
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompeting C–H and C–F bond activation reactions of a fluorinated olefin at Rh: a fluorido vinylidene complex as an intermediate in an unprecedented dehydrofluorination step†
Maria
Talavera
 and
Thomas
Braun
*
and
Thomas
Braun
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: thomas.braun@cms.hu-berlin.de
First published on 12th January 2022
Abstract
The hydrofluoroolefin Z-1,3,3,3-tetrafluoropropene has been activated via an initial C–F bond activation and subsequent C–H bond activation using [Rh(H)(PEt3)3] (1) or via C–H bond activation at [Rh(CH3)(PEt3)3] (8). In both cases the formation of [Rh{(E)-CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCF3}(PEt3)3] (3) was observed. Importantly, the C–F activation product [Rh{(E)-CHCHCF3}(PEt3)3] (2) reacts in the presence of Z-1,3,3,3-tetrafluoropropene into 3. The latter converted into [Rh(C
CHCF3}(PEt3)3] (3) was observed. Importantly, the C–F activation product [Rh{(E)-CHCHCF3}(PEt3)3] (2) reacts in the presence of Z-1,3,3,3-tetrafluoropropene into 3. The latter converted into [Rh(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCF3)(PEt3)3] (6) by an unprecedented dehydrofluorination reaction, presumably via a vinylidene complex as intermediate. When the carbonyl complex [Rh(CCCF3)(CO)(PEt3)3] (12) was treated with an excess of NEt3·3HF or HBF4 at low temperature, the formation of the phosphonioalkenyl compounds [Rh{(Z)-C(PEt3)CHCF3}(CO)(PEt3)2]X (X = F(HF)x, BF4) (13) was observed. The formation of 13 can be explained by an attack of PEt3 at the electrophilic α-carbon atom of an intermediate vinylidene complex. The employment of PiPr3 derivatives as model compounds allowed for the isolation of the unique fluorido vinylidene complex trans-[Rh(F)(CCHCF3)(PiPr3)2] (16), which in the presence of PEt3 transforms into [Rh(CCCF3)(PEt3)3] (6).
CCF3)(PEt3)3] (6) by an unprecedented dehydrofluorination reaction, presumably via a vinylidene complex as intermediate. When the carbonyl complex [Rh(CCCF3)(CO)(PEt3)3] (12) was treated with an excess of NEt3·3HF or HBF4 at low temperature, the formation of the phosphonioalkenyl compounds [Rh{(Z)-C(PEt3)CHCF3}(CO)(PEt3)2]X (X = F(HF)x, BF4) (13) was observed. The formation of 13 can be explained by an attack of PEt3 at the electrophilic α-carbon atom of an intermediate vinylidene complex. The employment of PiPr3 derivatives as model compounds allowed for the isolation of the unique fluorido vinylidene complex trans-[Rh(F)(CCHCF3)(PiPr3)2] (16), which in the presence of PEt3 transforms into [Rh(CCCF3)(PEt3)3] (6).
