
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe smallest near-infrared fluorescence complementation system for imaging protein–protein and RNA–protein interactions†
Minghai
Chen
*a,
Chuang
Yan
a,
Luping
Zheng
a and
Xian-En
Zhang
 *bc
*bc
aCAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: mh.chen1@siat.ac.cn
bFaculty of Synthetic Biology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: zhangxe@ibp.ac.cn
cNational Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China
First published on 25th December 2021
Abstract
Bimolecular fluorescence complementation (BiFC) and its derivative molecular biosensor systems provide effective tools for visualizing biomolecular interactions. The introduction of red and near-infrared fluorescence emission proteins has expanded the spectrum of signal generating modules, enabling BiFC for in vivo imaging. However, the large size of the signal module of BiFC can hinder the interaction between proteins under investigation. In this study, we constructed the near-infrared BiFC and TriFC systems by splitting miRFP670nano, the smallest cyanobacteriochrome-evolved phytochrome available. The miRFP670nano-BiFC sensor system identified and enabled visualization of protein–protein interactions in living cells and live mice, and afforded a faster maturation rate and higher photostability and cellular stability when compared with those of reported near-infrared BiFC systems. We used the miRFP670nano-BiFC sensor system to identify interactions between the nucleocapsid (N) protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and cellular stress granule proteins in living cells and found that the N protein downregulated the expression level of granule protein G3BP1. With the advantages of small size and long wavelength emission of the signal module, the proposed molecular biosensor system should be suitable for various applications in cell imaging studies.
1. Introduction
Protein–protein interactions (PPIs) and RNA–protein interactions play pivotal roles during cell physiological and biochemical processes, such as virus–host interactions,1,2 gene expression and regulation3 and disease development.4 Monitoring these interactions in living subjects aids our understanding of the mechanisms responsible for biological events. Bimolecular fluorescence complementation (BiFC), which relies on the reconstruction of a fluorescent protein from two nonfluorescent parts fused with interacting proteins, has become a valuable and attractive method to investigate PPIs under physiological conditions because of its simplicity, high sensitivity and non-invasiveness.5,6 BiFC reporters have been developed from different fluorescent proteins and their mutants to study various PPIs in living cells or in live mice.5,7–10 In particular, the introduction of red, far-red and near-infrared fluorescent emission proteins (i.e., green fluorescence protein (GFP)-like and phytochromes) has expanded the spectrum of signal modules acting as molecular sensors and facilitated in vivo applications6–8,11–14 because the mammalian body has low tissue autofluorescence, low light scattering and minimal absorbance of melanin, water and hemoglobin in the near-infrared spectral region of ∼600–1200 nm, which is termed the “optical window” of mammalian tissue.15Currently, BiFC systems are primarily developed from GFP-like proteins or near-infrared phytochromes, which are large macromolecules (27–35 kDa).6,13,14 The fusion of large proteins has raised the question of whether the function of each protein is affected by its fusion partner. If this occurred in BiFC, the detection results would be questionable. The trimolecular fluorescence complementation (TriFC) to study the RNA–protein interactions derived from the BiFC also has similar problems.8,16,17 Intriguingly, Oliinyk et al. developed a near-infrared fluorescent protein from a single-domain of cyanobacteriochrome through directed molecular evolution, named miRFP670nano, which could efficiently bind the endogenous biliverdin chromophore and produced bright fluorescence in mammalian cells and live mice.18 miRFP670nano is a monomer with a molecular weight of 17 kDa, which is 2-fold smaller than bacterial phytochrome (BphP)-based near-infrared fluorescent proteins and 1.6-fold smaller than GFP-like fluorescent proteins. miRFP670nano also displays a faster maturation rate and higher photostability and cellular stability when compared with BphP-based near-infrared fluorescent proteins. Such properties make miRFP670nano an ideal tag for protein labeling both in living cells and in vivo.18
Taking advantage of these attributes, we created BiFC and TriFC sensing systems using miRFP670nano as the signal module. After intracellular and in vivo characterization, the systems were used to investigate interactions of N protein from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and host cell target proteins. The results are presented herein.
