
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biocompatible hydrogel as a template for oxidative decomposition reactions: a chemodosimetric analysis and in vitro imaging of hypochlorite†
Dipen
Biswakarma
ac,
Nilanjan
Dey
d and
Santanu
Bhattacharya
 *abc
*abc
aDepartment of Organic Chemistry, Indian Institute of Science, Bangalore, Karnataka 560012, India
bSchool of Applied & Interdisciplinary Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: sb@iisc.ac.in; director@iacs.res.in; Tel: +91 8022932664
cJawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru 560064, Karnataka, India
dDepartment of Chemistry, BITS Pilani, Hyderabad Campus, Jawahar Nagar, Shameerpet Mandal, Hyderabad-500078, India
First published on 3rd February 2022
Abstract
The self-assembly properties of new biocompatible, thermoreversible fluorescent hydrogels, composed of amino acid residues, e.g., L-phenylalanine (PyL-PheOx) and L-tyrosine (PyL-TyrOx), have been reported. Spectroscopic investigations indicate that PyL-PheOx forms π-stacked ‘compact’ aggregates, while ‘loose’ aggregates with stronger CT characteristics are observed for PyL-TyrOx. Both the compounds showed the presence of fibrous networks in the self-assembled state. Circular dichroism spectral studies indicate the formation of M-helical and P-helical structures for PyL-PheOx and PyL-TyrOx, respectively. A striking gel-to-sol transition, caused by oxidative decomposition, is explicitly noticed in the presence of hypochlorite. A mechanistic investigation reveals the oxidation of the acyl aroyl hydrazine core of the gelators in the presence of ClO−. In addition to this, change in the fluorescence emission intensity of the hydrogel in the presence of ClO− is utilized for the analysis of commercial bleach samples. Gel-coated paper strips are also developed for the on-site detection of ClO−. Furthermore, the system is utilized for imaging hypochlorite in live mammalian cells.
Introduction
Bio-responsive supramolecular hydrogels derived from biocompatible low-molecular weight amphiphiles have recently emerged as advanced photo-responsive materials, due to their applications in cell imaging, tissue engineering, biosensing and drug delivery.1–10 Fluorescent hydrogels composed of naturally occurring hydrophobic amino acid residues are of particular interest, since they can form 3D-chiral nanostructures depending upon the H-bonding and π-stacking ability of the side-chain residues.11–22 On the other hand, as a signalling moiety, pyrene remains as the fluorophore of choice due to its excellent photostability, high quantum yield and ability to undergo monomer to excimer transformation depending upon the microenvironment.23–28A well-known Reactive Oxygen Species (ROS), hypochlorite (ClO−), is found in living organisms as a result of catalytic conversion. Though it takes an active part in developing the innate immune system, exposure to excess hypochlorite may cause serious tissue injuries by disrupting the structure of nucleic acids, proteins, and cellular lipid bilayers.29–41 Also, ClO− is regularly used as a bleaching agent and widely employed for wastewater management or even for sanitizing Covid-19 infected indoors.32,42–48 Thus, the development of an effective monitoring device for ClO− is important for quality control and diagnostic purposes. Though recently a few hydrogel systems capable of optical sensing have appeared in the literature,23,49 reports on ‘reaction-based’ biosensing using hydrogels however remains relatively fewer.49–56 However, detection strategies based on mild reactions are considered to be more effective than host–guest interactions due to their high specificity. Moreover, unlike conventional reaction-based probes (which work on a one-to-one detection strategy), gel-based sensors can amplify the response signal due to long-range intermolecular interactions.57–63
Considering this, herein we demonstrate the design and self-assembly of amino acid functionalized novel fluorescent hydrotropes (PyL-PheOx and PyL-TyrOx) that form hydrogels (Fig. 1a). Such hydrogel systems are unique because they comprise three key components: (a) a gallic acid amide-based anchor with oligo oxyethylene chains that allow intermolecular H-bonding and adequate hydration; (b) a chiral aromatic amino acid amide-based core (L-phenylalanine for PyL-TyrOx/L-tyrosine for PyL-PheOx), which directs the self-assembly via a combination of H-bonding, hydrophobic interactions etc.; (c) a hydrophobic signal transduction unit (pyrene), which maintains a lipophilic and hydrophilic balance and controls the propagation of the gel network. Interestingly, both the hydrogels showed high mechanical strength, thermoreversibility and thixotropic behaviour. A sharp gel-to-sol transition accompanied by a change in fluorescence emission intensity was noticed when gel samples were exposed to a Reactive-Oxygen Species (ROS), e.g., ClO−. Thus, the present system offers a unique example where gel-to-sol transition is triggered by chemodosimetric interaction with a specific ROS. Hypochlorite-induced oxidative decomposition can also be monitored inside live mammalian cells.
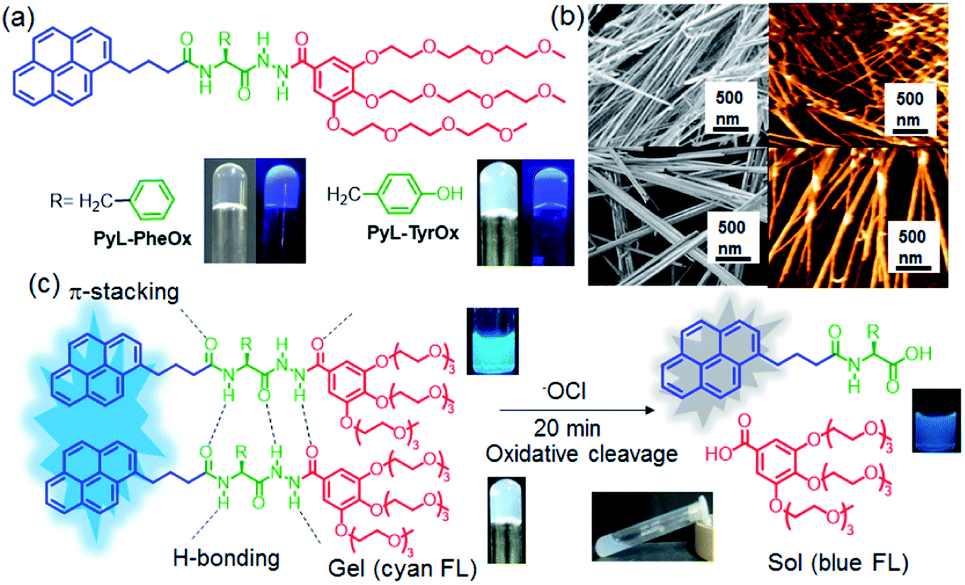 | ||
| Fig. 1 (a) Structures of the gelator molecules (PyL-PheOx and PyL-TyrOx) used in the present study and images of the gel as seen under normal and UV light (>365 nm). (b) SEM (left) and AFM (right) images of gel samples (top: PyL-PheOx, 1.5 mM and down: PyL-TyrOx, 1.5 mM). (c) Schematic representation of the interaction of PyL-PheOx and down: PyL-TyrOx gels with ClO−. | ||
Experimental section
Materials and methods
All chemicals, solvents and silica gel for TLC were obtained from well-known commercial sources and were used without further purification, as appropriate. Solvents were freshly distilled and dried by a standard procedure before use. Melting points were measured in open capillaries and were uncorrected. 1H-NMR and 13C-NMR spectra were recorded using a Bruker-400 Advance NMR spectrometer. Chemical shifts were reported in ppm downfield from the internal standard tetramethyl silane (TMS). Mass spectrometry of individual compounds was performed using a Micro Mass ESI-TOF MS instrument. Elemental analysis was performed using a Thermo Finnigan EA FLASH 1112 SERIES.Gelation studies
The formation of the gel was confirmed using the tube inversion method. In a typical experiment, PyL-PheOx and PyL-TyrOx were taken and to this mixture 1 mL of deionized water was added, followed by heating at 80 °C till the solution became transparent. The resultant mixture was sonicated for a few minutes and subsequently allowed them to cool at room temperature. If a gel was formed, it was evaluated quantitatively by determining the critical gelator concentration (CGC). For the evaluation of the CGC, a known amount of PyL-PheOx/PyL-TyrOx in pure water was heated to 75–80 °C, which resulted in a slightly turbid solution and then sonicated. The resulting clear solution transformed into an optically transparent/opaque robust hydrogel within 1–2 min. The gelation propensity of PyL-PheOx/PyL-TyrOx has been checked systematically by gradually lowering the amount of the gelator. In that way, the minimum concentration of the gelator that was required to immobilize 1 mL of water, i.e., the CGC value was determined as reported with other systems. The above measurement was carried out three times and the average CGC value has been reported. A glass tube with a capacity of 10 × 75 mm has been used for the CGC measurement. For thixotropic properties, the gel was vortexed for 60 s for complete gel-to sol transition and kept at rest for 10 min to revert to gel.FT-IR spectroscopy
The prepared solution and gels of individual samples were drop coated on a CaF2 cell and dried under vacuum and FT-IR spectra were recorded using a PerkinElmer Spectrum BX FT-IR system. For solution measurements, PyL-PheOx/PyL-TyrOx was dissolved in CHCl3 and was transferred to a constant path length cell (50 mm) containing CaF2 windows. For baseline correction, the spectrum of neat CHCl3 was subtracted from the sample spectrum.UV/Vis and fluorescence spectroscopy
