
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransparent TiO2 thin films with high photocatalytic activity for indoor air purification†
Jekaterina Sydorenko *a,
Arvo Merea,
Malle Krunksa,
Marina Krichevskaya*b and
Ilona Oja Acik*a
*a,
Arvo Merea,
Malle Krunksa,
Marina Krichevskaya*b and
Ilona Oja Acik*a
aLaboratory of Thin Films Chemical Technologies, Department of Materials and Environmental Technology, Tallinn University of Technology, Ehitajate tee 5, 19086 Tallinn, Estonia. E-mail: jekaterina.spiridono@taltech.ee; ilona.oja@taltech.ee
bLaboratory of Environmental Technology, Department of Materials and Environmental Technology, Tallinn University of Technology, Ehitajate tee 5, 19086 Tallinn, Estonia. E-mail: marina.kritsevskaja@taltech.ee
First published on 12th December 2022
Abstract
The development of low-material-quantity, transparent, anatase TiO2 nanoparticle free thin films as photocatalytic materials together with a profound understanding of their photocatalytic activity under ultraviolet (UV-A) and visible (VIS) light is crucial for environmentally friendly indoor air photocatalytic coatings. In this work, a TiO2 thin film modified by an increased amount of acetylacetone in the precursor solution with a material quantity of 0.2 mg cm−2 was successfully deposited on a borosilicate glass substrate by ultrasonic spray pyrolysis. VOC degradation as a single model pollutant and in mixtures under different operating conditions was studied in a multi-section continuous flow reactor. Under UV-A the reaction rate constants for heptane and toluene oxidation as individual pollutants were 1.7 and 0.9 ppm s−1, respectively. In 9 ppm VOC mixtures of acetaldehyde, acetone, heptane and toluene all the compounds were completely oxidized in a reaction time of less than 50 s. The TiO2 film showed moderately high photocatalytic activity under VIS light. The conversions of acetaldehyde, acetone, heptane and toluene in 9 ppm VOC mixtures under VIS light reached 100, 100, 78 and 31%, respectively. The synthesized TiO2 film shows promising ability in indoor air purification from VOCs. The results of this study give an extensive estimation of the thin film's photocatalytic efficiency and provide valuable data for future applications in environmental remediation.
1. Introduction
People used to spend most of their time indoors. The COVID-19 pandemic forced people to stay in their homes for months and to take more care about disinfection and cleaning. Detergents and antiseptics release volatile organic compounds (VOCs) into the air, which are the primary source of poor indoor air quality and sick building syndrome.1,2 Such syndromes as allergies, rhinitis, asthma and conjunctivitis could appear.3 Not all technologies are suitable for indoor air purification at home. Photocatalytic oxidation is an ideal technology for air treatment that mimics nature's photochemical process, which is capable of completely mineralizing almost all types of organic pollutant while being both energy and cost-efficient.1,4,5TiO2 is considered the most widely used photocatalyst. However, it must be mentioned that 99% of the TiO2 studies (according to the Web of Science database only 285 papers were found with the keywords ‘transparent film’ out of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 910 entries with the keywords ‘TiO2 photocatalysis’) focus on pre-fabricated nanopowders such as P25, PC500, and UV100. Nanopowder-based coatings with a thickness of several micrometres have a milky non-transparent surface.6 Despite this, several studies have been conducted on TiO2 thin films and coatings,3–13 and still higher photocatalytic activity has been achieved using thick coatings prepared from TiO2 nanopowders.7–9 In particular, Zou and co-authors obtained 55% conversion of 100 ppm of toluene after 60 min of ultraviolet (UV-A) irradiation on a 7 μm-thick TiO2 coating prepared from P25 powder.9 However, the thin films with a thickness less than 1 μm needed up to 30 hours for the degradation of a couple of ppm of the same pollutant.10,11
910 entries with the keywords ‘TiO2 photocatalysis’) focus on pre-fabricated nanopowders such as P25, PC500, and UV100. Nanopowder-based coatings with a thickness of several micrometres have a milky non-transparent surface.6 Despite this, several studies have been conducted on TiO2 thin films and coatings,3–13 and still higher photocatalytic activity has been achieved using thick coatings prepared from TiO2 nanopowders.7–9 In particular, Zou and co-authors obtained 55% conversion of 100 ppm of toluene after 60 min of ultraviolet (UV-A) irradiation on a 7 μm-thick TiO2 coating prepared from P25 powder.9 However, the thin films with a thickness less than 1 μm needed up to 30 hours for the degradation of a couple of ppm of the same pollutant.10,11
In the case of any photocatalytic application, special care should be taken over the material design so as not to be the source of extra pollution. The use of powders is not amenable since it leads to serious drawbacks related to the recovery and recycling of the photocatalyst. These are extremely significant issues, considering the risk of the release of nanosized particles of the material into the environment.12 Thus, the deposition of well-adhered photocatalysts in the form of thick or thin films onto a substrate, such as glass or stainless steel depending on its destined application, is the most viable strategy for the technological application of photocatalytic nanomaterials in any field, and especially in the environmental sector.12,13 Polycrystalline TiO2 thin films synthesized from a Ti-alkoxide precursor solution deposited by sol–gel spin/dip coating or spray pyrolysis methods is an old established route14 and it profits from lower quantities of materials (<1 mg cm−2), good adhesion properties and optical transparency in the visible range.10,13,15 Photocatalysts in the form of thin or thick films possess another advantage over nanopowders, making them applicable as multifunctional surfaces for photocatalytic VOC degradation: antimicrobial and self-cleaning properties.13 Thus, they are also applicable in medicine, e.g. to deactivate viruses.1
However, there is still a technological gap in the development of thin transparent films with high photocatalytic activity under both UV-A and VIS light, which can be applied as a windowpane coating for indoor air treatment.13,16 The loss in photocatalytic performance in the case of thin films compared to nanopowders can be effectively compensated for by using a convenient deposition technique, selecting suitable substrates and properly tuning preparative conditions.12 In order to increase the VIS light activity of a TiO2 photocatalyst, several strategies, such as doping with vanadium,17 silver,18 nitrogen19 or composite materials such as TiO2/ZrO220 and TiO2/carbon,21–23 have been applied. It has been found that with modification of TiO2 the conversion of toluene under VIS light was in the range 35–80%, while for pure TiO2 the conversion was below 10%.17–24 The incorporation of doping components into the TiO2 lattice results in the formation of new mid-gap energy states, promoting the absorption of VIS light.17–22
Moreover, when designing the photocatalyst, it must be taken into account that indoor air is a challenging source of pollution, as air in total could contain a wide range of VOCs in small concentrations (ppb). Many VOCs have similar structures, which makes it difficult to distinguish them in a gas mixture. Therefore, it is important to have a non-selective photocatalyst and to study the synergetic effects of VOC photocatalytic oxidation in the mixtures. There are only a few studies where photocatalytic oxidation of VOC mixtures in air was investigated, and most of these studies still consider nanopowders or nanopowder-based coatings.7,8,11,17,18,25 The use of transparent thin films is rather limited due to their lower activity. It should be highlighted that in this paper the term ‘thin film’ means a transparent layer of TiO2 less than 500 nm thick synthesized from chemicals, while ‘coating’ refers to a TiO2 layer prepared from pre-fabricated nanopowders. Peerakiatkhajorn et al. studied the oxidation of a 25 ppm VOC mixture (toluene, benzene, ethylbenzene, xylene) on Ag-doped TiO2 film with a thickness of 87 nm in a batch reactor under VIS light.18 After 300 min of irradiation they achieved ca. 80% VOC degradation.18 Pham and Lee studied the oxidation of hexane and butyl acetate individually and in a mixture in a continuous flow reactor on an Ag/V-co-doped TiO2 photocatalyst immobilized on polyurethane. They found that in the mixture stream, oxidation of butyl acetate was higher because of the higher polarity of the compound.17 Most studies are carried out in laboratory-scale batch reactors, which do not have the potential for practical application.7,18,25 However, the main problem in a continuous flow device design is the low contact time between the photocatalyst surface and pollutants.9,26 Therefore, in this paper we used a continuous flow multi-section reactor that allowed us to increase the reaction time by increasing the photocatalytic surface area. The area coated by photocatalytic thin film was increased five times, from 120 to 600 cm2, to follow the effects of reaction time and specific surface area. Additional sections of the reactor can be easily added to amplify the air purification. The specific construction of the reactor gives an opportunity to estimate the effectiveness of the performance of the obtained films in a real application to indoor air purification.
This paper builds on our previous research on the development of UV-A and VIS light active TiO2 thin films by systematically changing the precursor's molar ratio in the solution. In particular, experimental studies have shown that changing the titanium(IV) isopropoxide (TTIP) and acetylacetone (AcacH) molar ratio from 1:1 to 1:20 in the precursor solution results in a higher amount of adsorbed carbon on the film surface27 and enhanced photocatalytic activity of the obtained TiO2 films under UV-A and VIS light. For example, a ten-fold increase in the degradation rate constant of stearic acid was observed with a change of TTIP:AcacH molar ratio in the precursor solution from 1:3 to 1:8.27 TiO2 film with a molar ratio of 1:8 showed twice-as much activity compared to a film with a molar ratio of 1:5 in the gas-phase photodegradation of acetone and acetaldehyde under UV-A and VIS light.28 In the present study, TiO2 films deposited from a solution with a TTIP and AcacH molar ratio of 1:8 were further investigated by the photocatalytic degradation of refractory VOCs—heptane and toluene and their mixtures with relatively easily oxidized VOCs such as acetone and acetaldehyde—under UV-A and VIS light. A multi-section gas flow reactor was used to conduct VOC degradation. The quantum efficiency, by-product formation and synergistic effects of compounds with different polarities and hydrophobicities in a synthetic air mixture during photocatalytic oxidation under different operating conditions, such as air flow rate, humidity and light source, were studied and are discussed in this study.
