
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of isoquinolines and isoquinoline N-oxides via a copper-catalyzed intramolecular cyclization in water†
Lujun Zhang‡
b,
Wenfang Xiong‡c,
Biao Yaoa,
Haitao Liub,
Meng Lia,
Yu Qinb,
Yujian Yub,
Xu Lib,
Meng Chenb,
Wanqing Wu a,
Jianxiao Li*a,
Jinliang Wang*b and
Huanfeng Jiang*a
a,
Jianxiao Li*a,
Jinliang Wang*b and
Huanfeng Jiang*a
aSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: jianghf@scut.edu.cn; cejxli@scut.edu.cn
bInstitute of Chemistry Co. Ltd, Henan Academy of Sciences, Zhengzhou 450000, China. E-mail: 15936281283@163.com
cSchool of Pharmacy, Guangdong Medical University, Dongguan, 523808, China
First published on 27th October 2022
Abstract
A highly efficient method for the facile access of isoquinolines and isoquinoline N-oxides via a Cu(I)-catalyzed intramolecular cyclization of (E)-2-alkynylaryl oxime derivatives in water has been developed. This protocol was performed under simple and mild conditions without organic solvent, additives or ligands. By switching on/off a hydroxyl protecting group of oximes, the selective N–O/O–H cleavage could be triggered, delivering a series of isoquinolines and isoquinoline N-oxides, respectively, in moderate to high yields with good functional group tolerance and high atom economy. Moreover, the practicality of this method was further demonstrated by the total synthesis of moxaverine in five steps.
With the pressing need for the sustainability of human society, more environmentally acceptable processes have been playing an increasingly important role in the chemical industry, especially in the manufacture of fine chemicals and pharmaceuticals.1 In 1998, the twelve principles of green chemistry were put forward by Anastas and Warner. Among the twelve principles, safer and green solvents have been recognized as one of the most important and indispensable elements in the current chemical society.2,3 Compared with the commonly toxic and highly volatile solvents, some green and sustainable solvents such as supercritical fluids,4 ionic liquids,5 and water,6 have been intensively studied from the beginning of the 21st century. It is noteworthy that water as a green and sustainable reaction medium has received increasing attention from the synthetic community.7 However, the study of organic chemical reactions in water is still in its infancy, and most organic synthesis still needs to be achieved in strictly dry organic solvents, especially when these systems involve transition metal catalysis.8 Therefore, the development of diverse and practical approaches by the use of water as a reaction medium in organic synthetic chemistry is more desirable.
Isoquinolines and their derivatives, one of the most important nitrogen-containing heterocycles, have attracted tremendous attention in recent years owing to their widespread occurrence in many natural products,9 pharmaceuticals,10 organic materials,11 and chiral ligands.12 Due to their versatile applications, a plethora of synthetic strategies have been developed to construct these functionalized heterocyclic compounds under different transition-metal-catalyzed systems,13 especially the simplest and most direct intramolecular cyclization. In 2009, Zhang and co-workers reported an interesting work involving the product selectivity control reactions to afford isoquinolines and isoquinolin-1(2H)-ones by simple subtle structure modification of ortho-alkynylaryl aldehyde oxime derivatives.14 Subsequently, Li,15 Harrity,16 Shi17 and others18 disclosed a rhodium(III), platinum(0), silver(I) and gold(I)-catalyzed intramolecular cyclization reaction using the same or analogous starting materials for the synthesis of isoquinolines and their derivatives, respectively. However, despite these significant advances made in accessibility of isoquinolines and their derivatives, some challenging issues still remain in terms of the use of toxic organic solvents, precious metal catalysts, harsh reaction conditions, which lead to poor atom economy and are inconsistent with the principles of green chemistry. Therefore, the development of more efficient and environment-friendly methods for the assemble of functionalized isoquinolines and their derivatives is of great significance but extremely challenging. As part of our continuing studies on green chemistry,19 combined with our interest in the development of new synthetic methods for the construction of nitrogen-containing heterocycles based on oxime compounds,20 we herein present an unprecedented strategy for the synthesis of isoquinoline and their derivatives in water via a copper-catalyzed cyclization reaction of (E)-2-alkynylaryl oxime derivatives. Interestingly, the reaction could be performed in the absence of organic solvent, additives or ligands, and enabled the formation of isoquinolines and isoquinoline derivatives respectively by the selective N–O/O–H cleavage of oximes in the same reaction system (Scheme 1).