UV-vis spectroscopy and fluorescence spectroscopy of the described solutions/suspensions were performed on a Shimadzu model 2100 spectrophotometer and Cary-Eclipse spectrofluorimeter respectively, both equipped with a temperature controlled bath. The slit width for the fluorescence experiment was kept at 5 nm (excitation) and 5 nm (emission) and the excitation wavelength was set at 345 nm.Circular dichroism spectroscopy (CD)
All the CD spectra of the described solutions were recorded on a JASCO instrument, Model J-815-150S. Experiments were performed by purging dry N2 gas continuously. Data were collected in a quartz cuvette of 1 mm path length between 200 and 450 nm.Atomic force microscopy (AFM)
A dilute solution of the sample of gel was deposited on a freshly cleaved mica surface and then allowed carefully to freeze-dry. Each of the samples was analysed using a JPK 00901 AFM instrument and Nano-Wizard software: tapping mode off 10 nm tip radius, silicon tip, 292 kHz resonant frequency, 0.7–1 Hz scan speed, 256 × 256 and 512 × 512 pixels.Scanning electron microscopy (SEM)
The gels or the specified solution was carefully drop coated onto brass stubs and was allowed to freeze-dry. The samples were then coated with gold vapor and analysed on a Quanta 200 SEM operated at 10–15 kV.X-ray diffraction (XRD)
The gel samples were frozen using liquid N2, followed by further drying in a lyophilizer to obtain a xerogel for X-ray diffraction study. The dried powder was then placed on a glass plate. These samples were examined using a Bruker D8 Advance instrument (θ, 2θ geometry with scintillation detectors). The X-ray beam generated with a rotating Cu anode at a wavelength of the KR beam of 1.5418 was directed toward the film edge and scanning was done up to a 2θ value of 30°. Data were then analysed and interpreted using the Bragg equation.Energy minimization
The energy minimization of the hydrogelator molecules was performed using the B3LYP/6-31G* level of computations.Rheological studies
Rheology experiments using the hydrogels were carried out using an Anton Paar MCR 52 with a cone and plate geometry (CP 25-2) having an adjustable Peltier temperature controlling system. The distance between the cone and plates was kept fixed at 0.105 mm for all the measurements. The gels were taken on the plate of the rheometer. An oscillatory strain amplitude sweep experiment was performed at a constant oscillation frequency of 1 Hz for the applied strain range 0.01–100% at 20 °C. The rheometer has inbuilt software that converts torque measurements into either G′ (the storage modulus) and G′′ (the loss modulus), which is represented either as strain or shear stress. Oscillatory frequency sweep experiments were performed in the linear viscoelastic region (strain 0.01%) to ensure that calculated parameters correspond to an intact network structure. The thixotropic properties were studied to examine the alteration of the rheological properties of the hydrogel material under the application and the release of shear stress. This process includes two steps: (i) deformation of the hydrogel with increasing shear stress from 0.01 to 80 Pa for 6.14 min and (ii) recovery of the sol state to the gel state under low shear stress (0.01 Pa) for 6 min. In these two steps, the frequency was kept constant at 1 Hz. Four consecutive cycles were applied on the hydrogels to compare the extent of gel strength recovery.Measurement of fluorescence lifetime
Fluorescence lifetimes were measured by using a time-correlated single-photon-counting fluorimeter (Horiba Jobin Yvon). The system was excited with nano-LED light (Horiba-Jobin Yvon) with a pulse duration of 1.2 ns (slit width: 2/2 and λem = 345 nm). The average fluorescence lifetimes (τav) for the exponential iterative fitting were calculated from the decay times (τi) and the relative amplitudes (αi) by using eqn (1), where α1–3 are the relative amplitudes and τ1–3 are the lifetime values.| τav = (α1τ12 + α2τ22 + α3τ32)/(α1τ1 + α2τ2 + α3τ3) | (1) |
Preparation of the analyte (reactive oxygen species)
A freshly prepared NaOCl solution (4 wt% stock solution) was used as the ClO− source, an aliquot of which was directly added to the solution. The solution of each oxidant was freshly prepared from their respective aqueous solutions of NaHCO3 (1 mM), NaNO3 (1 mM), NaNO2 (1 mM), NaCl (1 mM), NaBr (1 mM), NaI (1 mM) and Na2SO4 (1 mM). Glutathione (GSH) was freshly prepared by dissolving concentrated GSH (1 mM) in Milli-Q water. A donor source for NO was prepared by dissolving sodium nitroferricyanide(III) dehydrate in Milli-Q water. Hydrogen peroxide (H2O2) was diluted freshly prior to use from a stabilized 30% solution. The hydroxyl radical (˙OH) was generated by adding ferrous chloride to 10 equiv. of H2O2 such that the concentration of ˙OH was maintained equal to the Fe(II) concentration (via Fenton reaction).64,65 Singlet oxygen (1O2) was generated from ClO− and H2O2.66 Peroxynitrate (ONOO−) was generated as reported in the literature67 and ROO˙ was generated in situ from 2,2′-azobis(2-amidinopropane)dihydrochloride.81 An O2˙− solution was prepared by vigorously stirring 1 mg of KO2 in dimethyl sulfoxide (1 mL) for 10 min.68Preparation of test strips
For the preparation of gel-coated paper strips, filter paper (Whatman 40) strips were cut into a square shape (1 × 2 cm). To these square shaped paper strips, 30 μL of the pre-prepared gel sample (2.5 mM) was applied on the surface so that it could spread uniformly. The gel coated on the paper strip was completely absorbed within 15 min under an ambient open atmosphere and then it was air-dried naturally. These gel coated paper strips were then used for checking ClO−.Analysis of hypochlorite in different water samples
Tap water samples were collected from the Department of Organic Chemistry Laboratory, IISc. Pool water samples were collected from the swimming pool of the Indian Institute of Science campus, Bangalore, India. Both tap water and pool water samples were separately used for analysis as received. Seawater samples were collected from the Arabian Sea water (near Mangalore). To remove the insoluble and suspended dirt particles from the sea water, it was filtered through a 0.22 μm membrane filter cartridge. To these water samples, different amounts of ClO− were added more than 30 min before the analysis.Cell culture
HeLa cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, CELL clone, India) containing 10% FBS (Invitrogen, USA) and antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin). The cells were incubated in a humidified 5% CO2 incubator (Sanyo, UK) at 37 °C.Cell viability assay
The cytotoxicity of the compounds was evaluated in HeLa and MCF7 cells by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In a typical experiment, the cells were seeded at a density of ∼1 × 104 cells per well in 96 well plates and incubated for 24 h. Then, the cells were treated with different concentrations of the probe. Finally, the cells were washed, and 20 μL of MTT (5 mg mL−1 in DMEM) was added into each well and incubated for 3 h in the dark. Finally, the whole medium was removed from the wells, and DMSO (100 μL) was added. The cells were incubated for 5 min in the dark, and then absorbance was measured at 560 nm.Cell imaging
For blank reading, the HeLa cells were incubated with 20 μM PyL-TyrOx for 30 min at 37 °C. Then, the medium was removed, and the cells were washed multiple times with pH 7.4 DMEM buffer.Synthesis of PyL-PheOx
