
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-dependent compression of threaded alkyldiphosphate in head to head cyclodextrin [3]pseudorotaxanes†
Jérémy
Scelle‡
,
Hugo
Vervoitte‡
,
Laurent
Bouteiller
 ,
Lise-Marie
Chamoreau
,
Matthieu
Sollogoub
,
Guillaume
Vives
* and
Bernold
Hasenknopf
*
,
Lise-Marie
Chamoreau
,
Matthieu
Sollogoub
,
Guillaume
Vives
* and
Bernold
Hasenknopf
*
Sorbonne Université, UMR CNRS 8232, Institut Parisien de Chimie Moléculaire, 4 Place Jussieu, 75005, Paris, France. E-mail: guillaume.vives@sorbonne-universite.fr; bernold.hasenknopf@sorbonne-universite.fr
First published on 17th January 2022
Abstract
The encapsulation of guests in a confined space enables unusual conformations and reactivities. In particular, the compression of akyl chains has been obtained by self-assembled molecular capsules but such an effect has not been reported in solution for pseudorotaxane architectures. By exploiting the tendency of cyclodextrin (CD) to form head to head [3]pseudorotaxanes and the hydrogen bonding abilities of phosphate groups, we have studied the effect of the CD dimer cavity on the conformation of threaded α,ω-alkyl-diphosphate axles. The formation of [2]pseudorotaxanes and [3]pseudorotaxanes was investigated by a combination of NMR, ITC and X-ray diffraction techniques. In the solid state, the [3]pseudorotaxane with a C8 axle presents a fully extended conformation with both terminal phosphate groups interacting with hydroxyl groups of the primary rim of CDs. Such hydrogen bonding interactions are also present with the C9 and C10 axles resulting in a compression of the alkyl chain with gauche conformations in the solid state. NMR studies have shown that this effect is maintained in solution resulting in a size-dependent progressive compression of the alkyl chain by the CD [3]pseudorotaxane architecture for C9, C10 and C11 axles.
Introduction
The study of small molecules in a confined environment is of prime interest to understand biological systems where substrates are isolated from solution in enzyme or receptor cavities.1 This confinement in pockets optimized by evolution enables multiple phenomena essential to life such as catalysis, signaling or replication. Recently, supramolecular chemistry has opened a way to understand the fundamentals of how molecules can pack within a confined nanospace by fostering the development of synthetic hosts such as self-assembled capsules or cages.2 Flexible guests can dynamically fold in the small space of a capsule and adopt high energy conformations that are not observed in solution. Such conformations are reminiscent of fatty acids bound to fatty acid-binding proteins that present compact conformations for their alkyl chain.3 In a seminal study, Rebek has shown that the cavity of a hydrophobic cavitand can stabilize the helical folding of alkanes by a combination of hydrophobic effects and C–H/π interactions.4 The encapsulation of n-alkane chains in self-assembled capsules has been further studied by Rebek5 and Gibb (Fig. 1).6 They have shown that a linear alkyl chain inside a water-soluble cavitand adopts a coiled conformation and can be compressed inside resorcinarene or pyrogallolarene capsules with dimensions smaller than their total linear length. Such coiled or helicoidal conformations result from the minimization of the solvatophobic effect, the optimization of the space filling and stabilizing interactions with the walls. Recently, Barboiu has reported the compression of alkyl chains inside a self-assembled “pyrene box”7 by a combination of hydrogen bonding, electrostatic interactions and van der Waals contacts.Such self-assembled capsules can pre-form in the absence of the guest, and induce their compression through the optimization of their packing inside a closed cavity. On the other hand, cyclodextrins (CDs) which are cone-shaped macrocycles tend to form head to head [3]rotaxanes due to the strong hydrogen bonding between their secondary rim resulting in a supramolecular open capsule around the axle that is formed only in the presence of a guest.8 It has been observed that the