2. Results
Construction of the miRFP670nano based near-infrared BiFC system
We referred to the principles of BiFC construction using the crystal structure of miRFP670nano (PDB ID: 6MGH) because there are no reports of a BiFC system based on splitting the single domain of the near-infrared phytochrome. Six sites within unstructured loops of miRFP670nano were chosen to test the possibility of splitting miRFP670nano for use in BiFC (Fig. 1a). The fragment information is shown in Table S1.† The coding sequences of these reporter fragments in split miRFP670nano were inserted into the pcDNA3.1(+) vector. The well-known strongly interacting protein pair bJun and bFos was used as the interaction model to test the BiFC systems.5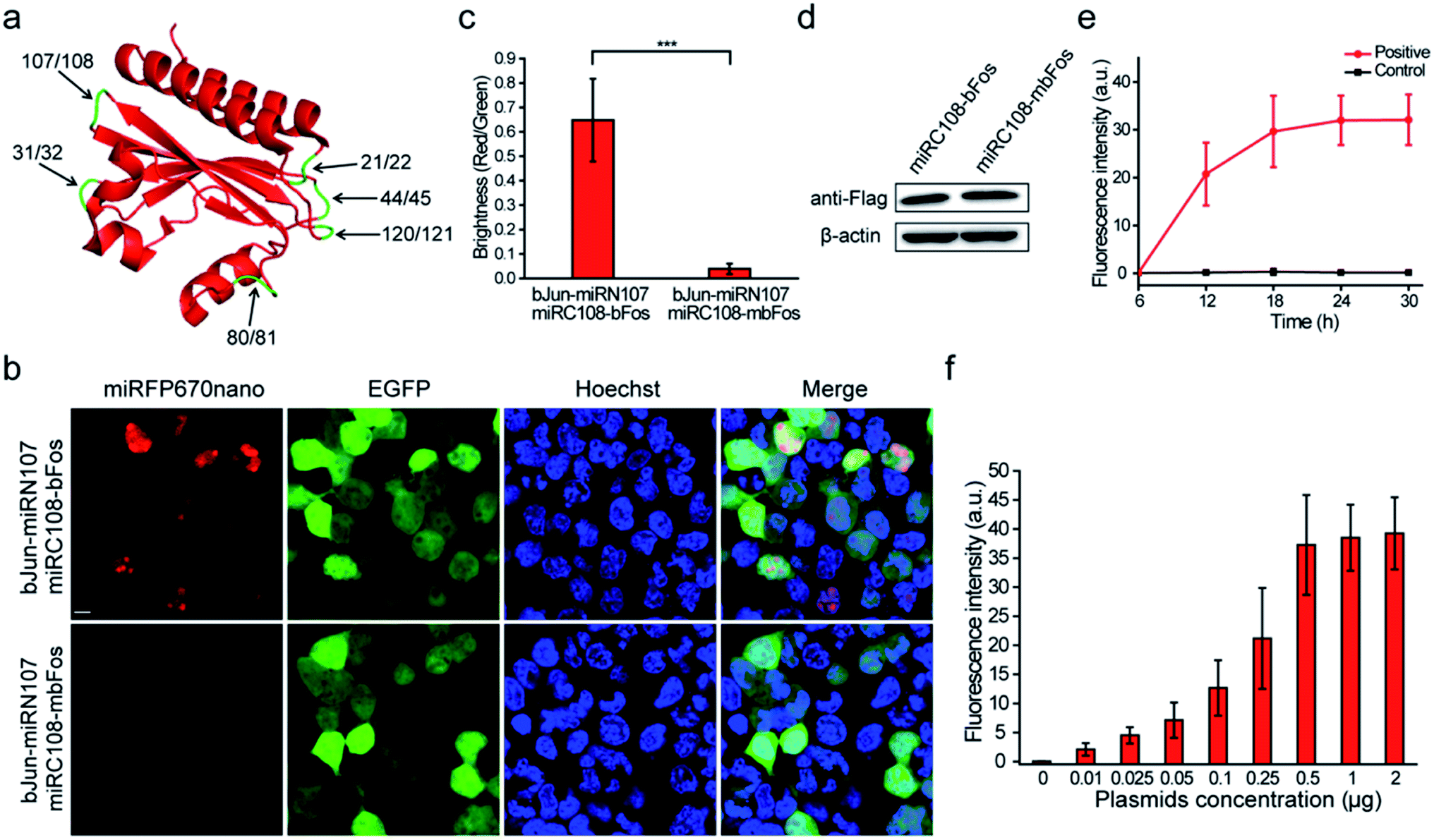 | ||
| Fig. 1 Construction of the miRFP670nano-BiFC system. (a) The split sites (arrows) in the unstructured loop regions of miRFP670nano are indicated. (b) BiFC signals (miRFP670nano channel) were detected in HEK 293T cells due to the bJun–bFos interaction. bJun–mbFos interaction (bJun and mbFos do not interact) was selected as the negative control. EGFP was also co-expressed in the cells as an internal control. (c) Quantitative analysis of the BiFC efficiency of the combinations of split miRFP670nano fragments fused with bJun–bFos or bJun–mbFos was calculated by dividing the red fluorescence intensity by green fluorescence intensity. (d) Expression of the miRFP670nano C-terminal split fragment miRC108 fused with bFos or mbFos determined by western blotting with an anti-Flag antibody. β-Actin was used as the loading control. (e) BiFC assay of bJun–bFos (positive) or bJun–mbFos (control) at different post-transfection time points. (f) BiFC assay of the bJun–bFos interaction in living HEK 293T cells when co-transfected with different amounts of pbJun-miRN107 and pmiRC108-bFos. Nuclei were stained with Hoechst 33342. Scale bar: 10 μm. All data are given as the mean ± S.D. (n = 30). The statistical significance was evaluated using a two-tailed Student's t-test. *** indicates p < 0.01. | ||
When the six sets of bJun-N and C-bFos proteins were co-expressed in HEK 293T cells, the combination of bJun-miRN107 and miRC108-bFos yielded a strong BiFC signal from the reconstitution of miRFP670nano (Fig. 1b). As a control, we also fused mbFos to miRC108 and co-expressed this protein with bJun-miRN107; mbFos is a mutated form of bFos that no longer interacts with bJun.5 As shown in Fig. 1b, no BiFC signal was detected in this control combination. During these experiments, pEGFP-N1 was also co-transfected into cells to express EGFP as an internal control. Statistical analysis of the complementary efficiency using the fluorescence intensity ratio of the miRFP670nano-BiFC system/-EGFP (i.e., red fluorescence/green fluorescence) produced in these assays confirmed significant differences in the BiFC signals between the bJun–bFos and bJun–mbFos groups (Fig. 1c). Flow cytometry analysis also acquired consistent results with the above fluorescence image analysis (Fig. S1†). Western blot analysis showed that the expression of bFos and mbFos was at the same level (Fig. 1d). We also co-transfected into cells the split fragments without fusion to bJun and bFos. No complementary signal was detected (Fig. S2†). The BiFC-reconstituted miRFP670nano had similar photophysical properties to native miRFP670nano (Fig. S3†). We also characterized the biophysical properties of this miRFP670nano-BiFC system with the native miRFP670nano, which are shown in Table S2.† Unlike some previously reported BiFC assays, which required pretreatment at low temperatures (e.g., 27 or 30 °C),8,9 the miRFP670nano-based BiFC system recovered its native fluorescence at 37 °C (physiological condition). For the other five splitting sites in miRFP670nano, the combination of bJun and bFos fusions produced no detectable BiFC signals (Fig. S4†). These findings showed that miRFP670nano can be split between amino acids (aa) 107 and 108 to build the smallest near-infrared BiFC system for imaging PPIs in living cells. This miRFP670nano-based BiFC system included a 12.5 kDa polypeptide fragment (termed miRN107) and a 4.6 kDa smaller peptide fragment (termed miRC108). The gene sequences of the new constructs of the miRFP670nano-BiFC system are shown in Fig. S5.†