Our focus is to add substantial data on photocatalytic oxidation, namely to examine a UV-A and VIS light active transparent TiO2 thin film fabricated in one step without any additional doping agents. Moreover, a multi-section gas flow reactor design allows mimicking of real indoor conditions and tests the performance of the obtained films. The results of this study bring photocatalysis closer to real application.
2. Experimental data
2.1 TiO2 thin film preparation
TiO2 thin films were prepared by an ultrasonic spray pyrolysis technique in ambient atmosphere (Fig. SI-1 in ESI†); more detailed information of film preparation has been published elsewhere.27 Borosilicate glass heated up to 350 °C was used as a substrate. Then, 0.2 M titanium(IV) isopropoxide (TTIP) (C12H28O4Ti 98+%, Acros Organics) was used as a titanium source for solution preparation, and acetylacetone (AcacH) (C5H8O2 > 99%, Sigma-Aldrich) was added as a stabilizing agent. A molar ratio of TTIP:AcacH of 1:8 was used for solution preparation as it was found to be an optimal molar ratio to obtain photocatalytically active film.27,28 The as-deposited films were annealed at 500 °C for one hour in air atmosphere. The films possess good adhesion properties and cannot be washed away by solvents e.g. water or ethanol.
2.2 Gas-phase photocatalytic activity measurements
A continuous multi-section plug flow photocatalytic reactor was used to study the gas-phase oxidation of VOCs over TiO2 film. A detailed description of the set-up used for the experiments (Fig. SI-2 in ESI†) was published elsewhere.8 The multi-section reactor has five sections connected in series with a volume of 130 mL each. The TiO2 catalyst in the form of a thin film on the glass substrate was located in each section of the reactor. Simplification was undertaken—the photocatalytic surface is the same as the glass substrate. The surface coated with photocatalytic thin film in one section of the reactor was 120 cm2, which gave 600 cm2 for all five sections. However, the surface of catalyst per reactor volume did not change during the experiments and was 0.92 cm2 cm−3. Ultraviolet29 (UV Philips Actinic BL 15 W, irradiance 3.5 mW cm−2 with reflector integrated in the range of 180–400 nm, with maximum emission at 365 nm, UV-B/UV-A ratio < 0.2%) or visible30 (VIS Philips TL-D 15 W, irradiance 3.3 mW cm−2 with reflector integrated in the range of 180–700 nm, UV/UV-VIS ratio < 5%) lights were located over each section of the reactor at a distance of 6 cm.The photocatalytic oxidation of heptane (C7H16 ≥ 99%, Honeywell) and toluene (C7H8 ≥ 99%, Sigma-Aldrich) in the reactor was studied separately in conjunction with their oxidation in a mixture with acetone (C3H6O ≥ 99.5%, Sigma-Aldrich) and acetaldehyde (C5H8O2 ≥ 99.5%, Sigma-Aldrich). The inlet concentrations of heptane and toluene in the polluted air varied from 5 to 40 ppm, when their oxidation was studied separately. The concentration in the polluted air mixtures was set to 9 ppm (3 ppm acetone, 3 ppm acetaldehyde and 3 ppm heptane or toluene). To simulate the air polluted with VOCs, a pre-calculated amount of the pollutants was injected into the feed tank under vacuum and after the evaporation of compounds, the tank was filled with compressed air. The temperature in the reactor was measured using a temperature controller with a thermocouple (Omega, CN9000A) and was 40 ± 2 °C. Two mass flow controllers were used: one for regulation of the flow rate of polluted air and one for that of diluent air. The gas flow rates varied from 0.5 to 2.5 L min−1, which resulted in a change in the reaction time in the reactor from 15.6 to 3.12 s. The reaction time was calculated as the residence time for an ideal plug flow reactor under steady-state conditions (not taking into account the residence time distribution) and does not reproduce the real contact time between the pollutant and the catalyst surface. Air was moving in the reactors in a laminar way; however, due to it passing through narrow piping between sections, the air flow was changed to turbulent and intensive mixing took place before reaching the new section of the reactor (Fig. SI and S2 in ESI†). Intensive mixing provided a uniform concentration of the polluted air after passing each section. Relative humidities of the air of 6 ± 1% and 40 ± 5% (measured at 20 °C) were used. In the experiments with toluene, ozone was generated by a UV-C lamp (LSE Lighting, GPH287T5VH/4) emitting at 254 and 185 nm.
An Interspec 200-X FTIR infrared spectrometer with an 8 m gas cell (Specac Tornado) was used to measure the concentration of heptane and toluene; a 20 m gas cell (Specac Atmos) was used to measure the concentration of VOCs in the mixtures. The air spectra were collected in-line using an Interspec version 3.40 Pro and processed with Essential FTIR software. For quantitative analysis, the heptane bond was measured at the IR bands 3020–2790 cm−1, toluene at the IR bands 2975–2850 cm−1, acetone at the IR bands 1250–1780 cm−1 and acetaldehyde at the IR bands 1170–1060 cm−1. The concentration was calculated based on one typical FTIR band for each organic pollutant. The limits of detection were 0.5 ppm and 0.2 ppm for Tornado and Atmos gas cells, respectively. The limits of quantification were 1 ppm and 0.4 ppm for the Tornado and Atmos gas cells, respectively. Control experiments for photochemical degradation and adsorption were carried out. No degradation of pollutants was detected in the presence of UV-A without a catalyst. No adsorption of the pollutants on the catalyst was detected in the dark where the limit of detection was 0.5 ppm. All spectra were recorded at least three times with a standard deviation lower than 5%, indicating good reproducibility of the results. The same TiO2 film was used in all photocatalytic experiments. The film showed high stability and repeatability of the results after a short 30 min period of regeneration with UV-A light.
3. Results and discussion
3.1 TiO2 thin film properties
TiO2 thin film prepared by ultrasonic spray pyrolysis on a borosilicate glass substrate from 0.2 M titanium(IV) isopropoxide (TTIP) solution with the addition of acetylacetone (AcacH) in a molar ratio TTIP:AcacH of 1:8 was used in this paper for VOC degradation. The reaction between TTIP and AcacH has been well studied in the literature.31–33 However, the effect of an increased amount of AcacH in the precursor on the photocatalytic properties of the film needs further investigation.27
The obtained TiO2 thin film with a thickness ca. 370 nm and a material quantity of ca. 0.2 mg cm−2 consists of crystalline anatase TiO2 (Fig. 1a), with a mean crystallite size in the range 30–40 nm. Optical transparency in the visible spectral range is ca. 80% and the bandgap is 3.4 eV (Fig. 1b). Surface morphology was studied using scanning electron microscopy27 and atomic force microscopy34 (Fig. SI-3 in ESI†). Studies showed that the synthesized film had a homogeneous, smooth non-porous surface with a root mean square roughness of 1.4 nm.
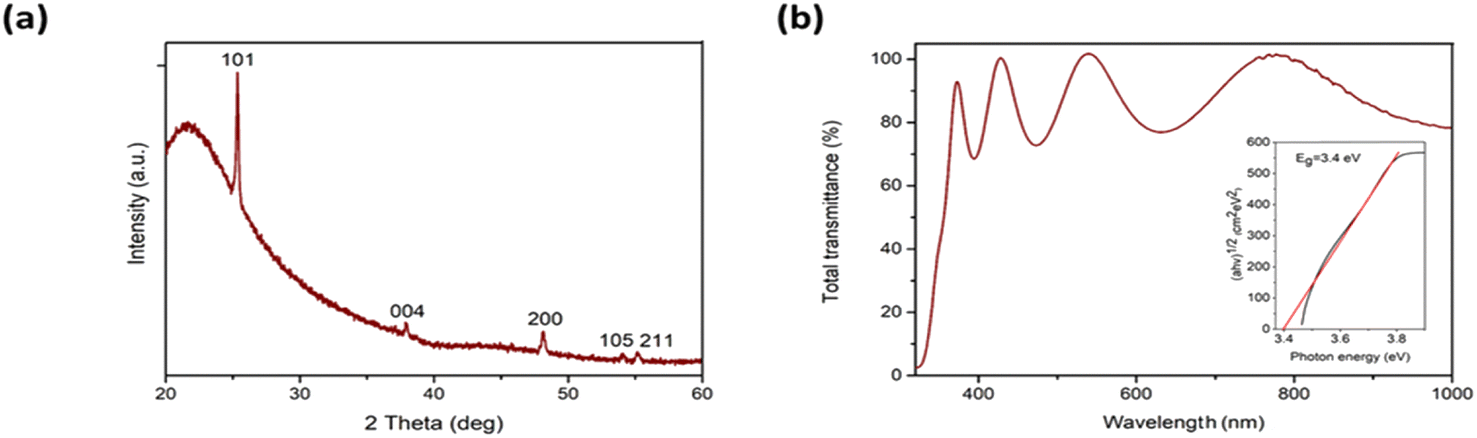 | ||
| Fig. 1 X-ray diffraction pattern (a) and total transmittance spectrum (b) of TiO2 thin film. The inset in the Fig. 1b shows the band gap value of TiO2 thin film. | ||
It was found by XPS studies that an increased AcacH molar ratio in the precursor solution promoted a larger number of carbon species on the surface of the film.21,27 Di Valentin et al.,35 in their study found the formation of localized midgap states for carbon-modified TiO2, in agreement with the findings of our previous study.28 Moreover, surface photovoltage (SPV) measurements showed that passivation of electron traps at the surface takes place with an increase in organic additive, which means preferential separation of photogenerated holes. The SPV signal changed from negative to positive with an increase in AcacH molar ratio, which allows us to control the charge transfer toward the TiO2 surface during the synthesis of the film.36 The recombination of the electron–hole pairs is reduced and more reactive oxygen species (ROS) are produced, which enhance the photocatalytic activity.27,28,35,36 The generation of ROS plays an essential role in photocatalytic reactions since ROS can degrade a wide range of organic pollutants.37–39
The film obtained in the current study shows superhydrophilic properties after 15 min of UV-A irradiation;27 thus a short ca. 30 min pre-treatment of the catalyst with UV-A was used before the photocatalytic experiments to restore the surface. The film oxidized gas-phase pollutants such as acetone and acetaldehyde with reaction rate constants of 2.3 and 2.4 ppm s−1, respectively. It was found that the films show high photocatalytic activity and stability. The same photocatalytic films were used for several months of study with no decrease in efficiency. Repeatability tests showed that the difference between the parallel series did not exceed 5%.28 More detailed analysis of the material and its properties is given in the ref. 27 and 28.