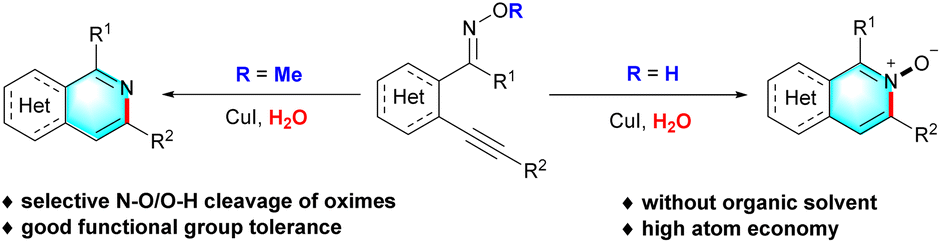 | ||
| Scheme 1 Synthesis of isoquinolines and isoquinoline N-oxides. | ||
Initially, (E)-1-(2-(phenylethynyl)phenyl)ethanone O-methyl oxime 1a was selected as model substrate for the optimization of reaction conditions (Table 1). When 1a was reacted in the presence of 10 mol% CuI in water at 80 °C under an air atmosphere, 1-methyl-3-phenylisoquinoline 2a was obtained in 92% yield (entry 1). Prolonging the reaction time led to a slightly higher yield (95%) (entry 2). In the absence of a copper catalyst, no product was observed (entry 3), which indicating that copper catalyst played an important role for the transformation. Subsequently, screening of other copper salts, including CuBr, CuCl, CuBr2 and Cu(OAc)2, showed that CuI was the best catalyst for the reaction (entries 4–7). Moreover, we observed the reaction temperature had great impact on the reaction. Decreasing the temperature to 70 °C led to lower yield (entry 8). Further optimization of solvents revealed that water was more efficient than other solvents, such as 1,4-dioxane, toluene, EtOH and AcOH (entries 9–12). Based on these results, the optimal reaction conditions were established as follows: 1a (0.5 mmol), CuI (10 mol%), H2O (2 mL) at 80 °C for 15 h in air.
|
||||
|---|---|---|---|---|
| Entry | [Cu] | Solvent | Time (h) | Yieldb (%) |
| a Reaction condition: 1a (0.5 mmol), [Cu] (10 mol%), and solvent (2 mL) at 80 °C in a sealed tube, and reaction time as specified.b Isolated yields.c 70 °C. | ||||
| 1 | CuI | H2O | 12 | 91 |
| 2 | CuI | H2O | 15 | 95 |
| 3 | — | H2O | 15 | NR |
| 4 | CuBr | H2O | 15 | 73 |
| 5 | CuCl | H2O | 15 | 46 |
| 6 | CuBr2 | H2O | 15 | 43 |
| 7 | Cu(OAc)2 | H2O | 15 | NR |
| 8c | CuI | H2O | 15 | 48 |
| 9 | CuI | 1,4-Dioxane | 12 | 87 |
| 10 | CuI | Toluene | 12 | 28 |
| 11 | CuI | EtOH | 12 | 65 |
| 12 | CuI | AcOH | 12 | Trace |
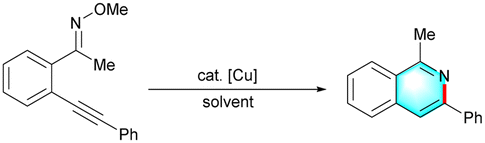
With the optimized conditions in hand, we further studied the substrate scope of this Cu(I) catalyzed intramolecular cyclization reaction, and the experimental results were displayed as follows (Table 2). First, the (E)-2-alkynylaryl ketone O-methyl oximes 1 bearing electron-donating substituents (p-Me, p-tBu, p-OMe, and p-Ph) proceeded well in this reaction, affording the corresponding products (2b–2e) in good to excellent yields. The structure of 2e was confirmed by X-ray crystallography analysis (CCDC 2095902, see the ESI† for details). While substrates possessing electron-withdrawing groups (1f–1i) showed lower reactivity than those with electron-rich groups, delivering the desired isoquinolines in 62–89% yields. Other functional groups, such as TMS (1j) and NH2 (1k) groups, were also tolerated under standard conditions, allowing for efficient synthesis of isoquinolines. In particular, the isoquinoline product 2k could be utilized as a bidentate ligand.21 1-Ethyl-3-phenylisoquinoline 2l and 1-benzyl-3-phenylisoquinoline 2m were obtained in 28% and 63% yield, respectively. Substrates 1n–1t bearing thienyl, naphthyl, n-pentyl, cycloprophyl, cyclopentyl and pyridyl groups were also applicable to this transformation, producing the corresponding products 2n–2t in moderate to good yields. Especially, this strategy was demonstrated to be efficient for the construction of polycyclic isoquinoline 2u. And, N-acetoxyl imine 1v was successfully converted to isoquinolines 2v in acceptable yield, which expanded the diversity of this method. In addition, the 3-phenylisoquinolines 2aa–2ah were also obtained in 73–96% yields.