The bJun–bFos interaction was also used to optimize the experimental conditions for the newly developed miRFP670nano-BiFC system. BiFC signals of the bJun–bFos interaction in living HEK 293T cells transfected with pbJun-miRN107 and pmiRC108-bFos were detected at different post-transfection time points. As shown in Fig. 1e, the BiFC signal was easily discriminated from the background at ∼12 h after transfection, and the signal increased from 12 to 24 h after transfection. The BiFC signal plateaued at about 24 h, which should be suitable for signal detection. We also tested plasmid use for the new miRFP670nano-BiFC system. Different amounts of pbJun-miRN107 and pmiRC108-bFos plasmids were cotransfected into living HEK 293T cells to image the bJun–bFos interaction. As shown in Fig. 1f, when low amounts of plasmids (∼0.01 μg) were used, detectable fluorescence was acquired. The corresponding western blot result of the protein expression levels is shown in Fig. S6.† All of these indicated that this novel miRFP670nano-BiFC system can be used to study PPIs at low expression levels.
In vivo imaging of PPIs using the miRFP670nano-BiFC system
We then tested the newly constructed miRFP670nano-BiFC system for imaging PPIs in live mice. Initially, the plasmid combination bJun-miRN107/miRC108-bFos/EGFP or bJun-miRN107/miRC108-mbFos/EGFP was co-expressed in HEK 293T cells and analyzed ex vivo using the IVIS Spectrum Imaging System. EGFP was also co-expressed in the cells as an internal control. The complementary fluorescence of the cells expressing bJun-miRN107/miRC108-bFos was higher when compared with cells expressing bJun-miRN107/miRC108-mbFos (Fig. 2a, left), while the corresponding cell lines exhibited similar EGFP signals (Fig. 2a, right). Statistical analysis of the miRFP670nano-based BiFC was consistent with the microscopy analysis. The BiFC/EGFP (i.e., red/green) fluorescence intensity ratio of the bJun–bFos group was significantly higher than that of the bJun–mbFos group (Fig. 2b).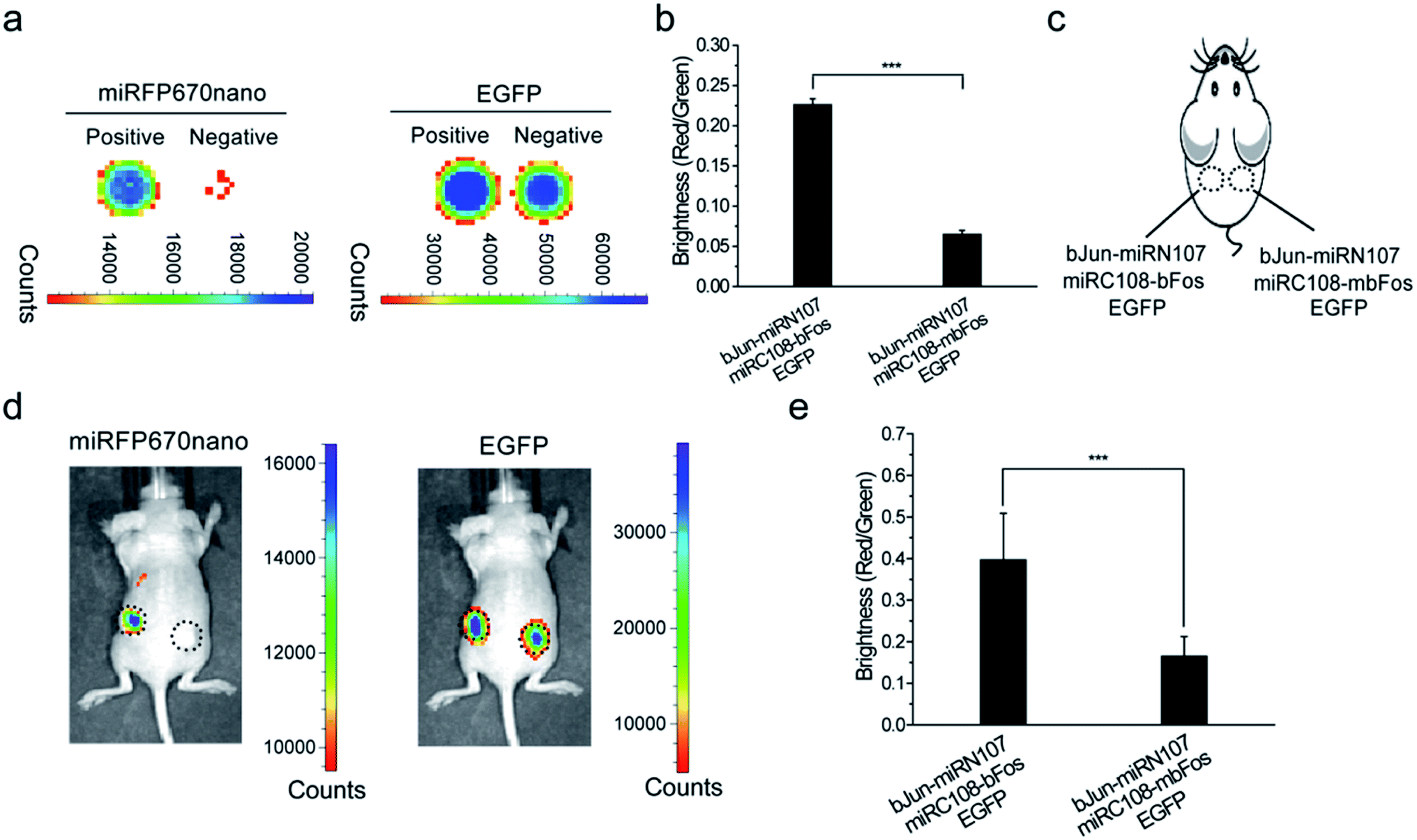 | ||
| Fig. 2 In vitro and in vivo imaging of PPIs using the miRFP670nano-BiFC system. (a) BiFC signals (miRFP670nano channel) and GFP signals (EGFP channel) of HEK 293T cells in plates co-expressing the protein interaction pair bJun–bFos (positive) or bJun–mbFos (negative). EGFP was also co-expressed in the cells as an internal control. (b) Quantitative analysis of BiFC signals in (a) based on the fluorescence intensity ratio BiFC/EGFP. (c) Implantation locations in mice of HEK 293T cell samples transiently co-expressing combinations of fusion proteins. (d) Imaging of the interaction of bJun and bFos with the miRFP670nano-BiFC system in live mice subcutaneously injected with cell samples. BiFC signals for the interactions of bJun-miRN107 and miRC108-bFos or miRC108-mbFos (mouse on the left), and EGFP signals corresponding to the cells at the same locations (mouse on the right). (e) Quantitative analysis of the BiFC signals arising from the interaction between bJun and bFos or mbFos in live mice based on the fluorescence intensity ratio BiFC/EGFP in (d). Quantitative data are given as the mean ± SD (n = 6). The statistical significance was evaluated using a two-tailed Student's t-test. *** indicates p < 0.01. | ||
Cells co-expressing each combination of the split proteins were then subcutaneously injected into mice for imaging (Fig. 2c). In live mice, the combination of bJun-miRN107/miRC108-bFos produced higher complementary fluorescence than the combination of bJun-miRN107/miRC108-mbFos (Fig. 2d, left), while each corresponding combination displayed similar EGFP signals (Fig. 2d, right). Quantitative analysis of the fluorescence intensity ratio of BiFC/EGFP (red/green) in mice also revealed that the fluorescence intensity of the bJun–bFos group was significantly higher when compared with the fluorescence intensity of the bJun–mbFos group (Fig. 2e).
Performance of miRFP670nano-BiFC compared with reported near-infrared BiFC systems in living cells
Following intracellular and in vivo characterization of the miRFP670nano-BiFC assay for imaging PPIs, we compared the performance of the miRFP670nano-BiFC assay against recently reported near-infrared BiFC systems. Phytochrome IFP1.4-, IFP2.0- and iRFP-based BiFC systems were used for comparison.12–14 Fluorescence intensities were detected 8 h after transfection of the cells expressing different BiFC systems. The fluorescence intensity of miRFP670nano-BiFC was 20.8-, 1.5- and 2.5-fold higher when compared with that of the IFP1.4-, IFP2.0- and iRFP-based BiFC systems (Fig. 3a and b), which suggests that the single-domain structure accelerates miRFP670nano-BiFC folding. Flow cytometry analysis also confirmed the above results (Fig. S7a†). In addition, western blot analysis showed that the fusion protein expressions of these systems were at a similar level (Fig. S7b†). miRFP670nano-BiFC also exhibited 3.0-, 2.1- and 1.4-fold higher photostability than IFP1.4-, IFP2.0- and iRFP-based BiFC in living cells, respectively (Fig. 3c). Furthermore, miRFP670nano-BiFC is highly stable in living cells. Protein degradation analysis showed that after 4 h incubation with a protein synthesis inhibitor cycloheximide, miRFP670nano-BiFC expressing cells retained ∼93% of their fluorescence (Fig. 3d). In contrast, cells expressing IFP1.4-, IFP2.0- and iRFP-based BiFC retained only ∼61%, ∼85% and ∼81% fluorescence, respectively (Fig. 3d). Overall, the fast maturation and high photostability and cellular stability make miRFP670nano-BiFC a favorable near-infrared signal module for imaging PPIs under physiological conditions.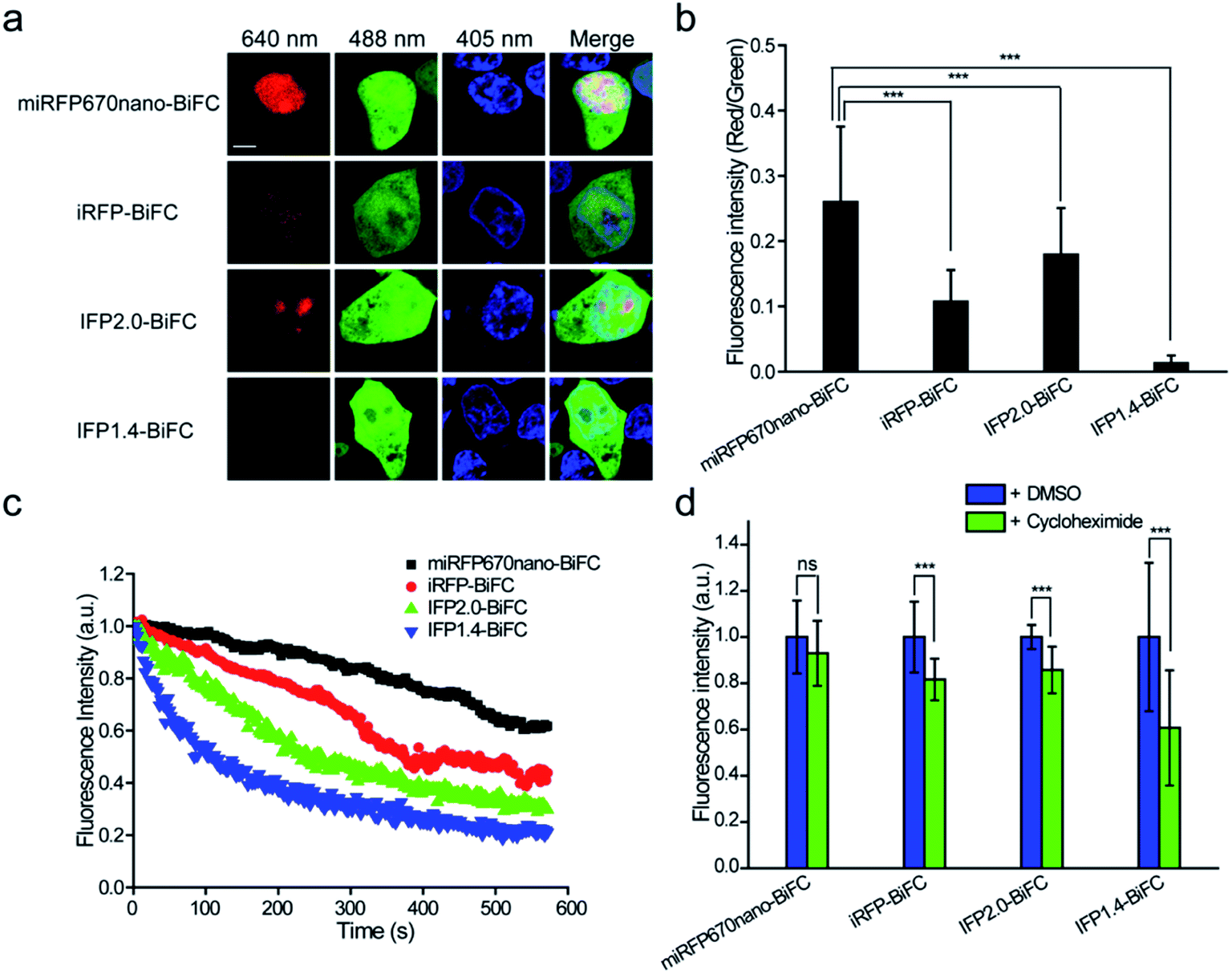 | ||