In this paper we present a systematic study of the synthesized TiO2 film with an optimized TTIP:AcacH molar ratio applied for the degradation of refractory compounds such as heptane and toluene. In addition to their more complex structures, these pollutants are both nonpolar and hydrophobic compounds compared to previously studied model pollutants such as acetone and acetaldehyde. Moreover, to mimic real indoor air pollution, the degradation of mixtures of VOCs with different polarities and hydrophilicities on TiO2 thin films was monitored.
3.2 Photocatalytic oxidation of heptane
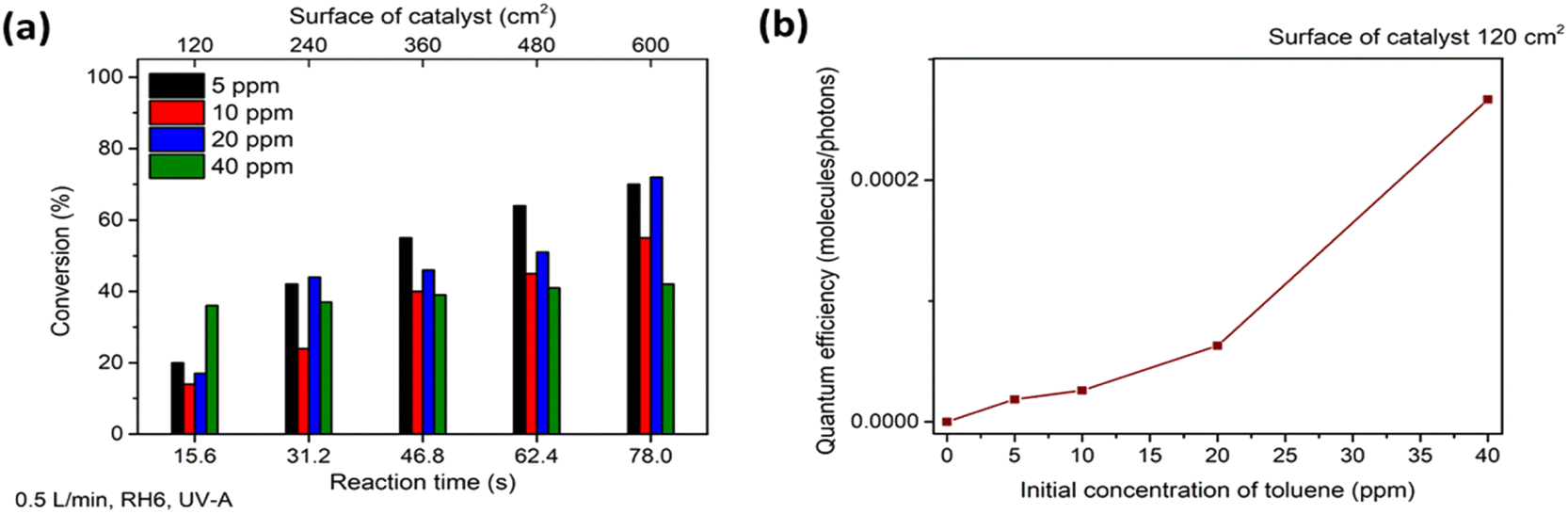
The photocatalytic oxidation of heptane at different initial concentrations, air flow rates, relative humidity (RH) and irradiation on the TiO2 thin film was studied.First, in order to estimate the initial concentration effect on heptane degradation on the TiO2 film under ultraviolet (UV-A) light, the photocatalytic oxidation of heptane at initial concentrations of 5, 10 and 40 ppm was studied (air flow rate 0.5 L min−1 and RH 6%) (Fig. 2a). At an initial concentration of 5 ppm complete oxidation of heptane occurred with a catalyst surface of 240 cm2, but an additional section of the reactor (catalyst surface of 360 cm2) was needed for the degradation of 10 ppm and a catalyst surface of 600 cm2 coped with the oxidation of 40 ppm of heptane (Fig. 2a). For comparison, in the case of sprayed TiO2 films from TTIP:AcacH 1:4 solution, only 20% conversion for 10 ppm of heptane was achieved at a catalyst surface of 360 cm2 under the same experimental conditions, indicating 5 times greater photocatalytic activity of TiO2 1:8 film.40 The effect of an increase in initial concentration on reaction performance was studied by calculating the quantum efficiency (QE) at initial heptane concentrations of 5, 10 and 40 ppm at a catalyst surface of 120 cm2 (reaction time 15.6 s) (eqn (1) and Fig. 2b).41
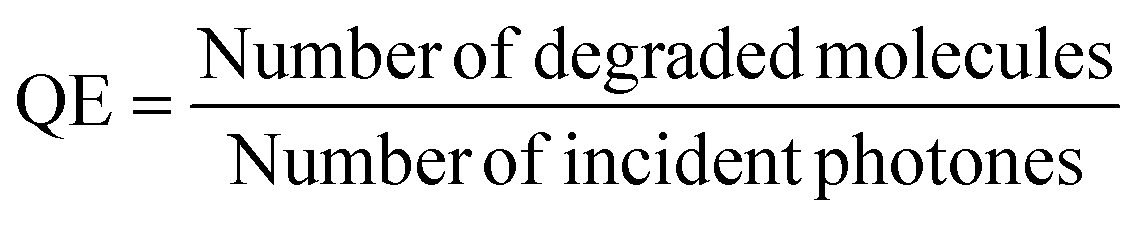 | (1) |
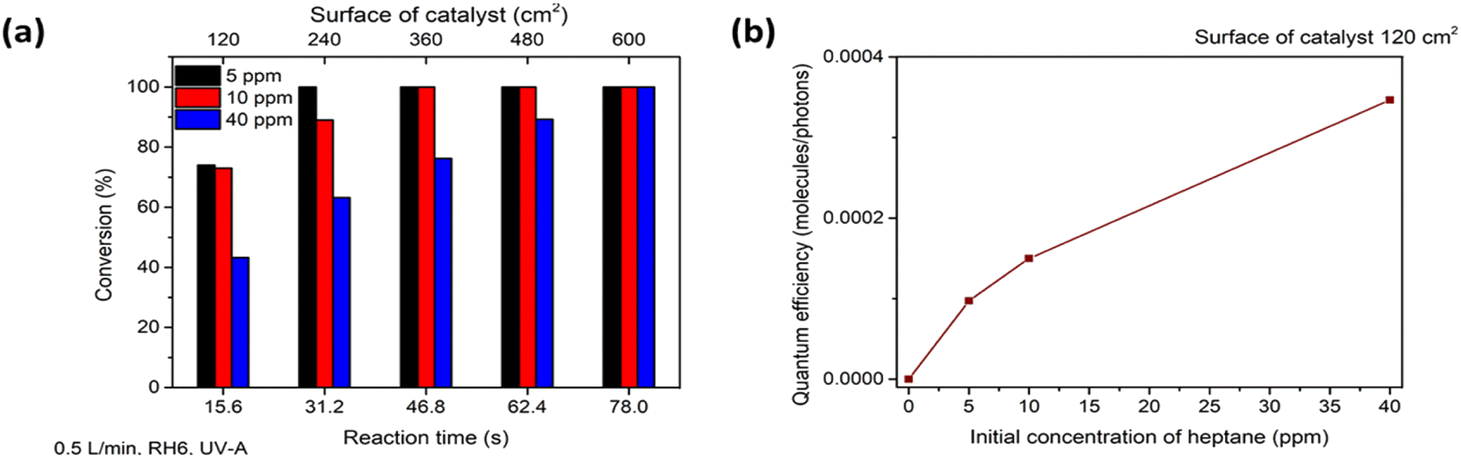 | ||
| Fig. 2 The effect of initial concentration of heptane on photocatalytic oxidation for different photocatalyst surface areas (a) and quantum efficiency (QE) of heptane degradation at different initial concentrations with a photocatalyst surface of 120 cm2 (b). The average values of heptane conversion are shown. The standard deviation was <5%. | ||
At a wavelength 365 nm in the case of UV-A, the measured incident photon energy in one section of the reactor at 1 s corresponded to 7.717 × 1017 J. The illuminated surface of the film in one section was 120 cm2 and in the calculations it was assumed that all photons impinge on the catalyst surface (the calculations are shown in ESI†).
Fig. 2b shows that under the studied conditions, QE increased from 9.74 × 10−5 to 1.5 × 10−4 molecules per photons when the concentration increased from 5 to 10 ppm. The increase is not linear, indicating that at some initial concentration the amount of ROS will start to limit the efficiency of the process. In contrast, TiO2 films deposited from TTIP:AcacH 1:4 solution showed a decrease in QE from 1.73 × 10−5 to 5.78 × 10−6 molecules per photons when the concentration of heptane increased from 5 to 10 ppm under similar experimental conditions.40 This demonstrates that the concentration of ROS produced on the surface of TiO2 thin film from the TTIP:AcacH 1:8 solution used in the current study does not limit oxidation under the studied conditions, unlike the case of TiO2 films deposited from 1:4 solution.