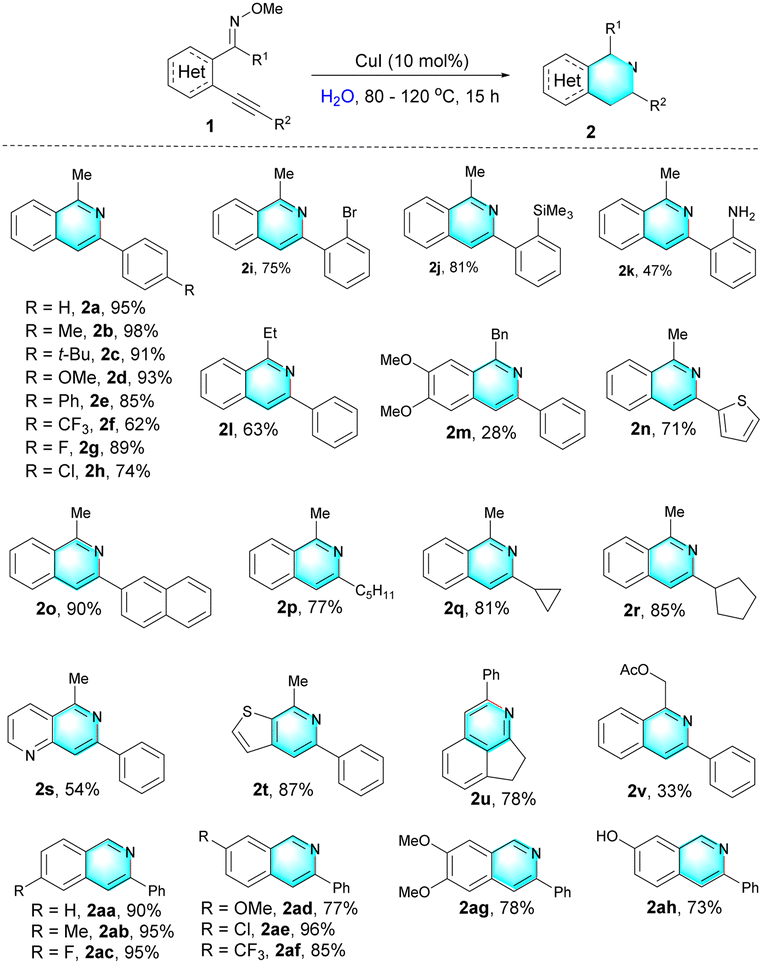
Interestingly, the reaction of (E)-2-alkynylaryl oximes 3 (Table 3) is quite different under standard reaction conditions, providing various 3-phenylisoquinoline N-oxide 4 as products. The structure of 4a was characterized by X-ray crystallography analysis (CCDC 2128901, see the ESI† for details). Substrates possessing either electron-donating (3b–3e, 3p–3q, and 3u) or electron-withdrawing groups (3f–3i, and 3r–3t) all underwent well in this system, producing the corresponding products in moderate to excellent yields. A series of important functional groups attached on the triple bond, including naphthyl (3j), thienyl (3k), n-pentyl (3m), cycloprophyl (3n), cyclopentyl (3o) groups, were also well tolerant, affording the desired products in 74–90% yields. The terminal alkyne (3l) and internal alkyne (the TMS substituent) (3y) gave the same product. Hydroxyl group (3v) was also compatible with the standard reaction conditions. Arene-fused nitrocycles 4w and 4x could also be obtained using this method, and the formula of 4w was established by X-ray crystal structure analysis (CCDC 2142641, see the ESI† for details). o-Alkynylaryl ketoxime 3z was also found to be tolerated in this reaction, and the corresponding product 4z were also obtained in a 35% yield at 50 °C.
To investigate the practicality of this method, gram-scale reactions and further transformations were carried out (Scheme 2). On a 10.0 mmol scale, the desired isoquinoline 2a and isoquinoline N-oxide 4a were isolated in 94% and 85% yield, respectively. Subsequently, treating isoquinoline N-oxides with ammonium chloride solution of zinc, isoquinoline N-oxides (4a, 4l, and 4p) could be converted into the corresponding isoquinolines 5a, 5l and 5p in excellent yields. Furthermore, the reaction of 4a, 4l, and 4p with TMSCN took place at room temperature, providing the desired cyanoisoquinolines 6a, 6l and 6p in 86–98% yields. More importantly, chiral ligand 7l could be further successfully prepared in a moderate yield from cyanoisoquinolines 6l.
