion
The above observations clearly show that our PCR-based SERS assay can easily distinguish between the presence and absence of a specific sequence in multiple genes distinctly, resulting in the detection of particular indels in the tomato genome. It can also differentiate between homozygous and heterozygous alleles. Furthermore, we demonstrated that by using different dyes, it is possible to perform multiplexing to study indel-based resistance in the crop, allowing us to learn the same about unknown DNA samples quickly. In addition, the assay has also been able to differentiate between two types of alleles with SNPs. Given the ability of Raman spectroscopy to read the molecular fingerprints of the dyes being used, this technique can be adapted to allow the usage of more than two dyes and thus be fine-tuned to do multiplexing to search for multiple SNPs in a single experiment. The simplicity of this assay also leaves space for automated detection by machines using well plates, which will reduce not only the manual errors but also the overall time of the assay. Therefore, this technique has the potential to be a highly dependable detection tool for indels and SNPs in the agricultural industry, not only for diseases like TYLCV and root-knot nematodes but also for the selection of desirable lines and thereby promoting overall genome improvement, given the advantages it has over the other conventionally used methods. Although RT-PCR is the most effective technology for detecting mutations today, SERS has come a long way in this regard. The most effective technology for detecting mutations currently is unquestionably RT-PCR, but SERS has the potential to be another means of detection owing to its excellent multiplexing capabilities and lack of requirement for a quencher. In the future to make it a profitable technology in the market. This opens the door to the development of customized Raman markers with distinct Raman signatures for multiplexing applications. Furthermore, with further refinements, this technique can be used to detect mutations associated with severe diseases in both animals and humans.Author contributions
S. S. and D. C. have contributed to the methodology, validation, formal analysis, investigation, data curation, writing-original draft, and visualization. J. T. has contributed to conceptualization, writing review and editing, resources, supervision, and validation. C. N. has contributed to conceptualization, validation, resources, writing – review and editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Plant DNA samples for this study were provided by Welcome Crop Science Pvt Ltd, Bengaluru. D. C. and S. S. acknowledge INSPIRE-DST and DST-DDP (DST/TDT/DDP-11/2018(G)) for fellowship respectively. N. C., S. S., and D. C. thank JNCASR for experimental facilities. NC thanks DST/TDT/DDP-11/2018(G) for funding. We thank Arun Panchapakesan for providing E. coli samples.References
- B. N. Ames, in The Biological Revolution: Applications of Cell Biology to Public Welfare, ed. G. Weissmann, Springer US, Boston, MA, 1979, pp. 117–148, DOI:10.1007/978-1-4684-3569-6_7.
- A. G. Knudson, in Advances in Cancer Research, ed. G. Kleln, S. Weinhouse and A. Haddow, Academic Press, 1973, vol. 17, pp. 317–352 Search PubMed.
- S. Wasserman, L. A. Urry, P. V. Minorsky and J. B. Reece, Campbell Biology Series, Pearson Education, Inc., 2017, p. 11 Search PubMed.
- A. Petitjean, M. I. W. Achatz, A. L. Borresen-Dale, P. Hainaut and M. Olivier, Oncogene, 2007, 26, 2157–2165 CrossRef CAS PubMed.
- V. Zappulli, G. De Zan, B. Cardazzo, L. Bargelloni and M. Castagnaro, J. Dairy Res., 2005, 72, 98–106 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- T. Jehan and S. Lakhanpaul, Indian J. Biotechnol., 2006, 5, 435–459 CAS.
- K.-B. G. Scholthof, S. Adkins, H. Czosnek, P. Palukaitis, E. Jacquot, T. Hohn, B. Hohn, K. Saunders, T. Candresse, P. Ahlquist, C. Hemenway and G. D. Foster, Mol. Plant Pathol., 2011, 12, 938–954 CrossRef CAS PubMed.
- H. Czosnek and H. Laterrot, Arch. Virol., 1997, 142, 1391–1406 CrossRef CAS PubMed.
- A. S. Thongrit D and T. Sutabutra, FFTC Book Ser., 1986, 33, 60–63 Search PubMed.
- Y. Levy, E. Glick and Y. Gafni, Plant Prot. Sci., 2009, 45, 81–97 CrossRef.
- S.-W. Ding and O. Voinnet, Cell, 2007, 130, 413–426 CrossRef CAS PubMed.
- V. M. Williamson and R. S. Hussey, Plant Cell, 1996, 8, 1735–1745 CAS.
- J. N. Sasser and C. C. Carter, An Advanced Treatise on Meloidogyne – Biology, and Control, 1985, vol. 1, pp. 19–24 Search PubMed.
- Z. Devran, A. Göknur and L. Mesci, Hortic., Environ. Biotechnol., 2016, 57, 156–160 CrossRef.
- B. Sharma, R. R. Frontiera, A.-I. Henry, E. Ringe and R. P. Van Duyne, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. Nie and R. Emory Steven, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- D. A. Stuart, K. B. Biggs and R. P. Van Duyne, Analyst, 2006, 131, 568–572 RSC.