Gas-phase photocatalytic reactions take place primarily at the interface between adsorbed molecules of pollutants and the surface of the photocatalyst. Thus, both the adsorption and activation of reactants play a significant role in the process.38 To determine the reaction rate constants of heptane oxidation on TiO2 film the reaction kinetics were studied. The Langmuir–Hinshelwood kinetic model was assumed (eqn (2)).7
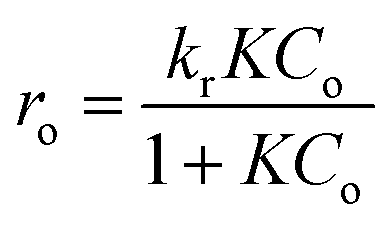 | (2) |
The reaction rate constant (kr) and adsorption constant (K) were found using the Langmuir–Hinshelwood kinetic plot 1/ro versus 1/Co, which showed linear dependence (Fig. SI-4 in ESI†).28 In Table 1, the kinetic constants of heptane oxidation are compared to those of acetone, which were obtained in our previous study for the same sprayed TiO2 film (TTIP:AcacH 1:8).28
| Pollutant | kr (ppm s−1) | K (ppm−1) | Ref. |
|---|---|---|---|
| Heptane | 1.7 | 0.05 | This study |
| Acetone | 2.3 | 0.03 | 28 |
The reaction rate constant of heptane decreased by about 30% compared to acetone, when the same film and operating parameters of the reactor were used. This can be explained by the more complex molecular structure of heptane, so it needs more time for oxidation. At the same time, the adsorption constant for heptane oxidation increased almost two times. This could be because the molecular weight of heptane is almost twice as high and the vapour pressure at 20 °C is almost five times lower than that of acetone. Moreover, the surface affinity of the photocatalyst could play a certain role in the adsorption process; pollutants prefer to interact with a photocatalyst of similar polarity. Carbon-species-containing TiO2 film was found to have a nonpolar structure.42 Acetone is a polar compound, while heptane is nonpolar. Adsorption of the pollutant molecules is an important step in the photocatalysis process since the molecule should reach the surface for oxidation. However, for a continuous flow reactor with a high surface area a high amount of ROS is beneficial for process efficiency. In gas-phase photocatalytic reactions, species such as ·OH and ·O2− play a dominant role. Air contains enough molecular oxygen and water, which can be converted to ROS on the surface of photoinduced TiO2.38,39
Heptane degradation with initial concentrations of 4.15–16.6 ppm in a 300 mL batch reactor was studied for TiO2 powders prepared via sol–gel by Shang et al.7 At a TiO2 load of 0.1 g, they obtained a reaction rate constant of 0.005 ppm s−1. Due to the high specific surface area of TiO2 particles, the adsorption constant in their study was 0.3 ppm−1; nevertheless, 360 min of irradiation was needed to achieve 99.7% mineralization.7 In the current study, 40 ppm of heptane was completely oxidized on the transparent TiO2 film with a material quantity of 0.2 mg cm−2 in a time of less than 2 minutes (Fig. 2a). A detailed comparison of the thin film prepared in this study with other thin films to oxidize heptane is given in ESI Table SI-I.† The construction of the reactor used in the present study allows the pollutant to continuously meet fresh catalyst, slowing down the deactivation. The results demonstrate that the effective continuous purification of polluted air by the obtained thin film in a multi-section reactor is more practical than that with powder in periodic mode reactors. Next, the effect of reaction time and mass transfer on heptane oxidation under UV-A was studied. The gas flow rate was changed from 0.5 to 2.5 L min−1, while the concentration of heptane was set to 10 ppm and the RH of the air was 6% (Fig. 3a).
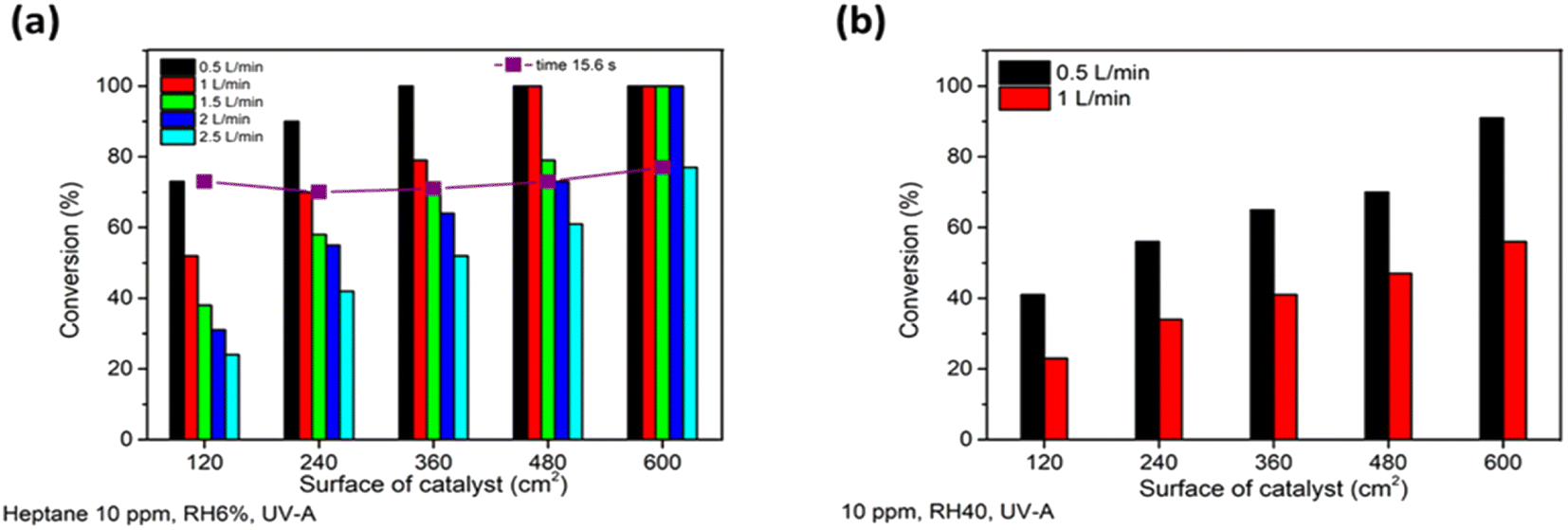 | ||
| Fig. 3 The effect of air flow rate on photocatalytic oxidation of heptane at different photocatalyst surface areas of TiO2 thin film at RH 6% (a) and the effect of increased relative humidity (RH 40%) on heptane oxidation at different photocatalyst surfaces. The average values of heptane conversion are shown. The standard deviation was < 5%. | ||
An increase in air flow rate resulted in faster oxidation of heptane (Fig. 3a and Table 2). As can be seen in the last row in Table 2, complete oxidation of 10 ppm of heptane at an air flow rate of 2 L min−1 occurred in twice as short a time interval (reaction time between 15.6 and 19.5 s) compared to the air flow rate of 0.5 L min−1 (reaction time between 31.2 and 46.8 s) (Fig. 3a and Table 2). Although it should be taken into account that the applied photocatalytic surface area is higher at an air flow rate of 2.0 L min−1 and air passed more sections of the reactors, which means more frequent mixing took place at the same reaction time. A decrease in reaction time means less time for adsorption and oxidation on the catalyst surface. However, on the other hand, if the amount of ROS does not limit the surface reaction, then increased mass transfer could promote the photocatalytic oxidation.43 As mentioned above, heptane degradation at an air flow rate of 0.5 L min−1 was not limited by the concentration of ROS, which proves that a higher air flow rate should enhance the overall reaction rate.
| Number of reactor sections | Surface of catalyst (cm2) | Reaction time of pollutant (s) at different air flow rates | ||||
|---|---|---|---|---|---|---|
| 0.5 L min−1 | 1 L min−1 | 1.5 L min−1 | 2 L min−1 | 2.5 L min−1 | ||
| 1 | 120 | 15.6 | 7.8 | 5.2 | 3.9 | 3.12 |
| 2 | 240 | 31.2 | 15.6 | 10.4 | 7.8 | 6.24 |
| 3 | 360 | 46.8 | 23.4 | 15.6 | 11.7 | 9.36 |
| 4 | 480 | 62.4 | 31.2 | 20.8 | 15.6 | 12.48 |
| 5 | 600 | 78 | 39 | 26 | 19.5 | 15.6 |
| Specific reaction time (s cm−2) | 0.13 | 0.065 | 0.043 | 0.033 | 0.026 | |
| Reaction time for complete degradation of 10 ppm of heptane (s) | 31.2–46.8 | 23.4–31.2 | 20.8–26 | 15.6–19.5 | Not achieved | |
An increase in the air flow rate from 0.5 to 2.5 L min−1 decreased the reaction time of the pollutant in the reactor from 15.6 to 3.12 s (Table 2), while it intensified the mass transfer. For the current reactor design, with an increase in air flow rate from 0.5 to 2.5 L min−1 air was still flowing in a laminar way: i.e. air layers slide in parallel with no swirls or currents normal to the catalyst surface (the Reynolds number changed from 19 to 97; calculations are shown in ESI†). Laminar air flow results in non-optimal mass transfer of pollutants to the catalyst surface. However, this represents a real indoor application in the form of windowpanes, when air is moving in the room by natural convection with periodic mixing by a fan, for example.
Moreover, the design of the multi-section reactor used in the current study allows us to obtain the same reaction time of 15.6 seconds by increasing the air flow rate by 0.5 L min−1 and the surface of the catalyst by 120 cm2 simultaneously (adding one more section to the reactor) (Table 2). This means that the reaction time of a pollutant molecule per unit of catalyst, i.e. the specific reaction time (Table 2), decreases, while the reaction time stays the same. In Fig. 3a it can be seen that at the same reaction time of 15.6 seconds (the purple line in Fig. 3a) at different air flow rates the conversion of 10 ppm of heptane was about 70%. This suggests that a higher volume of air could be purified by increasing the surface of the catalyst and by mass transfer. Applying a higher catalyst surface helps to overcome the challenge of the low contact time between the pollutants and the catalyst surface in continuous flow reactors and gives a good representation of TiO2 film application in indoor air purification.