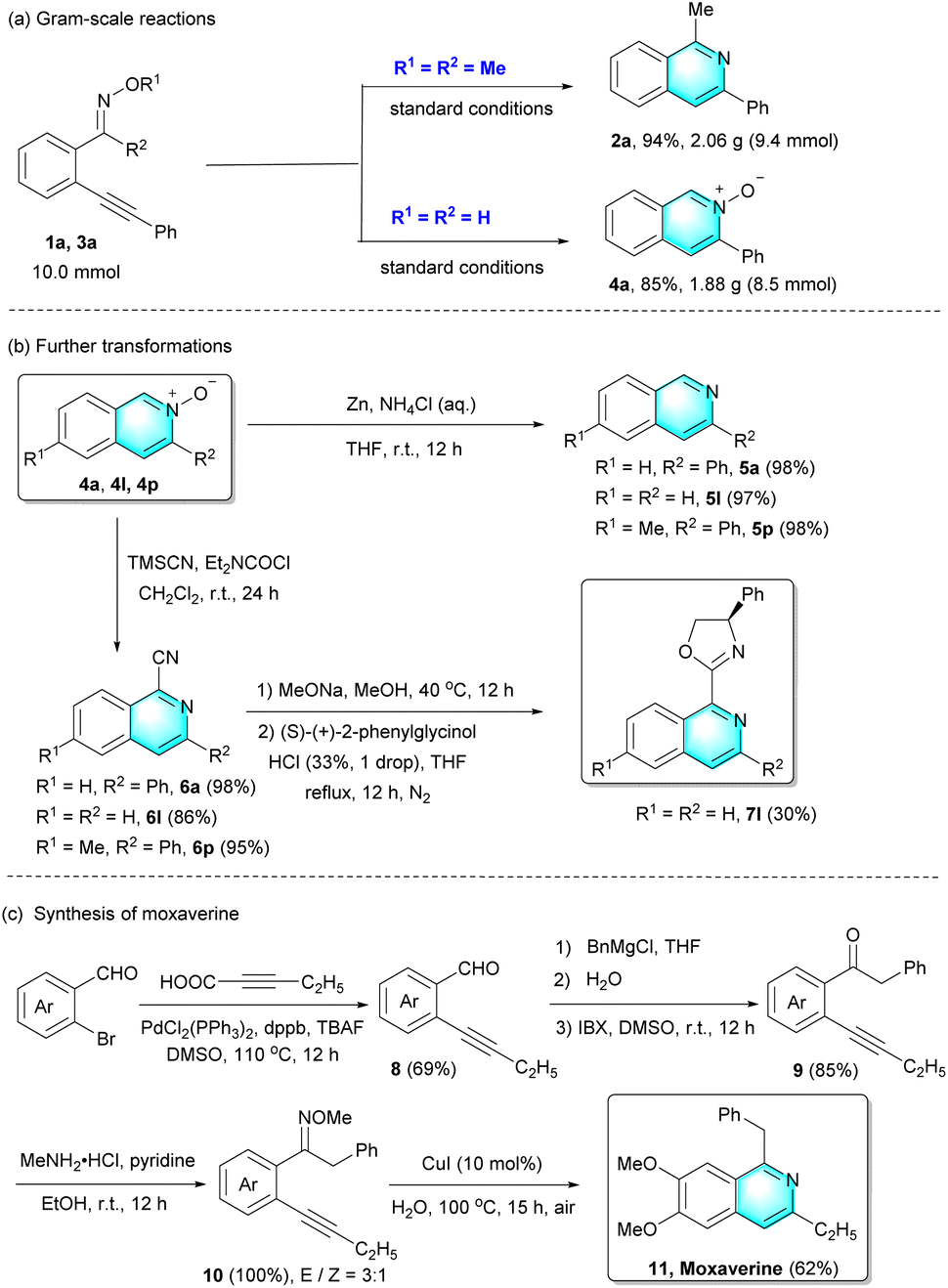 | ||
| Scheme 2 Gram-scale reactions (a), further transformations (b) and synthesis of moxaverine (c). | ||
On the other hand, this Cu(I)-catalyzed intramolecular cyclization could also be applied to the synthesis of isoquinoline alkaloid moxaverine 11 (Scheme 2(c)), which is employed as a famous drug to treat functional gastrointestinal disorders. Initially, the commercially available 6-bromoveratraldehyde and 2-pentynoic acid were used to synthesize the internal alkyne 8 with palladium catalysis. The internal alkyne 8 reacting with benzylmagnesium chloride was converted to the product containing alcohol functionality, which was then oxidized to the intermediate product 9. Further transformation of ketone to oxime derivative 10 underwent well, albeit the two configurations (E/Z = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) could not be completely purified. Finally, moxaverine 11 was easily prepared in 62% yield by employing this novel cyclization.
:1) could not be completely purified. Finally, moxaverine 11 was easily prepared in 62% yield by employing this novel cyclization.
In order to further understand the reaction mechanism, some control experiments (Scheme 3) were conducted. Firstly, the reaction of the Z-isomer of oxime ethers 1b (Scheme 3(i)) did not take place to give product 2a under the standard condition. Subsequently, we found that the reaction of E-oxime ethers could be carried out under N2 atmosphere to yield the desired heterocycle 2a in 96% yield (Scheme 3(ii)), showing that oxygen might not be involved in the intramolecular cyclization reaction. The addition of TEMPO (Scheme 3(iii)) did not prevent the reaction, which indicates that the reaction might not proceed via a radical pathway. Finally, the reaction of (E)-1-(2-(phenylethynyl)phenyl)ethanone O-(4-methoxybenzyl) oxime ether 1w (Scheme 3(iv)) was conducted under the standard reaction conditions, and we found that the corresponding product 2a and p-anisaldehyde 12 could be produced in 93% and 75% yield, respectively, which shows that the methoxy on the N atom might convert to formaldehyde.