Introduction
Hydrofluoroolefins (HFOs) have attracted a lot of attention during the last decade due to both their industrial use and their role as sources of fluorinated building blocks.1–5 In this regard, both C–H and C–F bond activation reactions of HFOs have been described using transition metal complexes, main group elements or heterogeneous catalysts.6–12 Commonly, the C–F bond activation is promoted by the formation of stable bonds such as H–F, B–F, Si–F or Al–F bonds among others.13–18At rhodium, the complexes [Rh(E)(PEt3)3] (E = H, F, boryl, silyl, germyl) exhibit distinct reaction pathways for the C–F or C–H bond activation of HFOs.19–25 For example, the activation of 2,3,3,3-tetrafluoropropene using [Rh(F)(PEt3)3] in the presence of fluorosilane resulted in the C–H bond activation and a concomitant 1,2-fluorine shift.24 On the other hand, E-1,3,3,3-tetrafluoropropene reacted with [Rh(E)(PEt3)3] (E = H, silyl, germyl) by C–F bond activation via two different reaction pathways, which impart either insertion into the M–E bond followed by a β-fluoride elimination step to yield [Rh(F)(PEt3)3] or the release of FE leading to a vinyl complex.23
Tetrafluoropropenes are used as refrigerants and blowing agents. They have zero ozone depletion potential and a very low global warming potential. On the other hand, there is a certain concern, because of depletion processes which can involve the generation of HF or environmentally persistent depletion products such as trifluoroacetate.26–28 Despite the broad research on HFOs,3,8,19 the studies regarding the reactivity of the isomer Z-1,3,3,3-tetrafluoropropene (Z-HFO-1234ze) are very scarce, but of interest, because it can be a source for more valuable building blocks. So far, only Crimmin and co-workers reported its C–F bond activation via an oxidative addition at an Al(I) complex.29
Hydrodefluorination is a very well-known reaction pathway at transition metal complexes for both stoichiometric and catalytic activation of fluorinated derivatives.15,18,30–34 On the contrary, dehydrofluorination (DHF) reactions at fluoroalkanes are rare, but can be catalysed by solid materials such as magnesium-35 or aluminium-based catalysts,36,37 and germylium ions as the only examples for homogeneous catalysts.38 Metal-mediated dehydrofluorination reactions of fluoroalkyl or fluoroalkenyl moieties to yield HF and the corresponding alkenyl or alkynyl entity have not been described previously. A report at scandium complexes involves a β-fluorine elimination step after a hydrogen atom abstraction, but this will always produce metal fluorido complexes.39 Hughes et al. discussed, as part of a mechanistic proposal, the defluorination of a perfluoroalkyl ligand bound at a iridium half-sandwich complex.40 A proposed intermediate [IrCp*(H)(CH2CF2CF3)(PMe3)] was suggested to convert into [IrCp*(H)(CHCFCF3)(PMe3)] and HF.
Herein, the reactivity of Z-1,3,3,3-tetrafluoropropene towards rhodium(I) complexes is reported. The studies include consecutive bond activation reactions of C–F and C–H bonds. The unprecedented dehydrofluorination of a fluorinated vinyl complex to yield an alkynyl complex is described and model studies suggest a fluorido vinylidene complex as an intermediate.
Results and discussion
A reaction of [Rh(H)(PEt3)3] (1) with Z-1,3,3,3-tetrafluoropropene resulted after 30 minutes in the formation of a mixture of the complexes [Rh{(E)-CHCHCF3}(PEt3)3] (2),24 [Rh{(E)-CFCHCF3)}(PEt3)3] (3), [Rh{(Z)-CHCHCF3}(PEt3)3] (4) and [Rh{F(HF)2)}(PEt3)3] (5)41,42 in a 3.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:4.4 ratio, as well as the release of 3,3,3-trifluoropropene (Scheme 1). After 5 hours complex 2 was completely consumed, while higher amounts of the complexes 3 and 5 were obtained; and the formation of the rhodium(I) alkynyl complex [Rh(CCCF3)(PEt3)3] (6)43 was also observed. After one day, the complexes 5 and 6 were detected in a 1:1 ratio (Scheme 1). Overall, this suggests that initially, from [Rh(H)(PEt3)3] (1), the vinyl complexes [Rh{(E)-CHCHCF3}(PEt3)3] (2) and [Rh{(Z)-CHCHCF3}(PEt3)3] (4) are formed by C–F bond activation, followed by a C–H activation step in the presence of the olefin to form [Rh{(E)-CFCHCF3}(PEt3)3] (3) together with 3,3,3-trifluoropropene. The latter complex converts into 5 and 6.
:2:1:4.4 ratio, as well as the release of 3,3,3-trifluoropropene (Scheme 1). After 5 hours complex 2 was completely consumed, while higher amounts of the complexes 3 and 5 were obtained; and the formation of the rhodium(I) alkynyl complex [Rh(CCCF3)(PEt3)3] (6)43 was also observed. After one day, the complexes 5 and 6 were detected in a 1:1 ratio (Scheme 1). Overall, this suggests that initially, from [Rh(H)(PEt3)3] (1), the vinyl complexes [Rh{(E)-CHCHCF3}(PEt3)3] (2) and [Rh{(Z)-CHCHCF3}(PEt3)3] (4) are formed by C–F bond activation, followed by a C–H activation step in the presence of the olefin to form [Rh{(E)-CFCHCF3}(PEt3)3] (3) together with 3,3,3-trifluoropropene. The latter complex converts into 5 and 6.