| Fig. 3 The performance of miRFP670nano-BiFC when compared with the performance of reported near-infrared BiFC systems. (a) The BiFC signals of miRFP670nano-BiFC, iRFP-BiFC, IFP2.0-BiFC and IFP1.4-BiFC systems were detected 8 h after transfection of the HEK 293T cells and arise from the bJun–bFos interaction. EGFP was co-expressed as the internal control. (b) Quantitative analysis of the complementary fluorescence efficiencies of the miRFP670nano-BiFC, iRFP-BiFC, IFP2.0-BiFC and IFP1.4-BiFC systems in (a). (c) Photostabilities of miRFP670nano-BiFC, iRFP-BiFC, IFP2.0-BiFC and IFP1.4-BiFC systems in living HEK 293 T cells. (d) Mean fluorescence intensities of HEK 293T cells transiently transfected with miRFP670nano-BiFC, iRFP-BiFC, IFP2.0-BiFC and IFP1.4-BiFC systems after 4 h incubation with cycloheximide or DMSO before imaging. Nuclei were stained with Hoechst 33342. Scale bar: 5 μm. Data are given as the mean ± S.D. (n = 20). The statistical significance was evaluated using a two-tailed Student's t-test. *** indicates p < 0.01. ns indicates not significant. | ||
Construction of the miRFP670nano-based TriFC system
Split miRFP670nano fragments were then tested as the reporter to construct the smallest near-infrared TriFC system in which fluorescence was recovered by the formation of RNA–protein interactions. The schematic principles of the TriFC system are shown in Fig. S8.† Here, the interactions between influenza A virus nonstructural protein 1 (NS1) and the 5′ untranslated region (UTR) of the matrix protein (M) and nucleoprotein (NP) mRNAs were used to validate the miRFP670nano-based TriFC system.8 The C-terminal fragment of miRFP670nano (miRC108) was fused to the bacteriophage MS2 coat protein (MCP) and tethered to its stem-loop RNA operator (ms2) by an MCP–ms2 interaction. A fusion between the N-terminal fragment of miRFP670nano (miRN107) and NS1 was also generated. The pECFP-C1 backbone plasmid was used to link the ms2 sequence and M or NP 5′ UTR. If the split site at aa 107–108 of miRFP670nano is suitable for the construction of the TriFC assay, the two fragments of miRFP670nano should come together and reconstitute to produce a TriFC signal based on the NS1-M/NP 5′ UTR interaction.(GGGGS)2 was used as the linker to build the plasmids pNS1-L-miRN107 and pmiRC108-L-MCP. The TriFC plasmids were co-transfected into HEK 293T cells. No complementary TriFC fluorescence signal was detected in the transfected cells (Fig. 4a). The length of the linker was extended to (GGGGS)4, which we denote as 2*L, and the plasmids were co-transfected into HEK 293T cells. Detectable complementary fluorescence was observed in the interaction assays of both NS1-M 5′ UTR and NS1-NP 5′ UTR (Fig. 4b). No TriFC signal was detected when the 5′ UTR of either the M or NP mRNA was absent (Fig. 4b). Quantitative analysis based on the fluorescence intensity ratio of miRFP670nano/ECFP demonstrated that the TriFC signal of cells co-expressing NS1 and the 5′ UTR of the M or NP mRNA was significantly higher when compared with that of cells expressing NS1 but no 5′ UTR of the M or NP mRNA (Fig. 4c). We also extended the length of the linker to (GGGGS)6, which is denoted as 3*L, in an effort to optimize the TriFC system. No detectable complementary fluorescence was detected for this system (Fig. 4d). Flow cytometry analysis also acquired similar results (Fig. S9†). These results indicated that miRFP670nano can be split at the aa 107–108 site with (GGGGS)4 as the fusion linker to yield the smallest near-infrared TriFC system.
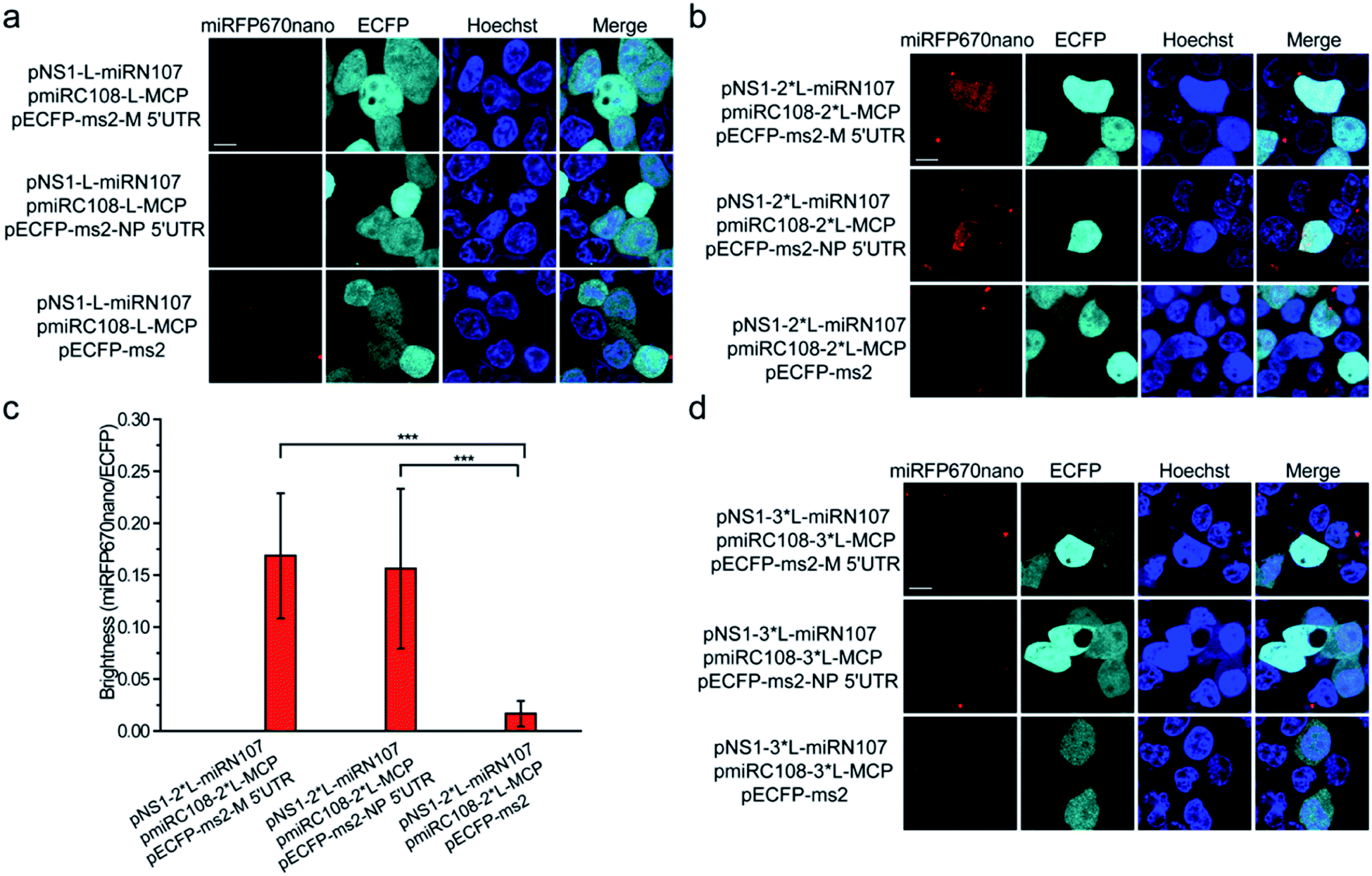 | ||