The effect of water vapour was studied by increasing the relative humidity (RH) of air from 6 to 40% at an initial heptane concentration of 10 ppm at air flow rates of 0.5 and 1 L min−1 under UV-A (Fig. 3b). Fig. 3 implies that an increase in water vapour content in air from 6 (Fig. 3a) to 40% (Fig. 3b) inhibited the oxidation of heptane. The initial QE of heptane decreased by 35 and 57% at air flow rates of 0.5 and 1 L min−1, respectively, with an RH increase from 6 to 40%. The negative impact of an RH increase was intensified 1.6 times with an air flow rate increase from 0.5 to 1 L min−1; thus the oxidation of heptane by active sites of the photocatalyst is limited because of the adsorption of water molecules. A similar tendency of a negative water vapour effect on heptane photooxidation due to competitive adsorption on the surface was observed in other studies.7,40
The obtained TiO2 film showed promising performance in heptane photocatalytic oxidation under different operating conditions under UV-A light. However, for real applications in indoor conditions, oxidation under visible (VIS) light is more essential. To study TiO2 film activity under VIS light, heptane oxidation at concentrations of 5 and 10 ppm at an air flow rate 0.5 L min−1 and RH 6% was studied (Fig. 4).
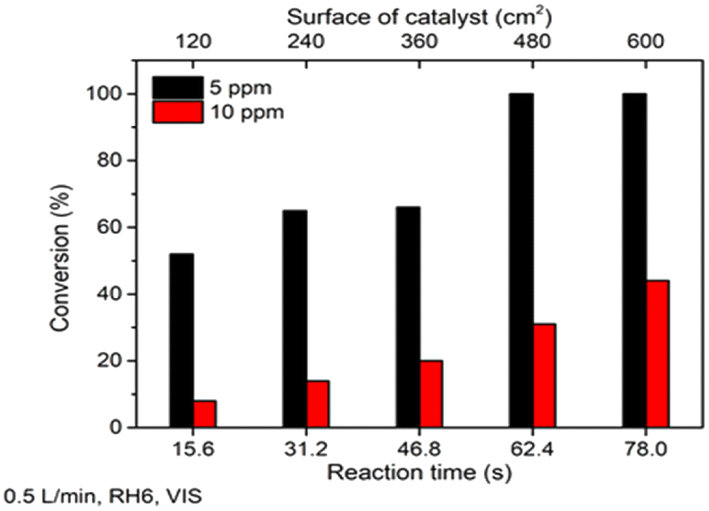 | ||
| Fig. 4 The effect of initial concentration on photocatalytic oxidation of heptane under visible light at different photocatalyst surfaces. The average values of heptane conversion are shown The standard deviation was <5%. | ||
Fig. 4 shows that TiO2 film could completely oxidize 5 ppm of heptane at a catalyst surface of 480 cm2 (reaction time 62.4 s) under VIS light. An increase in heptane initial concentration from 5 to 10 ppm led to 45% conversion of the pollutant at a catalyst surface of 600 cm2 (reaction time 78 s), which means oxidation of ca. 4.5 ppm of heptane (Fig. 4). These results indicate that the prepared TiO2 film is active under VIS light. The VIS light photocatalytic activity of the TiO2 film could be explained by a synergistic effect between carbon species and TiO2. That is, carbon absorbs VIS light and generates electrons, which are then transferred to the valence band of TiO2.20–22
As expected, the activity under VIS was lower than that under UV-A. QE for 5 ppm of heptane were 9.74 × 10−5 and 3.44 × 10−5 molecules per photons under UV-A and VIS light, respectively. At a concentration of 10 ppm under UV-A, QE increased to 1.50 × 10−4 molecules per photons, while under VIS light it decreased to 1.60 × 10−5 molecules per photons. If in the case of UV-A the oxidation process was not limited by the concentration of ROS, then under VIS light not enough ROS are produced and thus, the process is surface reaction limited. However, the results demonstrate that the film completely oxidized heptane at an initial concentration of 5 ppm under VIS light, which makes its application suitable for indoor air treatment for VOCs.
According to the literature, the formation of propanal, butanal, 3-heptanone, 4-heptanone and CO could follow the oxidation of heptane by TiO2.7 However, in the current study no gaseous by-products except CO2 and H2O were detected on the FTIR spectra of purified air during the performed experiments. Moreover, long-term experiments with 40 ppm of heptane at a catalyst surface of 120 cm2 (air flow rate 0.5 L min−1, RH 6%, UV-A) were performed to study the deactivation of the photocatalyst. The conversion of heptane was ca. 43% and did not change during 180 min of continuous use. This means that at a residence time of 15.6 s and catalyst surface of 120 cm2, TiO2 film can continuously oxidize 17 ppm of heptane out of 40 ppm with no deactivation during the experimental run time up to 180 min.
To summarise this section, TiO2 film synthesized in the current study can continuously oxidize such model pollutants as heptane under both UV-A and VIS light under different experimental conditions without forming any toxic by-products.
3.3 Photocatalytic oxidation of toluene
The photocatalytic oxidation of toluene at different initial concentrations and RH and with the addition of ozone on the TiO2 thin film was studied. Similar to heptane oxidation, no by-products except CO2 and H2O were detected on the FTIR spectra of purified air during the experiments with toluene.The photocatalytic oxidation of toluene at initial concentrations of 5, 10, 20 and 40 ppm at an air flow rate of 0.5 L min−1 and RH 6% was studied under UV-A (Fig. 5). Fig. 5a reveals that it was possible to partially oxidize toluene at initial concentrations in the range 5–40 ppm on the TiO2 thin film. 3.5 ppm out of 5 ppm was converted after 78 s in the reactor; 16.8 ppm was oxidized at the same time when the initial concentration was increased to 40 ppm (Fig. 5a). The maximum conversion of ca. 70% was achieved at a catalyst surface of 600 cm2 at an initial toluene concentration of 20 ppm (Fig. 5a). Toluene photocatalytic oxidation is more complicated compared to straight-chain compounds such as heptane. Moreover, the toluene oxidation pathway contains intermediates, which can block the active site of the photocatalyst and deactivate it.8,44 During photocatalytic toluene decomposition, gas-phase oxidation by-products are generated. Furthermore, during the toluene gas-phase oxidation, solid-phase partial oxidation products can form and adsorb on the surface of the photocatalyst causing its deactivation. The by-products of toluene monitored in the gas phase are benzaldehyde, methanol, acetaldehyde, acetone, acetic acid, butyraldehyde, benzene. Benzoic acid, benzene, acrylaldehyde, butyraldehyde and pentanal could adsorb on the surface of TiO2 during the oxidation of toluene.44
 | ||
| Fig. 5 The effect of initial concentration on the photocatalytic oxidation of toluene at different catalyst surfaces (a) and QE of toluene degradation at different initial concentrations at a photocatalyst surface of 120 cm2 (b). The average values of toluene conversion are shown. The standard deviation was <5%. | ||
The increase in conversion with an increase in the surface of the catalyst (reaction time) was negligible (Fig. 5); most of the toluene was converted after the first section of the reactor (reaction time 15.6 s). At the highest initial concentration (40 ppm), the conversion at all sections of the reactor was almost the same at ca. 40% (Fig. 5a). This could indicate that toluene and its intermediate products are absorbed on the surface of TiO2, blocking the active sites. Hence, no intermediate products of toluene were detected in the FTIR spectra of treated air during all the experimental runs, so it could be assumed that the formation of mostly solid-phase partial oxidation products takes place.
The QE at the first section of the reactor increased with the growth in the initial concentration (Fig. 5b). Thus, the film is capable of oxidizing some amount of toluene at a catalyst surface of 120 cm2. Then ROS produced by additional surface of the catalyst mostly compensate for the number of ROS which were involved in the oxidation of toluene and blocked by solid intermediate products in the previous section of the reactor.
The possible deactivation of a catalyst surface by toluene by-products has been reported in the literature.8,20,45,46 For example it was found by M. D. Hernández-Alonso et al. in the case of TiO2 films deposited by the sol–gel method that the adsorption of a toluene by-product benzoic acid decreased the initial reaction rate from 0.14 to 0.01 ppm s−1 after 180 min of measurements.20
Several studies on powdered materials with high material quantity have reported a color change of the photocatalyst and a decrease in toluene conversion.8,45,46 Kask et al., in their study achieved complete degradation of 20 ppm of toluene at a catalyst surface of 240 cm2 under similar experimental conditions to those in the current study for 1 μm P25 TiO2-covered glass. They observed the yellowish surface of the catalyst after a 190 min experimental run, which probably indicates the deactivation of the catalyst by intermediates of toluene oxidation.8 Einaga et al. observed a decrease in toluene conversion from 66 to 9% after 120 min of continuous use with P25 powder applied to the wall of a flow-type reactor. They suggested that deactivation takes place due to the formation of less reactive intermediates, which can be detected as carbon deposits on the surface of the catalyst.45
In the current study, no changes in film color were observed, which could be due to the low material quantity and transparency of the synthesized material. However, no decrease in photocatalytic activity could mean that the intermediates of toluene oxidation adsorbed on the film surface are degradable. The toluene oxidation rate was lower than that of heptane (Section 3.2, Fig. 2). The reaction rate (k = 0.895 ppm s−1) and adsorption (K = 0.028 ppm−1) constants of toluene decreased by almost two times compared to heptane. However, this is still higher than other kinetic constants reported in the literature for toluene. For example, Sleiman et al. reported a reaction rate constant of 3.55 ppb s−1 for toluene and an adsorption constant of 0.0025 ppb−1 for an annular-flow-through reactor with PC500 TiO2 immobilized on fiber paper.47 In another study by Garlisi and Palmisano of TiO2 thin film with a thickness of 20 nm, the photocatalytic oxidation of toluene with an initial concentration of 0.5 ppm obtained an initial reaction rate of 3.6 × 10−6 ppm s−1.10 Ku and co-authors obtained 26% conversion of toluene with an initial concentration of 41.13 ppm on P25 TiO2 powder spin coated onto Pyrex glass with a material quantity of 1.3 mg cm−2 at an air flow rate of 0.4 L min−1 (reaction time ca. 2 min) and RH 5%.48 A detailed comparison of the thin film prepared in this study with other thin films to oxidize toluene is presented in ESI Table SI-I.†
The film synthesized in the current study has a material quantity of about 0.2 mg cm−2, whereas the initial reaction rate for 40 ppm of toluene oxidation is more than 10 times higher compared to the study by Ku and co-authors, who obtained an initial reaction rate of 0.1 ppm s−1. This confirms that the activity of the thin film under study is competitive with coatings prepared by powder immobilization.