solid state aliphatic α,ω-diacids with 12 to 16 carbons form [3]pseudorotaxanes with β-CD and position both their carboxylic groups at the primary rims of the β-CD dimer.9 This results in a bend in the middle of the aliphatic chain that increases with the length of the carbon chain. A similar bending has been observed for 1,12-dodecanediol in the crystallographic structure of its β-CD [3]pseudorotaxane.10 All of these compressed rotaxanes were observed only in the solid state. Their existence in solution has not been reported as aliphatic axles in CD-based pseudorotaxanes usually stick out of the rims of the CD. This is probably due to the absence of packing stabilization in solution, to the weak hydrogen bonding between the terminal groups and the hydroxyl groups of the CD primary rim and to the solvation of these polar groups that result in a decompression of the chain. Indeed, particularly in water, the solvation of the end groups of the axle plays an important role. It has been recently shown that pseudorotaxanes composed of a 3D macrocycle threaded onto an axle favor extended conformations for the guest to allow the protruding polar ends to interact with the solvent, instead of a compressed conformation to maximize interactions with both rims of the macrocycle.11 However, during the course of our work on cyclodextrin rotaxanes and polyrotaxanes for imaging applications,12 we have serendipitously observed an unexpected compression of α,ω-alkyldiphosphate axles by CD [3]pseudorotaxanes in solution. This points out the delicate balance between interactions of the CDs and the axle inside, and solvation of the axle end groups outside the CDs. Phosphate groups, due to their strong hydrogen acceptor properties and their negative charge that enables water solubility of α,ω-alkyl-diphosphate axles, are good candidates to achieve strong interactions with the primary rim of the CD head to head dimer (Fig. 1). We have thus investigated here, by a combination of NMR, ITC and X-ray diffraction techniques, how hydrogen bonding of the polar ends of the axle with the CD yields a compression of the axle in the solid state and in solution. Herein, we wish to disclose the formation of [2] or [3]pseudorotaxanes depending on the axle length and the effect of the self-assembly process on the axle compression.
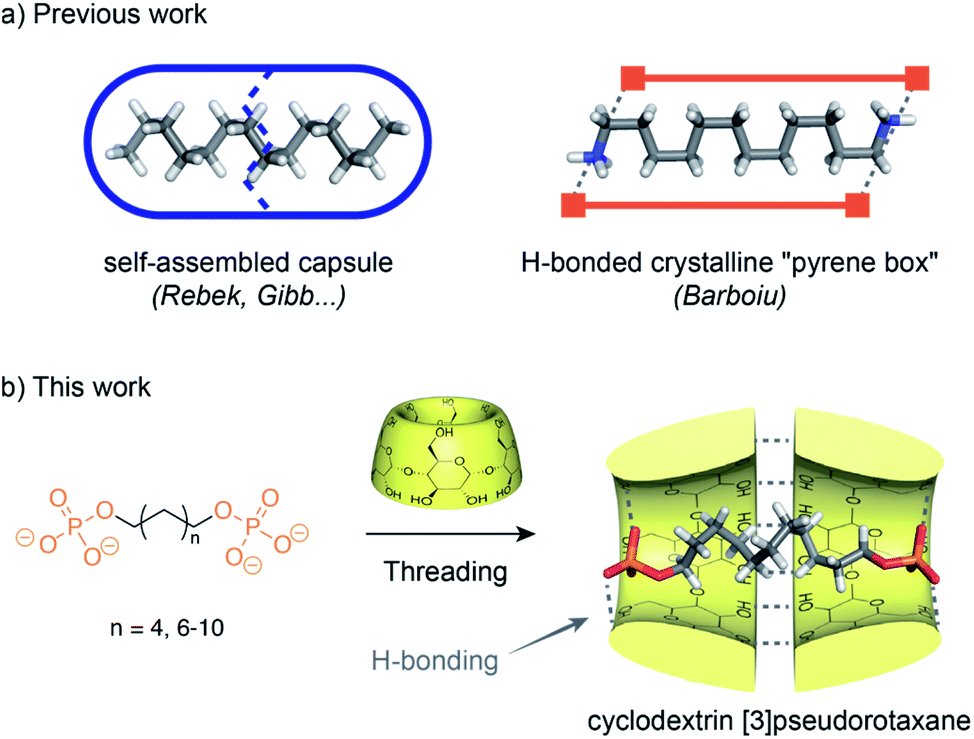 | ||
| Fig. 1 (a) Previous work on compression of alkyl chains by self-assembled capsules or boxes. (b) Alkyldiphosphate axles used in this study and corresponding CD [3]pseudorotaxane with potential compression resulting from hydrogen bonding and hydrophobic interactions between the threaded axle and head to head CDs. | ||
Results and discussion
Threading studies at acidic pH
The threading abilities of α,ω-alkyldiphosphate guests with α-CD were first studied in solution by NMR. Titration experiments were performed at pH 5.0 in an acetate buffer to ensure a monoanionic form for both phosphate terminal groups since their first and second acidity constants (pKa) are around 2 and 7, respectively (see Table S1†). Upon addition of CD to a solution of the C6 axle, a progressive appearance of a new set of peaks for the axle and the CD was observed (Fig. 2). This indicates the formation of a new species in slow exchange compared to the acquisition time of NMR. However, the equilibrium is reached within the time necessary to prepare and record the NMR spectra as no evolution was observed over time. Upon addition of CD the signals of the alkyl chain protons in the 1.30–1.85 ppm range are split into two sets indicating a desymmetrization of the axle in agreement with the threading of one CD and the formation of a [2]pseudorotaxane. Moreover, the CD protons display two sets of signals corresponding to the free and threaded CDs in slow exchange. In particular H6a and H4 are clearly deshielded and shielded, respectively, upon threading compared to free CD. By fitting the integration of selected CD protons with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model an association constant of logK = 2.6 was determined (see Fig. S6† and Table 1).