| Fig. 4 Construction of the miRFP670nano-TriFC system. (a) Detection of the influenza virus NS1-M 5′ UTR and NS1-NP 5′ UTR interaction in living cells. Cells were transfected with plasmids expressing MCP fused to the C-terminal complementing portion of miRFP670nano (pmiRC108-L-MCP) and the NS1 protein fused to the N-terminal complementing portion of miRFP670nano (pNS1-L-miRN107). The reporter mRNAs contained the ms2 operator and the 5′ UTR sequences of the M or NP mRNA. (GGGGS)2 was used as the linker to build the pNS1-L-miRN107 and pmiRC108-L-MCP plasmids and is denoted as “L”. (b) Cells were transfected with pmiRC108-2*L-MCP and pNS1-2*L-miRN107 plasmids and the reporter mRNAs. (GGGGS)4 was used as the linker to build pNS1-2*L-miRN107 and pmiRC108-2*L-MCP plasmids and is denoted as “2*L”. (c) Quantitative analysis of the TriFC system for the M/NP 5′ UTR and NS1 interaction. Statistical analysis was based on the fluorescence intensity ratio of TriFC/ECFP (miRFP670nano/ECFP). Data are given as the mean ± S.D. (n = 20). The statistical significance was evaluated using a two-tailed Student's t-test. *** indicates p < 0.01. (d) Cells were transfected with pmiRC108-3*L-MCP and pNS1-3*L-miRN107 plasmids and the reporter mRNAs. (GGGGS)6 was used as the linker to build pNS1-3*L-miRN107 and pmiRC108-3*L-MCP plasmids and is denoted as “3*L”. Nuclei were stained with Hoechst 33342. Scale bars: 10 μm. | ||
Identification and visualization of the interaction between the N protein of SARS-CoV-2 and cellular stress granule proteins in living cells
Next, the miRFP670nano-based BiFC system was used to identify and visualize the interaction between the N protein of SARS-CoV-2 and cellular stress granule proteins in living cells. The SARS-CoV-2 N protein is an RNA binding protein found in abundance that plays a variety of roles in the viral life cycle including replication, transcription, and genome packaging.19–21 Previous studies also found that manipulation of the stress granule and related RNA biology was common among Coronaviridae22–24 and stress granule formation was thought to be a primarily antiviral response. So, it is meaningful to investigate the interaction between N protein and cellular stress granule proteins in living cells to resolve the underlying mechanisms of SARS-CoV-2. Here, the cellular stress granule proteins CSNK2B, G3BP1 and G3BP2 were selected for identifying interactions with N protein. We first used Co-immunoprecipitation (Co-IP) and immunofluorescence (IF) assays to detect these interactions. As presented in Fig. 5a, all three proteins have specific interactions with N protein, as observed in the Co-IP assay. The IF assay also revealed that CSNK2B, G3BP1 and G3BP2 co-localize with N protein (Fig. 5b), and the Pearson correlation coefficient (PCC) was 0.87, 0.77 and 0.77 for CSNK2B, G3BP1 and G3BP2 with N protein, respectively. The graphics showing the quantification of the co-localization are shown in Fig. S10.† We also fused miRC108 to the N protein, and fused miRC107 to CSNK2B, G3BP1 and G3BP2 to identify interactions using our newly developed miRFP670nano-BiFC system. All three protein combinations produced strong complementary fluorescence of miRFP670nano (Fig. 5c), indicating specific interactions between these three proteins and N protein. The miRFP670nano-based BiFC system is a powerful tool for the detection of these interactions because of its simplicity, high sensitivity and non-invasiveness when compared with Co-IP and IF assays.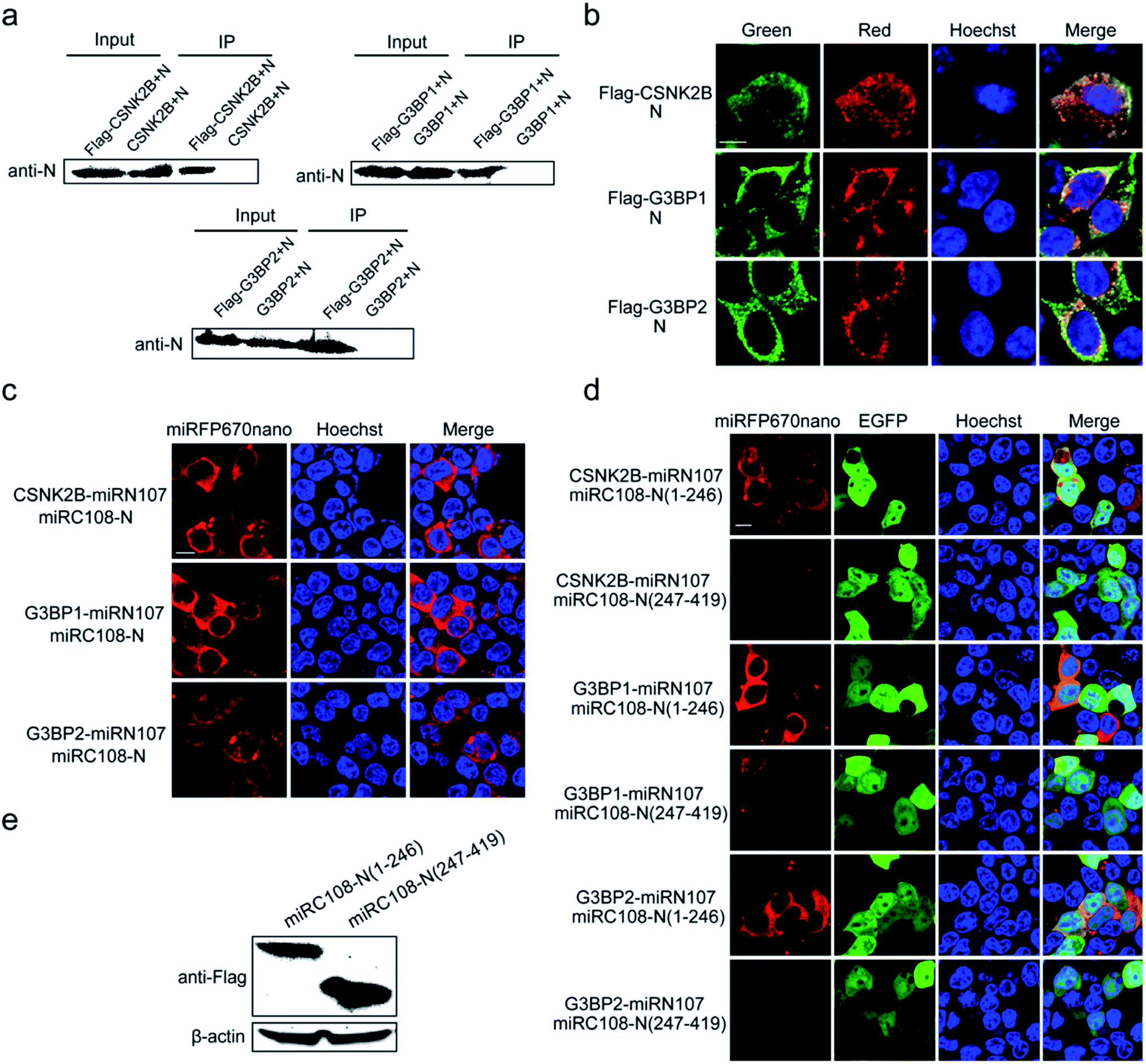 | ||
| Fig. 5 Identification of the N protein of SARS-CoV-2 interacting with cellular stress granule proteins. (a) Co-immunoprecipitation (Co-IP) assays were used to verify the interactions between cellular stress granule proteins and N protein. (b) Immunofluorescence (IF) assays were used to detect the co-localization of cellular stress granule proteins with N protein. Green represents the Flag-tag fused protein channel. Red represents the N protein channel. (c) Visualization of the interactions of cellular stress granule proteins CSNK2B, G3BP1 and G3BP2 with N protein by using the smallest miRFP670nano-BiFC system. (d) Identification and visualization of the regions of N protein that mediate interactions with CSNK2B, G3BP1 and G3BP2. EGFP was co-expressed as the internal control. (e) Expression levels of N(1–246) and N(247–419) fusion proteins were determined by western blotting with an anti-Flag antibody. β-Actin was used as the loading control. Nuclei were stained with Hoechst 33342. Scale bars: 10 μm. | ||
We then wanted to further identify the regions of N protein that mediate these interactions. According to the structure of N protein, which includes an RNA-binding domain and a dimerization domain,25 we generated two split region fragments named N(1–246) and N(247–419), which represent the aa sequences 1–246 and 247–419 of N protein, respectively. Bright red BiFC fluorescence signals were observed with the combinations of pCSNK2B-miRN107 and pmiRC108-N(1–246), pG3BP1-miRN107 and pmiRC108-N(1–246) and pG3BP2-miRN107 and pmiRC108-N(1–246) (Fig. 5d) but a negligible or weak fluorescence signal was detected with pmiRC108-N(247–419) combinations. Western blot analysis showed that the expression levels of N(1–246) and N(247–419) fusion proteins were similar (Fig. 5e). The above results confirmed that the aa sequence 1–246 of N protein mainly mediated interactions with CSNK2B, G3BP1 and G3BP2.