 | ||
| Scheme 1 Reactivity of [Rh(H)(PEt3)3] (1) with Z-1,3,3,3-tetrafluoropropene. | ||
NMR studies at low temperature support this assumption and give further insight. Thus, treatment of [Rh(H)(PEt3)3] (1) with Z-1,3,3,3-tetrafluoropropene at 233 K led initially to the formation of complex fac-[Rh(H)(CHFCHCF3)(PEt3)3] (7) due to the coordination of the olefin at rhodium (Scheme 1). Complex 7 displays characteristic spectroscopic data revealing a syn-configuration of the CF3 group at the coordinated olefin and the hydrido ligand (see ESI† for DFT calculations), as previously reported for the coordination of fluoroolefins at 1.22–25 After warming up the reaction mixture to 273 K, 90% of complex 7 converted to give the C–F bond activation product [Rh{(E)-CHCHCF3}(PEt3)3] (2) together with HF. The preference of complex 1 towards C–F bond activation over C–H bond activation is in accordance with the reactivity of other fluoroolefins, but in contrast to the observed C–H bond activation reactions of partially fluorinated aromatics.22–25,44,45 Finally, at room temperature after 10 min in the presence of Z-1,3,3,3-tetrafluoropropene, a 45% conversion of complex 2 was observed towards a mixture of the complexes [Rh{(E)-CFCHCF3}(PEt3)3] (3) and [Rh{F(HF)2}(PEt3)3] (5) in a 1:4 ratio (Scheme 1). Note that [Rh{(E)-CFCHCF3}(PEt3)3] (3) appeared upon consumption of [Rh{(E)-CHCHCF3}(PEt3)3] (2), whereas the amount of 3,3,3-trifluoropropene increased accordingly. The reaction monitoring at 273 K also suggests that complex 5 is mainly formed from complex 2 – presumably by protonation with HF – and not from complex 7. However, it cannot be ruled out that small amounts of complex 7 react by insertion of the olefin into the Rh–H bond and a subsequent β-F-elimination to furnish [Rh(F)(PEt3)3] and 3,3,3-trifluoropropene, which is a common pathway in the chemistry of fluorinated olefins.11,22,23,25,46–49
Further studies on model reactions give an insight into particular reaction steps. Thus, an independent synthesis of complex 2 by C–H bond activation of 3,3,3-trifluoropropene at [Rh(CH3)(PEt3)3] (8) was developed. Indeed, complex 2 allowed for the C–H bond activation of Z-HFO-1234ze to give [Rh{(E)-CFCHCF3}(PEt3)3] (3) and 3,3,3-trifluoropropene in less than one hour (Scheme 2). In addition, complex 2 reacted instantly with the HF source NEt3·3HF to yield complex [Rh{F(HF)2}(PEt3)3] (5) and 3,3,3-trifluoropropene (Scheme 2).
 | ||
| Scheme 2 Synthesis and reactivity of the complexes 2 and 3. | ||
Alternatively complex 3 can also be synthesized by C–H bond activation of Z-HFO-1234ze at [Rh(CH3)(PEt3)3] (8) which gave after 50 minutes complex 3 and methane (Scheme 3). When the reaction was followed at variable temperature, the coordination of the olefin at rhodium fac-[Rh(CH3)(CHFCHCF3)(PEt3)3] (9) was observed at 223 K, however, upon warming up to 253 K, complex 8 was regenerated, to convert into [Rh{(E)-CFCHCF3}(PEt3)3] (3) at 273 K, which suggests that 9 is not an intermediate in the formation of 3 (Scheme 2). The nature of complex 9 was determined by its spectroscopic similarities to complex 7 and the presence of the resonance for the methyl ligand in the 1H NMR spectrum to δ −0.74 ppm.