To the solution of PyL-PheOH (1 g, 2.29 mmol) in DCM (30 mL), a gallate derivative (1.72 g, 2.75 mmol), DCC (0.7 g, 3.43 mmol) and HOBt (0.53 g, 3.43 mmol) were added, followed by the addition of dry Et3N (1.18 mL, 9.16 mmol) at 0 °C under an inert atmosphere. The resultant mixtures were kept stirring at room temperature for 24 h. After the completion of the reaction, the solvent was evaporated, and the residual reaction mixtures were taken in ethyl acetate (40 mL). This mixture contained the suspension of DCU (dicyclohexylurea), which was filtered off. The filtrate thus obtained was dried and purification was done with silica column chromatography using 4% MeOH/CHCl3 as the eluent. The product thus obtained was a pale yellowish solid in 78% yield. Mp 148 °C; IR (Neat cm−1): 3258, 3180, 2940, 2867, 1641, 1540, 1463, 1333, 1242, 1092; 1H NMR (400 MHz, CDCl3) δ 2.06–2.11 (m, 2H), 2.23–2.29 (m, 2H), 3.00–3.06 (m, 1H), 3.19–3.25 (m, 3H), 3.35 (s, 9H), 3.47–3.53 (m, 6H), 3.57–3.69 (m, 18H), 3.74–3.77 (m, 6H), 4.10–4.19 (m, 6H), 4.88–4.93 (q, J = 7.32 Hz, 1H), 6.43–6.45 (d, J = 8.04 Hz, 1H), 7.13–7.14 (m, 3H), 7.21–7.24 (m, 4H), 7.73–7.75 (d, J = 8 Hz, 1H), 7.94–7.99 (m, 3H), 8.03–8.14 (m, 4H), 8.19–8.21 (d, J = 8 Hz, 1H), 8.89 (s, 1H), 9.36 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 27.09, 32.57, 35.73, 37.56, 58.91, 58.97, 68.96, 69.70, 70.35, 70.55, 70.64, 71.88, 72.37, 107.63, 119.34, 123.36, 124.76, 124.95, 125.04, 125.82, 126.42, 126.67, 127.05, 127.36, 128.66, 129.27, 130.89, 131.39, 135.73, 136.27, 142.11, 152.48, 164.90, 169.41, 173.30; HRMS: m/z calcd for C57H73N3O15 (M + Na)+: 1062.4939, found: 1062.4940; elem. anal.: calcd for C57H73N3O15: C 65.82; H 7.07; N 4.04; found: C 65.59; H 6.98; N 4.13.Synthesis of PyL-TyrOx
To the solution of PyL-TyrOH (1 g, 2.21 mmol) in DCM (30 mL), a gallate derivative (1.65 g, 2.65 mmol), DCC (0.68 g, 3.31 mmol) and HOBt (0.51 g, 3.31 mmol) were added, followed by the addition of dry Et3N (1.14 mL, 8.85 mmol) at 0 °C under an inert atmosphere. The resultant mixtures were kept stirring at room temperature for 24 h. After the completion of the reaction, the solvent was evaporated, and the residual reaction mixtures were taken in ethyl acetate (40 mL). This mixture contained the suspension of DCU (dicyclohexylurea), which was filtered off. The filtrate thus obtained was dried and purification was done with silica column chromatography using 4% MeOH/CHCl3 as the eluent. The product thus obtained was a pale yellowish solid in 76% yield. Mp 152 °C; IR (Neat cm−1): 3428, 3291, 2923, 2861, 1645, 1516, 1429, 1338, 1246, 1111; 1H NMR (400 MHz, CDCl3) δ 1.98–2.96 (m, 2H), 2.19–2.23 (m, 2H), 2.78–2.83 (m, 1H), 3.06–3.15 (m, 3H), 3.35 (s, 9H), 3.41–3.44 (m, 6H), 3.49–3.51 (m, 18H), 3.55–3.77 (m, 6H), 3.90–4.13 (m, 6H), 4.75–4.98 (q, J = 7.02 Hz, 1H), 6.59–6.61 (d, J = 8 Hz, 2H), 6.89–6.91 (m, J = 8 Hz, 2H), 7.09 (s, 2H), 7.64–7.66 (d, J = 8.2 Hz, 1H), 7.91–7.93 (m, 4H), 8.01–8.07 (m, 3H), 8.11–8.13 (d, J = 8.4 Hz, 1H), 9.67 (s, 1H), 9.91 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 27.19, 32.57, 35.74, 37.12, 52.91, 58.86, 68.56, 69.55, 70.28, 70.59, 71.79, 72.27, 107.09, 115.58, 119.75, 123.33, 124.73, 124.79, 124.95, 125.78, 126.59, 127.29, 127.34, 127.45, 128.60, 129.82, 130.85, 131.33, 135.80, 136.22, 141.55, 152.31, 155.66, 165.43, 170.44, 173.51; HRMS: m/z calcd for C57H73N3O16 (M + Na)+: 1078.4889, found: 1078.4890; elem. anal.: calcd for C57H73N3O16: C 64.82; H 6.97; N 3.98; found: C 64.39; H 7.08; N 3.65.Results and discussion
Design and synthesis of fluorescent hydrotropes
Herein, we have synthesized two different fluorescent hydrotropes, attached with a gallate derivative (Fig. 1) and linked via aromatic amino acids namely phenylalanine (PyL-PheOx) and tyrosine (PyL-TyrOx). Both the compounds were synthesized following a conventional peptide coupling protocol and characterized thoroughly using 1H-NMR, 13C NMR, ESI-MS, FT-IR, and elemental analysis (for details see the Experimental section and ESI†).Self-assembly and gelation properties in aqueous medium
Both PyL-PheOx and PyL-TyrOx belong to the class of supergelators as the former formed a stable transparent hydrogel (CGC: 0.52 mM) in water, while an opaque hydrogel (CGC: 0.82 mM) was found with the latter (Fig. 1a). The strong gelation ability of PyL-PheOx may presumably have originated from the enhanced π–π stacking interactions attributed to unsubstituted phenyl residues. As expected, FT-IR analysis indicated that in both cases, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Δλ ∼ 15 cm−1) and N–H (Δλ ∼ 45 cm−1) stretching frequencies got shifted to higher energy regions upon the formation of the hydrogel (in comparison to that in CHCl3). The extent of shift was found to be larger for PyL-PheOx, and this might be due to the stronger H-bonding interactions between the acyl aroyl hydrazine (AAH) units, promoted by enhanced π–π stacking interactions (Fig. S1†). Owing to the presence of AAH units, both the gel samples were found to be susceptible to pH change as well (Fig. S2†). Interestingly, on applying mechanical force (shaking), a gel-to-sol transformation was observed, which on keeping at rest reverted to the native gel state in a few minutes (thixotropic behavior, Fig. S2†).
O (Δλ ∼ 15 cm−1) and N–H (Δλ ∼ 45 cm−1) stretching frequencies got shifted to higher energy regions upon the formation of the hydrogel (in comparison to that in CHCl3). The extent of shift was found to be larger for PyL-PheOx, and this might be due to the stronger H-bonding interactions between the acyl aroyl hydrazine (AAH) units, promoted by enhanced π–π stacking interactions (Fig. S1†). Owing to the presence of AAH units, both the gel samples were found to be susceptible to pH change as well (Fig. S2†). Interestingly, on applying mechanical force (shaking), a gel-to-sol transformation was observed, which on keeping at rest reverted to the native gel state in a few minutes (thixotropic behavior, Fig. S2†).
Spectroscopic studies of hydrogelators in the solution phase
Concentration-dependent UV-visible studies indicate the broadening of the pyrene-characteristic absorption peaks in the higher concentration range (Fig. S3†), probably due to the π-stacking interactions between such residues.69,70 Similarly, concentration-dependent fluorescence emission spectral studies showed the formation of a broad band in the red-shifted region (at 422 nm for PyL-PheOx and at 460 nm for PyL-TyrOx) in addition to pyrene-characteristic vibronic bands (Fig. S4†). Excitation spectral analysis indicates that this newly formed structureless band on the longer-wavelength side is originated from the aggregated species (ground-state interaction) and not due to the formation of photo-excited dimers (Fig. S5a†).The larger fluorescence intensity of the aggregated band apparently indicates the effective prevalence of π-stacking interactions in the case of PyL-PheOx compared to the case of PyL-TyrOx. However, a closer look reveals that the aggregate band positioning was rather red shifted for PyL-TyrOx (Fig. 2a). We presume that the relatively poorer alignment of the pyrenyl residues in PyL-TyrOx might have impeded the formation of a compact packing structure, but it could initiate charge-transfer interactions due to the presence of the electron-donating –OH group in it.71 The time-dependent fluorescence decay profile shows a shorter decay time for PyL-TyrOx (τav = 0.85 ± 0.03 ns) in comparison to that of PyL-PheOx (τav = 1.32 ± 0.02 ns). The formation of such short-lived excited state species further confirms the existence of intermolecular CT interactions in the self-assembled state of PyL-TyrOx (Fig. 2a, inset). Similarly, the large steady-state fluorescence anisotropy values indicate a rather rigid microenvironment around the pyrene residues in both the cases (Fig. S5b†). When PyL-PheOx (Fig. S6a†) and PyL-TyrOx (Fig. S6b†) were subjected to variable temperature fluorescence spectral studies, the diminution of the aggregate emission band was observed, probably due to the heat-induced ‘dissolution’ of the aggregate species. Similarly, a urea-induced shift in the aggregate to monomer emission was observed for both PyL-PheOx and PyL-TyrOx perhaps due to the dissociation of the hydrogen-bonded networks.