N protein of SARS-CoV-2 regulates the expression level of G3BP1 in cells
The N protein expression plasmid was transfected into HEK 293T cells to further explore the roles that these interactions play during the SARS-CoV-2 infection process. The mock vector, pcDNA3.1(+), was also transfected as the control. After 24 h transfection, cells were collected and western blot assays were performed to detect the expression levels of CSNK2B, G3BP1 and G3BP2. As shown in Fig. 6a, the expression levels of CSNK2B and G3BP2 in the N protein transfected cells were similar to the levels in the mock vector transfected cells. In contrast, there was a significant reduction of the G3BP1 expression level of N protein transfected cells when compared with that of mock vector transfected cells. Quantitative PCR (Q-PCR) experiments were carried out to detect the mRNA levels of these three proteins with NADPH used as the control. The mRNA level of G3BP1 in N protein transfected cells was significantly lower than that in the mock vector transfected cells, whereas the mRNA levels of CSNK2B and G3BP2 in N protein transfected cells were similar to those in the mock vector transfected cells (Fig. 6b). These results are consistent with the western blot results (Fig. 6a). SARS-CoV-2 may escape clearance of the host through the interaction between N protein and G3BP1 because cellular stress granule proteins play a key role in the antiviral process.26 This possible interaction and role in avoiding clearance requires further examination.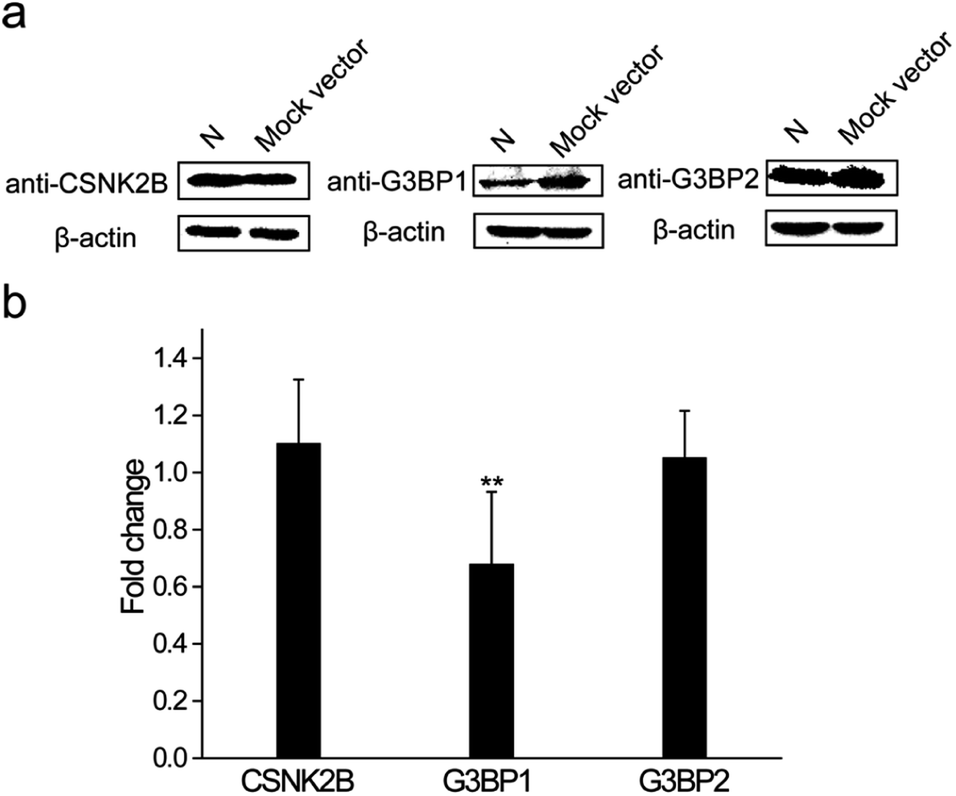 | ||
| Fig. 6 N protein of SARS-CoV-2 regulates the expression level of G3BP1 in cells. (a) Western blotting was used to determine the expression levels of cellular stress granule proteins CSNK2B, G3BP1 and G3BP2 in N protein or mock vector transfected cells. β-Actin was used as the loading control. (b) Q-PCR assays were used to determine the expression levels of cellular stress granule proteins CSNK2B, G3BP1 and G3BP2 in N protein or mock vector transfected cells. GAPDH was used as the control. The statistical significance was evaluated using a two-tailed Student's t-test. ** indicates p < 0.05. | ||
3. Discussion
In this study, we systematically screened for suitable split sites of miRFP670nano for the construction of BiFC and TriFC systems. The selected split sites were positioned in unstructured loops to ensure restoration of the 3D structure of miRFP670nano. Because miRFP670nano has a distinctly different structure to that of GFP-like and iRFP-like proteins, the selected split sites can be used as a reference for constructing similar types of molecular biosensor systems using analogous phytochromes.The newly constructed miRFP670nano-BiFC system represents the smallest genetically encoded fluorescence complementation assay and was found to have similar photophysical properties when compared with those of native phytochrome miRFP670nano. The faster maturation and higher photostability and cellular stability when compared with those of reported near-infrared BiFC systems make miRFP670nano-BiFC a favorable near-infrared signal module for imaging of PPIs under physiological conditions. The miRFP670nano-BiFC system has several major advantages over existing BiFC systems. Firstly, this small signal module ensures the miRFP670nano-BiFC system has a reduced influence on the interaction between target proteins and enables acquisition of data that match more closely with natural physiological conditions. Secondly, the application of the miRFP670nano-BiFC system in PPIs showed advantages over traditional biochemical methods such as Co-IP and IF despite having to introduce a genetic tag to both target and putative binding partner or transfect cells, thus it may facilitate PPI inhibitor screening and drug evaluation in living cells and animals. Furthermore, the split site for the development of the miRFP670nano-BiFC system was used successfully to construct the miRFP670nano-TriFC system, which should enable the detection of RNA–protein interactions under physiological conditions.
As the first application of this new BiFC system, we investigated interactions between the N protein of SARS-CoV-2 and cellular stress granule proteins CSNK2B, G3BP1 and G3BP2 in living cells. Recent studies have revealed that N protein impairs stress granule assembly;27,28 however, the exact mechanisms remain unresolved. Our results showed that the N-terminal domain of N protein mediates these interactions and the N protein of SARS-CoV-2 regulated the expression level of G3BP1 in cells. Taking into consideration the roles that cellular stress granule proteins play in the antiviral process, SARS-CoV-2 may escape clearance of the host through interaction between N protein and G3BP1. Further analysis of this interaction should provide more information on how SARS-CoV-2 escapes host clearance.
In summary, we developed the smallest near-infrared BiFC and TriFC systems using a cyanobacteriochrome-evolved phytochrome, miRFP670nano. The systems can be used to detect biomolecular interactions in living cells and animals. The optical performance of the systems is better than existing similar near-infrared molecular sensing systems and has a low risk of interfering with native interactions. The first application of the BiFC system was demonstrated by investigating the interactions between the N protein of SARS-CoV-2 and cellular stress granule proteins. The findings provide new insights for studying viral infection mechanisms. The miRFP670nano-based BiFC and TriFC systems should be useful tools in cellular imaging.
4. Materials and methods
Construction of plasmids
The miRFP670nano protein was divided into fragments at six positions to develop the miRFP670nano-based BiFC systems. The fragment information is shown in Table S1.† To generate the plasmids pbJun-miRNs and pmiRCs-bFos, bJun was amplified using plasmid pbJun-iRN97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 as the template, bFos was amplified using plasmid piRC98-bFos13 as the template. The acquired split fragment pairs were named miRN21/miRC22, miRN31/miRC32, miRN44/miRC45, miRN80/miRC81, miRN107/miRC108 and miRN120/miRC121 respectively. These split fragments were amplified using the miRFP670nano sequence (Genewiz, Suzhou) as the template. Overlap-PCR was used to produce bJun-miRN21, bJun-miRN31, bJun-miRN44, bJun-miRN80, bJun-miRN107, bJun-miRN120, miRC22-bFos, miRC32-bFos, miRC45-bFos, miRC81-bFos, miRC108-bFos and miRC121-bFos fragments. pmiRC108-mbFos was produced by replacing bFos in pmiRC108-bFos. mbFos was generated using plasmid piRC98-mbFos13 as the template. (GGGGS)2 was used as the linker between all fusion fragments.