:1 binding model an association constant of logK = 2.6 was determined (see Fig. S6† and Table 1).
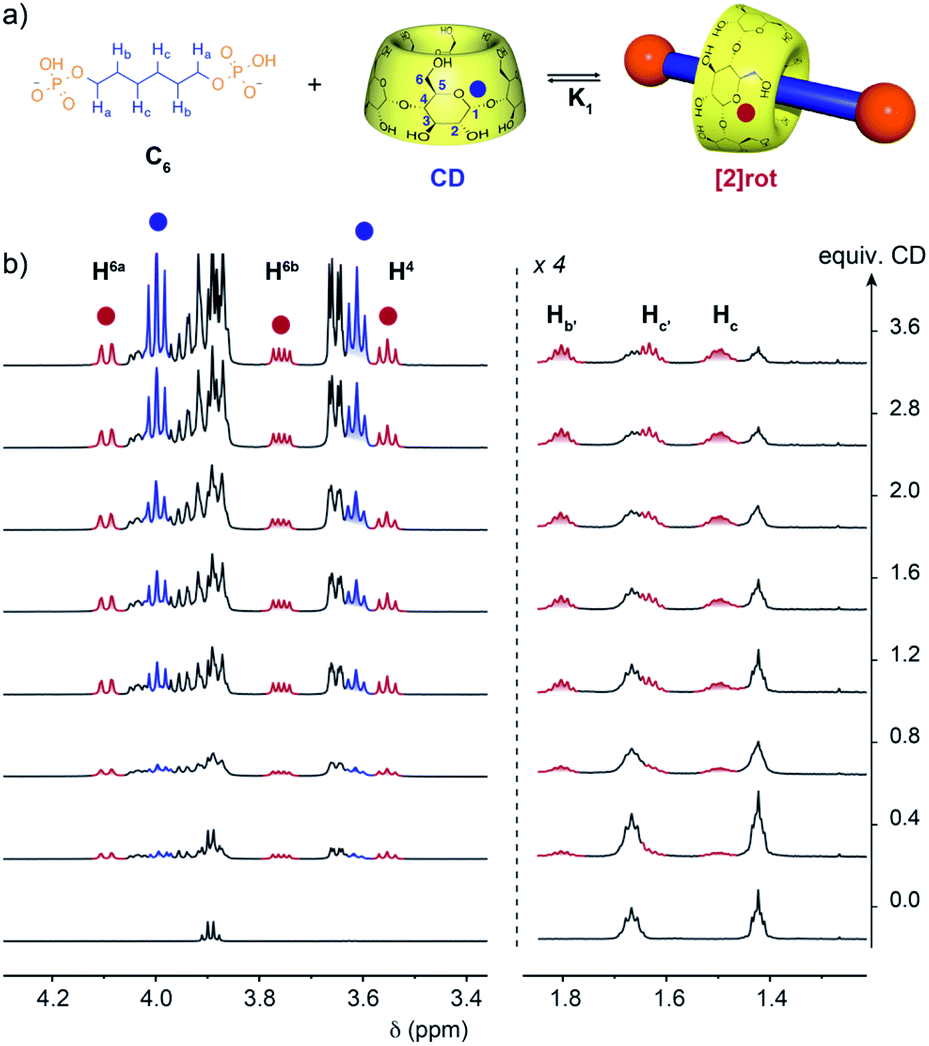 | ||
| Fig. 2 (a) Equilibrium of formation of [2]pseudorotaxane between α-CD and the C6 axle. (b) 1H NMR (600 MHz, D2O, 300 K) titration of the C6 axle (2.8 mM) with CD in acetate buffer at pH = 5 showing the progressive appearance of [2]rot (red) and free CD (blue). | ||
| Axle | C6 | C8 | C9 | C10 | C11 | C12 |
|---|---|---|---|---|---|---|
| log(K1) NMR | 2.62 ± 0.08 | 3.21 ± 0.08 | 3.41 ± 0.03 | 3.72 ± 0.03 | 3.68 ± 0.03 | 3.72 ± 0.07 |
| log(K2) NMR | — | 3.18 ± 0.03 | 2.97 ± 0.01 | 2.77 ± 0.01 | 3.09 ± 0.01 | 3.54 ± 0.04 |
| α (NMR) | — | 3.7 | 1.4 | 0.45 | 1.0 | 2.6 |
| log(K1) ITC | 2.40 ± 0.02 | 3.45 ± 0.03 | 3.51 ± 0.02 | 3.50 ± 0.02 | 3.56 ± 0.04 | 3.72 ± 0.02 |
| log(K2) ITC | — | 3.04 ± 0.02 | 3.00 ± 0.01 | 2.79 ± 0.02 | 3.05 ± 0.03 | 3.50 ± 0.02 |
| α (ITC) | — | 1.5 | 1.2 | 0.8 | 1.2 | 2.4 |
| ΔH1; ΔH2 (kcal mol−1) | −7.30; — | −4.20; −13.7 | −4.60; −11.7 | −5.50; −9.70 | −5.00; −14.1 | −4.70; −11.0 |
| ΔS1; ΔS2 (cal mol−1 K−1) | −15; — | 1.8; −32 | 0.6; −26 | −2.6; −20 | −0.43; −33 | 1.3; −21 |
| TΔS1; TΔS2 (kcal mol−1) | −4.3; — | 0.54; −9.6 | 0.18; −7.8 | −0.78; −6.0 | −0.13; −9.9 | 0.39; −6.3 |
| ΔG1; ΔG2 (kcal mol−1) | −3; — | −4.74; −4.10 | −4.78; −3.90 | −4.72; −3.70 | −4.87; −4.20 | −5.09; −4.70 |