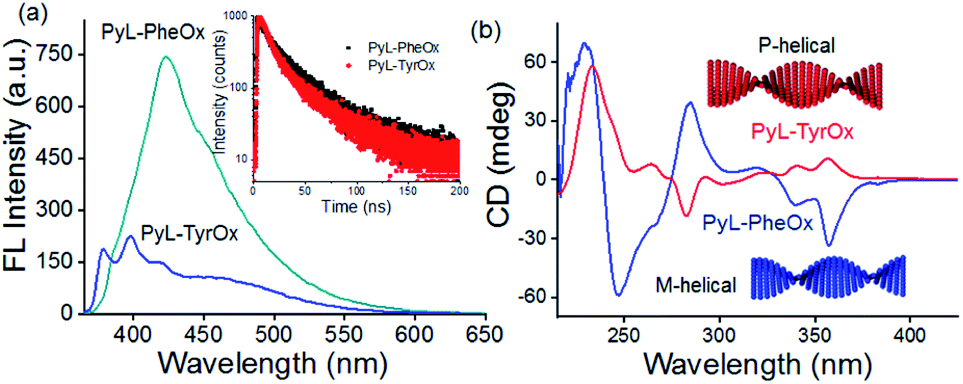 | ||
| Fig. 2 (a) Fluorescence spectra of PyL-PheOx and PyL-TyrOx (0.24 mM and λex = 345 nm) in aqueous media (inset shows the fluorescence decay profiles of PyL-PheOx and PyL-TyrOx at 425 and 460 nm respectively). (b) Circular dichroism spectra of PyL-PheOx (0.96 mM) and PyL-TyrOx (0.96 mM) in aqueous media. | ||
Amino acid-directed distinctive supramolecular chirality
To unravel whether chiral organizations from the self-assembled aggregates were formed in aqueous media, both PyL-PheOx and PyL-TyrOx were subjected to circular dichroism (CD) spectral analysis. The CD spectrum of PyL-PheOx showed a bisignated negative Cotton signal in the aggregated state, indicating the formation of a left-handed M-helical structure. In contrast, the self-assembly of PyL-TyrOx (Fig. 2b) exhibited a bisignated positive Cotton signal, suggesting the formation of a right-handed P-helical organisation.72–75 However, the gradual diminution of CD signals was observed in both the cases with rising temperature, leading to CD-silent situation beyond ∼70 °C (Fig. S7†). This indicates the ‘disintegration’ of the P and M-helical aggregates of PyL-PheOx and PyL-TyrOx respectively at higher temperatures, attributed to thermal-agitation.Viscoelastic properties of fluorescent hydrogels
The nanostructures formed via 3D-assembly of the gelator molecules were also studied using both Scanning Electron Microscopy (SEM) and Atomic Force Microscopy (AFM). In both the cases, the presence of a fibrillar network was observed indicating a strong intermolecular interaction. However, the fibrils appeared to be more ‘densely-packed’ in the case of PyL-PheOx, confirming the formation of ‘compact’ aggregates as described earlier (Fig. 1b).The mechanical strengths of the hydrogel samples (PyL-PheOx and PyL-TyrOx) were evaluated using rheological studies.76–79 Oscillatory frequency (Fig. S8a†) and amplitude (Fig. S8b†) sweep experiments suggest that these hydrogels are highly viscoelastic in nature, and the viscoelasticity increases with increasing gelator concentration. Also, the viscoelasticity of the hydrogels was maintained over a frequency range of 1–100 Hz under a constant applied strain of 0.01%. Interestingly, the hydrogel sample derived from PyL-PheOx exhibited greater mechanical stiffness than the PyL-TyrOx based hydrogel.
The critical stress required to disturb the solid-like behavior was found to be larger (∼53 Pa) in the former case than what was needed for the latter (∼41 Pa). To confirm the thixotropic behavior of the hydrogels, we performed the hysteresis loop test. The mechanical stiffness could be recovered to >98% of the original value within 30 s upon removal of the induced strain (Fig. 3a and S8c†). Keeping this in mind, we had further explored the injectability of the hydrogel sample, where shape-persistent alphabetical letters could be written using the gel samples (inset Fig. 3a).
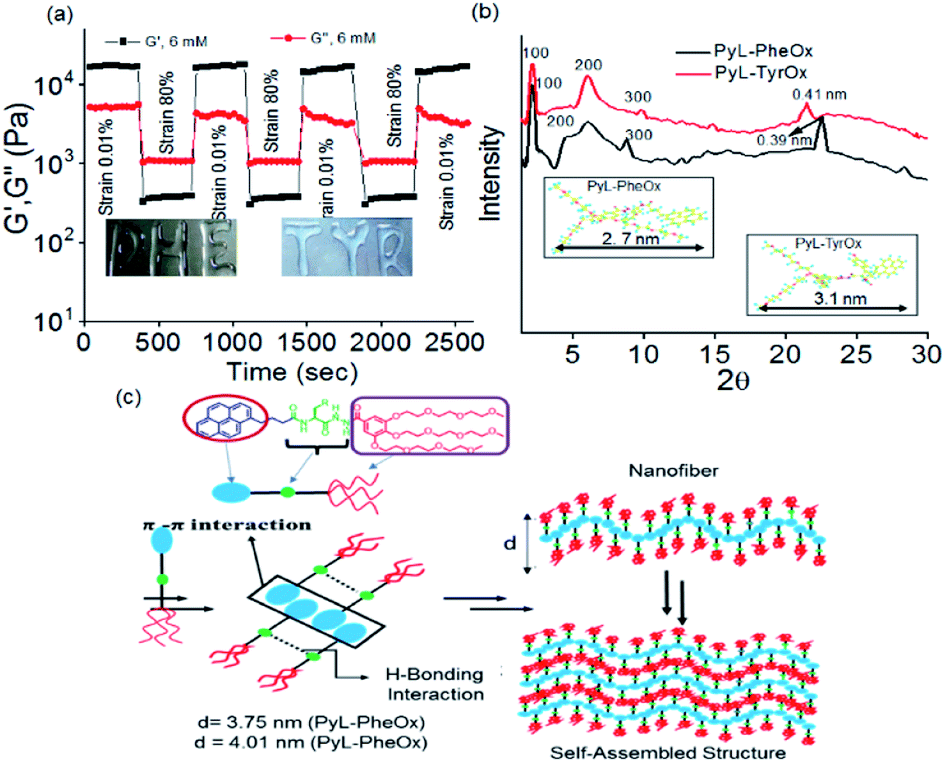 | ||
| Fig. 3 (a) Hysteresis loop test of the PyL-PheOx (6 mM) hydrogel. (b) pXRD of xerogels of PyL-PheOx and PyL-TyrOx. (c) Nano-structural molecular model for the self-assembly of PyL-PheOx and PyL-TyrOx in water. | ||
3D arrangement and proposed nanomolecular model
The XRD studies of dried gel of PyL-PheOx (Fig. 3b) showed the presence of reflection peaks at 2θ = 2.35°, 4.94° and 8.78° with the corresponding d-spacing values of d1 = 3.75 nm, d2 = 1.78 nm and d3 = 0.98 nm respectively. The reciprocal ratio of these sets of reflection peaks is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/2:1/3, which suggests that the gelator molecules are arranged in an ordered lamellar pattern in the 3D network with a periodicity of 3.75 nm. The dried gel of PyL-TyrOx also showed a similar lamellar packing with a layer thickness of 4.01 nm. Also, the XRD spectra of PyL-PheOx and PyL-TyrOx showed the presence of broad reflection peaks in the wide-angle region, centred at 22.74° and 21.75°, indicating d-spacings of 0.39 nm and 0.41 nm, respectively. This observation again confirms the formation of more compact structures for PyL-PheOx.