It is generally known that toluene photocatalytic oxidation could be limited due to the low reactivity of the aromatic ring and deactivation of the catalyst by toluene intermediate products.8,45–48 Several studies claim that water molecules could help to protect the photocatalyst from deactivation since they prevent the adsorption of intermediate products.44,47
To increase the efficiency of toluene degradation, initially the RH of polluted air was increased from 6 to 40%; the study was carried out with an initial toluene concentration of 10 ppm and air flow rate of 0.5 L min−1 (Fig. 6).
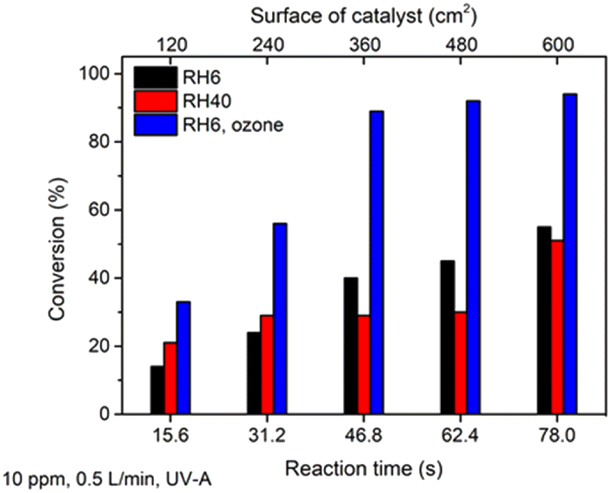 | ||
| Fig. 6 The effect of an increase in RH and addition of ozone on the photocatalytic oxidation of toluene at different photocatalyst surface areas. The average values of toluene conversion are shown. The standard deviation was <5%. | ||
Fig. 6 shows that an increase in RH from 6 to 40% resulted in slight changes in toluene conversion (not more than 15%). If in the first two sections of the reactor (reaction times 15.6 and 31.2 s) conversion was increased at RH 40%, then starting from a reaction time of 46.8 s the conversion was higher in drier air. It can be concluded that no significant effect of an increase in water vapour content in air on toluene oxidation was detected in the current study. However, it was reported by Sleiman et al. that for toluene with a high initial concentration (499 ppm) an increase in RH from 0 to 70% enhanced the decomposition rate.47
Another common additive to promote photocatalytic toluene degradation is a strong oxidant like ozone. The positive effect of ozone on the photocatalytic degradation of toluene for a P25 TiO2 photocatalyst has been studied by several authors.8,26 Therefore, the ozone was generated by a UV-C lamp and mixed with polluted air, giving an initial concentration of ozone in the polluted air of ca. 50 ppm. The results showed that addition of small amount of ozone as a strong oxidant in combination with TiO2 photocatalysis increased the conversion of 10 ppm of toluene from 51 to 94% at a catalyst surface of 600 cm2 (Fig. 6). Applying only 50 ppm of ozone or ozone combined with UV-A without a TiO2 photocatalyst did not result in toluene decomposition. Thus, it could be concluded that the photocatalytic oxidation of toluene by ROS on the TiO2 surface is the main degradation mechanism. Ozone is depleted on the surface of TiO2 film and forms additional radicals (O·) to protect the catalytic surface from fast deactivation. Probably ozone helps to avoid rapid blocking of the active sites by toluene degradation intermediates, thus enhancing the oxidation ability of the catalyst. According to eqn (3) and (4) for toluene oxidation, 36 OH· are needed, while the number of O· molecules needed for toluene oxidation is 18.
| 36OH· + C7H8 → 7CO2 + 22H2O | (3) |
| 18O· + C7H8 → 7CO2 + 4H2O | (4) |
Prepared by spray pyrolysis, the TiO2 thin film used in the current study is able to significantly reduce the amount of toluene in air with an initial concentration up to 40 ppm without forming any detectable gas-phase intermediate products, which makes it a suitable candidate for indoor air purification.
3.4 Photocatalytic oxidation of air mixtures
Indoor air could contain hundreds of different organic pollutants in very small concentrations. To mimic the real conditions of indoor air pollution, the degradation of two synthetic air mixtures of 9 ppm of heptane–acetone–acetaldehyde (Fig. 7a) and 9 ppm of toluene–acetone–acetaldehyde (Fig. 7b) were studied. The multicomponent mixtures of synthetic air were analysed to investigate how components interfere with each other. Moreover, how compounds in a multicomponent mixture with different polarities and hydrophilicities affect the photocatalytic performance was studied under different operating conditions.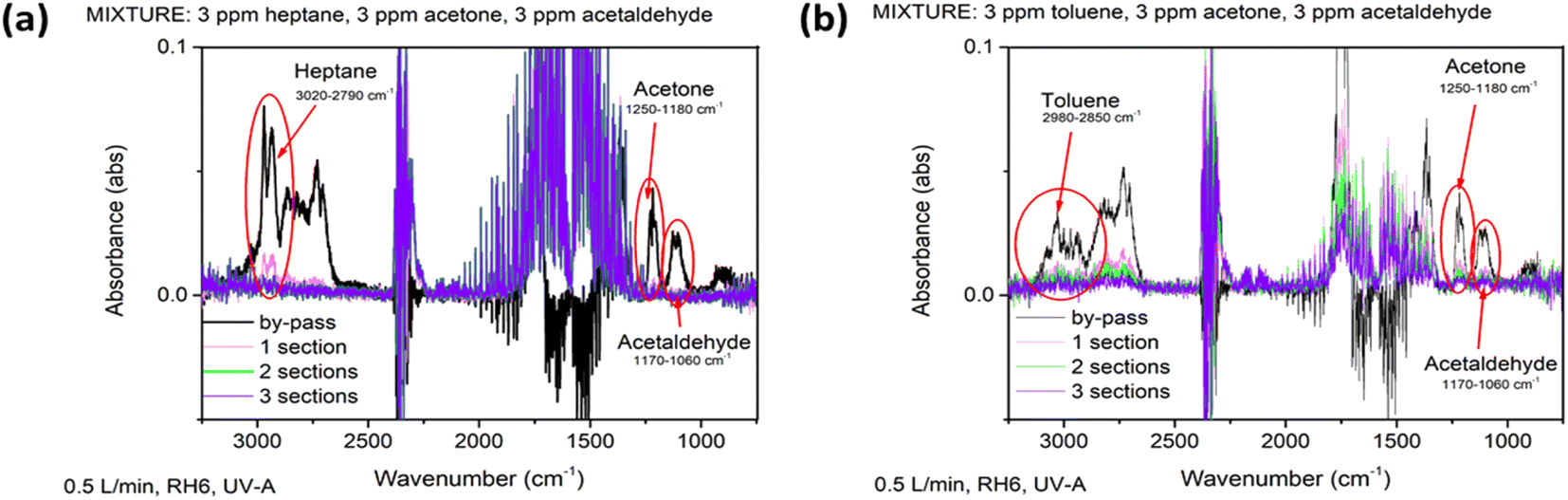 | ||
| Fig. 7 FTIR spectra of heptane–acetone–acetaldehyde (a) and toluene–acetone–acetaldehyde (b) mixtures passing through the by-pass line and through the reactors under UV-A irradiation on the TiO2 film. | ||
All pollutants in both studied mixtures were completely oxidized at RH 6% and an air flow rate of 0.5 L min−1 under UV-A, if three sections of the reactor were applied (catalyst surface of 360 cm2) (Fig. 7 and ESI Tables SI-2 and SI-3†). Complete oxidation of acetaldehyde had already been achieved after the polluted air had passed through the first section of the reactor (catalyst surface of 120 cm2), when it was in the mixture with heptane, while the conversion of acetone and heptane were 93 and 77%, respectively (Fig. 7a and Table SI-2†). The conversions of acetaldehyde, acetone and toluene in the mixture with toluene at the same catalyst surface (120 cm2) were 78, 70 and 71%, respectively (Fig. 7b and Table SI-3†). The degradation rate of compounds in the mixture with heptane followed the order: acetaldehyde, acetone, heptane with QE of 5.6, 5.2, 4.3 × 10−5 molecules per photons, respectively (Table 3). Acetaldehyde and acetone having simpler molecular structures are expected to be oxidized faster in the mixture. However, the QE of acetone and acetaldehyde degradation decreased 25 and 22%, respectively, when these compounds were in the mixture with toluene (Tables 3 and 4). The degradation of acetone and acetaldehyde molecules in the mixture with toluene probably decreased due to competitive adsorption on the surface of the photocatalyst. Toluene by-products oxidize more slowly than the initial compound, so these could adsorb on the surface of the photocatalyst inhibiting the adsorption of acetone and acetaldehyde.