 | ||
| Scheme 3 Reactivity of complex 3 towards CO. | ||
Complex [Rh{(E)-CFCHCF3}(PEt3)3] (3) is only stable for about 2 h in solution or after work-up by removing all the volatiles as an oil. As observed in the reactions described above, it transformed into a mixture of [Rh(CCCF3)(PEt3)3] (6), and [Rh{F(HF)2}(PEt3)3] (5) (≈15%) as well as into Z-HFO-1234ze. If around two equivalents of NEt3/Cs2CO3 are added to complex 3 to trap HF, the dehydrofluorination required two days, but no formation of complex 5 was observed (Scheme 2). This suggests that the HF, which is released in the dehydrofluorination step of the vinyl ligand, can react further with complex 3 to yield the fluorido complex 5 and Z-1,3,3,3-tetrafluoropropene. Note that an independent reaction of [Rh{(E)-CFCHCF3}(PEt3)3] (3) with NEt3·3HF provided indeed complex 5 as the only rhodium complex and several organic derivatives including Z-HFO-1234ze.
When an excess of NEt3/CsCO3 was added to a solution of [Rh{(E)-CFCHCF3}(PEt3)3] (3), conversion into 6 by dehydrofluorination was also observed, although it took up to several weeks. Similarly, addition of free triethylphosphine hampered the dehydrofluorination reaction. This behaviour suggests that the creation of a coordination vacant site might be necessary to allow for the HF elimination at the vinyl ligand in 3 and that the presence of NEt3 or PEt3 might block it. Literature data indicate that a phosphine dissociation in the trans position to the organyl ligand is a likely reaction step,22,25,50–53 but the coordination site can be blocked by a CO ligand. To further confirm this hypothesis, [Rh{(E)-CFCHCF3}(PEt3)3] (3) was treated with CO to give initially trans,cis-[Rh{(E)-CFCHCF3}(CO)2(PEt3)2] (10) together with two unknown minor complexes54 (Scheme 3). Complex 10 exhibits a trigonal bipyramidal structure based on DFT calculations (see ESI†). The 31P{1H} NMR spectrum showed signals for two inequivalent phosphines. The 31P nuclei coupled to two equivalent carbonyl ligands for the 13CO isotopologue of 10. Complex 10 is stable in solution, however, after removing all the volatiles of the mixture containing 10 and the unknown complexes, one of the CO ligands in 10 is released and trans-[Rh{(E)-CFCHCF3}(CO)(PEt3)2] (11) was obtained as sole product (Scheme 3). In contrast to complex 3, complex 11 is stable in solution or after work-up as an oil for weeks and HF elimination was not observed. Therefore, it can be assumed that the initial phosphine dissociation to create a vacant site in the trans position to the vinyl ligand is necessary for the dehydrofluorination reaction.
After phosphine dissociation in 3, a vinylidene intermediate can be proposed in the dehydrofluorination step at [Rh{(E)-CFCHCF3}(PEt3)3] (3) (Scheme 2). Indeed, there is literature precedence for the generation of alkynyl complexes from vinylidene complexes in presence of a base.55–58 Thus, a vacant coordination site can allow for a 1,2-fluorine shift to yield a putative vinylidene complex [Rh(F)(CCHCF3)(PEt3)2] bearing a rhodium-bonded fluorido ligand (Scheme 2). The latter complex would eliminate HF followed by an association of PEt3 and form [Rh(CCCF3)(PEt3)3] (6). Note that compounds providing Lewis-acidity, such as BF3 or LiBF4, were capable to facilitate the dehydrofluorination reaction at [Rh{(E)-CFCHCF3}(PEt3)3] (3) within a few minutes, but no cationic vinylidene intermediates where observed even at low temperatures.
Hence, the independent synthesis of a CF3 group containing vinylidene similar to [Rh(F)(CCHCF3)(PEt3)2] was attempted. Addition of acids such as HOTf or HBF4 to the alkynyl complex [Rh(CCCF3)(PEt3)3] (6) led only to cationic species like [Rh(PEt3)4]+ or [Rh(toluene)(PEt3)2]+. The conversions resemble an independent reaction of 6 with NEt3·3HF to form the rhodium fluorido complex 5 and the hydrofluorination product, Z-HFO-1234ze, among other organic derivatives. Interestingly, when [Rh(CCCF3)(CO)(PEt3)3] (12)43 was treated with an excess of NEt3·3HF or HBF4 at 243 K, the formation of [Rh{(Z)-C(PEt3)CHCF3}(CO)(PEt3)2]X (X = F(HF)x, BF4) (13) was observed as main product (Scheme 4, see ESI† for details).