:1/2:1/3, which suggests that the gelator molecules are arranged in an ordered lamellar pattern in the 3D network with a periodicity of 3.75 nm. The dried gel of PyL-TyrOx also showed a similar lamellar packing with a layer thickness of 4.01 nm. Also, the XRD spectra of PyL-PheOx and PyL-TyrOx showed the presence of broad reflection peaks in the wide-angle region, centred at 22.74° and 21.75°, indicating d-spacings of 0.39 nm and 0.41 nm, respectively. This observation again confirms the formation of more compact structures for PyL-PheOx.
The molecular lengths calculated along the long molecular axis using the B3LYP/6-31G* level of theory were found to be 2.7 nm and 3.1 nm (inset Fig. 3b) for PyL-PheOx and PyL-TyrOx, respectively. The repetitive distance obtained for the lamellar structure from the XRD studies was less than double the full lengths of the minimized structure. The observation suggests that the gelator molecules undergo substantial interdigitation via the pyrenyl unit in the gel state. Thus, we believe that in the gel state the hydrophobic pyrene residues will form π-stacked arrays, while the oxyethylene units, being hydrophilic, remain exposed to bulk water. On the other hand, the AAH units of the adjacent molecules participate in the complementary H-bonding interactions. The formation of fibre like structures, as proposed in this model, will presumably undergo entanglement further to eventually form gels (Fig. 3c).70,80
Chemodosimetric analysis of hypochlorite using a hydrogel nano-template
Considering the presence of redox sensitive AAH units, PyL-PheOx and PyL-TyrOx were exposed to various reactive oxidative species in aqueous media. In both the cases, the addition of ClO− could specifically lead to a change in the solution color from colorless to yellow, which eventually became transparent soon. At the same time, the enhancement of fluorescence intensity in the ∼390–420 nm region was observed at the expense of the aggregate emission (ratiometric response) (Fig. 4a and S9a†). Irrespective of the nature of the amino acid residue, in both the cases, the saturation in the optical signal required an incubation time of ∼20 min for the completion of the event (Fig. S9b†). Time-course studies indicate that it followed a pseudo-first-order kinetic behavior in the presence of excess ClO−. As expected, both at elevated temperature (Fig. S10†) and at higher concentration (Fig. S9b–d†) of ClO−, the reaction rates accelerated. Selectivity is another key parameter to evaluate the performance of a new probe. Accordingly, the responses to the fluorescence spectra of both the compounds were tested in the presence of various other reactive oxygen species (such as NO˙, H2O2, ˙OH, ONOO−, ROO˙, O2˙−, and 1O2), biologically relevant anions (such as Cl−, Br−, I−, CO32−, HCO3−, NO2−, NO3−, and SO42−) and intracellular bio-reductants (GSH). No analyte, other than ClO−, could produce any detectable change in the fluorescence signal (Fig. 4b and S11†). Although a weak signal in the presence of ROO˙ could be observed, it was negligible in comparison to that observed in the presence of ClO− (Fig. S12b†). Titrimetric studies further indicate that the present systems could detect ClO− as low as 0.04 ppm with PyL-TyrOx and 0.12 ppm with PyL-PheOx gel (Fig. 4a). A comparison table for different hypochlorite sensors with their detection limits (especially for the hydrazone moiety, which underwent a chemodosimetric reaction on addition of hypochlorite) is also given in Table S1.†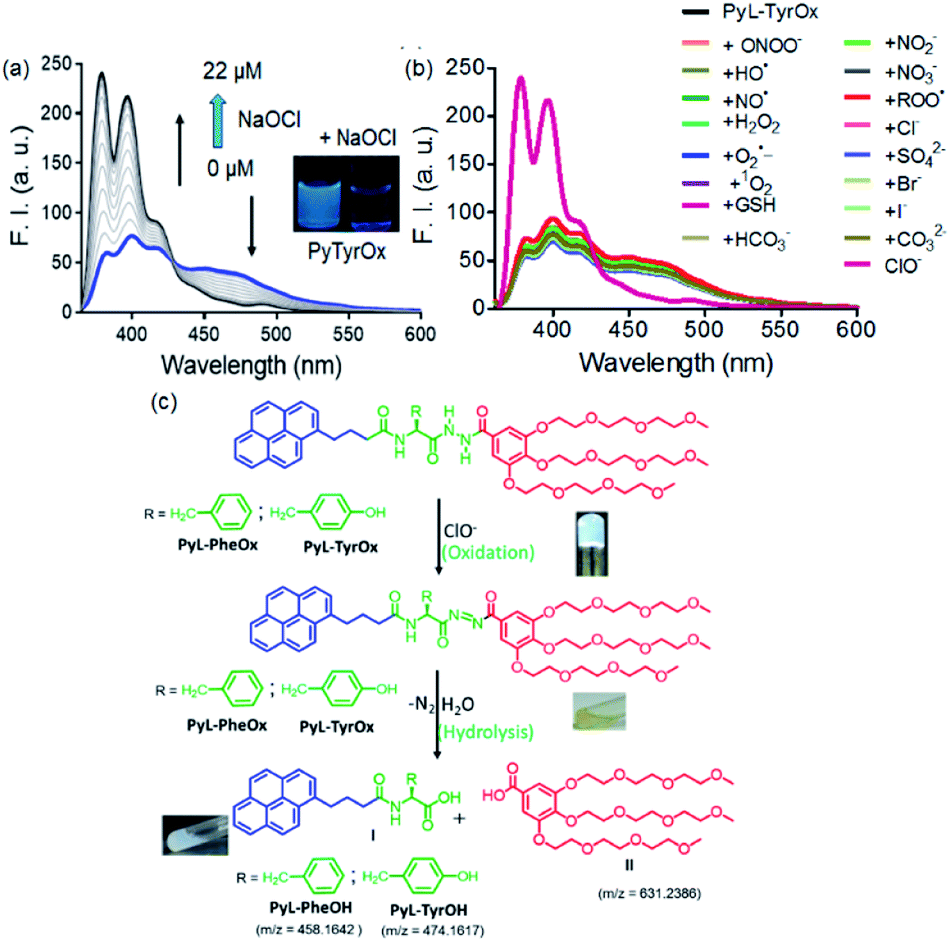 | ||
| Fig. 4 (a) Fluorescence titration spectra of PyL-TyrOx (20 μM and λex = 345 nm) in the presence of ClO−. (b) Change in the emission spectra of PyL-TyrOx (20 μM and λex = 345 nm) upon addition of various competitive analytes, such as biologically relevant anions, reactive oxygen species (20 μM), etc. (c) Schematic representation of the oxidative cleavage (hydrolysis) by ClO− (inset sol picture shows the formation of PyL-TyrOH). | ||
Hypochlorite triggered oxidative decomposition reaction
The hypochlorite anion (ClO−) is known to selectively oxidize dibenzoyl hydrazine to dibenzoyl diimide, which undergoes further decomposition in nucleophilic solvents like water. Analogously considering the presence of AAH units, both in PyL-PheOx and PyL-TyrOx molecules, these are susceptible to get oxidized to acyl aroyl diimides on exposure to ClO−. These intermediates thus formed undergo spontaneous hydrolysis in water to the corresponding acids I and II with the concomitant release of N2 (Scheme is given in Fig. 4c).44,81 Though we couldn't detect the presence of diimide derivative in the reaction media, one can assume that it might be responsible for the immediate yellow coloration. However, to ascertain and confirm the identity of various products upon treatment of ClO− with the hydrogel of PyL-TyrOx, the reaction mixture was subjected to ESI-MS analysis. The experimental figure (see Fig. S13 in the ESI†) shows that, besides the peak due to the probe PyL-TyrOx (m/z: 1078.4889), there are two more peaks that appear at m/z 474.1617 (peak I, calculated M + Na+) and 631.2368 (peak II, calculated M + Na+) respectively (Fig. S13a and b†). Also, the fluorescence spectra of PyL-PheOx and PyL-TyrOx in the presence of ClO− showed a close resemblance to the fluorescence spectra of free PyL-PheOH and PyL-TyrOH amino acid conjugates (synthesized separately, Fig. 5b and S13c†). Based on our data and previous observation reported in the literature, we conclude that