| Operating parameters (air flow rate, RH, irradiation source) | Quantum efficiency, × 10−5 molecules per photons | ||
|---|---|---|---|
| Heptane | Acetone | Acetaldehyde | |
| 0.5 L min−1, RH6, UV-A | 4.282 | 5.170 | 5.560 |
| 1 L min−1, RH6, UV-A | 12.46 | 14.01 | 14.01 |
| 0.5 L min−1, RH40, UV-A | 1.683 | 1.703 | 2.568 |
| 0.5 L min−1, RH6, VIS | 0.797 | 1.791 | 2.085 |
| Operating parameters (air flow rate, RH, irradiation source) | Quantum efficiency, × 10−5 molecules per photons | ||
|---|---|---|---|
| Toluene | Acetone | Acetaldehyde | |
| 0.5 L min−1, RH6, UV-A | 3.970 | 3.895 | 2.232 |
| 1 L min−1, RH6, UV-A | 10.59 | 8.934 | 8.946 |
| 0.5 L min−1, RH40, UV-A | 2.056 | 1.790 | 2.322 |
| 0.5 L min−1, RH6, VIS | 1.514 | 1.318 | 1.077 |
To study the synergetic effect of the VOC mixtures on photocatalytic oxidation in more detail, the decomposition of VOC mixtures under different experimental conditions was studied. The effects of air flow rate, water vapour and irradiation source on the QE and conversions of VOCs in the mixtures are presented in Tables 3 and 4 and in ESI Tables SI-2 and SI-3.† In the heptane–acetone–acetaldehyde mixture, the degradation reaction rate under all operating conditions followed the order (from the highest to lowest): acetaldehyde, acetone, heptane (Tables 3 and SI-2†). In the toluene–acetone–acetaldehyde mixture, toluene affected the degradation rates of acetone and acetaldehyde and the values of the QE of the components were very close (Tables 4 and SI-3†).
An increase in the air flow rate from 0.5 to 1 L min−1 resulted in higher QE for all compounds in both mixtures (Tables 3 and 4). However, the increase in QE of heptane and toluene degradation with increased mass transfer was higher than for acetone and acetaldehyde. At an air flow rate of 1 L min−1, RH 6%, under UV-A, the QE of toluene was even higher than that of acetone and acetaldehyde (Table 4). This indicates that for less polar compounds, such as heptane and toluene, compared to oxygen-containing compounds, the intensification of mass transfer favors their oxidation. The same was found for individual pollutants as RH increases from 6 to 40%: the QE of all compounds in the mixtures decreased by about twice (Tables 3 and 4). This also confirms the competitive adsorption of compounds on the surface of TiO2 with water vapor. The QE of all compounds decreased drastically under VIS light (Tables 3 and 4), showing that the amount of ROS produced is limited under VIS light. However, with increased residence time in the reactor (catalyst surface of 600 cm2) the average conversion of the pollutants in the mixture with heptane and toluene under VIS light reached 92 and 45%, respectively (Tables SI-2 and SI-3†). That is, 3 ppm of acetaldehyde, 3 ppm of acetone and 2.3 ppm of heptane were oxidized in the first mixture at a catalyst surface of 600 cm2 (air flow rate 0.5 L min−1, RH6%) (Table SI-2†). In the second mixture, 1.6 ppm of acetaldehyde, 1.6 ppm of acetone and 0.9 ppm of toluene were oxidized under the same conditions (Table SI-3†).
There are only a few studies, where photocatalytic oxidation of VOCs in air mixtures is investigated and most use modified TiO2 powders under UV-A.7,8,11,25 For, example, the photocatalytic oxidation of toluene, decane and trichloroethylene separately and in mixtures was studied by Debono et al. in a batch reactor with 100 mg of P25 TiO2 powder load. In general, the oxidation of pollutants in the mixture was up to twice as slow as its degradation as a single compound. As in the current study, they observed sequential degradation of VOCs, which related to the competitive adsorption of different compounds on the surface of the photocatalyst.25 Liang et al. studied the degradation of 11 ppb of acetone, toluene and p-xylene in a mixture and separately on sol–gel TiO2 film. They found that degradation of p-xylene and acetone occurred at lower rates in the mixture, and the conversion of acetone and p-xylene decreased more than 15% in the mixtures. The opposite trend was observed for toluene, where the conversion increased by ca. 20%.8,11 Chen and Zhang studied the oxidation of a VOC mixture, which consisted of 16 different compounds, in a honeycomb reactor coated with a TiO2-based catalyst. The oxidation efficiency for all components in the mixture was lower compared to the single compounds. It was found that the interference effect is obvious and the molecules with higher affinity to the catalyst are more easily adsorbed on its active sites and oxidize faster.26
The TiO2 thin film prepared in this study degraded 9 ppm of VOCs in the mixture at a reaction time of 46.8 s (catalyst surface of 360 cm2, RH 6%, UV-A). A reaction time of 31.2 s was needed to oxidize 10 ppm of acetone or acetaldehyde, when these compounds were studied separately under the same experimental conditions.28 A reaction time of 46.8 s was needed for 10 ppm of heptane oxidation as a separate compound (Section 3.2, Fig. 2a) and ca. 4 ppm of toluene (initial concentration 10 ppm, Section 3.3, Fig. 5a) was oxidized at a reaction time of 46.8 s. The results of the current study demonstrate that TiO2 films could significantly decrease or completely photocatalytically oxidize the variety of VOCs under different operating conditions.
4 Conclusions
A sprayed TiO2 thin film prepared from a solution with a TTIP:AcacH molar ratio of 1:8 was thoroughly studied for its photocatalytic activity in the continuous flow gas-phase degradation of heptane and toluene as single compounds and in mixtures with acetone and acetaldehyde. Under UV-A light, heptane degradation was not hindered by an increase in its initial concentration up to 40 ppm and air flow rate up to 2.5 L min−1; only an increased content of water molecules in the air decreased the reaction rate. Under VIS light, 5 ppm of heptane were completely oxidized at a reaction time of 62.4 s; however in the case of 10 ppm, the photocatalytic oxidation was limited by the number of ROS produced. In the case of toluene, 70% of its 5 ppm initial concentration was degraded at a reaction time of 78 s under UV-A light. Toluene oxidation was hindered by the insufficient amount of ROS and the adsorption of less reactive degradation intermediates on the catalyst surface. The performance of the photocatalytic air treatment device could be augmented by decreasing the reaction time with a simultaneous increase in the surface area of the film. Variations in relative humidity from 6 to 40% were found not to have an effect on toluene degradation. However, the addition of 50 ppm of ozone increased the conversion of 10 ppm of toluene from 51 to 94%.
The degradation rate of compounds in mixtures with heptane followed the order (from the highest to lowest): acetaldehyde, acetone, heptane. However, in a mixture with toluene, the degradation of acetone and acetaldehyde was affected by toluene. The presence of aromatic compounds in the VOC mixture retards the degradation of otherwise easily degraded aliphatic compounds. An increase in air flow rate promotes the degradation of all compounds in both mixtures. The degradation rate of less polar poorly adsorbed VOCs could be enhanced by intensifying the mass transfer in the reactor. Higher humidity and visible light decreased the initial reaction rate of all compounds in both mixtures. This indicates the formation of a limiting amount of reactive species and competitive adsorption between the pollutants and their intermediate products on the surface of the catalyst.
Under UV-A (irradiance 3.5 mW cm−2), the synthesized TiO2 thin film at catalyst surfaces of 120–600 cm2 degrades 9 ppm of VOCs in the mixtures at a reaction time of 46.8 s. As individual compounds 10 ppm of acetone and acetaldehyde degrade at a reaction time of 31.2 s, 10 ppm of heptane at a reaction time of 46.8 s and ca. 4 ppm of toluene out of 10 ppm was oxidized at a reaction time of 46.8 s. Under VIS light in a mixture with heptane, 3 ppm of acetaldehyde, 3 ppm of acetone and 2.3 ppm of heptane were oxidized at a reaction time of 78 s. In a mixture with toluene under VIS light, oxidation of 1.6 ppm of acetaldehyde, 1.6 ppm of acetone and 0.9 ppm of toluene oxidation was achieved. TiO2 film prepared in the current study demonstrated excellent capability for successful application for the treatment of indoor air from low concentrations of VOC mixtures causing sick building syndrome.
This study confirms that TiO2 thin films obtained by a simple and cost-effective method of chemical spray pyrolysis are highly effective for indoor air purification from VOCs. Transparent TiO2 film with a material quantity of ca. 0.2 mg cm−2 shows competitive photocatalytic activity compared with a coating prepared from P25 powder and could continuously oxidize 9 ppm of VOC mixture. The obtained film can be widely applied as an innovative air purifying material in buildings or devices.
Author contributions
Jekaterina Sydorenko: formal analysis, investigation, writing—original draft, visualization. Arvo Mere: writing – review & editing. Malle Krunks: writing – review & editing. Marina Krichevskaya: conceptualization, methodology, writing – review & editing, supervision. Ilona Oja Acik: conceptualization, methodology, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Estonian Ministry of Education and Research, Estonian Research Council project PRG627 “Antimony chalcogenide thin films for next-generation semi-transparent solar cells applicable in electricity producing windows”, European Union's Horizon 2020 programme under the ERA Chair project 5GSOLAR, grant agreement No. 952509 and the Estonian Centre of Excellence project TK141 (TAR16016EK) “Advanced materials and high-technology devices for energy recuperation systems”.Notes and references
- W. Abou Saoud, A. Kane, P. Le Cann, A. Gerard, L. Lamaa, L. Peruchon, C. Brochier, A. Bouzaza, D. Wolbert and A. A. Assadi, Chem. Eng. J., 2021, 411, 128622, DOI:10.1016/j.cej.2021.128622.