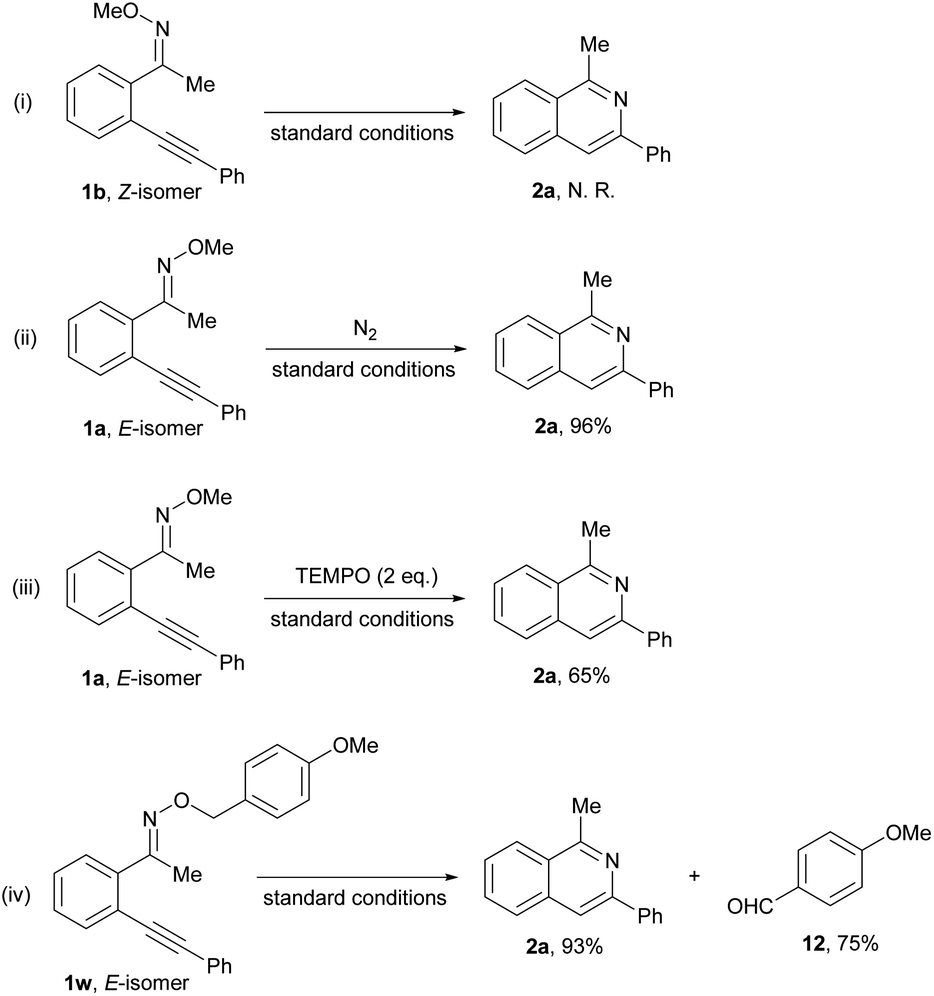 | ||
| Scheme 3 Control experiments (i–iv). | ||
Based on the above results and the previous work,14,17,18 a possible reaction pathway is proposed for the formation of isoquinolines 2 and isoquinoline N-oxides 4 (Scheme 4). Initially, ortho-alkynylaryl oxime derivatives 1 or 3 were easily converted to intermediates A by a Cu(I)-catalyzed intramolecular cyclization. When R1 is a methyl group, the cleavage of the N–O bond could give the intermediate C with the losing of one molecular of CH2O. The subsequent protonation of intermediate C would afford the isoquinolines 2. When R1 is a hydrogen atom, the cleavage of the O–H bond of intermediate D could afford the isoquinoline N-oxides 4 with the assistance of one molecule of H2O.