ClO− selectively oxidizes acyl aroyl hydrazine of PyL-TyrOx to the corresponding acyl aroyl diimide, which then undergoes spontaneous decomposition in aqueous media. We propose that the fluorescence turn on reaction in this system proceeds primarily through the route that is depicted in Fig. 4c. These two observations confirm the oxidative dissociation reaction in the presence of ClO−. However, the extent of fluorescence enhancement in the ∼395 nm region was different for PyL-TyrOxvs.PyL-PheOx, and thus an ∼10-fold rise was observed for the former and only ∼4-fold for the latter. It is possible since the stability of the diimide derivative is known to be dependent on the nature of the adjacent aromatic residues. The formation of PyL-PheOH or PyL-TyrOH was also confirmed by HPLC studies (Fig. 5a). As shown in Fig. S13d,† the chromatographic peak due to PyL-TyrOx appeared at 12.3 min. After reaction with ClO−, two additional chromatographic peaks emerged at ∼8.2 and 4.8 min, respectively. The new chromatographic peak that appeared at ∼8.2 min was assigned to PyL-TyrOH, formed via oxidative hydrolysis of PyL-TyrOx. This was also confirmed by injecting authentic PyL-TyrOH into the above mentioned HPLC column.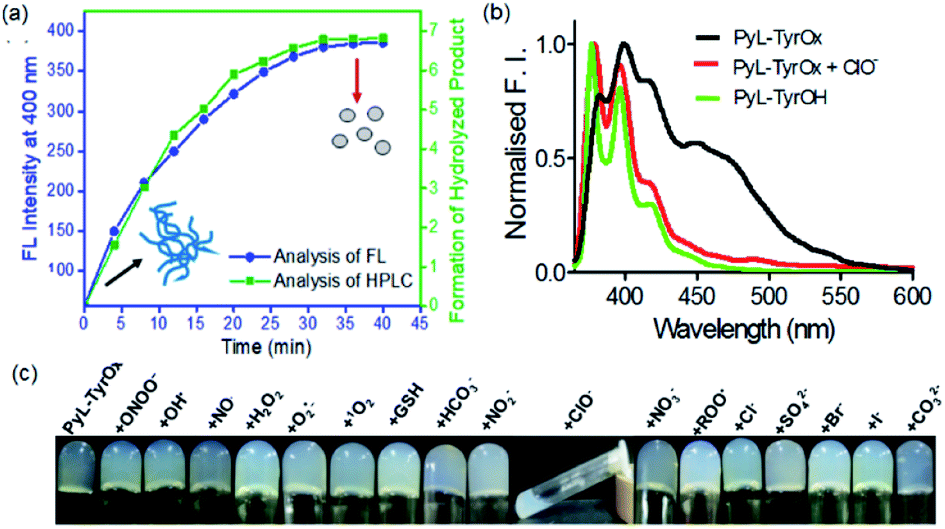 | ||
| Fig. 5 (a) Time-dependent evolution of the hydrolyzed product (PyL-TyrOH) upon interaction of PyL-TyrOx with ClO− as detected by fluorescence spectral analysis and reversed phase HPLC. (b) Fluorescence emission spectra of PyL-TyrOx (20 μM and λex = 345 nm) in the presence and absence of ClO− (20 μM). (c) Photographs of inverted tubes containing gels of PyL-TyrOx (2.5 mM) in the presence of different ROSs (1 mM sequence as mentioned in the figure). | ||
Gel-to-sol transition induced by hypochlorite
Furthermore, we studied the effect of ClO− on the stability of hydrogel samples. In both the cases, the mechanical strength of the hydrogels decreased remarkably in the presence of ClO− (Fig. 6c and S14b†). Finally, when the ClO− concentration exceeded ∼2 mM, a visible transition from solid gel to fluid sol could be seen (Fig. 5c and S12a†). As expected, such phase transition led to changes in the microscopic structures, and the fibrillar morphology of gels changed to irregularly shaped particles in the presence of ClO− (Fig. 6b and S14a†). A decrease in the hydrodynamic diameter and lowering of the fluorescence anisotropy values with ClO− further confirm the dissociation of the preformed self-assembled structure (Fig. S14c, d and S15†). Also, the analysis of CD reveals the loss in helical chirality upon addition of ClO− (Fig. 6a).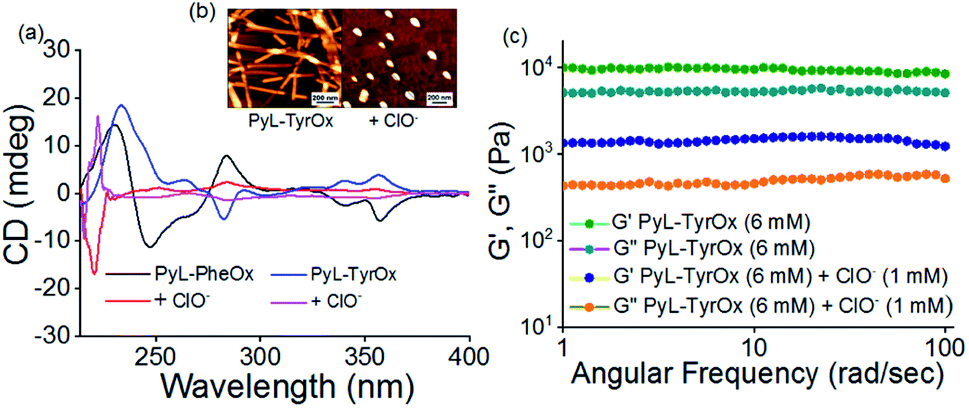 | ||
| Fig. 6 (a) CD spectra of PyL-PheOx and PyL-TyrOx (0.24 mM) in the presence of ClO− (25 μM). (b) AFM images of PyL-TyrOx (1.5 mM) in the presence of ClO− (25 μM). (c) Oscillatory frequency rheology data of PyL-TyrOx (6 mM) in the presence of ClO− (1 mM). | ||
Real-life application of hypochlorite sensing
Due to superior sensitivity, further we employed PyL-TyrOx in the detection of ClO− in different natural water sources such as tap, pond, sea, etc.82 In all the cases, one could observe almost quantitative recovery values with relatively small standard deviations (<5%) (Fig. 7a and Table S2†). Furthermore, the estimation of ClO− was performed in commercial bleach samples with a high degree of accuracy (Fig. S16a†). We have calculated % recovery values and relative standard deviations for the estimation of ClO− in diluted bleach solution. The nearly 100% recovery values with a relatively small standard deviation (<5%) indicate that our system is indeed suitable for analyzing hypochlorite in real-life samples, such as bleach (Table S3†).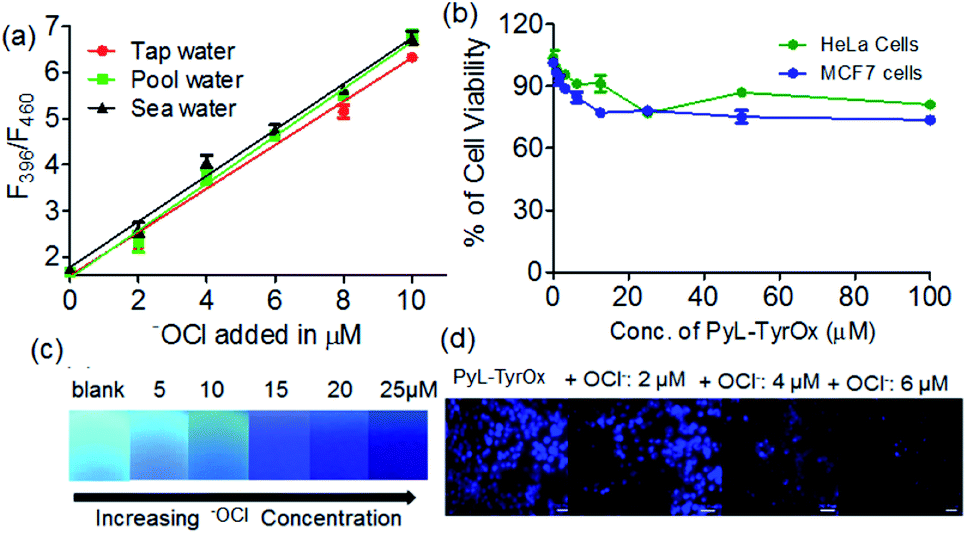 | ||
| Fig. 7 (a) Changes in the fluorescence emission intensity of PyL-TyrOx (20 μM and λex = 345 nm) upon interaction with ClO− in different types of water samples. (b) MTT assay of PyL-TyrOx (20 μM) in HeLa and MCF7 cell lines. (c) Picture captured (UV lamp 365 nm) after the addition of ClO− onto pre-coated filter paper with the PyL-TyrOx hydrogel. (d) Intracellular imaging of ClO− with PyL-TyrOx (20 μM) using HeLa cells as a model system. | ||
Gel-coated portable paper strips83 were developed for the detection of ClO− at remote locations without involving sophisticated analytical tools; for this a filter paper (Whatman 40) strip was cut into a rectangular shape (1 × 2 cm). 30 μL of the prepared gel sample (2.5 mM) was slowly applied on the surface of the paper strips using a micropipette such that it spreads uniformly. The gel coated on the paper strip was apparently totally absorbed within 15 min and then it was kept overnight to air-dry naturally. The paper strips coated with PyL-TyrOx showed strong cyan fluorescence under a >365 nm UV lamp. Before the sensing experiments, we also checked the stability of the paper strips under ambient conditions. No detectable change in the fluorescence emission intensity was observed even after two weeks. This suggests that the strips could easily be stored at room temperature. After this, the gel-coated paper strips were exposed to different amounts of ClO− (0 to 25 μM). A concentration-dependent gradual quenching of cyan fluorescence was observed (Fig. 7c). The changes in intensity could be quantified using readily available image processing software, Image J (Fig. S16b†). Because of the operational procedure mentioned here, this is very simple, and even the users with no technical knowledge can use them without any difficulty.