13 as the template, bFos was amplified using plasmid piRC98-bFos13 as the template. The acquired split fragment pairs were named miRN21/miRC22, miRN31/miRC32, miRN44/miRC45, miRN80/miRC81, miRN107/miRC108 and miRN120/miRC121 respectively. These split fragments were amplified using the miRFP670nano sequence (Genewiz, Suzhou) as the template. Overlap-PCR was used to produce bJun-miRN21, bJun-miRN31, bJun-miRN44, bJun-miRN80, bJun-miRN107, bJun-miRN120, miRC22-bFos, miRC32-bFos, miRC45-bFos, miRC81-bFos, miRC108-bFos and miRC121-bFos fragments. pmiRC108-mbFos was produced by replacing bFos in pmiRC108-bFos. mbFos was generated using plasmid piRC98-mbFos13 as the template. (GGGGS)2 was used as the linker between all fusion fragments.
The miRFP670nano protein was divided into fragments at the site between amino acids 107 and 108 to construct the miRFP670nano-based TriFC system. The protein expression plasmids were generated based on mIrisFP and iRFP TriFC systems.17,29 The fragments of miRN107 and miRC108 were produced using the pUC57-miRFP670nano plasmid (Genewiz, Suzhou) as the template. The pNS1-L-miRN107 plasmid was then generated by replacing the mIrisFP fragment mIN165 in pNS1-mIN165 with miRN107 using restriction enzymes KpnI and XbaI.17 The pmiRC108-L-MCP plasmid was produced by replacing the iRFP fragment iRC124m in piRC124m-MCP with miRC108 using restriction enzymes NheI and HindIII.29 The pNS1-2*L-miRN107/pNS1-3*L-miRN107 plasmids and pmiRC108-2*L-MCP/pmiRC108-3*L-MCP plasmids with different linker lengths within the fused proteins were constructed in a similar manner to the pNS1-L-miRN107 and pmiRC108-L-MCP plasmids.
To measure the expression levels of the fusion proteins of the different NIR-BiFC assays, Myc tag was fused to the N-terminal split fragments of different near-infrared BiFC systems, and Flag tag was fused to the C-terminal split fragments of different near-infrared BiFC systems. Restriction enzymes NheI and HindIII were used to build these plasmids. Polyhistidine tag was fused to the N termini of native miRFP670nano and miRFP670nano-BiFC constructs to purify the proteins.
pCSNK2B-miRN107, pG3BP1-miRN107 and pG3BP2-miRN107 plasmids were produced using cDNA from HEK 293T cells as the template to amplify the CSNK2B, G3BP1 and G3BP2 sequences first. The amplified sequences were then cloned into pbJun-miRN107. The pmiRC108-N plasmid was generated as follows: the N protein of SARS-CoV-2 was amplified from the N protein sequence (Genewiz, Suzhou) and cloned into pmiRC108-bFos. For identifying regions of N protein that mediate interactions with cellular stress granule proteins, we constructed two N protein deletion mutants based on the structure of N protein. N(1–246) and N(247–419) were generated by exchanging the N protein coding region in pmiRC108-N for the 1–246 or 247–419 coding sequences of N protein, respectively.
The primers used in this study are shown in Table S3.† All sequences of the plasmid constructs developed were verified by DNA sequencing.
Cell culture and transfection
HEK 293T cells were cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. The culture temperature was set as 37 °C, and 5% CO2 was added to the humidified incubator. The cells were inoculated in 35 mm glass-bottomed wells the day before transfection. When the cells reached 70–80% confluency, each of the plasmids was transfected into cells using Lipofectamine 2000 (Invitrogen; U.S.) as the reagent. Before imaging, the transfected cells were cultured at 37 °C (5% CO2) for 24 h.Fluorescent imaging and data analysis
Cells were imaged with a Nikon TiE inverted microscope using a 60× oil immersion objective lens. The green fluorescence of the EGFP channel and cyan fluorescence of the ECFP channel were excited by using a 488 nm laser. The red fluorescence of the miRFP670nano-BiFC/-TriFC channel was excited by using a 640 nm laser. The fluorescence of the reported near-infrared BiFC systems, including IFP1.4-, IFP2.0- and iRFP-BiFC, was excited by using a 640 nm laser. The immunofluorescence (IF) of the N protein channel was excited by using a 561 nm laser. Hoechst 33342 was used to stain cell nuclei and excited by using a 405 nm laser. In this study, all complementary fluorescence signals were detected either in living cells or in live mice.For quantitative analysis of the complementary fluorescence for the miRFP670nano-BiFC system, the green and red fluorescence intensities were acquired from their original green or red images by subtracting background fluorescence respectively. Background fluorescence is defined as the average fluorescence intensity of a region (50 × 50 pixels) without red fluorescence in the miRFP670nano channel and the corresponding region in the green fluorescence channel. The red-to-green fluorescence ratio was calculated by dividing the intensity of the red fluorescence by that of the green fluorescence. For each group, the average fluorescence intensity of about 30 fluorescent cells, which were randomly selected from 6–8 images was used to calculate the relative complementary fluorescence efficiency for the miRFP670nano-BiFC system. Quantitative analysis of the complementary fluorescence for the miRFP670nano-TriFC system was conducted in a similar manner to that used for the miRFP670nano-BiFC system. The red-to-cyan fluorescence ratio was calculated by dividing the intensity of the red fluorescence by that of the cyan fluorescence.
Flow cytometry
The flow cytometry experiment was conducted as described previously.18 Briefly, the samples were analyzed using a CytoFLEX S flow cytometer. Prior to acquisition, cell pellets were washed with PBS and diluted in cold PBS to a density of 100000 cells ml−1. At least 10000 cells per sample were recorded. To analyze the miRFP670nano-BiFC and reported near-infrared BiFC systems, EGFP fluorescence was detected by the FITC channel, near-infrared fluorescence was detected by the APC channel. To analyze the miRFP670nano-TriFC system, ECFP fluorescence was detected by the PB450 channel, reconstructed miRFP670nano fluorescence was detected by the PC5.5 channel. The data were analyzed using a FlowJo software.
Biophysical properties characterization
Absorbance and fluorescence spectroscopy data were collected by using a monochromator-based BioTek Synergy Mx Microplate Reader. The extinction coefficients of miRFP670nano and miRFP670nano-BiFC were determined as a ratio between the absorbance value for the protein at the main peak and the absorbance value at the 391 nm peak assuming the latter to have the extinction coefficient of 39900 M−1 cm−1,30 which has been introduced in the original miRFP670nano paper. The fluorescence quantum yields of miRFP670nano and miRFP670nano-BiFC were determined using fluorescein as the reference.31
Western blotting
Cells were subjected to western blot analysis to determine the expression levels of fusion proteins. The experiment was conducted as described previously.13 Briefly, cell lysates were cleared by centrifugation at 10000×g for 10 min and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred onto polyvinylidene fluoride membranes. After blocking at 4 °C overnight with phosphate-buffered saline (PBS) supplemented with 5% (w/v) skim milk, the membranes were incubated with specific antibodies, including anti-Flag antibody (1:1000 dilutions, cat#66008-2-Ig, Proteintech), anti-Myc antibody (1:5000 dilutions, cat#A02060, Abbkine) or anti-β-actin antibody (1:1000 dilutions, cat#3700S, Cell Signaling Technology). The membranes were then incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (1:3000 dilutions, cat#7076S, Cell Signaling Technology) at 37 °C for 2 h, and bands were detected using a chemiluminescence detection system (BioRad).
Co-immunoprecipitation (Co-IP) assay
HEK 293T cells co-transfected with N protein and cellular stress granule proteins that were Flag tagged or untagged were collected and lysed. Cell lysates were cleared by centrifugation at 10000×g for 10 min and then incubated with anti-Flag M2 magnetic beads (Sigma) overnight at 4 °C. Lysis buffer was used to wash the magnetic beads several times. SDS loading buffer was then added to the samples and boiled at 100 °C for 5 min. Samples were then subjected to SDS-PAGE analysis. Proteins were then transferred onto polyvinylidene fluoride membranes. After blocking at 37 °C for 2 h with PBS supplemented with 5% (w/v) skim milk, the membranes were incubated with SARS-CoV-2 nucleoprotein rabbit pAb (1:1000 dilutions, cat#A18797, Abclonal) at 4 °C overnight. The membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:3000 dilutions, cat#7074S, Cell Signaling Technology) at 37 °C for 2 h, and bands were detected using a chemiluminescence detection system (BioRad).