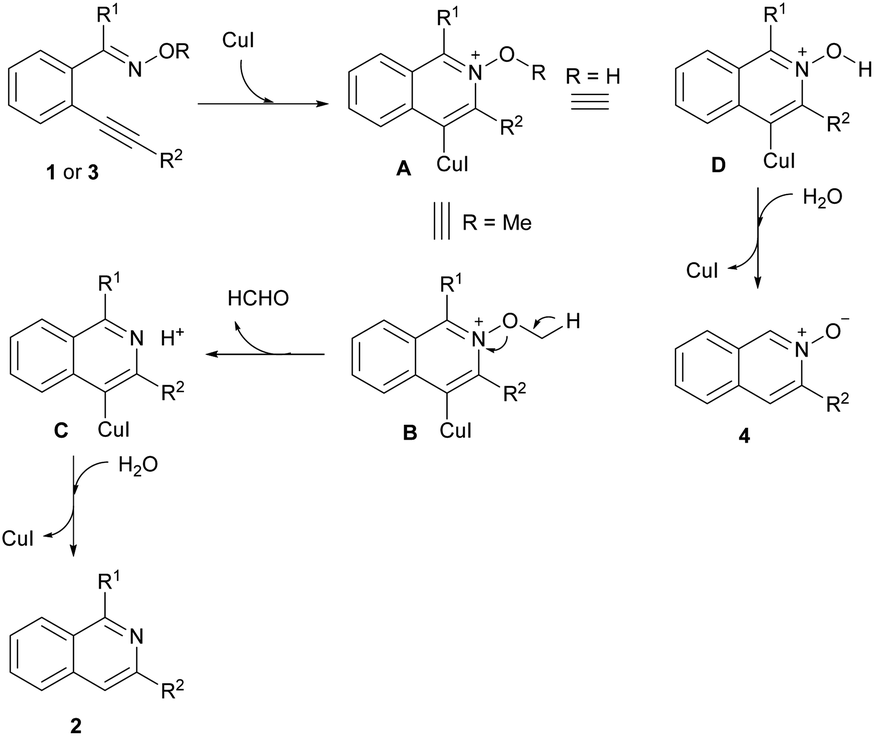 | ||
| Scheme 4 Plausible mechanism. | ||
In conclusion, we have developed an environment-friendly cyclization reaction for the synthesis of isoquinolines and isoquinoline N-oxides by selective N–O/O–H cleavage of oximes. This reaction featured in the use of green solvent, high atom economy, broad substrate scope and good functional group tolerance. The diversity of isoquinoline derivatives has been realized by subtle structure modification under mild reaction conditions with simple operations. More importantly, Moxaverine could be efficiently prepared in five steps employing this new method. Further investigations of the reaction mechanism and applications are ongoing in our lab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Henan Academy of Sciences Project (210803004, 220403012, and 220603062), the Key-Area Research and Development Program of Guangdong Province (2020B010188001), and the National Natural Science Foundation of China (22001075).Notes and references
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- S. L. Y. Tang, R. L. Smith and M. Poliakoff, Green Chem., 2005, 7, 761–762 RSC.
- (a) R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC; (b) M. O. Simon and C. J. Li, Green Chem., 2012, 41, 267–278 Search PubMed; (c) M. Deetlefs and K. R. Seddon, Green Chem., 2010, 12, 17–30 RSC; (d) S. Protti, D. Dondi, M. Fagnoni and A. Albini, Green Chem., 2009, 11, 239–249 RSC.
- (a) W. Leitner, Top. Curr. Chem., 1999, 206, 107–132 CrossRef; (b) W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef CAS PubMed; (c) Green Chemistry Using Liquid and Supercritical Carbon Dioxide, ed. J. M. DeSimone and W. Tumas, Oxford University Press, New York, 2003 Search PubMed; (d) E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- (a) R. Sheldon, Chem. Commun., 2001, 2399–2407 RSC; (b) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed; (c) C. E. Song, Chem. Commun., 2004, 1033–1043 RSC.
- (a) C.-J. Li, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS PubMed; (b) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC; (c) A. Chatterjee and T. R. Ward, Catal. Lett., 2016, 146, 820–840 CrossRef CAS; (d) X.-Y. Jin, L.-J. Xie, H.-P. Cheng, A.-D. Liu, X.-D. Li, D. Wang, L. Cheng and L. Liu, J. Org. Chem., 2018, 83, 7514–7522 CrossRef CAS PubMed; (e) C.-L. Ren, Y. Wang, D. Wang, Y.-J. Chen and l. Liu, Sci. China: Chem., 2010, 53, 1492–1496 CrossRef CAS.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed; (b) L. Liu and D. Wang, Handbook of Green Chemistry: Green Solvent. Reaction in Water, ed. C. J. Li, WILEY-VCH, Weinheim, 2010, vol. 5, pp. 207–228 Search PubMed.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- (a) Z. X. Qing, J. L. Huang, X. Y. Yang, J. H. Liu, H. L. Cao, F. Xiang, P. Cheng and J. G. Zeng, Curr. Med. Chem., 2018, 25, 5088–5114 CrossRef CAS PubMed; (b) M. Dastmalchi, M. R. Park, J. S. Morris and P. Facchini, Phytochem. Rev., 2018, 17, 249–277 CrossRef CAS; (c) A. Diamond and I. Desgagné-Penix, Plant Biotechnol. J., 2016, 14, 1319–1328 CrossRef CAS PubMed; (d) A. Y. Khan and G. S. Kumar, Biophys. Rev., 2015, 7, 407–420 CrossRef CAS PubMed; (e) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC.