- J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834–5840 CrossRef CAS PubMed.
- H. Flood Amar, J. F. Stoddart, W. Steuerman David and R. Heath James, Science, 2004, 306, 2055–2056 CrossRef CAS PubMed.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- D. S. Grubisha, R. J. Lipert, H. Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS PubMed.
- M. Y. Sha, H. Xu, M. J. Natan and R. Cromer, J. Am. Chem. Soc., 2008, 130, 17214–17215 CrossRef CAS PubMed.
- H. T. Beier, C. B. Cowan, I. H. Chou, J. Pallikal, J. E. Henry, M. E. Benford, J. B. Jackson, T. A. Good and G. L. Coté, Plasmonics, 2007, 2, 55–64 CrossRef CAS.
- J. H. An, W. A. El-Said, C. H. Yea, T. H. Kim, and J. W. Choi, 2011.
- Y. Wu, H. Dang, S.-G. Park, L. Chen and J. Choo, Biosens. Bioelectron., 2022, 197, 113736 CrossRef CAS.
- H. Chen, S.-G. Park, N. Choi, J.-I. Moon, H. Dang, A. Das, S. Lee, D.-G. Kim, L. Chen and J. Choo, Biosens. Bioelectron., 2020, 167, 112496 CrossRef CAS PubMed.
- S. M. Z. A. Naqvi, Y. Zhang, S. Ahmed, M. I. Abdulraheem, J. Hu, M. N. Tahir and V. Raghavan, Talanta, 2022, 236, 122823 CrossRef CAS PubMed.
- L. Mandrile, S. Rotunno, L. Miozzi, A. M. Vaira, A. M. Giovannozzi, A. M. Rossi and E. Noris, Anal. Chem., 2019, 91, 9025–9031 CrossRef CAS.
- D. van Lierop, K. Faulds and D. Graham, Anal. Chem., 2011, 83, 5817–5821 CrossRef CAS PubMed.
- D. Macdonald, E. Smith, K. Faulds and D. Graham, Analyst, 2020, 145, 1871–1877 RSC.
- Y. Wu, N. Choi, H. Chen, H. Dang, L. Chen and J. Choo, Anal. Chem., 2020, 92, 2628–2634 CrossRef CAS PubMed.
- H. G. Lee, W. Choi, S. Y. Yang, D.-H. Kim, S.-G. Park, M.-Y. Lee and H. S. Jung, Sens. Actuators, B, 2021, 326, 128802 CrossRef CAS.
- N. Lyu, V. K. Rajendran, J. Li, A. Engel, M. P. Molloy and Y. Wang, Analyst, 2021, 146, 5714–5721 RSC.
- Y. Liu, N. Lyu, V. K. Rajendran, J. Piper, A. Rodger and Y. Wang, Anal. Chem., 2020, 92, 5708–5716 CrossRef CAS PubMed.
- M. Kim, Y. Park, J. Lee and S.-C. Sim, Sci. Hortic., 2020, 265, 109230 CrossRef CAS.
- J. Jung, H. J. Kim, J. Lee, C.-S. Oh, H.-J. Lee and I. Yeam, Euphytica, 2015, 205 Search PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- H. S. Basu and L. J. Marton, Biochem. J., 1987, 244, 243–246 CrossRef CAS PubMed.
- C. Lee and J. B. Choo, Bull. Korean Chem. Soc., 2011, 32, 2003–2007 CrossRef CAS.
- D. Graham, W. E. Smith, A. M. T. Linacre, C. H. Munro, N. D. Watson and P. C. White, Anal. Chem., 1997, 69, 4703–4707 CrossRef CAS.
- J. C. Jones, C. McLaughlin, D. Littlejohn, D. A. Sadler, D. Graham and W. E. Smith, Anal. Chem., 1999, 71, 596–601 CrossRef CAS PubMed.
- C. Cao YunWei, R. Jin and A. Mirkin Chad, Science, 2002, 297, 1536–1540 CrossRef PubMed.
- K. Faulds, W. E. Smith and D. Graham, Anal. Chem., 2004, 76, 412–417 CrossRef CAS PubMed.
- J. Hu, P.-C. Zheng, J.-H. Jiang, G.-L. Shen, R.-Q. Yu and G.-K. Liu, Analyst, 2010, 135, 1084–1089 RSC.
- L. Wang, A. Roitberg, C. Meuse and A. K. Gaigalas, Spectrochim. Acta, Part A, 2001, 57, 1781–1791 CrossRef CAS PubMed.
- H. Sato, M. Kawasaki, K. Kasatani and M.-a. Katsumata, J. Raman Spectrosc., 1988, 19, 129–132 CrossRef CAS.
- R. F. V. Medrano and C. A. de Oliveira, Mol. Biotechnol., 2014, 56, 599–608 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06044b |
| ‡ Shared equal first authorship. |
| This journal is © The Royal Society of Chemistry 2022 |