Immunofluorescence (IF) assays
For IF assays, cells grown in 35 mm glass-bottomed wells were fixed by incubation with 4% fresh formaldehyde in PBS for 20 min, washed with PBS and permeabilized with 0.5% TritonX-100. Cells were then blocked in 10% FBS to prevent nonspecific staining and incubated with anti-Flag (with a dilution of 1:500, cat#66008-2-Ig, Proteintech) and N antibodies (1:500 dilutions, cat#A18797, Abclonal), followed by Alexa Fluor 488-conjugated goat anti-mouse (with a dilution of 1:1000, cat#4408S, Cell Signaling Technology) and Alexa Fluor 555-conjugated goat anti-rabbit (with a dilution of 1:1000, cat#4413S, Cell Signaling Technology) antibodies. We used the Colocalization Finder function of Image J software to calculate the PCC of different channels. A threshold of PCC > 0.5 has been stated to indicate a meaningful colocalization.32,33
Fluorescence imaging of live mice
In vivo fluorescent images were acquired using an IVIS Spectrum Imaging System (Caliper). HEK 293T cells transiently expressing the miRFP670nano-BiFC system were first analyzed ex vivo in plates using the IVIS Spectrum Imaging System. In each corresponding combination, equal amounts of cells were subcutaneously injected into 6–8 week-old BALB/c-nude mice. The fluorescence intensities of the regions of interest were measured using Living Image Version 4.5 Software to quantify the relative complementary fluorescence signals.All of the animal experiments were performed in strict accordance with ethical standards in Laboratory Animal-Guideline for Ethical Review of Animal Welfare (The National Standard of the People's Republic of China GB/T 35892-2018) and were approved by the Institutional Animal Care and Use Committee of Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences (Shenzhen, China).
Author contributions
M. H. C. and X. E. Z. conceived and supervised the project. M. H. C. designed the experiments. M. H. C., C. Y. and L. P. Z. performed the experiments and analyzed the data. M. H. C. and X. E. Z. wrote the paper.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data related to this paper may be requested from the authors.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2020YFC0861100, 2017YFA0205503), Strategic Priority Research Program of the Chinese Academy of Sciences, China (XDB29050100), National Natural Science Foundation (21890743, 32101210), and Guangdong Basic and Applied Basic Research Foundation (2021A1515011279).References
- G. Maertens, P. Cherepanov, W. Pluymers, K. Busschots, E. De Clercq, Z. Debyser and Y. Engelborghs, J. Biol. Chem., 2003, 278, 33528–33539 CrossRef CAS PubMed
.
- P. Zhou, X. L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. D. Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed
- S. Kishore, S. Luber and M. Zavolan, Briefings Funct. Genomics, 2010, 9, 391–404 CrossRef CAS PubMed
- C. F. Calkhoven, C. Muller and A. Leutz, Trends Mol. Med., 2002, 8, 577–583 CrossRef CAS PubMed
- C. D. Hu, Y. Chinenov and T. K. Kerppola, Mol. Cell, 2002, 9, 789–798 CrossRef CAS PubMed
- X. E. Zhang, Z. Q. Cui and D. B. Wang, Biosens. Bioelectron., 2016, 76, 243–250 CrossRef CAS PubMed
- J. Y. Fan, Z. Q. Cui, H. P. Wei, Z. P. Zhang, Y. F. Zhou, Y. P. Wang and X. E. Zhang, Biochem. Biophys. Res. Commun., 2008, 367, 47–53 CrossRef CAS PubMed
- Y. Han, S. F. Wang, Z. P. Zhang, X. H. Ma, W. Li, X. W. Zhang, J. Y. Deng, H. P. Wei, Z. Y. Li, X. E. Zhang and Z. Q. Cui, Nucleic Acids Res., 2014, 42, e103 CrossRef CAS PubMed
- M. H. Chen, S. Y. Liu, W. Li, Z. P. Zhang, X. W. Zhang, X. E. Zhang and Z. Q. Cui, ACS Nano, 2016, 10, 8482–8490 CrossRef CAS PubMed
- M. H. Chen, W. Li, Z. P. Zhang, J. D. Pan, Y. H. Sun, X. W. Zhang, X. E. Zhang and Z. Q. Cui, Anal. Chem., 2018, 90, 13299–13305 CrossRef CAS PubMed
- G. S. Filonov and V. V. Verkhusha, Chem. Biol., 2013, 20, 1078–1086 CrossRef CAS PubMed
- E. Tchekanda, D. Sivanesan and S. W. Michnick, Nat. Methods, 2014, 11, 641–644 CrossRef CAS PubMed
- M. H. Chen, W. Li, Z. P. Zhang, S. Y. Liu, X. W. Zhang, X. E. Zhang and Z. Q. Cui, Biomaterials, 2015, 48, 97–107 CrossRef CAS PubMed
- M. H. Chen, C. Yan, Y. X. Ma and X. E. Zhang, Biomaterials, 2021, 268, 120544 CrossRef CAS PubMed
- B. J. Tromberg, N. Shah, R. Lanning, A. Cerussi, J. Espinoza, T. Pham, L. Svaasand and J. Butler, Neoplasia, 2000, 2, 26–40 CrossRef CAS PubMed
- O. Rackham and C. M. Brown, EMBO J., 2004, 23, 3346–3355 CrossRef CAS PubMed
- M. H. Chen, S. T. Li, W. Li, Z. P. Zhang, X. W. Zhang, X. E. Zhang, F. Ge and Z. Q. Cui, ACS Chem. Biol., 2021, 16, 1003–1010 CrossRef CAS PubMed
- O. S. Oliinyk, A. A. Shemetov, S. Pletnev, D. M. Shcherbakova and V. V. Verkhusha, Nat. Commun., 2019, 10, 279 CrossRef PubMed
- J. Cubuk, J. J. Alston, J. J. Incicco, S. Singh, M. D. Stuchell-Brereton, M. D. Ward, M. I. Zimmerman, N. Vithani, D. Griffith, J. A. Wagoner, G. R. Bowman, K. B. Hall, A. Soranno and A. S. Holehouse, Nat. Commun., 2021, 12, 1936 CrossRef CAS PubMed
- M. H. Chen and X. E. Zhang, Int. J. Biol. Sci., 2021, 17, 1574–1580 CrossRef CAS PubMed
- M. H. Chen, C. Yan, F. J. Qin, L. P. Zheng and X. E. Zhang, Int. J. Biol. Sci., 2021, 17, 3889–3897 CrossRef CAS PubMed
- K. Nakagawa, K. Narayanan, M. Wada and S. Makino, J. Virol., 2018, 92, e00902–18 CAS
- M. Raaben, M. J. A. G. Koerkamp, P. J. M. Rottier and C. A. M. de Haan, Cell. Microbiol., 2007, 9, 2218–2229 CrossRef CAS PubMed
- P. Ivanov, N. Kedersha and P. Anderson, Cold Spring Harbor Perspect. Biol., 2019, 11, a032813 CrossRef CAS PubMed
- S. Lu, Q. Z. Ye, D. Singh, Y. Cao, J. K. Diedrich, J. R. Yates, E. Villa, D. W. Cleveland and K. D. Corbett, Nat. Commun., 2021, 12, 502 CrossRef CAS PubMed
- S. S. Naz, A. Aslam and T. Malik, Infect. Disord.: Drug Targets, 2021 DOI:10.2174/1871526521666210317161329
- A. Savastano, A. I. de Opakua, M. Rankovic and M. Zweckstetter, Nat. Commun., 2020, 11, 6041 CrossRef CAS PubMed
- S. M. Cascarina and E. D. Ross, FASEB J., 2020, 34, 9832–9842 CrossRef CAS PubMed
- S. Li, Q. Xiong, M. Chen, B. Wang, X. Yang, M. Yang, Q. Wang, Z. Cui and F. Ge, Life Sci. Alliance, 2021, 4, e202101139 CrossRef CAS PubMed
- G. S. Filonov, K. D. Piatkevich, L. M. Ting, J. H. Zhang, K. Kim and V. V. Verkhusha, Nat. Biotechnol., 2011, 29, 757–761 CrossRef CAS PubMed
- M. H. Chen, S. Zhang, Y. Xing, X. Y. Li, Y. He, Y. Wang, J. Oberholzer and H. W. Ai, Anal. Chem., 2019, 91, 12212–12219 CrossRef CAS PubMed
- J. Adler and I. Parmryd, Cytometry, Part A, 2010, 77, 733–742 CrossRef PubMed
- A. Rana, M. Yen, A. M. Sadaghiani, S. Malmersjo, C. Y. Park, R. E. Dolmetsch and R. S. Lewis, J. Cell Biol., 2015, 209, 653–669 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04839b |
| This journal is © The Royal Society of Chemistry 2022 |