- S. Domínguez-amarillo, J. Fernández-agüera, S. Cesteros-garcía and R. A. González-lezcano, Int. J. Environ. Res. Public Health, 2020, 17, 7183, DOI:10.3390/ijerph17197183.
- Y. Huang, S. S. H. Ho, R. Niu, L. Xu, Y. Lu, J. Cao and S. Lee, Molecules, 2016, 21, 56, DOI:10.3390/ijerph17197183.
- A. Maudhuit, C. Raillard, V. Héquet, L. Le Coq, J. Sablayrolles and L. Molins, Chem. Eng. J., 2011, 170, 464–470, DOI:10.1016/j.cej.2011.02.040.
- Y. Ji, A. Mattsson, G. A. Niklasson, C. G. Granqvist and L. Österlund, Joule, 2019, 3, 2457–2471, DOI:10.1016/j.joule.2019.06.024.
- S. Obregón and V. Rodríguez-González, J. Sol-Gel Sci. Technol., 2022, 102, 125–141, DOI:10.1007/s10971-021-05628-5.
- J. Shang, Y. Du and Z. Xu, Chemosphere, 2002, 46, 93–99, DOI:10.1016/S0045-6535(01)00115-1.
- M. Kask, J. Bolobajev and M. Krichevskaya, Chem. Eng. J., 2020, 399, 125815, DOI:10.1016/j.cej.2020.125815.
- T. Zou, C. Xie, Y. Liu, S. Zhang, Z. Zou and S. Zhang, J. Alloys Compd., 2013, 552, 504–510, DOI:10.1016/j.jallcom.2012.11.061.
- C. Garlisi and G. Palmisano, Appl. Surf. Sci., 2017, 420, 83–93, DOI:10.1016/j.apsusc.2017.05.077.
- W. J. Liang, J. Li and Y. Q. Jin, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2010, 45, 1384–1390, DOI:10.1080/10934529.2010.500925.
- M. Dell'Edera, C. Lo Porto, I. De Pasquale, F. Petronella, M. L. Curri, A. Agostiano and R. Comparelli, Catal. Today, 2021, 380, 62–83, DOI:10.1016/j.cattod.2021.04.023.
- A. H. Navidpour, A. Hosseinzadeh, J. L. Zhou and Z. Huang, Catal. Rev.–Sci. Eng., 2021, 1–52, DOI:10.1080/01614940.2021.1983066.
- S. D. Burnside, V. Shklover, C. Barbé, P. Comte, F. Arendse, K. Brooks and M. Grätzel, Chem. Mater., 1998, 10, 2419–2425, DOI:10.1021/cm980702b.
- V. Rodríguez-González, M. Sasaki, J. Ishii, S. Khan, C. Terashima, N. Suzuki and A. Fujishima, Chemosphere, 2021, 275, 129992, DOI:10.1016/j.chemosphere.2021.129992.
- F. He, W. Jeon and W. Choi, Nat. Commun., 2021, 12, 6259, DOI:10.1038/s41467-021-26541-z.
- T. D. Pham and B. K. Lee, Chem. Eng. J., 2017, 307, 53–73, DOI:10.1016/j.cej.2016.08.068.
- P. Peerakiatkhajorn, C. Chawengkijwanich, W. Onreabroy and S. Chiarakorn, Mater. Sci. Forum, 012, 712, 133–145, DOI:10.4028/www.scientific.net/MSF.712.133.
- Y. Y. Kannangara, R. Wijesena, R. M. G. Rajapakse and K. M. N. de Silva, Int. Nano Lett., 2018, 8, 31–39, DOI:10.1007/s40089-018-0230-x.
- M. D. Hernández-Alonso, I. Tejedor-Tejedor, J. M. Coronado and M. A. Anderson, Appl. Catal., B, 2011, 101, 283–293, DOI:10.1016/j.apcatb.2010.09.029.
- M. Marszewski, J. Marszewska, S. Pylypenko and M. Jaroniec, Chem. Mater., 2016, 28, 7878–7888, DOI:10.1021/acs.chemmater.6b03429.
- Y. Yuan, Z. H. Ruan, X. Huang, Y. Q. Jiang and H. P. Tan, J. Catal., 2017, 348, 246–255, DOI:10.1016/j.jcat.2016.12.022.
- L. Hua, Z. Yin and S. Cao, Catalysts, 2020, 10, 1431, DOI:10.3390/catal10121431.
- D. M. Tobaldi, D. Dvoranová, L. Lajaunie, N. Rozman, B. Figueiredo, M. P. Seabra, A. S. Škapin, J. J. Calvino, V. Brezová and J. A. Labrincha, Chem. Eng. J., 2021, 405, 126651, DOI:10.1016/j.cej.2020.126651.
- O. Debono, V. Hequet, L. Le Coq, N. Locoge and F. Thevenet, Appl. Catal., B, 2017, 218, 359–369, DOI:10.1016/j.apcatb.2017.06.070.
- W. Chen and J. S. Zhang, Build. Environ., 2008, 43, 246–252, DOI:10.1016/j.buildenv.2006.03.024.
- J. Spiridonova, A. Katerski, M. Danilson, M. Krichevskaya, M. Krunks and I. Oja Acik, Molecules, 2019, 24, 4326, DOI:10.3390/molecules24234326.
- J. Spiridonova, A. Mere, M. Krunks, M. Rosenberg, A. Kahru, M. Danilson, M. Krichevskaya and I. O. Acik, Catalysts, 2020, 10, 1011, DOI:10.3390/catal10091011.
- Philips, conventional lamps and tubes, special lamps, insect trap, actinic BL, Actinic BL TL(-K)/TL-D(-K) spec sheet. https://www.lighting.philips.com/api/assets/v1/file/PhilipsLighting/content/fp928024801029-pss-global/ADAM-20151211113552500%40en_AA.pdf. (accessed on 12 October 2022).
- Philips, conventional lamps and tubes, fluorescent lamps and starters, TL-D, TL-D 15W/827 1PP/10 spec sheet, https://www.lighting.philips.com/api/assets/v1/file/PhilipsLighting/content/fp927922282766-pss-global/ADAM-20151211035306769%40en_AA.pdf. (accessed on 12 October 2022) Search PubMed.
- J. Moon, T. Li, C. A. Randall and J. H. Adair, J. Mater. Res., 1997, 12, 189–197, DOI:10.1016/j.cej.2021.132766.
- A. O. Juma, I. O. Acik, V. Mikli, A. Mere and M. Krunks, Thin Solid Films, 2015, 594, 287–292, DOI:10.1016/j.tsf.2015.03.036.
- A. Léaustic, F. Babonneau and J. Livage, Chem. Mater., 1989, 1, 240–247, DOI:10.1021/cm00002a015.
- T. Olukan, J. Sydorenko, A. Katerski, M. Al Mahri, C.-Y. Lai, A. Al-Hagri, M. Chiesa and S. Santos, Appl. Phys. Lett., 2022, 121, 031901, DOI:10.1063/5.0098788.
- C. Di Valentin, G. Pacchioni and A. Selloni, Chem. Mater., 2015, 17, 6656–6665, DOI:10.1021/cm051921h.
- T. Dittrich, J. Sydorenko, N. Spalatu, N. H. Nickel, A. Mere, M. Krunks and I. Oja Acik, ACS Appl. Mater. Interfaces, 2022, 14, 43163–43170, DOI:10.1021/acsami.2c09032.
- R. Wang, M. Shi, F. Xu, Y. Qiu, P. Zhang, K. Shen, Q. Zhao, J. Yu and Y. Zhang, Nat. Commun., 2020, 11, 4465, DOI:10.1038/s41467-020-18267-1.
- R. Chen, J. Li, H. Wang, P. Chen, X. Dong, Y. Sun, Y. Zhou and F. Dong, J. Mater. Chem. A, 2021, 9, 20184, 10.1039/d1ta03705f.
- Z. Rao, G. Lu, L. Chen, A. Mahmood, G. Shi, Z. Tang, X. Xie and J. Sun, Chem. Eng. J., 2022, 430, 132766, DOI:10.1016/j.cej.2021.132766.
- I. Dundar, M. Krichevskaya, A. Katerski, M. Krunks and I. O. Acik, Catalysts, 2019, 9, 915, DOI:10.3390/catal9110915.
- N. Serpone, J. Photochem. Photobiol., A, 1997, 104, 1–12, DOI:10.1016/S1010-6030(96)04538-8.
- X. Dai, Y. Wang, X. Wang, S. Tong and X. Xie, Appl. Surf. Sci., 2019, 485, 255–265, DOI:10.1016/j.apsusc.2019.04.221.
- L. Zhong and F. Haghighat, Build. Environ., 2015, 91, 191–203, DOI:10.1016/j.buildenv.2015.01.033.
- J. Mo, Y. Zhang, Q. Xu, Y. Zhu, J. J. Lamson and R. Zhao, Appl. Catal., B, 2009, 89, 570–576, DOI:10.1016/j.apcatb.2009.01.015.
- H. Einaga, S. Futamura and T. Ibusuki, Appl. Catal., B, 2002, 38, 215–225, DOI:10.1016/S0926-3373(02)00056-5.
- S. Jõks, D. Klauson, M. Krichevskaya, S. Preis, F. Qi, A. Weber, A. Moiseev and J. Deubener, Appl. Catal., B, 2012, 111–112, 1–9, DOI:10.1016/j.apcatb.2011.09.007.
- M. Sleiman, P. Conchon, C. Ferronato and J. M. Chovelon, Appl. Catal., B, 2009, 86, 159–165, DOI:10.1016/j.apcatb.2008.08.003.
- Y. Ku, J. S. Chen and H. W. Chen, J. Air Waste Manage. Assoc., 2007, 57, 279–285, DOI:10.1080/10473289.2007.10465335.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06488j |
| This journal is © The Royal Society of Chemistry 2022 |