In vitro imaging of hypochlorite
Since both PyL-PheOx and PyL-TyrOx gelators showed high biocompatibility even at ∼30 μM concentration, the gelators could be used for the imaging of ClO− in HeLa and MCF7 cells (Fig. 7b and S17a†).84 Dose-dependent kinetic studies indicated that with 20 μM PyL-TyrOx a 30 min incubation time (at 37 °C) was enough for the complete cellular uptake. The stability of the gelator molecules was then monitored from time to time using HPLC and ESI-MS mass spectrometry. No significant degradation of PyL-TyrOx was observed even when incubated in the presence of proteinase K (5 mg mL−1) for more than 35 h at physiological pH 7.4 (Fig. S17b†). As expected, the cells treated with PyL-TyrOx showed blue fluorescence in the 450–600 nm range due to cellular internalization (Fig. S17c†). However, upon exposure to aqueous ClO−, the cells exhibited dose-dependent diminution of fluorescence, which might be due to the oxidative-dissociation reactions inside the living cells (Fig. 7d). These results demonstrate that PyL-TyrOx could also be used for the ratiometric fluorescence imaging of ClO− in living cells.Conclusions
In conclusion, we have demonstrated the design and synthesis of two novel pyrene-based biocompatible hydrotropes, PyL-PheOx and PyL-TyrOx, which can readily form biocompatible, thermoreversible thixotropic hydrogels in aqueous media. Spectroscopic investigations indicate the formation of π-stacked ‘compact’ aggregates in the case of PyL-PheOx, while the aggregates with stronger CT characteristics were formed from PyL-TyrOx. Though both the compounds showed the presence of fibrous networks in the self-assembled state, CD spectral investigations indicate the formation of M-helical and P-helical structures for PyL-PheOx and PyL-TyrOx respectively. Furthermore, the compounds showed clearly visible solid gel-to-fluid sol transition in the presence of ClO−via oxidative hydrolysis. The change in the fluorescence intensity of the hydrogel in the presence of ClO− was utilized for the analysis of commercial bleach samples and live-cell imaging. Finally, gel-coated paper strips were also developed for the on-site detection of ClO−. Such findings may inspire widespread analytical use of hydrogels and related soft matter and also encourage developing newer designs or assays of reactive oxygen species and other analytes.Author contributions
S. B. conceptualized the project; experimental investigation and data collection were done by DB and ND under supervision of SB. The original draft of the manuscript was prepared by DB, its review, and editing were done by DB, ND and SB. The infrastructure and funding of the research were provided by SB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This is dedicated to the memory of Professor Har Gobind Khorana on his birth centenary. Prof. S. Bhattacharya thanks the Department of Science and Technology for the J. C. Bose Fellowship. We thank JNCASR for the research associateship to Dr D. Biswakarma.References
- C. M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef PubMed.
- X. Wang, T. He, L. Yang, H. Wu, R. Zhang, Z. Zhang, R. Shen, J. Xiang, Y. Zhang and C. Wei, Nanoscale, 2016, 8, 6479–6483 RSC.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141(12), 4886–4899 CrossRef CAS PubMed.
- X. Qi, M. Zhang, T. Su, W. Pan, X. Tong, Q. Zeng, W. Xiong, N. Jiang, Y. Qian, Z. Li, X. He, L. Shen, Z. Zhou and J. Shen, J. Agric. Food Chem., 2020, 68, 3770–3778 CrossRef CAS PubMed.
- J. Chen, J. Huang, H. Zhang and Y. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 38647–38654 CrossRef CAS PubMed.
- C. Wang, A. Fischer, A. Ehrlich, Y. Nahmias and I. Willner, Chem. Sci., 2020, 11, 4516–4524 RSC.
- Z. Deng, R. Yu and B. Guo, Mater. Chem. Front., 2021, 5, 2092–2123 RSC.
- T. Su, Z. Tang, H. He, W. Li, X. Wang, C. Liao, Y. Sun and Q. Wang, Chem. Sci., 2014, 5, 4204–4209 RSC.
- M. Vázquez-González and I. Willner, Angew. Chem., Int. Ed., 2019, 59, 15342–15377 CrossRef PubMed.
- C. C. Piras, A. G. Kay, P. G. Genever and D. K. Smith, Chem. Sci., 2021, 12, 3958–3965 RSC.
- N. Mehwish, X. Dou, Y. Zhao and C. L. Feng, Mater. Horiz., 2019, 6, 14–44 RSC.
- P. R. A. Chivers, R. S. Dookie, J. E. Gough and S. J. Webb, Chem. Commun., 2020, 56, 13792–13795 RSC.
- D. M. Ryan, S. B. Anderson and B. L. Nilsson, Chem. Commun., 2011, 47, 475–477 RSC.
- Y. Xia, B. Xue, M. Qin, Y. Cao, Y. Li and W. Wang, Sci. Rep., 2017, 7, 9691 CrossRef PubMed.
- C. H. Zhang, Y. Li, X. Xue, P. Chu, C. Liu, K. Yang, Y. Jiang, W. Q. Chen, G. Zou and X. J. Liang, Chem. Commun., 2015, 51, 4168–4171 RSC.
- S. Wei, Z. Li, W. Lu, H. Liu, J. Zhang, T. Chen and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 17, 8608–8624 CrossRef PubMed.
- Y. Li, D. J. Young and X. J. Loh, Mater. Chem. Front., 2019, 3, 1489–1502 RSC.
- A. Brito, Y. M. Abul-Haija, D. S. da Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem. Sci., 2019, 10, 2385–2390 RSC.
- J. Wu, A. Chen, M. Qin, R. Huang, G. Zhang, B. Xue, J. Wei, Y. Li, Y. Cao and W. Wang, Nanoscale, 2015, 7, 1655–1660 RSC.
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819–2822 CrossRef CAS PubMed.
- K. Y. Kim, M. Ok, J. Kim, S. H. Jung, M. L. Seo and J. H. Jung, Gels, 2020, 6, 16 CrossRef CAS PubMed.
- M. Y. Yeh, C. W. Huang, J. W. Chang, Y. T. Huang, J. H. Lin, S. M. Hsu, S. C. Hungad and H. C. Lin, Soft Matter, 2016, 12, 6347–6351 RSC.
- D. Biswakarma, N. Dey and S. Bhattacharya, Chem. Commun., 2020, 56, 7789–7792 RSC.
- S. Bartocci, J. A. Berrocal, P. Guarracino, M. Grillaud, L. Franco and M. Mba, Chem.–Eur. J., 2018, 24, 2920–2928 CrossRef CAS PubMed.
- S. Mukherjee, T. Kar and P. K. Das, Chem.–Asian J., 2014, 9, 2798–2805 CrossRef CAS PubMed.
- D. P. Hickey, K. Lim, R. Cai, A. R. Patterson, M. Yuan, S. Sahin, S. Abdellaouia and S. D. Minteer, Chem. Sci., 2018, 9, 5172–5177 RSC.
- N. Kapuria, V. Sharma, P. Kumar and A. L. Koner, J. Mater. Chem. C, 2018, 6, 11328–11335 RSC.
- J. Chen, Y. Zhang, Z. Meng, L. Guo, X. Yuan, Y. Zhang, Y. Chai, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2020, 11, 6275–6282 RSC.
- M. Y. Li, K. Li, Y. H. Liu, H. Zhang, K. K. Yu, X. Liu and X. Q. Yu, Anal. Chem., 2020, 92, 3262–3269 CrossRef CAS PubMed.