- (a) E. Prchalová, N. Hin, A. G. Thomas, V. Veeravalli, J. Ng, J. Alt, R. Rais, C. Rojas, Z. Li, H. Hihara, M. Aoki, K. Yoshizawa, T. Nishioka, S. Suzuki, T. Kopajtic, S. Chatrath, Q. Liu, X. Z. Dong, B. S. Slusher and T. Tsukamoto, J. Med. Chem., 2019, 62, 8631–8641 CrossRef PubMed; (b) W. Yang, L. X. Li, Y. L. Wang, X. W. Wu, T. T. Li, N. Yang, M. B. Su, L. Sheng, M. Y. Zheng, Y. Zang, J. Li and H. Liu, Bioorg. Med. Chem., 2015, 23, 5881–5890 CrossRef CAS PubMed; (c) D. B. Khadka, H. Woo, S. H. Yang, C. Zhao, Y. F. Jin, T. N. Le, Y. Kwon and W. J. Cho, Eur. J. Med. Chem., 2015, 92, 583–607 CrossRef CAS PubMed; (d) S. C. Wu, D. Yoon, J. Chin, K. van Kirk, R. Seethala, R. Golla, B. He, T. Harrity, L. K. Kunselman, N. N. Morgan, R. P. Ponticiello, J. R. Taylor, R. Zebo, T. W. Harper, W. Y. Li, M. M. Wang, L. Zhang, B. G. Sleczka, A. Nayeem, S. Sheriff, D. M. Camac, P. E. Morin, J. G. Everlof, Y. X. Li, C. A. Ferraro, K. Kieltyka, W. Shou, M. B. Vath, T. A. Zvyaga, D. A. Gordon and J. A. Robl, Bioorg. Med. Chem. Lett., 2011, 21, 6693–6698 CrossRef CAS PubMed; (e) P. Ray, J. Wright, J. Adam, J. Bennett, S. Boucharens, D. Black, A. Cook, A. R. Brown, O. Epemolu, D. Fletcher, A. Haunso, M. Huggett, P. Jones, S. Laats, A. Lyons, J. Mestres, J. de Man, R. Morphy, Z. Rankovic, B. Sherborne, L. Sherry, N. van Straten, P. Westwood and G. Z. R. Zaman, Bioorg. Med. Chem. Lett., 2011, 21, 97–101 CrossRef CAS PubMed; (f) C. E. Gutteridge, M. M. Hoffman, A. K. Bhattacharjee, W. K. Milhous and L. Gerena, Bioorg. Med. Chem. Lett., 2011, 21, 786–789 CrossRef CAS PubMed; (g) A. L. Smith, F. F. DeMorin, N. A. Paras, Q. Huang, J. K. Petkus, E. M. Doherty, T. Nixey, J. L. Kim, D. A. Whittington, L. F. Epstein, M. R. Lee, M. J. Rose, C. Babij, M. Fernando, K. Hess, Q. Le, P. Beltran and J. Carnahan, J. Med. Chem., 2009, 52, 6189–6192 CrossRef CAS PubMed; (h) H. Morita, Y. Tomizawa, J. Deguchi, T. Ishikawa, H. Arai, K. Zaima, T. Hosoya, Y. Hirasawa, T. Matsumoto, K. Kamata, W. Ekasari, A. Widyawaruyanti, T. S. Wahyuni, N. C. Zaini and T. Honda, Bioorg. Med. Chem., 2009, 17, 8234–8240 CrossRef CAS PubMed; (i) A. Graulich, S. Dilly, A. Farce, J. Scuvée-Moreau, O. Waroux, C. Lamy, P. Chavatte, V. Seutin and J. F. Liégeois, J. Med. Chem., 2007, 50, 5070–5075 CrossRef CAS PubMed.
- (a) S. J. Liu, Q. Zhao, R. F. Chen, Y. Deng, Q. L. Fan, F. Y. Li, L. H. Wang, C. H. Huang and W. Huang, Chem.–Eur. J., 2006, 12, 4351–4361 CrossRef CAS PubMed; (b) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed; (c) D. Collado, E. Perez-Inestrosa, R. Suau, J. P. Desvergne and H. Bouas-Laurent, Org. Lett., 2002, 4, 855–858 CrossRef CAS PubMed.
- (a) R. Hrdina, I. Valterová, J. Hodačová, I. Císařová and M. Kotora, Adv. Synth. Catal., 2007, 349, 822–826 CrossRef CAS; (b) A. V. Malkov, L. Dufková, L. Farrugia and P. Kočovský, Angew. Chem., Int. Ed., 2003, 42, 3674–3677 CrossRef CAS PubMed.