 | ||
| Scheme 4 Formation of a fluorinated phosphoniopropenyl complex 13. | ||
The compound exhibited in the 31P{1H} NMR spectrum two correlating resonances with a 2 Hz coupling constant between the phosphines. One signal for the rhodium bonded phosphine ligand appears at δ 14.7 ppm and a second one for the phosphonioalkenyl unit at δ 37.1 ppm with a rhodium coupling of only 4.8 Hz. A doublet of quartets of pseudo quartets in the 1H NMR spectrum for the vinyl proton at δ 6.68 ppm reveals a 36 Hz coupling to the phosphonio moiety, which is typical for a cis substitution,55,59,60 a 6 Hz coupling to the CF3 group which is typical for the geminal arrangement,24,25,29 and 3 Hz coupling to rhodium and the phosphine ligands.
Phosphonioalkenyl complexes are commonly obtained by nucleophilic addition of a phosphine to a η2-coordinated alkyne.59–62 Other methodologies such as nucleophilic substitution of a fluorine atom at the β-position of a vinyl ligand63 or nucleophilic attack at the α-position of a vinylidene ligand have been scarcely reported.55 Noteworthy, only in the latter case the phosphonio moiety is bonded to the α-carbon. Therefore, mechanistically, the formation of complex 13 can be explained by the attack of PEt3 at the electrophilic α-carbon atom of an intermediate vinylidene complex. Obviously, the polyfluoride anion is not nucleophilic enough to react with the vinylidene and remains as counter anion. A reaction of the square planar derivative [Rh(CCCF3)(CO)(PEt3)2] with NEt3·3HF does not result in the generation of any vinylidene complex. Instead, a mixture of unknown products and cationic complexes is obtained, suggesting a low stability of a putative vinylidene ligand.
Werner and co-workers described the synthesis of the vinylidene rhodium complex trans-[Rh(F)(CCH(Ph)(PiPr3)2)] by reaction of the binuclear complex [Rh(F)(PiPr3)2]2 (14a) with phenylacetylene.64 Though, treatment of [Rh(F)(PEt3)2]2 (14b)65 with two equivalents of 3,3,3-trifluoropropyne led to a mixture of products where only [Rh(H)(CCCF3)2(PEt3)3] and [Rh{(E)-CHCHCF3}(CCCF3)2(PEt3)3] were identified.43 Similarly, the use of phenylacetylene or pentafluorophenylacetylene did not provide any vinylidene complex.
Interestingly, when complex 14a was treated with 3,3,3-trifluoropropyne the formation of η2-alkyne complex trans-[Rh(F)(HCCCF3)(PiPr3)2] (15) was observed (Scheme 5). In contrast to trans-[Rh(F)(HCCPh)(PiPr3)2],64 complex 15 is stable for days. It is remarkable that after 3 weeks in the presence of phosphine or another base such as trimethylamine, 15 transformed into the vinylidene complex trans-[Rh(F)(CCHCF3)(PiPr3)2] (16) (Scheme 5). In 19F NMR spectrum, complex 15 exhibits a rhodium fluorido resonance at δ = −242.9 ppm which shifts to δ = −208.0 ppm for complex 16. Similarly, in 1H NMR, the signal for the coordinated alkyne proton in 15 at δ = 4.52 ppm shifts to δ = 0.67 ppm for the vinylidene complex 16. Finally, the 13C{1H} NMR shifts confirmed the η2-alkyne ligand of 15 with two resonances at δ = 61 and 83 ppm, while complex 16 displays the typical signals for carbon atoms at rhodium vinylidene complexes at δ = 104 and 282 ppm.64,66
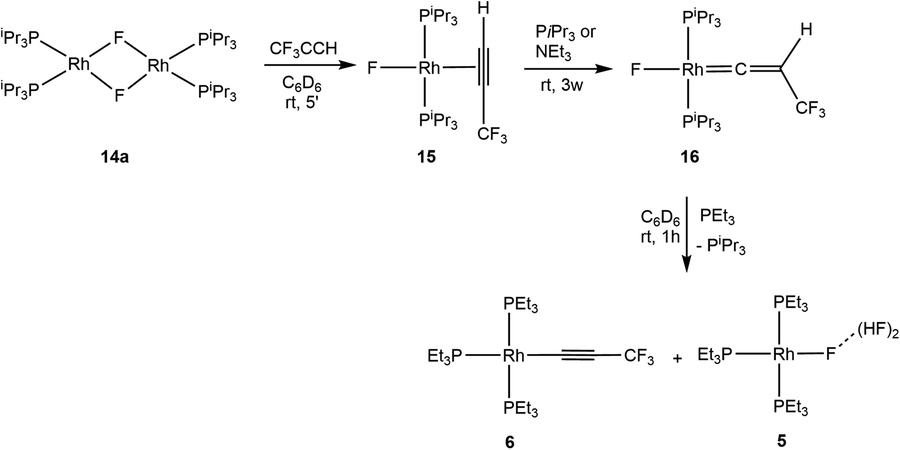 | ||
| Scheme 5 Reactivity of rhodium(I) fluorido dimer 14 towards 3,3,3-trifluoropropyne. | ||
Complex 16, which represents the first example for a CF3-containing vinylidene complex described, is also stable in solution and any release of HF was not observed within one week. Remarkably, upon addition of triethylphosphine, HF elimination, i.e. dehydrofluorination, took place together with phosphine exchange and the formation of the alkynyl complex [Rh(CCCF3)(PEt3)3] (6) and complex 5 (Scheme 5).
Conclusions
In conclusion, the selective C–H bond activation of Z-HFO-1234ze using a fluorinated vinyl rhodium complex has been described. This reaction was preceded by the initial C–F bond activation of the olefin at a rhodium hydrido complex. The transformation of a fluorovinyl complex into an alkynyl complex by dehydrofluorination is unprecedented. Whereas hydrodefluorination and dehydrofluorination play a crucial role for the depletion of fluororganyl compounds, the latter had not been directly observed at transition metals. Mechanistic studies strongly support a vinylidene complex as an intermediate. Complex trans-[Rh(F)(CCHCF3)(PiPr3)2] was synthesized independently and HF elimination could be triggered by PEt3 addition (i.e. phosphine exchange). In general, the reactivity patterns demonstrate versatile activation pathways at rhodium(I) complexes, which can be useful tools for the study of catalytic transformations.
Data availability
All experimental data as well as DFT details are provided in the ESI.†Author contributions
Conceptualization, M. T. and T. B.; investigation, M. T.; writing—original draft preparation, M. T.; writing—review and editing, M. T. and T. B.; supervision and funding acquisition, T. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the DFG (Deutsche Forschungsgemeinschaft, BR 2065/13-1) for financial support.Notes and references
- P. Javidmand and K. A. Hoffmann, Int. J. Refrig., 2016, 69, 114–135 CrossRef CAS.
- B. Ameduri and H. Sawada, Fluorinated Polymers: Applications, Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- B. Varga, J. T. Csenki, B. L. Tóth, F. Béke, Z. Novák and A. Kotschy, Synthesis, 2021, 45 Search PubMed.
- B. J. Murray, L. T. Boulton and G. Sandford, J. Fluorine Chem., 2021, 245, 109774 CrossRef CAS.
- B. J. Murray, T. G. F. Marsh, D. S. Yufit, M. A. Fox, A. Harsanyi, L. T. Boulton and G. Sandford, Eur. J. Org. Chem., 2020, 2020, 6236–6244 CrossRef CAS.
- M. Ohashi and S. Ogoshi, Top. Organomet. Chem., 2015, 52, 197–216 CrossRef CAS.
- J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224–10239 RSC.
- G. Coates, F. Rekhroukh and M. R. Crimmin, Synlett, 2019, 30, 2233–2246 CrossRef CAS.
- T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS.
- T. Braun, G. Meißner, E. Kemnitz and K. Kretschmar, Angew. Chem., Int. Ed., 2017, 56, 16338–16341 CrossRef PubMed.
- N. O. Andrella, K. Liu, B. Gabidullin, M. Vasiliu, D. A. Dixon and R. T. Baker, Organometallics, 2018, 37, 422–432 CrossRef CAS.
- N. V. Pavlenko, S. Peng, V. Petrov, A. Jackson, X. Sun, L. Sprague and Y. L. Yagupolskii, Eur. J. Org. Chem., 2020, 2020, 5425–5435 CrossRef CAS.
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS.
- L. Keyes and J. A. Love, C-H and C-X Bond Functionalization: Transition Metal Mediation, The Royal Society of Chemistry, 2013 Search PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS.
- O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed.
- N. O. Andrella, N. Xu, B. M. Gabidullin, C. Ehm and R. T. Baker, J. Am. Chem. Soc., 2019, 141, 11506–11521 CrossRef CAS.
- M. Talavera and T. Braun, Synlett, 2020, 31, 1760–1774 CrossRef CAS.
- T. Braun, D. Noveski, B. Neumann and H.-G. Stammler, Angew. Chem., Int. Ed., 2002, 41, 2745–2748 CrossRef CAS PubMed.
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed.
- T. Ahrens, M. Teltewskoi, M. Ahrens, T. Braun and R. Laubenstein, Dalton Trans., 2016, 45, 17495–17507 RSC.
- M. Talavera, R. Müller, T. Ahrens, C. N. von Hahmann, B. Braun-Cula, M. Kaupp and T. Braun, Faraday Discuss., 2019, 220, 328–349 RSC.
- M. Talavera, C. N. vonHahmann, R. Müller, M. Ahrens, M. Kaupp and T. Braun, Angew. Chem., Int. Ed., 2019, 58, 10688–10692 CrossRef CAS.
- M. Talavera and T. Braun, Chem.–Eur. J., 2021, 27, 11926–11934 CrossRef CAS.
- E. J. K. Nilsson, O. J. Nielsen, M. S. Johnson, M. D. Hurley and T. J. Wallington, Chem. Phys. Lett., 2009, 473, 233–237 CrossRef CAS.
- D. Fleet, J. Hanlon, K. Osborne, M. La Vedrine and P. Ashford, Study on environmental and health effects of HFO refrigerants, RPA, London, 2017 Search PubMed.
- H. Flerlage, G. J. M. Velders and J. de Boer, Chemosphere, 2021, 283, 131208 CrossRef CAS PubMed.
- M. R. Crimmin, C. Bakewell and A. White, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef PubMed.
- H. Iwamoto, H. Imiya, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 19360–19367 CrossRef CAS PubMed.
- C. Yao, S. Wang, J. Norton and M. Hammond, J. Am. Chem. Soc., 2020, 142, 4793–4799 CrossRef CAS PubMed.
- J.-Y. Hu and J.-L. Zhang, in Organometallic Fluorine Chemistry, ed. T. Braun and R. P. Hughes, Springer International Publishing, Cham, 2015, pp. 143–196 Search PubMed.
- M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152–3159 CrossRef CAS.
- G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS PubMed.
- J.-D. Song, T.-Y. Song, T.-T. Zhang, Y. Wang, M.-F. Luo and J.-Q. Lu, J. Catal., 2018, 364, 271–281 CrossRef CAS.
- M.-C. Kervarec, T. Braun, M. Ahrens and E. Kemnitz, Beilstein J. Org. Chem., 2020, 16, 2623–2635 CrossRef CAS PubMed.
- X.-X. Fang, Y. Wang, W.-Z. Jia, J.-D. Song, Y.-J. Wang, M.-F. Luo and J.-Q. Lu, Appl. Catal., A, 2019, 576, 39–46 CrossRef CAS.
- M. Talavera, G. Meißner, S. G. Rachor and T. Braun, Chem. Commun., 2020, 56, 4452–4455 RSC.
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS PubMed.
- R. P. Hughes, I. Kovacik, D. C. Lindner, J. M. Smith, S. Willemsen, D. Zhang, I. A. Guzei and A. L. Rheingold, Organometallics, 2001, 20, 3190–3197 CrossRef CAS.
- The amount of HF was determined by addition of ClSiEt3 to form FSiEt3 in presence of a CF3Ph standard.
- D. Noveski, T. Braun and S. Krückemeier, J. Fluorine Chem., 2004, 125, 959–966 CrossRef CAS.
- C. N. von Hahmann, M. Talavera, C. Xu and T. Braun, Chem.–Eur. J., 2018, 24, 11131–11138 CrossRef CAS PubMed.
- D. Noveski, T. Braun, B. Neumann, A. Stammler and H.-G. Stammler, Dalton Trans., 2004, 4106–4119 RSC.
- S. I. Kalläne, M. Teltewskoi, T. Braun and B. Braun, Organometallics, 2015, 34, 1156–1169 CrossRef.
- N. Bramananthan, M. Carmona, J. P. Lowe, M. F. Mahon, R. C. Poulten and M. K. Whittlesey, Organometallics, 2014, 33, 1986–1995 CrossRef CAS.
- T. Ichitsuka, T. Fujita, T. Arita and J. Ichikawa, Angew. Chem., Int. Ed., 2014, 53, 7564–7568 CrossRef CAS PubMed.
- M. Hu, Z. He, B. Gao, L. Li, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 17302–17305 CrossRef CAS PubMed.
- H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855–12862 CrossRef CAS PubMed.
- B. M. Trost and R. J. Kulawiec, J. Am. Chem. Soc., 1992, 114, 5579–5584 CrossRef CAS.
- H. Xu and W. H. Bernskoetter, J. Am. Chem. Soc., 2011, 133, 14956–14959 CrossRef CAS PubMed.
- P. Zhao and J. F. Hartwig, Organometallics, 2008, 27, 4749–4757 CrossRef CAS.
- C. Xu, M. Talavera, S. Sander and T. Braun, Dalton Trans., 2019, 48, 16258–16267 RSC.
- The unknown products present the same vinyl ligand and the amount of free phosphine suggests that two equivalent phosphines are present in one and three equivalent phosphines in the other. However, due to the broadening of the resonances which could be not resolved at 173 K, the arrangement of the phosphine and CO ligands was not determined.
- D. R. Senn, A. Wong, A. T. Patton, M. Marsi, C. E. Strouse and J. A. Gladysz, J. Am. Chem. Soc., 1988, 110, 6096–6109 CrossRef CAS PubMed.
- L. M. Hall, D. P. Tew, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and J. M. Slattery, Angew. Chem., Int. Ed., 2017, 56, 7551–7556 CrossRef CAS PubMed.
- M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS.
- D. Touchard, P. Haquette, A. Daridor, A. Romero and P. H. Dixneuf, Organometallics, 1998, 17, 3844–3852 CrossRef CAS.
- V. Cadierno, M. P. Gamasa, J. Gimeno, C. González-Bernardo, E. Pérez-Carreño and S. García-Granda, Organometallics, 2001, 20, 5177–5188 CrossRef CAS.
- C. S. Chin, M. Lee, M. Oh, G. Won, M. Kim and Y. J. Park, Organometallics, 2000, 19, 1572–1577 CrossRef CAS.
- J. C. Jeffery, P. A. Jelliss, E. Psillakis, G. E. A. Rudd and F. G. A. Stone, J. Organomet. Chem., 1998, 562, 17–27 CrossRef CAS.
- Y. Nishimura, Y. Arikawa, T. Inoue and M. Onishi, Dalton Trans., 2005, 930–937 RSC.
- T. Braun, B. Blöcker, V. Schorlemer, B. Neumann, A. Stammler and H.-G. Stammler, J. Chem. Soc., Dalton Trans., 2002, 2213–2218 RSC.
- J. Gil-Rubio, B. Weberndörfer and H. Werner, J. Chem. Soc., Dalton Trans., 1999, 1437–1444 RSC.
- L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 54, 10652–10656 CrossRef PubMed.
- D. Schneider and H. Werner, Angew. Chem., Int. Ed., 1991, 30, 700–702 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization, NMR spectra, DFT details and XYZ coordinates. See DOI: 10.1039/d1sc06713c |
| This journal is © The Royal Society of Chemistry 2022 |