- Y. Zhan, F. Luo, L. Guo, B. Qiu, Y. Lin, J. Li, G. Chen and Z. Lin, ACS Sens., 2017, 2, 1684–1691 CrossRef CAS PubMed.
- S. Goswami, S. Paul and A. Manna, Dalton Trans., 2013, 42, 10097–10101 RSC.
- K. Yin Zhang, J. Zhang, Y. Liu, S. Liu, P. Zhang, Q. Zhao, Y. Tanga and W. Huang, Chem. Sci., 2015, 6, 301–307 RSC.
- X. Meng, Y. Shi, Z. Chen, L. Song, M. Zhao, L. Zou, S. Liu, W. Huang and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 35838–35846 CrossRef CAS PubMed.
- Q. Xu, K. A. Lee, S. Lee, K. M. Lee, W. J. Lee and J. Yoon, J. Am. Chem. Soc., 2013, 135, 9944–9949 CrossRef CAS PubMed.
- J. T. Hou, M. Y. Wu, K. Li, J. Yang, K. K. Yu, Y. M. Xie and X. Q. Yu, Chem. Commun., 2014, 50, 8640–8643 RSC.
- X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. James, Chem. Sci., 2014, 5, 3368–3373 RSC.
- H. Ma, B. Song, Y. Wang, D. Cong, Y. Jiang and J. Yuan, Chem. Sci., 2017, 8, 150–159 RSC.
- L. Long, Y. Wu, L. Wang, A. Gong, F. Hu and C. Zhang, Chem. Commun., 2015, 51, 10435–10438 RSC.
- D. Andina, J. C. Leroux and P. Luciani, Chem.–Eur. J., 2017, 23, 13549–13573 CrossRef CAS PubMed.
- V. Brega and S. W. Thomas, ACS Appl. Mater. Interfaces, 2021, 13, 13658–13665 CrossRef CAS PubMed.
- X. Chen, X. Wang, S. Wang, W. Shi, K. Wang and H. Ma, Chem.–Eur. J., 2008, 14, 4719–4724 CrossRef CAS PubMed.
- Y. Guo, Q. Ma, F. Cao, Q. Zhao and X. Ji, Anal. Methods, 2015, 7, 4055–4058 RSC.
- X. Lou, Y. Zhang, Q. Li, J. Qina and Z. Li, Chem. Commun., 2011, 47, 3189–3191 RSC.
- S. P. Goswami, A. K. Das, A. Manna, A. K. Maity, P. Saha, C. K. Quah, H. K. Fun and H. A. Abdel-Aziz, Anal. Chem., 2014, 86, 6315–6322 CrossRef CAS PubMed.
- A. Gangopadhyay, S. S. Ali, U. N. Guria, S. K. Samanta, R. Sarkar, P. Datt and A. K. Mahapatra, New J. Chem., 2018, 42, 15990–15996 RSC.
- X. Zhong, Q. Yang, Y. Chen, Y. Jiang and Z. Dai, J. Mater. Chem. B, 2020, 8, 7375–7381 RSC.
- L. Jin, M. Xu, H. Jiang, W. Wang and Q. Wang, Anal. Methods, 2018, 10, 4562–4569 RSC.
- Z. M. Prokopowicz, F. Arce, R. Biedron, C. L. L. Chiang, M. Ciszek, D. R. Katz, M. Nowakowska, S. Zapotoczny, J. Marcinkiewicz and B. M. Chain, J. Immunol., 2010, 184, 824–835 CrossRef CAS PubMed.
- D. Biswakarma, N. Dey and S. Bhattacharya, Soft Matter, 2020, 16, 9882–9889 RSC.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740–750 CrossRef CAS PubMed.
- S. Akama, T. Makia and M. Yamanaka, Chem. Commun., 2018, 54, 8814–8817 RSC.
- H. Fang, W.-J. Qu, H.-H. Yang, J. X. He, H. Yao, Q. Lin, T. B. Wei and Y.-M. Zhang, Dyes Pigm., 2020, 174, 108066 CrossRef CAS.
- U. Baruaha and U. Manna, Chem. Sci., 2021, 12, 2097–2107 RSC.
- T. Yoshii, S. Onogi, H. Shigemitsu and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 3360–3365 CrossRef CAS PubMed.
- T. Sugiura, T. Kanada, D. Mori, H. Sakai, A. Shibata, Y. Kitamura and M. Ikeda, Soft Matter, 2020, 16, 899–906 RSC.
- H. R. Culver, J. R. Clegg and N. A. Peppas, Acc. Chem. Res., 2017, 50, 170–178 CrossRef CAS PubMed.
- J. H. Tang, Y. Li, Q. Wu, Z. Wang, S. Hou, K. Tang, Y. Sun, H. Wang, H. Wang, C. Lu, X. Wang, X. Li, D. Wang, J. Yao, C. J. Lambert, N. Tao, Y. W. Zhong and P. J. Stang, Nat. Commun., 2019, 10, 4599 CrossRef PubMed.
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., Int. Ed., 2014, 53, 365–386 CrossRef CAS PubMed.
- H. Fang, G. Cai, Y. Hu and J. Zhang, Chem. Commun., 2018, 54, 3045–3048 RSC.
- M. A. Beatty, J. Borges-Gonzalez, N. J. Sinclair, A. T. Pye and F. Hof, J. Am. Chem. Soc., 2018, 140, 3500–3504 CrossRef CAS PubMed.
- B. Pramanik, N. Singha and D. Das, ACS Appl. Polym. Mater., 2019, 1, 833–843 CrossRef CAS.
- K.-I. Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. J. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef CAS PubMed.
- S. Bhattacharya and S. S. Mandal, J. Chem. Soc., Chem. Commun., 1995, 2489–2490 RSC.
- X. Li, G. Zhang, H. Ma, D. Zhang, J. Li and D. Zhu, J. Am. Chem. Soc., 2004, 126, 11543–11548 CrossRef CAS PubMed.
- J. W. Reed, H. H. Ho and W. L. Jolly, J. Am. Chem. Soc., 1974, 96, 1248–1249 CrossRef CAS.
- L. K. Bancroft, J. R. Lupton, L. A. Davidson, S. S. Taddeo, M. E. Murphy, R. J. Carroll and R. S. Chapkin, Free Radical Biol. Med., 2003, 35, 149–159 CrossRef CAS PubMed.
- S. R. Nelli, R. D. Chakravarthy, Y. M. Xing, J. P. Wenga and H. C. Lin, Soft Matter, 2017, 13, 8402–8407 RSC.
- S. Bhattacharjee and S. Bhattacharya, J. Mater. Chem. A, 2014, 2, 17889–17898 RSC.
- S. K. Samanta and S. Bhattacharya, Chem. Commun., 2013, 49, 1425–1427 RSC.
- B. Maiti, S. Bhattacharjee and S. Bhattacharya, Nanoscale, 2019, 11, 2223–2230 RSC.
- Z. Shen, T. Wang, L. Shi, Z. Tang and M. Liu, Chem. Sci., 2015, 6, 4267–4272 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- J. T. van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 3077–3083 CrossRef CAS PubMed.
- S. Bhattacharjee and S. Bhattacharya, Chem. Commun., 2015, 51, 6765–6768 RSC.
- J. H. Lee, J. Park, J. W. Park, H. J. Ahn, J. Jaworski and J. H. Jung, Nat. Commun., 2015, 6, 6650 CrossRef CAS PubMed.
- S. Bhattacharjee, B. Maiti, D. Biswakarma and S. Bhattacharya, Macromol. Symp., 2016, 369, 14–18 CrossRef CAS.
- M. Sponchioni, C. T. O'Brien, C. Borchers, E. Wang, M. N. Rivolta, N. J. W. Penfold, I. Canton and S. P. Armes, Chem. Sci., 2020, 11, 232–240 RSC.
- H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 4122–4126 CrossRef CAS PubMed.
- P. Chen, Z. Zheng, Y. Zhu, Y. Dong, F. Wang and G. Liang, Anal. Chem., 2017, 89, 5693–5696 CrossRef CAS PubMed.
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- D. Biswakarma, N. Dey, D. Bhagat and S. Bhattacharya, ACS Agric. Sci. Technol., 2021, 1, 322–328 CrossRef CAS.
- N. Dey, A. Ali, M. Kamra and S. Bhattacharya, J. Mater. Chem. B, 2019, 7, 986–993 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05424d |
| This journal is © The Royal Society of Chemistry 2022 |