- (a) R. Lavernhe, R. O. Torres-Ochoa, Q. Wang and J. P. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24028–24033 CrossRef CAS PubMed; (b) Y. M. Zhou and R. M. Hua, J. Org. Chem., 2021, 86, 8862–8872 CrossRef CAS PubMed; (c) W. Lin, X. X. Hu, C. W. Zhuang and Y. Z. Wang, Tetrahedron, 2019, 75, 3015–3023 CrossRef CAS; (d) Y. X. Tang, Y. J. Yu, X. Y. Wei, J. Yang, Y. T. Zhu, Y. H. Zhao, Z. L. Tang, Z. H. Zhou, X. F. Li and X. Y. Yu, Tetrahedron Lett., 2019, 60, 151187 CrossRef CAS; (e) F. Yang, J. J. Yu, Y. Liu and J. Zhu, Org. Lett., 2017, 19, 2885–2888 CrossRef CAS PubMed; (f) J. Li, M. Y. Tang, L. Zang, X. L. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742–2745 CrossRef CAS PubMed; (g) H. F. Jiang, J. D. Yang, X. D. Tang and W. Q. Wu, J. Org. Chem., 2016, 81, 2053–2061 CrossRef CAS PubMed; (h) A. B. Pawar, D. Agarwal and D. M. Lade, J. Org. Chem., 2016, 81, 11409–11415 CrossRef CAS PubMed; (i) J. Li, Z. Zhang, M. Y. Tang, X. L. Zhang and J. Jin, Org. Lett., 2016, 18, 3898–3901 CrossRef CAS PubMed; (j) J. Wang, S. K. Zha, K. H. Chen and J. Zhu, Org. Chem. Front., 2016, 3, 1281–1285 RSC; (k) X. B. Yang, J. Y. Jie, H. Y. Li and M. H. Piao, RSC Adv., 2016, 6, 57371–57374 RSC; (l) Z. Z. Zhu, X. D. Tang, X. W. Li, W. Q. Wu, G. H. Deng and H. F. Jiang, J. Org. Chem., 2016, 81, 1401–1409 CrossRef CAS PubMed; (m) Y. J. Liu, M. Gao, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2016, 7, 5436–5444 RSC; (n) R. J. Chen, J. F. Qi, Z. J. Mao and S. L. Cui, Org. Biomol. Chem., 2016, 14, 6201–6204 RSC; (o) H. Wang, J. L. Koeller, W. P. Liu and L. Ackermann, Chem.–Eur. J., 2015, 21, 15525–15528 CrossRef CAS PubMed; (p) S. Dhara, R. Singha, Y. Nuree and J. K. Ray, Tetrahedron Lett., 2014, 55, 795–798 CrossRef CAS; (q) K. R. Roesch, H. M. Zhang and R. C. Larock, J. Org. Chem., 2001, 66, 8042–8051 CrossRef CAS PubMed.
- H. Y. Gao and J. L. Zhang, Adv. Synth. Catal., 2009, 351, 85–88 CrossRef CAS.
- P. Zhao, F. Wang, K. Han and X. W. Li, Org. Lett., 2012, 14, 3400–3403 CrossRef CAS PubMed.
- H. Mora-Radó, L. Sotorríos, M. P. Ball-Jones, L. Bialy, W. Czechtizky, M. Méndez, E. Gómez-Bengoa and J. P. A. Harrity, Chem.–Eur. J., 2018, 24, 9530–9534 CrossRef PubMed.
- Q. Z. Li, R. X. Liu, Y. Wei and M. Shi, Adv. Synth. Catal., 2021, 363, 2664–2669 CrossRef CAS.
- (a) H. S. Yeom, Y. Lee, J. E. Lee and S. Shin, Org. Biomol. Chem., 2009, 7, 4744–4752 RSC; (b) S. Hwang, Y. Lee, P. H. Lee and S. Shin, Tetrahedron Lett., 2009, 50, 2305–2308 CrossRef CAS; (c) H. S. Yeom, S. Kim and S. Shin, Synlett, 2008, 6, 924–928 Search PubMed.
- (a) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736–1748 CrossRef CAS PubMed; (b) M. Hu, W. Wu and H. Jiang, ChemSusChem, 2019, 12, 2911–2935 CrossRef CAS PubMed.
- (a) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (b) D. S. Bolotin, N. A. Bokach, M. Y. Demakova and V. Y. Kukushkin, Chem. Rev., 2017, 117, 13039–13122 CrossRef CAS PubMed.
- (a) N. S. Gul, T. M. Khan, Y. C. Liu, M. I. Choudhary, Z. F. Chen and H. Liang, CCS Chem., 2020, 2, 1626–1641 Search PubMed; (b) N. S. Gul, T. M. Khan, M. Chen, K. B. Huang, C. Hou, M. I. Choudhary, H. Liang and Z. F. Chen, J. Inorg. Biochem., 2020, 213, 111260 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2095902, 2128901 and 2142641. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06097c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |