
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGa+-catalyzed hydrosilylation? About the surprising system Ga+/HSiR3/olefin, proof of oxidation with subvalent Ga+ and silylium catalysis with perfluoroalkoxyaluminate anions†‡
Antoine
Barthélemy
 ,
Kim
Glootz
,
Harald
Scherer
,
Annaleah
Hanske
and
Ingo
Krossing
*
,
Kim
Glootz
,
Harald
Scherer
,
Annaleah
Hanske
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
First published on 23rd November 2021
Abstract
Already 1 mol% of subvalent [Ga(PhF)2]+[pf]− ([pf]− = [Al(ORF)4]−, RF = C(CF3)3) initiates the hydrosilylation of olefinic double bonds under mild conditions. Reactions with HSiMe3 and HSiEt3 as substrates efficiently yield anti-Markovnikov and anti-addition products, while bulkier substrates such as HSiiPr3 are less reactive. Investigating the underlying mechanism by gas chromatography and STEM analysis, we unexpectedly found that H2 and metallic Ga0 formed. Without the addition of olefins, the formation of R3Si–F–Al(ORF)3 (R = alkyl), a typical degradation product of the [pf]− anion in the presence of a small silylium ion, was observed. Electrochemical analysis revealed a surprisingly high oxidation potential of univalent [Ga(PhF)2]+[pf]− in weakly coordinating, but polar ortho-difluorobenzene of E1/2(Ga+/Ga0; oDFB) = +0.26–0.37 V vs. Fc+/Fc (depending on the scan rate). Apparently, subvalent Ga+, mainly known as a reductant, initially oxidizes the silane and generates a highly electrophilic, silane-supported, silylium ion representing the actual catalyst. Consequently, the [Ga(PhF)2]+[pf]−/HSiEt3 system also hydrodefluorinates C(sp3)–F bonds in 1-fluoroadamantane, 1-fluorobutane and PhCF3 at room temperature. In addition, both catalytic reactions may be initiated using only 0.2 mol% of [Ph3C]+[pf]− as a silylium ion-generating initiator. These results indicate that silylium ion catalysis is possible with the straightforward accessible weakly coordinating [pf]− anion. Apparently, the kinetics of hydrosilylation and hydrodefluorination are faster than that of anion degradation under ambient conditions. These findings open up new windows for main group catalysis.
Introduction
Classical GaI-sources, e.g. “GaI”,1,2 Ga[GaX4] (X = Cl, Br, and I)3 or GaCp(*)4 do have some drawbacks in their applications: they undergo facile dis- or comproportionation reactions upon addition of σ-donating ligands,1,5,6 due to the presence of reactive and strongly coordinating counterions such as [GaX4]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 or, for Green's “GaI”, have a non-homogenous composition1,8 that hampered systematic studies of GaI chemistry for a long time. Subsequently, well-defined GaI compounds including GaI[DippNacNac] ([DippNacNac]− = [(Dipp)NC(Me)CHC(Me)N(Dipp)]−; Dipp = 2,6-diisopropylphenyl)9 or GaI[{(Dipp)N
7 or, for Green's “GaI”, have a non-homogenous composition1,8 that hampered systematic studies of GaI chemistry for a long time. Subsequently, well-defined GaI compounds including GaI[DippNacNac] ([DippNacNac]− = [(Dipp)NC(Me)CHC(Me)N(Dipp)]−; Dipp = 2,6-diisopropylphenyl)9 or GaI[{(Dipp)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH}2]10 allowed to investigate the interesting carbene-like reactivity of GaI (vide infra). Yet, they are no source for “naked” cationic Ga+ to be tested in any application.
CH}2]10 allowed to investigate the interesting carbene-like reactivity of GaI (vide infra). Yet, they are no source for “naked” cationic Ga+ to be tested in any application.
In this respect, the introduction of weakly coordinating anions (WCAs),11,12 for example, in [Ga2Cp*][B(ArF)4]13 (ArF = 3,5-(CF3)2C6H3) and [In2Cp*][B(C6F5)4]14 was another improvement in subvalent MI chemistry. However, the follow-up chemistry of these salts is complicated and the atom efficiency is limited because one excess equivalent of M(Cp*) (M = Ga or In) is released per M+ ion introduced. Therefore, employing the [pf]− anion ([pf]− = [Al(ORF)4]−; RF = C(CF3)3) in conjunction with weakly coordinating solvents now allows for the rational application of “naked” univalent gallium ions with the well-defined Ga+ source [Ga(PhF)2][pf].15,16 The respective indium salt [In(PhF)2][pf] was reported shortly thereafter.17,18 Both are suitable for coordination chemistry with classical σ-donor ligands.6 In addition, Wehmschulte has recently presented salts of the type [Ga(arene)x]A with A = [CHB11Cl11]− or [B(C6F5)4]−.19 Still, these carborate or borate salts are expensive and also difficult to synthesize, unlike the straightforward large-scale accessible [pf]− salts.20
Consequently, salts of the type [M(arene)x][pf] (M = Ga or In; x = 1–3) are increasingly employed as M+ sources in catalysis, for example, in C–C bond forming reactions, like hydroarylation, hydrogenative cyclization, alkene transfer hydrogenation or Friedel–Crafts reactions.21,22 Intriguingly, the univalent MI salts display equal or even superior activity to more traditional MIII compounds.22,23 In these reactions, the univalent metal ions presumably act as π-Lewis acids and coordinate to a CC double or triple bond. Confirming this hypothesis, recently the isolation of [Ga(1,5-COD)2]+[pf]− (1,5-COD = 1,5-cyclooctadiene) as the first homoleptic main group metal olefin complex was reported.24
Moreover, our group has previously shown that univalent gallium catalyzes the polymerization of isobutylene.25,26 DFT studies suggest that the reaction proceeds via oxidative addition of GaI, β-hydrides elimination and insertion of isobutylene units into the C–Ga bond. Chain growth could be terminated via reductive elimination from GaIII, thereby regenerating catalytically active GaI.25 Remarkably, the proposed reaction sequence is reminiscent of a coordinative polymerization mechanism, typically invoked for transition metals. In fact, spontaneous reductive H2 elimination has been reported for cationic [H2GaIII(PhF)2]+[CHB11Cl11]−, giving [GaI(PhF)2]+[CHB11Cl11]−.19 Additionally, it is well known that neutral and anionic GaI complexes readily add oxidatively to a variety of covalent element–element bonds of like and dislike elements, e.g. H–H,27 H–C,28 H–N,27 H–O,27 H–P,27 H–Sn,27 C–Cl29 and group 15 and 16 element E–E bonds,30inter alia.31,32 Only recently, a PPh3-supported cationic Ga complex has been reported to insert into a H–P bond of a phosphonium cation.124
Such transition metal- or silylene-like33 reactivity of univalent GaI results from the 4s24p0 electron configuration18 that potentially allows for oxidative addition and reductive elimination reactions in catalytic cycles. This encouraged us to investigate the catalytic potential of Ga+ in other usually transition metal-catalyzed reactions. In this paper, we present a systematic investigation of the [Ga(PhF)2][pf]-initiated hydrosilylation of olefinic double bonds, with a focus on mechanistic considerations. While working on this and independently of us, Wehmschulte reported that similarly the use of catalytic amounts of Ga+ salts with the WCAs [CHB11Cl11]− or [B(C6F5)4]− initiates hydrosilylation of 1-hexene and benzophenone.19 Yet, no mechanistic investigations were performed and the authors refrained from speculations.
Hydrosilylation of CC double bonds is an important Si–C bond forming reaction. It is widely used in industrial processes for the production of consumer goods, e.g. for the synthesis of silicone elastomers, resins or oils.34–38 Although addition of a H–Si bond across CC double bonds is exothermic by ca. 160 kJ mol−1, the reaction is kinetically hindered. Thus, suitable catalytic systems are required, with first reports dating back to 1947, using a radical initiator.39 The introduction of hexachloroplatinic acid [H2PtCl6]·H2O (Speier's catalyst)40 and, even more importantly, Karstedt's catalyst,41 a dinuclear Pt(0) complex containing unsaturated disiloxanes, is an important milestone in homogeneous catalysis. Today, complexes containing precious transition metals such as rhodium,42 iridium43 and especially platinum are most commonly employed as catalysts, but Karstedt's catalyst still serves as the benchmark system.35,36,38
Nevertheless, Pt-catalyzed hydrosilylation reactions also suffer from drawbacks, since they are often accompanied by side reactions such as olefin-oligomerization, -hydrogenation and -isomerization, resulting in yield loss.35 In some cases, the low selectivity of Pt-catalyzed hydrosilylation, as well as the high cost, insecurity of supply and environmental issues of platinum necessitate the search for alternative catalytic systems.34,36,44
Through extensive research in this field, it was found that hydrosilylation of multiple bonds can also be catalyzed by alkaline or alkaline earth metals,45 lanthanides46 and non-precious transition metals.36,47 Besides this, group 13-based Lewis acids such as boranes as well as neutral and cationic AlIII compounds were shown to efficiently catalyze hydrosilylation reactions of olefins,48–50 imines51–54 or carbonyl compounds.52,53,55–59 According to the Piers–Oestreich mechanism, the Lewis acid forms an adduct with the silane, thus polarizing the Si–H bond, increasing the electrophilicity of the silicon atom and facilitating the nucleophilic attack of the multiple bond.49,56,58,60,61 For the aluminum halide-catalyzed hydrosilylation of alkynes, a different mechanism was proposed, with the aluminum halide coordinating to the multiple bond.62 Only very few examples of GaIII catalysts in hydrosilylation reactions have been reported in the literature.44,63 They exclusively describe the hydrosilylation of carbonyl compounds64 or CO2.65 To the best of our knowledge, the Ga+ carborate and borate salts presented by Wehmschulte are the only gallium-based systems that have been employed to promote hydrosilylation of olefins so far, yet without any mechanistic investigation.19
Results and discussion
First, we turn to an overview of the hydrosilylation capacity of the [Ga(PhF)2][pf]/silane/olefin system, before turning to mechanistic issues and further experimental and theoretical studies to understand the mechanism of the reaction.Scope of the hydrosilylation reactions with [Ga(PhF)2][pf]
The scope of the hydrosilylation reaction was investigated by employing [Ga(PhF)2][pf] (1) as the Ga+ catalyst and using different organohydrosilanes HxSiR4−x (R = aryl or alkyl substituents) and olefin substrates, listed in Table 1. Reactions were carried out in ortho-difluorobenzene (oDFB) as NMR tube reactions. The yield was determined by NMR spectroscopy and was referred to the minimum substrate. Exemplary NMR spectra for all reactions as well as a detailed evaluation of NMR data are deposited in the ESI.‡| # | Silane | Olefin | Molar ratio silane:olefin:1 |
c (olefin) [M] | Reaction time (temperature) | Main products | Yielda |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy, referred to the deficit substrate. b rt = room temperature. c 2,5-Dimethyltetrahydrofuran (7%) formed as a side product. | |||||||
| 1 | HSiMe3 |
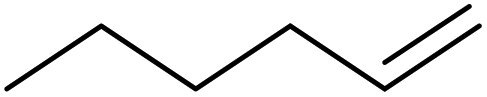
|
1.2:1.0:0.1 |
0.11 | 4 h (rtb) |
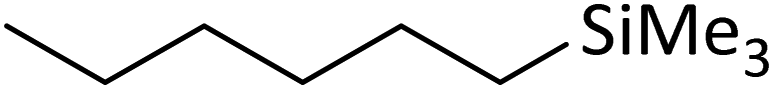
|
93% |
| 2 | HSiMe3 |
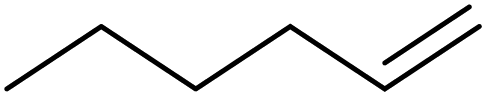
|
1.2:1.0:0.01 |
0.11 | 3 d (rt) |
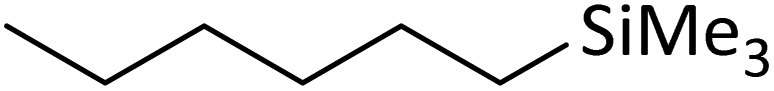
|
91% |
| 3 | HSiMe3 |
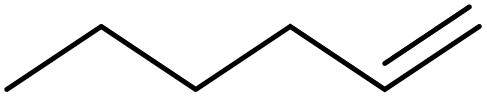
|
1.0:1.0:0.005 |
0.11 | 4.5 d (rt) |
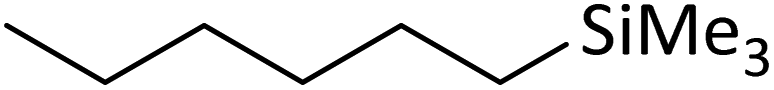
|
20% |
| 4 | HSiMe3 |
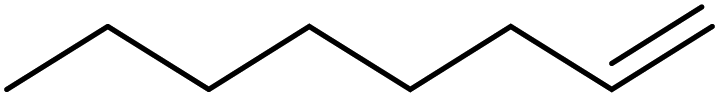
|
1.0:1.0:0.14 |
0.18 | 3 h (rt) |

|
>97% |
| 5 | HSiMe3 |
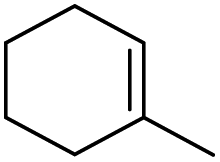
|
1.0:1.0:0.1 |
0.22 | 10 h (rt) |
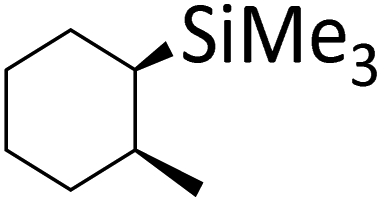
|
93% |
| 6 | HSiMe3 |
|
1.0:1.0:0.05 |
0.11 | 1 d (rt) |
|
95% |
| 7 | HSiMe3 |
|
2.2:1.0:0.1 |
0.20 | 10 h (rt) | Crude mixture of products; olefin oligomerization and addition of scrambling products | — |
| 8 | HSiMe2Et |
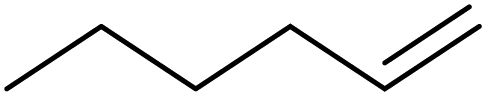
|
0.4:1.0:0.1 |
0.25 | 8 h (rt) |
 + oligomerized species + oligomerized species |
R = Me, R′ = Et: 38% |
| R = Me, R′ = Me: 1% | |||||||
| R = Et, R′ = Et: 2%; ca. 60% olefin oligomers | |||||||
| 100% of silane consumed | |||||||
| 9 | HSiMe2Et |
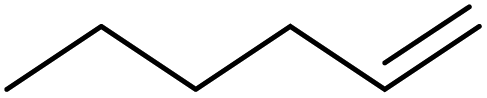
|
1.0:1.0:0.01 |
0.11 | 3.5 d (rt) + 9 h (60 °C) |
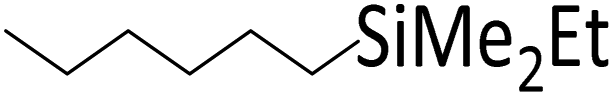
|
84% |
| 10 | HSiMe2Et |
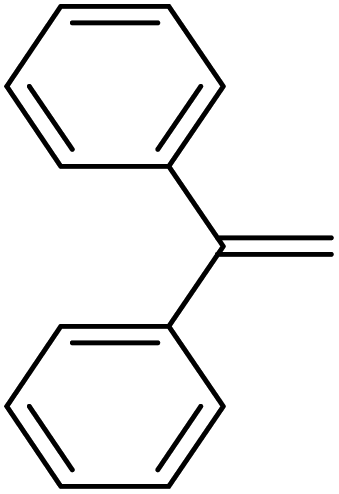
|
4.7:1.0:0.02 |
0.17 | 8 h (rt) |
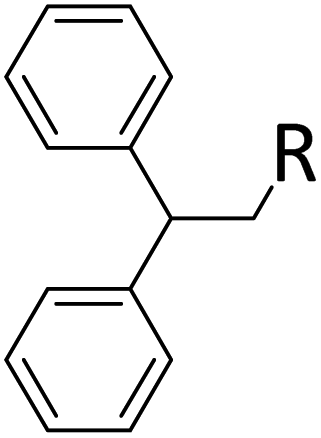
|
R = SiHMeEt: 48% |
| R = SiMe2Et: 31%; R = H: 6%; R = SiHMe2: 5%; traces of other addition products and silane scrambling products; 100% of olefin consumed | |||||||
| 11 | HSiMe2Et |
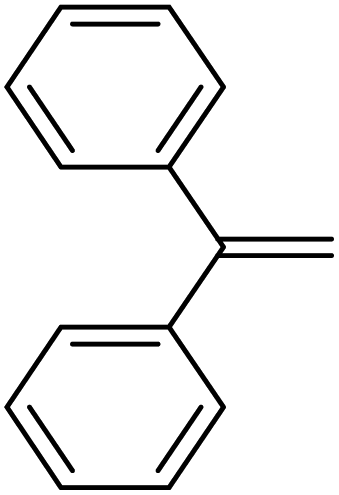
|
0.9:1.0:0.01 |
0.10 | 2 h(rt) + 1 d (60 °C) |
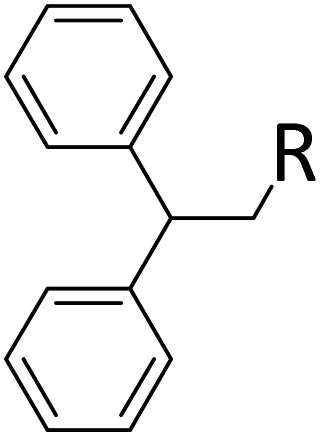
|
R = SiHMeEt: 74% |
| R = H: 4%; and silane scrambling products like Me3SiEt (7%) | |||||||
| 12 | HSiEt3 |
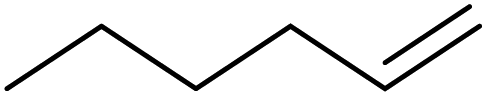
|
1.1:1.0:0.1 |
0.11 | 4 d (rt) + 1 d (60 °C) |
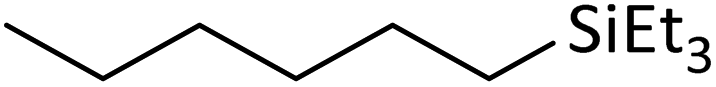
|
>97% |
| 13 | HSiEt3 |
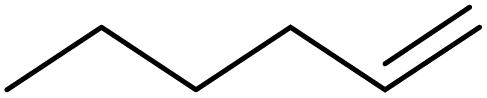
|
1.0:1.0:0.05 |
0.11 | 3.5 d (60 °C) |
|
91% |
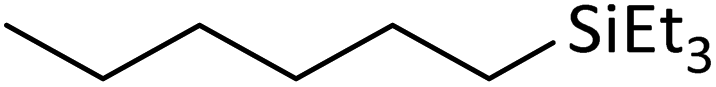
| 14 | HSiEt3 |
|
4.2:1.0:0.02 |
0.17 | 6 d (rt) + 4 h (60 °C) |
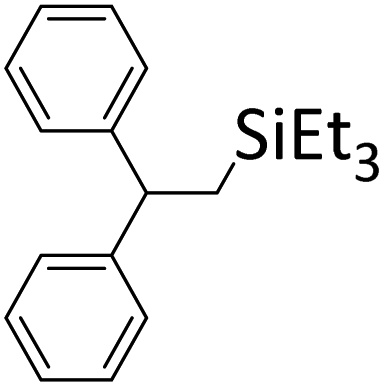
|
94% |
| 15 | HSiEt3 |
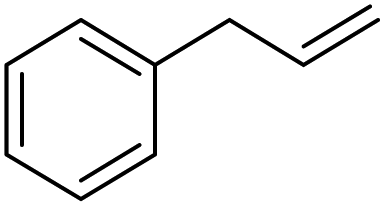
|
0.9:1.0:0.1 |
0.65 | 7 d (rt) + 2.5 d (60 °C) |
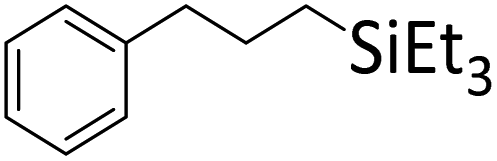
|
89% |
| 16 | HSiEt3 |
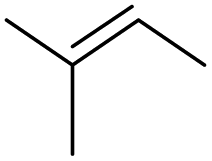
|
0.8:1.0:0.05 |
0.11 | 7 d (60 °C) + 2.5 d (80 °C) |
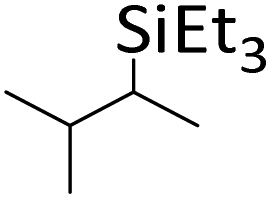
|
86% |
| 17 | HSiEt3 |
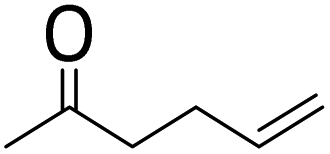
|
0.9:1.0:0.1 |
0.61 | 5 h (rt) |
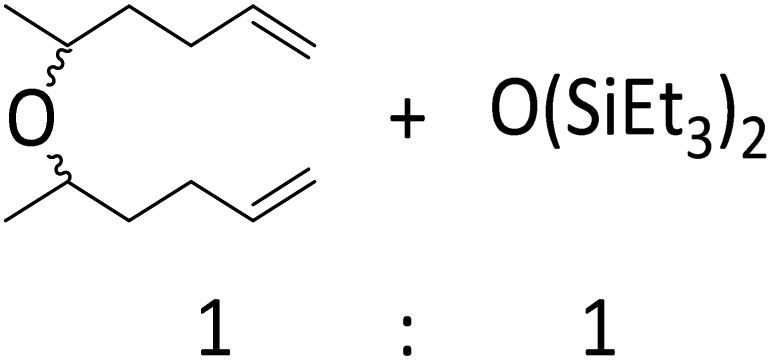
|
>90%c |
| 18 | HSiiPr3 |
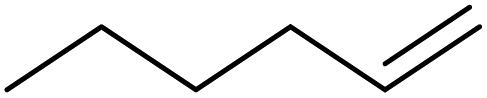
|
1.8:1.0:0.1 |
0.24 | 11 d (rt) + 2 d (60 °C) |

|
57% |
| 19 |
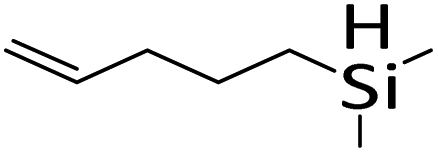
|
1.0:0.1 |
1.3 | 1 d (rt) |
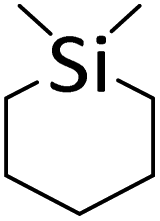
|
96% | |
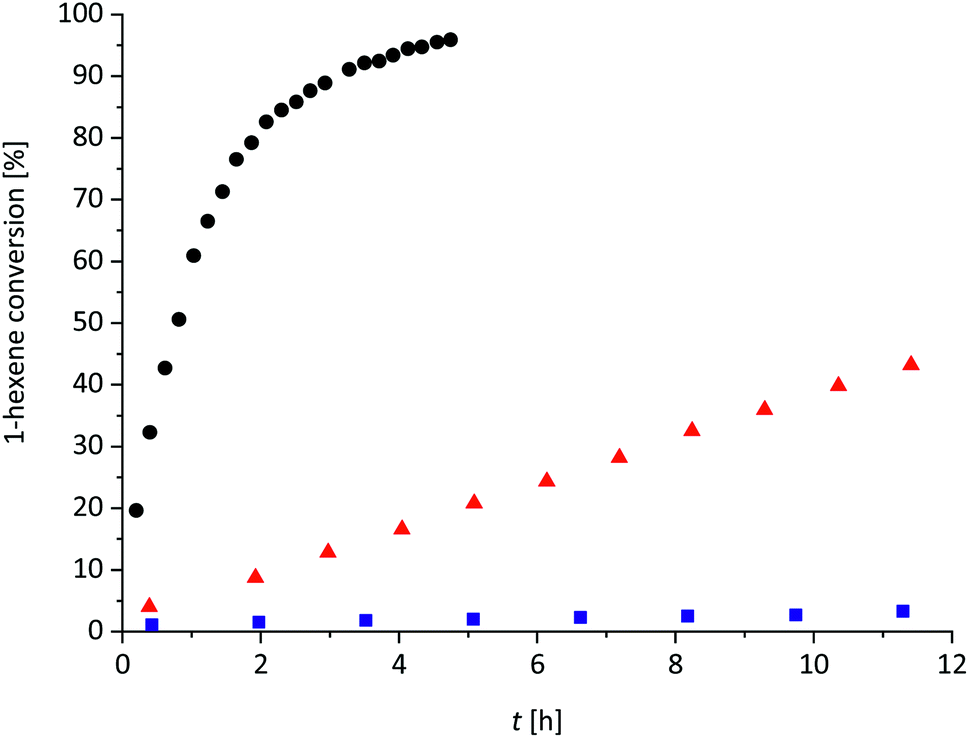 | ||
| Fig. 1 Plot of 1-hexene conversion versus time for the catalytic hydrosilylation reaction of 1-hexene with 1.2 eq. Me3SiH and 10 mol% 1 (black dots), with 1.2 eq. Me3SiH and 1 mol% 1 (red triangles) and with 1.0 eq. Me3SiH and 0.5 mol% 1 (blue squares) in oDFB (0.11 M for 1-hexene) at rt. The 1-hexene conversion was obtained by 1H NMR integration (1-hexene conversion = c (RH2C–H2C–SiMe3)/c (RH2C–H2C–SiMe3 + H2CCH–R); R = nBu). | ||
Obviously, the use of 10 mol% 1 allows for fast hydrosilylation and loadings of 1% or lower slow down the reaction, but still initiate hydrosilylation of the olefin at room temperature.
With excess HSiMe2Et, pronounced scrambling of the alkyl ligands is observed and the reaction with this silane is somewhat unselective (compare entries 10 and 14). In order to suppress these side reactions, HSiMe2Et and the olefin have to be mixed in a 1:1 stoichiometry. Probably, scrambling takes place with HSiMe3 and HSiEt3 as well. Yet, these silanes are more symmetrical and have only two different ligands, so that ligand scrambling is less pronounced in the addition product. However, if excess HSiMe3 is employed, the hydrosilane reacts with the hydrosilylation product RSiMe3 under formation of SiMe4 and RSiMe2H after completion of hydrosilylation. Obviously, ligand redistribution competes with the hydrosilylation reaction. Oligomerization of the olefin (entry 8) is another typical side reaction, especially when excess olefin is applied. The reactions with HSiEt3 usually require heating at 60 °C for several hours or days; a similar observation was reported by Wehmschulte.19 However, the hydrosilylation of trisubstituted olefins with HSiEt3 is complicated and rather slow (entry 16). The addition of bulkier HSiiPr3 is considerably slower than the reaction with less sterically hindered silanes, even with 1-hexene (entry 18).
Phenylsilanes H3SiPh and H2SiPh2 are no suitable substrates. With these silanes, extensive ligand redistribution under formation of silanes such as H–SiH3 and H–SiPh3 takes place, as well as the addition of these silanes (Section 2.1.11 in ESI‡). Obviously, ligand scrambling is faster than hydrosilylation for phenylsilanes.
C double bond does not react and instead formation of a symmetrical ether and a disiloxane is observed (entry 17). Similar results were reported for the reaction of ketones or aldehydes with a Ga(OTf)3/R3SiH system.57 Since electrophilic silicon atoms are oxophilic, this is a first indication that (stabilized) silylium ions may be present in the solution, as such species should preferably react with a CO bond rather than with a CC bond.
In some hydrosilylation reaction mixtures, the 71Ga signal is shifted downfield from −756 ppm (1 in oDFB). This probably results from interactions of the olefin or the silane with Ga+. Such interactions can possibly explain the observation that with HSiMe2Et and 1,1-diphenylethylene, the initiation of the reaction is delayed for 8 hours, most probably due to the coordination of the phenyl moieties to Ga+.7,26 Yet, once started, it proceeds within half an hour to full conversion at rt (entry 10; Section 2.1.7 in ESI‡).
The reaction with diolefins like 1,5-hexadiene (entry 7) or 1,5-COD resulted in the formation of a crude mixture of products, suggesting the presence of highly reactive intermediates.
66 as a solvent is crucial for the reaction. The addition of more coordinating solvents slowed down the reaction. For example, when the hydrosilylation of 1-hexene with HSiMe3 (10 mol% of 1) was repeated in oDFB with 10 vol% of slightly electron-richer PhF (= ca. 90 equivalents PhF referred to 1), it took more than 20 h until 90% of the olefin was hydrosilylated. The reaction was further slowed down to 40% conversion after 11 days at rt, when only 10 vol% toluene (= ca. 80 equivalents toluene referred to 1) was added to the reaction mixture.
As typical donor–acceptor complexes, the stability of [Ga(arene)x]+ complexes increases with the increase in π-basicity of the arene ligands. Consequently, in a mixture of aromatic solvents, the [Ga(arene)x]+ complex with the more π-basic ligand is always observed in solutions by NMR spectroscopy, as also supported by quantum chemical calculations.7,15 Evidently, the Ga+ ions have to be nearly “naked” in solution to initiate the hydrosilylation of olefins.
Mechanistic DFT investigation: Ga+-centered reaction?
The formation of anti-Markovnikov addition products is also typically observed with transition metal catalysts. This is rationalized by the widely accepted and thoroughly investigated Chalk–Harrod mechanism,67 involving oxidative addition of a transition metal into the H–Si bond, hydrometalation and reductive elimination. Thus, we first assumed that the Ga+-catalyzed hydrosilylation proceeds via a similar mechanism. This is plausible in light of the 4s24p0 electron configuration of GaI, principally allowing for transition metal or silylene33-like reactivity. In line with this, oxidative addition of neutral or anionic GaI species into covalent bonds has been reported for a multitude of different covalent bonds.31 However, to the best of our knowledge, oxidative addition of cationic, unsupported GaI arene complexes into element–element bonds has not been proven experimentally so far. In order to add oxidatively into a covalent bond, a narrow HOMO/LUMO gap and energetically high lying occupied frontier orbitals are required. Therefore, the use of anionic ligands, e.g. in GaI[DippNacNac], typically facilitates oxidative addition of the resulting neutral GaI compounds in confined environments.27,29,31 | ||
| Scheme 1 Energy landscape for Ga+-catalyzed hydrosilylation of propylene with HSiMe3, according to a Ga+-centered Chalk–Harrod mechanism (calculated at the RI-BP86(D3BJ)/def2-TZVPP level of theory; all values are expressed in kJ mol−1). | ||
With activation barriers surpassing 200 kJ mol−1, the computational study strongly suggests that the oxidative addition of oDFB-complexed Ga+ into the H–Si bond is not possible under ambient conditions. As expected, the reductive elimination of the cationic gallium species is slightly less disfavored, but activation barriers are still prohibitive, especially since single-point calculations with the gold standard CCSD(T) at the basis set limit and our model chemistry RI-BP86(D3BJ)/def2-TZVPP do not differ by more than 14 kJ mol−1 and also the effect of solvating the system with the COSMO model only changes the energetics by less than 10 kJ mol−1 (Section 6.2.1 in ESI‡).
Further experimental investigations on the mechanism
 | ||
| Scheme 2 The anti-addition product of HSiMe3 and 1-methylcyclohexene is formed exclusively instead of the syn product. Since the starting materials are achiral, the chiral reaction product is racemic. | ||
This implies that the H and SiMe3 moieties add in a stepwise reaction sequence, which effectively rules out the Chalk–Harrod mechanism and underscores the theoretical calculations.
This prompted us to examine a mixture of 1 and HSiMe3 in oDFB in some detail by NMR spectroscopy. The components were mixed at −40 °C in a 1.0:4.8 ratio, and the NMR spectrum at this temperature showed no direct sign of reaction between the components. Yet, the coupling constant 3JSiH,CH could not be resolved (vide infra). Slowly increasing the temperature allowed for reaction monitoring. 1H NMR spectra recorded at different temperatures are displayed in Fig. 2.
 | ||
| Fig. 2 From bottom to top: 1H NMR-spectra of HSiMe3 in oDFB at 298 K (300.18 MHz), HSiMe3/1 (4.8 : 1.0) after 3 h at 233 K, after 15 h at 253 K, after 2 h at 273 K, after 4 h at 298 K and after 3 d at 298 K (all 400.17 MHz). Signal intensities were normalized to the oDFB signal at 6.96 ppm (not shown). The signal at 0.35 ppm is caused by traces of Cl–SiMe3 in the HSiMe3 solution. | ||
Above and at 0 °C, the formation of H2SiMe2 and SiMe4 is observed. These species must be formed due to a ligand exchange of H and Me groups. 19F NMR spectra show that, at room temperature, the [pf]− anion is quantitatively converted into perfluorinated epoxide F2C(O)C(CF3)2 and Me3Si–F–Al(ORF)3 (Section 2.3.1 in ESI‡). These compounds are the typical decomposition products of the [pf]− anion in the presence of a [SiMe3]+ silylium ion.68,69 Additionally, the presence of silylium ions would easily account for the observed ligand redistribution.70–73 Note that the underlying mechanism has been investigated in detail.74,75 Consequently, the fact that aryl ligands display a greater migration tendency75 probably explains why the attempted hydrosilylation with H3SiPh or H2SiPh2 and 1 led to extensive ligand redistribution. In line with this, we isolated crystals of SiPh4 in a mixture of H2SiPh2 and 1.
Another evidence for the presence of silylium cations is the fact that the 3JH,H coupling constant in HSiMe3 in a mixture of HSiMe3 and 1 in oDFB is obviously reduced (Fig. 3). This is a general feature and also holds for a HSiEt3/1 mixture in oDFB (Section 2.3.2 in ESI‡).
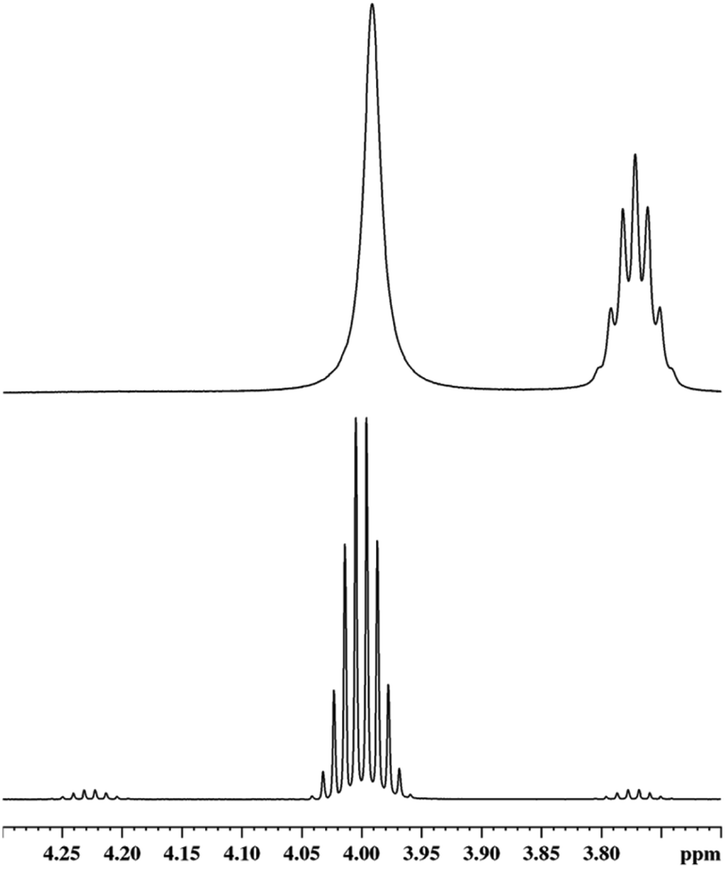 | ||
| Fig. 3
1H NMR signal (400.17 MHz, oDFB, 298 K) of the H–Si hydrogen atom in HSiMe3 (bottom) and in a HSiMe3/1 (4.8:1.0) mixture (top) in oDFB. On the top right, one notes the H–Si septet-signal of H2SiMe2; see text. | ||
The signal of the Si–H hydrogen atom in HSiMe3 is not only broadened, indicating chemical exchange, but its full width at half maximum of 8.0 Hz does not allow to cover fully the original multiplet, which is at least 11.5 Hz broad at the same height. Hence, the absolute value of the 3JH,H coupling constant must be reduced, which can only occur when the hydrogen atoms are exchanged between different silicon atoms. Although the splitting pattern in the resonance of the Si–H group of H2SiMe2 is still resolved, there is chemical exchange between H2SiMe2 and HSiMe3, which is demonstrated in 1H EXSY NMR spectra (Section 2.3.1 in ESI‡). In the same spectrum, in the area of the H3C–Si groups, additional exchange processes between Me3SiH and other species containing Me3Si groups, mainly Me3Si–F–Al(ORF)3, can be observed.
In addition, the 71Ga NMR signal disappears in HSiR3/1 (R = Me, Et) mixtures and a metallic mirror forms inside the NMR tube (Section 5 in ESI‡), indicating that the Ga+ ions were reduced to elemental gallium. In agreement with this, a new 1H NMR signal at 4.5 ppm could be ascribed to H2, in line with the results from gas chromatography (vide infra).76 In a mixture of 1 and HSiEt3, the analogous reactions were observed by NMR spectroscopy (Section 2.3.2 in ESI‡). Moreover, crystals of Et3Si–F–Al(ORF)3 were isolated from a concentrated solution of 1 and HSiEt3 in oDFB. A balanced reaction equation and molecular structure of Et3Si–F–Al(ORF)3 are shown in Scheme 3. The structural parameters are comparable to those found in Me3Si–F–Al(ORF)368 and tBu3Si–F–Al(ORF)3,77 identifying an “ion-like” silylium complex.78
 | ||
| Scheme 3 Formation and molecular structure of Et3Si–F–Al[OC(CF3)3]3 in a mixture of 1 and HSiEt3. All atoms were drawn with anisotropic thermal ellipsoids at the 50% probability level. Hydrogen atoms and a minor disorder in the Al(ORF)3-part were omitted for clarity. Selected bond lengths [pm] and angles [°] of the ordered sections of the molecule: F1–Si1: 173.18(17), F1–Al1: 178.82(16), Al1–O: 169.2(5)–170.80(19), Si1–F1–Al1: 157.67(9). Sum of C–Si–C angles: 346.44(14)°. | ||
 | ||
| Fig. 4 Gas chromatogram of the gas space above the reaction solution of HSiMe3 and 1 (5.8:1.0) in oDFB (a). Cyclic voltammograms of 1 and [Fc][pf] in oDFB (0.005 M, respectively) at rt and at a Pt working electrode (WE); measured with different scan rates. [NBu4][pf] (0.1 M) was used as a conducting salt (b). STEM element maps (fluorine, silicon and gallium) associated with the dark-field image of the residue of the HSiMe2Et/1-hexene/1 (2.2:1.0:0.1) reaction mixture (c) and background-corrected EDX line scan for the elements O, F, Al, Si, and Ga in the same sample across a Ga-rich particle (d). The F-rich area in (c) (top left) probably results from traces of non-vaporized oDFB. | ||
The gas formed upon mixing 1 and a silane was unambiguously identified as H2 by gas chromatography (Fig. 4a). Adding HSiMe3 or HSiEt3 to a solution of 1 in oDFB resulted in the almost immediate formation of H2, whereas addition of HSiEt3 to a mixture of 1 and 1-hexene in oDFB resulted in a slightly slower gas evolution (Section 3.1 in ESI‡). This is probably due to coordination of olefin molecules to Ga+, which have to be displaced by the silane. No H2 could be detected in solutions of 1 and an olefin in oDFB.
The cyclic voltammograms of a 0.005 M solution of 1 in oDFB (Fig. 4b) reveal that the redox potential of Ga+/Ga0 is more positive than the potential of [Fc]+/[Fc] in oDFB (Fc = ferrocene). The exact redox potential E1/2 is difficult to determine, since it depends on the scan rate (e.g. E1/2 = +0.26 V vs. Fc+/Fc for 20 mV s−1, and E1/2 ≈ +0.37 V vs. Fc+/Fc for 100 mV s−1). Thus, the conversion of Ga+ into Ga0 is electrochemically not fully reversible. For further experimental proof of this high and positive Ga+/Ga0 potential, 1 was added to the orange-yellow solution of ferrocene and the mixture turned blue immediately, indicating oxidation of neutral ferrocene to ferrocenium (Section 5 in ESI‡). Thus, we showed that Ga+, typically viewed as a subvalent reductant,79 can act as an oxidizing agent with a formal potential even higher than that of Fc+. Note that ferrocenium salts are typically used as chemical oxidants.80 Interestingly, no electrochemical oxidation of Ga+ to Ga3+ was observed (Section 3.2 in ESI‡).
Unfortunately, no cyclic voltammograms of HSiEt3 could be recorded under the same conditions. Yet, it has already been shown in 1958 that HSiEt3 can reduce inorganic halides with the formation of H2, elemental metal and XSiEt3 (X = Br and Cl).81 Silanes and related H–Si containing compounds have been employed as reducing agents for more oxidizing metal ions, e.g. for Rh3+,82 Pd2+,82,83 Pt4+,82,83 Cu2+,84 Au3+,82,83 [AuCl4]−,85 and Ag+83,86 ions, in order to obtain the respective metal nano-particles. Besides this, hydrosilanes act as reducing agents in redox-initiated cationic polymerization reactions.87 It is known that GaIII can oxidize organic compounds under H2 formation, however, without being reduced to elemental gallium.88 Yet, the use of naked “Ga+” as an oxidizing agent towards silanes is new. Moreover, in oDFB, HSiMe3 reacts with the oxidizing salts NO[pf] and Ag[pf] in a similar manner to 1, i.e. under H2 formation, ligand scrambling and [pf]− anion decomposition (Sections 2.3.3 and 2.3.4 in ESI‡). This supports the notion that Ga+, too, acts as an oxidizing agent towards silanes. The metallic precipitate formed during a hydrosilylation reaction was isolated in small amounts and was analyzed by STEM-analysis. It includes largely metallic gallium particles (Ga0 by STEM-analysis, Fig. 4c and d) embedded in a Ga-poor but O- and Si-rich matrix, confirming that a redox reaction between 1 and hydrosilanes takes place.
As already pointed out, the addition of toluene slows down the hydrosilylation reaction initiated by 1 in oDFB. Toluene is more electron-rich and the arene molecules may coordinatively saturate the Ga+ ions, thereby preventing the coordination of silane molecules and thus the suspected redox reaction between silane and univalent gallium. Moreover, the hydrosilylation with HSiiPr3 and initiated by 1 is extremely slow even in oDFB (entry 18 in Table 1). This is a hint that the reaction between silane and Ga+ is dependent on a coordinatively unsaturated Ga+ cation, and that the steric demand of ligands may also play a major role in the reaction kinetics. Possibly, in order to initiate the redox reaction, at least two silane molecules have to coordinate to Ga+. Therefore, it seems plausible that an inner sphere mechanism is operative and that the steric bulk of the iPr groups disfavors the redox reaction.
Computational analysis of the redox reaction between 1 and silane, catalytic cycle
The thermodynamics of the postulated redox reaction between Ga+ and HSiMe3 were examined by DFT methods. It was assumed that [Ga(oDFB)(HSiMe3)2]+ and, subsequently, [(Me3Si)2H]+ are formed. Optimized structures and their underlying thermodynamics are shown in Scheme 4. The species [(Me3Si)2H]+ was chosen as a silylium equivalent, since silylium ions [R3Si]+ are highly reactive electrophiles89,90 and already form Lewis acid–base adducts with moderate to weak nucleophiles like toluene.11,12,91 Such silylium-silane adducts, or bissilylhydronium ions, are well known92–94 and due to the great excess of silane and the non-resolved 3JH,H coupling in mixtures of 1 and a hydrosilane, it is plausible to assume that such species are present in an oDFB solution. Computational analysis suggests that the bissilylhydronium ion [Me3Si–H–SiMe3]+ is more stable than [Me3Si(oDFB)]+ adducts by ca. 50 kJ mol−1 (Section 6.2.3 in ESI‡), which is in agreement with previous experimental findings.90,93,94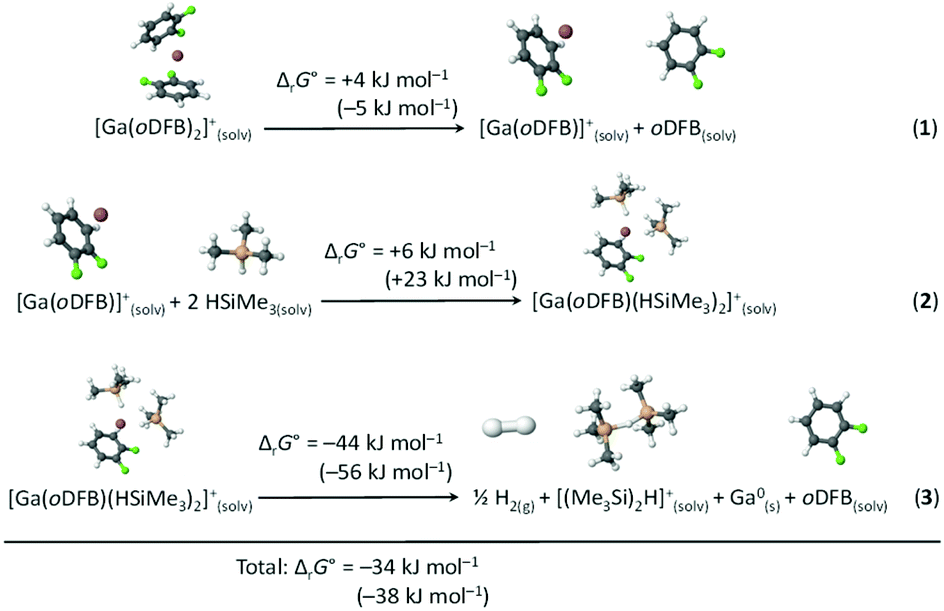 | ||
| Scheme 4 Calculated Gibbs free energies ΔrG° (oDFB solution, calculated with the COSMO model, εr = 13.3866) for the dissociation of [Ga(oDFB)2]+ (reaction (1)), subsequent addition of two HSiMe3 molecules to yield [Ga(oDFB)(HSiMe3)2]+ (reaction (2)), and its decomposition to give H2, Ga0, oDFB and [(Me3Si)2H]+ (reaction (3)). The Gibbs free energies were calculated at the RI-BP86(D3BJ)/def2-TZVPP level at 298 K (values in parentheses: RI-B3LYP(D3BJ)/def2-TZVPP). The optimized structures of the involved species are included. | ||
It follows from the computational analysis that the postulated reaction is thermodynamically possible, with the formation of gaseous H2 and elemental gallium clearly being the driving force. In addition, the oDFB/silane ligand exchange is expected to be a fast process in the solution. Moreover, only a smaller fraction of the silane molecules would have to react according to the reaction in Scheme 4, since we propose that the supported silylium ions are the genuine, catalytically very active, species.
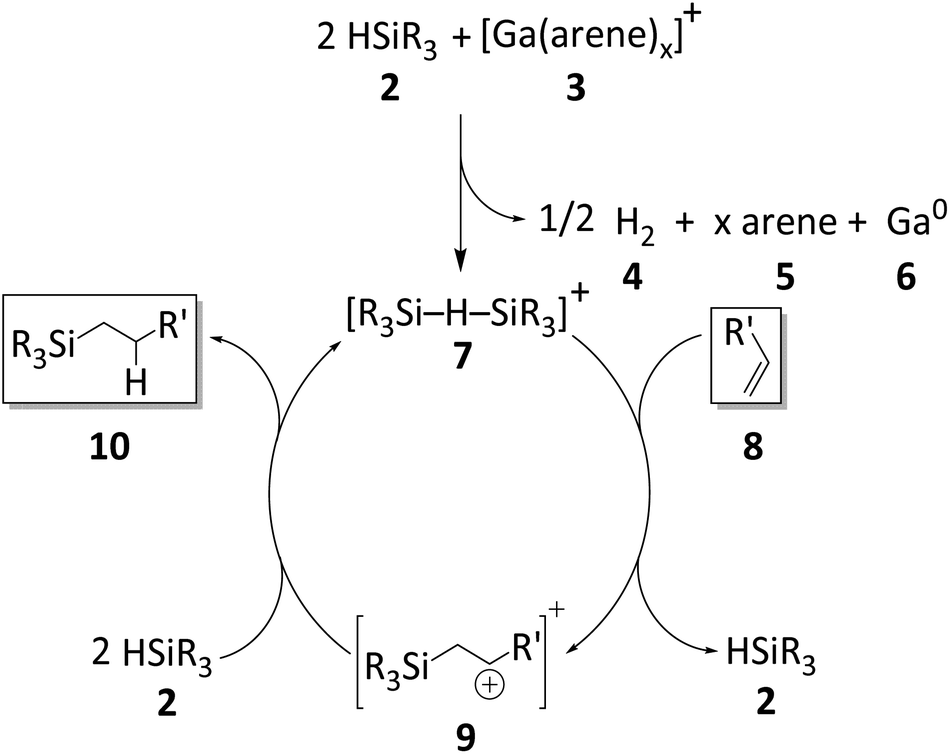 | ||
| Scheme 5 Proposed catalytic cycle for the hydrosilylation of olefins initiated by Ga+ (arene = oDFB or PhF). The olefin 8 and the hydrosilylation product 10 are highlighted. | ||
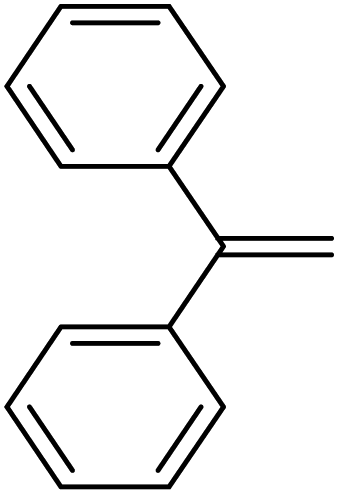
Since silylium ions are highly reactive species that usually cannot be observed in the solution,98 we attempted to observe β-silyl carbocations instead (9 in Scheme 5). We chose 1,1-diphenylethylene as a suitable substrate, due to the high stability of the intermediate β-silyl carbocation.95 Unfortunately, no intermediates were observed in a mixture of 1, HSiEt3 and 1,1-diphenylethylene (Section 2.1.9 in ESI‡), even below 0 °C. The accumulation of β-silyl carbocations is probably prevented by the fact that silylium ions are generated in situ together with excess silane that acts as an available hydride donor and reduces the lifetime of the carbocation.
The exact mechanism of the initial redox reaction is not entirely clear. For example, a direct one-electron reduction of Ga+ is conceivable as well as a Piers–Oestreich-like reaction. The Piers–Oestreich mechanism has been extensively studied and applies to hydrosilylation reactions of various substrates with neutral or cationic Lewis acids.36,49–51,55,56,60,99 If the Piers–Oestreich mechanism is applied to the herein investigated reaction, Ga+ and a silane molecule would form adducts of the type [Ga–H–SiR3]+, which are subsequently attacked by the olefin, forming β-silyl carbocations and “GaH”. The latter would decompose into elemental gallium and H2, while the β-silyl carbocations would initiate the reactions of the catalytic cycle shown in Scheme 5. Thus, a Piers–Oestreich-like mechanism and a direct initial redox reaction would essentially lead to the same outcome and both mechanisms account for the observations and experimental results presented herein. However, quantum chemical calculations suggest that, even when the formation of a Si–C bond in the β-silyl carbocation is considered, the formation of an intermediate gallium hydride is endergonic by ca. 150 kJ mol−1 in oDFB (Section 6.2.4 in ESI‡). This is ultimately due to the weakness of the Ga–H bond especially in weakly coordinating environments19,66 and due to the relative stability of Ga+ cations compared to silylium ions or carbocations. Besides this, the fact that Ga+ oxidizes ferrocene suggests that Ga+ acts as a one-electron oxidizing agent. Thus, even though it cannot be ruled out experimentally, it seems rather unlikely that a classical Piers–Oestreich mechanism is operative in the system 1/HSiR3/olefin.
Verification of silylium ion catalysis by initiation with trityl aluminate
The validity of the mechanism shown in Scheme 5 is further supported by the fact that catalytic amounts of [Ph3C][pf]100 instead of 1 also initiate hydrosilylation reactions at room temperature. The reaction between trityl salts and hydrosilanes is known as the Bartlett–Condon–Schneider reaction and is widely employed in order to generate silylium ions.70,73,75,95,96,101 As shown in Table 2, with 1.0 mol%, 0.5 mol% and 0.3 mol% of [Ph3C][pf], the reaction between 1-hexene and HSiMe3 (Scheme 6) is almost immediately completed and also yields the anti-Markovnikov product (cf. entries 1–3 in Table 1). Using 0.2 mol% is less reliable. When employing such low concentrations of [Ph3C][pf], the reactivity of this system is probably influenced by trace impurities, due to the high reactivity of both the trityl cation102 and silylium ions.71,89,90| # | Molar ratio silane:olefin:[Ph3C][pf] |
Reaction time | Yielda |
|---|---|---|---|
| a Determined by 1H NMR spectroscopy, referred to the deficit substrate. b The rate of the reaction with 0.2 mol% [Ph3C][pf] varies significantly and is somewhat erratic: full conversion was observed after 1 h to 5 d. | |||
| 1 | 1.1:1.0:0.01 |
<5 min | >97% |
| 2 | 1.1:1.0:0.005 |
<5 min | >97% |
| 3 | 1.1:1.0:0.003 |
8 min | >97% |
| 4 | 2.0:1.0:0.002 |
1 h | >97%b |
| 5 | 1.0:1.0:0.002 |
1 h | 93%b |
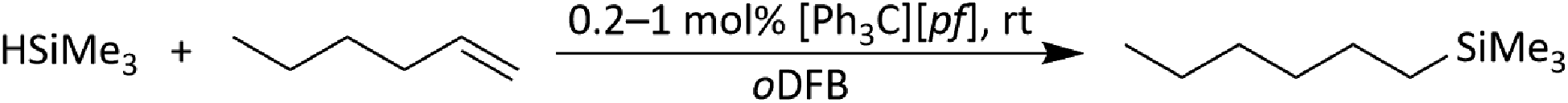 | ||
| Scheme 6 Hydrosilylation of 1-hexene with HSiMe3, initiated by [Ph3C][pf] and catalyzed by silylium ions. | ||
The fact that the hydrosilylation reaction is considerably faster with [Ph3C][pf] than with 1 is not surprising and indicates that [Ph3C]+ is more efficient in generating silylium ions in situ than Ga+. Partial anion decomposition to the perfluorinated epoxide F2C(O)C(CF3)2 and to Me3Si–F–Al(ORF)3 (Section 2.1.17 in ESI‡) again points to the presence of silylium ions, but does not affect the hydrosilylation reaction.
Hydrodefluorination with the 1/HSiEt3 and the [Ph3C][pf]/HSiEt3-system
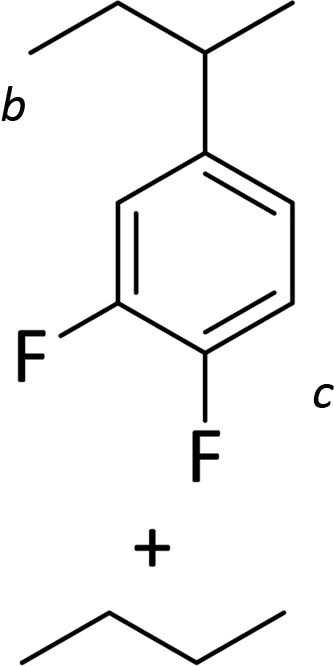
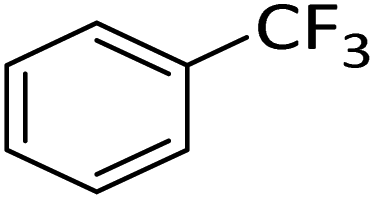
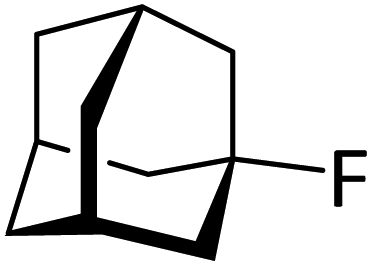
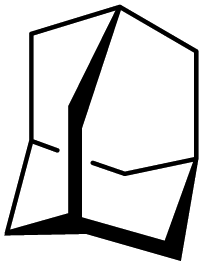
Silylium catalysis is a growing research field and has already become a powerful tool for various chemical transformations.71,89,90,103 For example, the concept of mild, [Et3Si][WCA]-catalyzed hydrodefluorination (WCA = [B(C6F5)4]− or carborate), i.e. the transformation of a C–F bond into a C–H bond was introduced by Ozerov.98,104 Such transformations are challenging, due to the strength of C–F bonds.105 In the systems presented by Ozerov, silylium ions abstract C(sp3)-bound fluorine atoms and stoichiometric amounts of hydrosilanes serve as hydride donors for the resulting carbocations, thus regenerating the catalytically active silylium ions.In order to further probe whether silylium ions are present in the mixture of 1 and a hydrosilane in oDFB, we tested whether hydrodefluorination reactions of C(sp3)–F bonds at room temperature are possible with this system. Considering the results presented in the previous sections, it is no surprise that the HSiEt3/1 mixture indeed induces hydrodefluorination. This was exemplarily demonstrated with four different, representative substrates, i.e. 1-fluorobutane, trifluorotoluene, 1-fluoroadamantane and n-perfluorohexane (Section 2.2 in ESI‡). With trifluorotoluene, a mixture of diphenylmethane derivatives was formed, whereas with 1-fluorobutane, the formation of butane and of an s-butylated oDFB derivate was observed (entries 1 and 2 in Table 3). The hydrodefluorination of 1-fluoroadamantane proceeded smoothly and quantitatively yielded adamantane (entries 3 and 4). We employed 1-fluoroadamantane since it serves as a benchmark substrate for hydrodefluorination reactions, in order to compare catalytic efficiencies of Lewis-acidic systems.106–118
| # | R–F | Molar ratio HSiEt3:R–F:1 |
c (R–F) [M] | Reaction time (temperature) | Main products | C–F conversiona |
|---|---|---|---|---|---|---|
|
a Determined by 19F NMR spectroscopy (C–F conversion = c (Et3Si−F)/c (R3C−F + Et3Si−F)).
b Additionally, traces of the regioisomer with the sBu group in 2 position of the aromatic ring were detected.
c The s-butylated oDFB derivateandnbutaneare formed in a 0.3:1.0 ratio.
d Anion decomposition was observed.
|
||||||
| 1 |
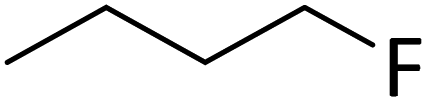
|
1.1:1.0:0.04 |
0.62 | 10 h (5 °C) |
|
>97% |
| 2 |
|
4.0:1.0:0.05 |
0.48 | 17 h (rt) |

|
96% (product mixture) |
| 3 |
|
2.8:1.0:0.05 |
0.18 | <3 min (rt) |
|
>97% |
| 4 |

|
2.0:1.0:0.001 |
0.26 | 14 h (rt) |
|
95% |
| 5 |
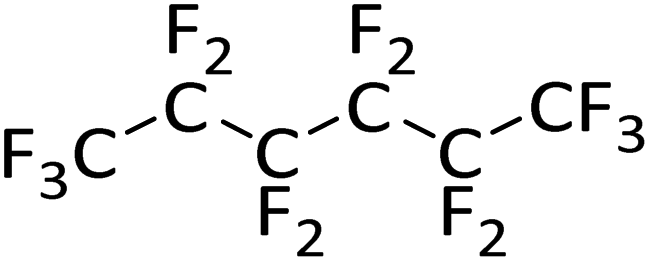
|
15:1.0:0.56 |
0.21 | 14 d (rt) | No reactiond | — |
The attempted hydrodefluorination of n-perfluorohexane with 1/HSiEt3 was unsuccessful (entry 5). The inertness of perfluorinated alkanes in silylium-catalyzed hydrodefluorination reactions is well documented98,119 and can probably be attributed to the strong –I effect of the adjacent fluorine atoms, which would destabilize intermediate alkylcarbocations.
The reaction products indicate that with 1-fluorobutane, trifluorotoluene and 1-fluoroadamantane, the intended hydrodefluorination reactions took place. Yet, the hydrodefluorination of trifluorotoluene was accompanied by Friedel–Crafts reactions and, for 1-fluorobutane, additionally by Wagner–Meerwein rearrangements. It is revealing that, in the reaction with 1-fluorobutane, the aromatic solvent is s-butylated instead of n-butylated (entry 1), since primary carbocationic species are usually less stable than secondary ones. Therefore, Friedel–Crafts reactions with alkylating agents often lead to unexpected products with rearranged alkyl substituents.120
The hydrodefluorination of trifluorotoluene yielded a mixture of diphenylmethane derivatives instead of the expected product, toluene (entry 2). However, toluene is most likely formed initially, but, as a reasonably electron-rich aromatic compound, reacts with the intermediate carbocations in Friedel–Crafts reactions under C–C bond formation. The reaction outcome is reminiscent of the results for [Et3Si][carborate]-catalyzed hydrodefluorination reactions with trifluorotoluene.104,119 Interestingly, as the reaction proceeds, only CH3–, CH2– and CF3 groups are present in the solution. No intermediates like Ar–CF2H or Ar–CFH2 were observed, even when only a 1.5-fold excess of triethylsilane was employed. Consequently, the abstraction of the first F-atom in trifluorotoluene is more energy-intensive than the abstraction of the next two F-atoms, in line with the decreasing C–F bond enthalpy of R–CFxH3−x for decreasing x.121 This is an important finding, since similar results were reported for the [Et3Si][B(C6F5)4]-catalyzed hydrodefluorination of PhCF3 by Ozerov.98 In a side reaction, the hydride source, HSiEt3, probably reacts with the protons released in the Friedel–Crafts reactions. This results in the formation of “[SiEt3]+”, and of H2, which is underpinned by an intense 1H NMR signal of H2 at ca. 4.50 ppm.
Gratifyingly, 1-fluoroadamantane was hydrodefluorinated in an almost immediate reaction at rt, yielding adamantane quantitatively (entry 3). It is noteworthy that the hydrodefluorination reaction with our herein presented system HSiEt3/1 is remarkably faster than the reaction with highly Lewis-acidic, but neutral, bis(catecholato)silanes recently presented by Greb,114 again indicating the presence of highly reactive species in the reaction solution. It is difficult to estimate turnover numbers (TON) or turnover frequencies (TOF) for our catalytic system, since the exact concentration of the silylium ions, the supposed catalysts, is not known. Even when assuming that every Ga+ (c = 8.4 mM) converts one silane molecule in a silylium ion, the TOF is greater than 0.1 s−1 at room temperature. This value is significantly higher than the TOF for the bis(catecholato)silanes in the analogous reaction (c = 7.5 mM; ca. 2.5 × 10−3 s−1 at 75 °C for the most active catalyst after 3 h). Typically, for catalytically active Lewis acid/hydride donor systems, TOF values between 1 × 10−4 s−1 and 7 × 10−2 s−1 for the hydrodefluorination of 1-fluoroadamantane are reported, underlining the high efficiency of the 1/HSiEt3 system in hydrodefluorination reactions.106–118 However, the hydrodefluorination of this substrate is considerably faster than any other GaI-initiated hydrosilylation or hydrodefluorination reaction presented herein. Thus, the reaction may follow a different mechanism with this particular substrate. Remarkably, the reaction is also catalyzed by 0.1 mol% of 1 (c = 0.29 mM; entry 4) at rt.
In this context, it has to be noted that the initiation reaction of the hydrodefluorination reaction sequence could similarly involve a fluoride abstraction by Ga+, resulting in the formation of “GaF” and a carbocation, which would subsequently react with a silane molecule to yield the hydrodefluorination product and a silylium ion. Either way, the results of the hydrodefluorination reactions with 1/HSiEt3 again imply that reactive cations, i.e. carbenium and silylium ions, are the reaction intermediates in Ga+-initiated hydrosilylation and hydrodefluorination reactions. In order to further support this thesis, we conducted another hydrodefluorination experiment with 1-fluoroadamantane, HSiEt3 and [Ph3C][pf] (c = 0.53 mM) in a 1.0:2.0:0.002 ratio. Complete hydrodefluorination was observed within 15 minutes, which corresponds to an exceptionally high TOF of at least 0.5 s−1.
These are important findings as it was often assumed that the use of carborate or borate anions is mandatory for silylium ion catalysis, since other anions are less robust towards these strong electrophiles.89,90 In line with this, to the best of our knowledge, the only alternative GaI species that initiate hydrosilylation reactions are a carborate and a borate salt.19 Gratifyingly, our results indicate that silylium catalysis is also possible with the straightforward and very large-scale accessible [pf]− anion (>100 g in one batch).20,122 For example, the GaI salt 115 and the trityl salt [Ph3C][pf]100 can easily be synthesized and the latter, obviously a very potent initiator for silylium-catalysis, is even commercially available.123
Conclusion
We demonstrated that the system [Ga(PhF)2][pf]/HSiR3 (R = alkyl) initiates hydrosilylation reactions of olefinic double bonds in oDFB under mild conditions. Pronounced ligand scrambling is observed with phenylsilanes and, if excess silane is applied, with the less symmetrical silane HSiMe2Et, which makes the hydrosilylation less selective with these silanes. A very slow reaction was observed with HSiiPr3. Additionally, efficient hydrodefluorination of C(sp3)–F bonds works with [Ga(PhF)2][pf]/HSiEt3 in oDFB. We proposed that the reaction sequence, for both hydrosilylation and hydrodefluorination, is initiated by a redox reaction between Ga+ and the silane, releasing Ga0, H2 and a HSiR3-masked silylium ion. The masked or supported silylium ions probably act as the actual catalytically active species and Ga+ as the initiator. To the best of our knowledge, this is the first systematic report of the use of subvalent gallium as an oxidizing agent, which adds a new exciting facet to the chemistry of Ga+. The surprisingly high oxidative potential of Ga+ in oDFB was confirmed by cyclic voltammetry, and we showed that Ga+ oxidizes ferrocene in oDFB. In addition, our results suggest that (masked) silylium ion catalysis is possible with the [pf]− anion. Consequently, highly efficient hydrosilylation of 1-hexene and hydrodefluorination of 1-fluoroadamantane were observed using only 0.2 mol% [Ph3C][pf]. We anticipate that the use of the [pf]− anion could simplify silylium catalysis in the future and promote the development of new silylium-catalyzed reactions. Sparked by this and other unusual chemistry, the application and understanding of [Ga(PhF)2][pf] in catalytic transformations is currently one of the main research interests in our laboratory.Data availability
Electronic supplementary information (ESI) is available. Full experimental details, 1D- and 2D NMR spectra of the reactions are deposited. Details to the quantum chemical calculations are given together with the results of gas chromatographic, cyclic voltammetry, STEM/EDX measurements and crystallographic details.Author contributions
I. K., A. B. and K. G. conceived the experiments. A. B., K. G. and A. H. performed the experiments. H. S. measured the NMR spectra, all authors analyzed and discussed the experimental data. A. B. and I. K. co-wrote the paper and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg and by the DFG in the Normalverfahren. We would like to thank Harald Scherer and Fadime Bitgül for the measurement of the NMR spectra, Dr Ralf Thomann for the collection of the STEM and EDX data and Dr Daniel Kratzert and Dr Burkhard Butschke for their support regarding single X-ray crystallography. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/467-1 and 575-1 FUGG (JUSTUS1 and 2 cluster). The use of the STEM-EDX set up, acquired through the BMBF project EDELKAT (FKZ 03X5524), is gratefully acknowledged.References
- R. J. Baker and C. Jones, Dalton Trans., 2005, 1341–1348 RSC.
- M. L. Green, P. Mountford, G. J. Smout and S. Speel, Polyhedron, 1990, 9, 2763–2765 CrossRef CAS.
- G. Garton and H. M. Powell, J. Inorg. Nucl. Chem., 1957, 4, 84–89 CrossRef CAS.
- (a) D. Loos, E. Baum, A. Ecker, H. Schnöckel and A. J. Downs, Angew. Chem., Int. Ed., 1997, 36, 860–862 CrossRef CAS; (b) P. Jutzi, B. Neumann, G. Reumann and H.-G. Stammler, Organometallics, 1998, 17, 1305–1314 CrossRef CAS; (c) C. Schenk, R. Köppe, H. Schnöckel and A. Schnepf, Eur. J. Inorg. Chem., 2011, 3681–3685 CrossRef CAS.
- (a) In Chemistry of aluminium, gallium, indium and thallium, A. J. Downs, Blackie Acad. & Professional, London, 1st edn, 1993 Search PubMed; (b) A. Schnepf and C. Doriat, Chem. Commun., 1997, 2111–2112 RSC.
- P. Dabringhaus, A. Barthélemy and I. Krossing, Z. Anorg. Allg. Chem., 2021, 647, 1660–1673 CrossRef CAS.
- H. Schmidbaur, Angew. Chem., Int. Ed., 1985, 24, 893–904 CrossRef.
- B. J. Malbrecht, J. W. Dube, M. J. Willans and P. J. Ragogna, Inorg. Chem., 2014, 53, 9644–9656 CrossRef CAS PubMed.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC.
- T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2015, 45, 789–899 RSC.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- (a) B. Buchin, C. Gemel, T. Cadenbach, R. Schmid and R. A. Fischer, Angew. Chem., Int. Ed., 2006, 45, 1074–1076 CrossRef CAS PubMed; (b) B. Buchin, C. Gemel, T. Cadenbach, R. Schmid and R. A. Fischer, Angew. Chem., Int. Ed., 2006, 45, 1674 CrossRef.
- J. N. Jones, C. L. Macdonald, J. D. Gorden and A. H. Cowley, J. Organomet. Chem., 2003, 666, 3–5 CrossRef CAS.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228–3231 CrossRef CAS PubMed.
- R. J. Wehmschulte, Angew. Chem., Int. Ed., 2010, 49, 4708–4709 CrossRef CAS PubMed.
- S. Welsch, M. Bodensteiner, M. Dušek, M. Sierka and M. Scheer, Chem.–Eur. J., 2010, 16, 13041–13045 CrossRef CAS PubMed.
- A. Higelin, U. Sachs, S. Keller and I. Krossing, Chem.–Eur. J., 2012, 18, 10029–10034 CrossRef CAS PubMed.
- R. J. Wehmschulte, R. Peverati and D. R. Powell, Inorg. Chem., 2019, 58, 12441–12445 CrossRef CAS PubMed.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490–502 CrossRef CAS.
- U. Schneider and S. Kobayashi, Acc. Chem. Res., 2012, 45, 1331–1344 CrossRef CAS PubMed.
- Z. Li, G. Thiery, M. R. Lichtenthaler, R. Guillot, I. Krossing, V. Gandon and C. Bour, Adv. Synth. Catal., 2018, 360, 544–549 CrossRef CAS.
- Z. Li, S. Yang, G. Thiery, V. Gandon and C. Bour, J. Org. Chem., 2020, 85, 12947–12959 CrossRef CAS PubMed.
- K. Glootz, A. Barthélemy and I. Krossing, Angew. Chem., Int. Ed., 2021, 60, 208–211 CrossRef CAS PubMed.
- M. R. Lichtenthaler, A. Higelin, A. Kraft, S. Hughes, A. Steffani, D. A. Plattner, J. M. Slattery and I. Krossing, Organometallics, 2013, 32, 6725–6735 CrossRef CAS.
- M. R. Lichtenthaler, S. Maurer, R. J. Mangan, F. Stahl, F. Mönkemeyer, J. Hamann and I. Krossing, Chem.–Eur. J., 2015, 21, 157–165 CrossRef CAS PubMed.
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem.–Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed.
- C. Jones, D. P. Mills and R. P. Rose, J. Organomet. Chem., 2006, 691, 3060–3064 CrossRef CAS.
- A. Kempter, C. Gemel and R. A. Fischer, Inorg. Chem., 2008, 47, 7279–7285 CrossRef CAS PubMed.
- (a) R. J. Baker, C. Jones and M. Kloth, Dalton Trans., 2005, 2106–2110 RSC; (b) R. J. Baker, C. Jones, D. P. Mills, D. M. Murphy, E. Hey-Hawkins and R. Wolf, Dalton Trans., 2006, 64–72 RSC; (c) G. Prabusankar, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Inorg. Chem., 2010, 49, 7976–7980 CrossRef CAS PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2015, 34, 2991–2996 CrossRef CAS.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- R. J. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9, 534 CrossRef PubMed.
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platinum Met. Rev., 1997, 41, 66–75 CAS.
- L. H. Sommer, E. W. Pietrusza and F. C. Whitmore, J. Am. Chem. Soc., 1947, 69, 188 CrossRef CAS.
- (a) J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974–979 CrossRef CAS; (b) R. A. Benkeser and J. Kang, J. Organomet. Chem., 1980, 185, C9–C12 CrossRef CAS.
- B. D. Karstedt, US Pat., US 3775452A, 1973 Search PubMed.
- (a) B. J. Truscott, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2013, 42, 270–276 RSC; (b) M. Xue, J. Li, J. Peng, Y. Bai, G. Zhang, W. Xiao and G. Lai, Appl. Organomet. Chem., 2014, 28, 120–126 CrossRef CAS.
- (a) J. A. Muchnij, F. B. Kwaramba and R. J. Rahaim, Org. Lett., 2014, 16, 1330–1333 CrossRef CAS PubMed; (b) X. Xie, X. Zhang, H. Yang, X. Ji, J. Li and S. Ding, J. Org. Chem., 2019, 84, 1085–1093 CrossRef CAS PubMed.
- S. Dagorne and R. Wehmschulte, ChemCatChem, 2018, 10, 2509–2520 CrossRef CAS.
- (a) F. Buch, J. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741–2745 CrossRef CAS PubMed; (b) P. Dabringhaus, M. Schorpp, H. Scherer and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 22023–22027 CrossRef CAS PubMed.
- (a) P.-F. Fu, L. Brard, Y. Li and T. J. Marks, J. Am. Chem. Soc., 1995, 117, 7157–7168 CrossRef CAS; (b) H. Schumann, M. R. Keitsch, J. Demtschuk and G. A. Molander, J. Organomet. Chem., 1999, 582, 70–82 CrossRef CAS.
- (a) Y. Chen, C. Sui-Seng, S. Boucher and D. Zargarian, Organometallics, 2005, 24, 149–155 CrossRef CAS; (b) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CrossRef CAS PubMed; (c) Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798–1801 CrossRef CAS PubMed.
- (a) K. Yamamoto and M. Takemae, Synlett, 1990, 1990, 259–260 CrossRef; (b) Y.-S. Song, B. R. Yoo, G.-H. Lee and I. N. Jung, Organometallics, 1999, 18, 3109–3115 CrossRef CAS; (c) J. Chen and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2015, 54, 6842–6846 CrossRef CAS PubMed.
- M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed.
- K. Jakobsson, T. Chu and G. I. Nikonov, ACS Catal., 2016, 6, 7350–7356 CrossRef CAS.
- J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed.
- J. Koller and R. G. Bergman, Organometallics, 2012, 31, 2530–2533 CrossRef CAS.
- D. T. Hog and M. Oestreich, Eur. J. Org. Chem., 2009, 2009, 5047–5056 CrossRef.
- M. Mewald and M. Oestreich, Chem.–Eur. J., 2012, 18, 14079–14084 CrossRef CAS PubMed.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- P. Bach, A. Albright and K. K. Laali, Eur. J. Org. Chem., 2009, 2009, 1961–1966 CrossRef.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- R. Kannan, R. Chambenahalli, S. Kumar, A. Krishna, A. P. Andrews, E. D. Jemmis and A. Venugopal, Chem. Commun., 2019, 55, 14629–14632 RSC.
- K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed.
- (a) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed; (b) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- N. Asao, T. Sudo and Y. Yamamoto, J. Org. Chem., 1996, 61, 7654–7655 CrossRef CAS PubMed.
- (a) K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC; (b) H.-J. Jung, Y. Cho, D. Kim and P. Mehrkhodavandi, Catal. Sci. Technol., 2021, 11, 62–91 RSC.
- A. J. Blake, A. Cunningham, A. Ford, S. J. Teat and S. Woodward, Chem.–Eur. J., 2000, 6, 3586–3594 CrossRef CAS.
- (a) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed; (b) M. Saleh, D. R. Powell and R. J. Wehmschulte, Organometallics, 2017, 36, 4810–4815 CrossRef CAS; (c) A. Caise, J. Hicks, M. Ángeles Fuentes, J. M. Goicoechea and S. Aldridge, Chem.–Eur. J., 2021, 27, 2138–2148 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, London, 93rd edn, 2016 Search PubMed.
- (a) A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16–21 CrossRef CAS; (b) G. Giorgi, F. de Angelis, N. Re and A. Sgamellotti, Future Gener. Comput. Syst., 2004, 20, 781–791 CrossRef.
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem.–Eur. J., 2014, 20, 1218–1222 CrossRef CAS PubMed.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058–7068 RSC.
- A. Schäfer, M. Reissmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef PubMed.
- A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 4526–4528 CrossRef CAS PubMed.
- (a) C. Eaborn, P. D. Lickiss, S. T. Najim and W. A. Stańczyk, J. Chem. Soc., Chem. Commun., 1987, 1461–1462 RSC; (b) N. Choi, P. D. Lickiss, M. McPartlin, P. C. Masangane and G. L. Veneziani, Chem. Commun., 2005, 6023–6025 RSC; (c) N. Lühmann, H. Hirao, S. Shaik and T. Müller, Organometallics, 2011, 30, 4087–4096 CrossRef; (d) K. Müther, P. Hrobárik, V. Hrobáriková, M. Kaupp and M. Oestreich, Chem.–Eur. J., 2013, 19, 16579–16594 CrossRef PubMed; (e) R. Labbow, F. Reiß, A. Schulz and A. Villinger, Organometallics, 2014, 33, 3223–3226 CrossRef CAS; (f) L. Albers, S. Rathjen, J. Baumgartner, C. Marschner and T. Müller, J. Am. Chem. Soc., 2016, 138, 6886–6892 CrossRef CAS PubMed.
- A. Schäfer, M. Reißmann, S. Jung, A. Schäfer, W. Saak, E. Brendler and T. Müller, Organometallics, 2013, 32, 4713–4722 CrossRef.
- K. Bläsing, R. Labbow, D. Michalik, F. Reiß, A. Schulz, A. Villinger and S. Walker, Chem.–Eur. J., 2020, 26, 1640–1652 CrossRef PubMed.
- L. Omann, B. Pudasaini, E. Irran, H. F. T. Klare, M.-H. Baik and M. Oestreich, Chem. Sci., 2018, 9, 5600–5607 RSC.
- J. Y.-C. Chen, A. A. Martí, N. J. Turro, K. Komatsu, Y. Murata and R. G. Lawler, J. Phys. Chem. B, 2010, 114, 14689–14695 CrossRef CAS PubMed.
- A. Budanow, M. Bolte, M. Wagner and H.-W. Lerner, Eur. J. Inorg. Chem., 2015, 2015, 2524–2527 CrossRef CAS.
- (a) Z. Xie, J. Manning, R. W. Reed, R. Mathur, P. D. W. Boyd, A. Benesi and C. A. Reed, J. Am. Chem. Soc., 1996, 118, 2922–2928 CrossRef CAS; (b) C. A. Reed, Acc. Chem. Res., 1998, 31, 325–332 CrossRef CAS.
- (a) L. Kloo, J. Rosdahl and M. J. Taylor, Polyhedron, 2002, 21, 519–524 CrossRef CAS; (b) F. Fetzer, C. Schrenk, N. Pollard, A. Adeagbo, A. Z. Clayborne and A. Schnepf, Chem. Commun., 2021, 57, 3551–3554 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- H. H. Anderson, J. Am. Chem. Soc., 1958, 80, 5083–5085 CrossRef CAS.
- N. Moitra, K. Kanamori, T. Shimada, K. Takeda, Y. H. Ikuhara, X. Gao and K. Nakanishi, Adv. Funct. Mater., 2013, 23, 2714–2722 CrossRef CAS.
- N. Moitra, K. Kanamori, Y. H. Ikuhara, X. Gao, Y. Zhu, G. Hasegawa, K. Takeda, T. Shimada and K. Nakanishi, J. Mater. Chem. A, 2014, 2, 12535–12544 RSC.
- M. Ohashi, R. Yaokawa, Y. Takatani and H. Nakano, ChemNanoMat, 2017, 3, 534–537 CrossRef CAS.
- A. Sugie, T. Somete, K. Kanie, A. Muramatsu and A. Mori, Chem. Commun., 2008, 3882–3884 RSC.
- Ö. Dag, E. J. Henderson, W. Wang, J. E. Lofgreen, S. Petrov, P. M. Brodersen and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 17454–17462 CrossRef PubMed.
- J. V. Crivello, Silicon, 2009, 1, 111–124 CrossRef CAS.
- F. Yonehara, Y. Kido, H. Sugimoto, S. Morita and M. Yamaguchi, J. Org. Chem., 2003, 68, 6752–6759 CrossRef CAS PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- (a) K. Hensen, T. Zengerly, P. Pickel and G. Klebe, Angew. Chem., Int. Ed., 1983, 22, 973–984 CrossRef; (b) G. K. S. Prakash, S. Keyaniyan, R. Aniszfeld, L. Heiliger, G. A. Olah, R. C. Stevens, H. K. Choi and R. Bau, J. Am. Chem. Soc., 1987, 109, 5123–5126 CrossRef CAS; (c) J. B. Lambert, S. Zhang, C. L. Stern and J. C. Huffman, Science, 1993, 260, 1917–1918 CrossRef CAS PubMed.
- (a) S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767–769 RSC; (b) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed; (c) C. Bolli, J. Derendorf, C. Jenne, H. Scherer, C. P. Sindlinger and B. Wegener, Chem.–Eur. J., 2014, 20, 13783–13792 CrossRef CAS PubMed.
- S. J. Connelly, W. Kaminsky and D. M. Heinekey, Organometallics, 2013, 32, 7478–7481 CrossRef CAS.
- M. Nava and C. A. Reed, Organometallics, 2011, 30, 4798–4800 CrossRef CAS PubMed.
- J. B. Lambert and Y. Zhao, J. Am. Chem. Soc., 1996, 118, 7867–7868 CrossRef CAS.
- J. B. Lambert, Y. Zhao and H. Wu, J. Org. Chem., 1999, 64, 2729–2736 CrossRef CAS PubMed.
- A. Martens, M. Kreuzer, A. Ripp, M. Schneider, D. Himmel, H. Scherer and I. Krossing, Chem. Sci., 2019, 10, 2821–2829 RSC.
- V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
- L. Süsse, J. Hermeke and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 6940–6943 CrossRef PubMed.
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluorine Chem., 2001, 112, 83–90 CrossRef CAS.
- (a) P. D. Bartlett, F. E. Condon and A. Schneider, J. Am. Chem. Soc., 1944, 66, 1531–1539 CrossRef CAS; (b) J. Y. Corey and R. West, J. Am. Chem. Soc., 1963, 85, 2430–2433 CrossRef CAS; (c) J. Y. Corey, J. Am. Chem. Soc., 1975, 97, 3237–3238 CrossRef CAS.
- (a) R. A. McClelland, N. Banait and S. Steenken, J. Am. Chem. Soc., 1986, 108, 7023–7027 CrossRef CAS; (b) V. R. Naidu, S. Ni and J. Franzén, ChemCatChem, 2015, 7, 1896–1905 CrossRef CAS.
- J. S. Siegel, Nat. Rev. Chem., 2020, 4, 4–5 CrossRef CAS.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS PubMed.
- J. H. W. LaFortune, T. C. Johnstone, M. Pérez, D. Winkelhaus, V. Podgorny and D. W. Stephan, Dalton Trans., 2016, 45, 18156–18162 RSC.
- S. Postle, V. Podgorny and D. W. Stephan, Dalton Trans., 2016, 45, 14651–14657 RSC.
- K. M. Szkop and D. W. Stephan, Dalton Trans., 2017, 46, 3921–3928 RSC.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS PubMed.
- S. S. Chitnis, F. Krischer and D. W. Stephan, Chem.–Eur. J., 2018, 24, 6543–6546 CrossRef CAS PubMed.
- D. B. Culver and M. P. Conley, Angew. Chem., Int. Ed., 2018, 57, 14902–14905 CrossRef CAS PubMed.
- D. Hartmann, M. Schädler and L. Greb, Chem. Sci., 2019, 10, 7379–7388 RSC.
- N. Kramer, H. Wadepohl and L. Greb, Chem. Commun., 2019, 55, 7764–7767 RSC.
- M. Mehta and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 2715–2719 CrossRef CAS PubMed.
- A. Hermannsdorfer and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 23132–23136 CrossRef CAS PubMed.
- K. I. Burton, I. Elser, A. E. Waked, T. Wagener, R. J. Andrews, F. Glorius and D. W. Stephan, Chem.–Eur. J., 2021, 27, 11730–11737 CrossRef CAS PubMed.
- C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- (a) G. Baddeley, Q. Rev., Chem. Soc., 1954, 8, 355–379 RSC; (b) V. Polito, C. S. Hamann and I. J. Rhile, J. Chem. Educ., 2010, 87, 969–970 CrossRef CAS.
- D. M. Lemal, J. Org. Chem., 2004, 69, 1–11 CrossRef CAS PubMed.
- (a) P. J. Malinowski, T. Jaroń, M. Domańska, J. M. Slattery, M. Schmitt and I. Krossing, Dalton Trans., 2020, 49, 7766–7773 RSC; (b) In Experiments in Green and Sustainable Chemistry, I. Raabe, A. Reisinger, I. Krossing, H. W. Roesky and D. K. Kennepohl, Wiley-VCH, Weinheim, 2009, pp. 131–144 Search PubMed.
- Can be purchased under https://www.iolitec.de.
- M. Schorpp, R. Tamim and I. Krossing, Dalton Trans., 2021, 50, 15103–15110 RSC.
Footnotes |
| † Dedicated to the occasion of the 60th birthday of Holger Braunschweig. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details, 1D- and 2D NMR spectra of the reactions are deposited. Details to the quantum chemical calculations are given together with the results of gas chromatographic, cyclic voltammetry, STEM/EDX measurements and crystallographic details. CCDC deposition number 2024333 (for Et3Si–F–Al[OC(CF3)3]3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05331k |
| § Since the reactions were carried out in oDFB and since the use of oDFB as a solvent is crucial for the reaction kinetics, [Ga(oDFB)]+ was chosen as a model complex instead of Ga+ or [Ga(PhF)]+. |
| This journal is © The Royal Society of Chemistry 2022 |