Upon increasing the axle length, the number of inclusion complexes also increases. Indeed, for the C8 axle, the NMR titration at pH = 5 showed the presence of three sets of signals indicating the formation of two different inclusion complexes (Fig. 3). The anomeric proton H1 of the CD around 5.1 ppm is particularly indicative to monitor the different species with three distinct signals at 5.08, 5.10, and 5.05 ppm corresponding to the free CD and the [2] and [3]pseudorotaxanes respectively. As expected, [2]rot appears first upon addition of CD, reaches a maximum concentration at around 1 equivalent and is then converted into [3]rot (Fig. S6†). The two binding equilibria are not successive but concomitant, with similar association constants K1 and K2 of logK = 3.21 and 3.18, respectively, determined by fitting with a 2:1 model (see Fig. S6† and Table 1). [3]rot was isolated by selective precipitation in DMF after switching to basic pH and was fully characterized by 1H and 31P NMR (see Fig. S16 and S17†). It presents a symmetrical spectrum for 1H and 31P NMR with only one singlet indicating that the two CDs are either in a head to head or tail to tail configuration. The inclusion of the axle inside the CDs was validated by 2D T-ROESY NMR and the observed correlations between the H5 and H3 protons pointing inside the cavity and the carbon chain protons (Hb, Hc/d). 2D HOESY 31P/1H experiments presented a correlation between the terminal P and H5and H6 protons of the CD indicating that the primary rims are oriented towards the phosphate groups and the secondary rims are able to interact with each-other through hydrogen bonds as usually observed in CD [3]rotaxane. Thus, the two additional carbons of the alkyl chain enabled the mechanostereoselective threading of two CDs12b forming a head to head dimeric capsule on a single axle.
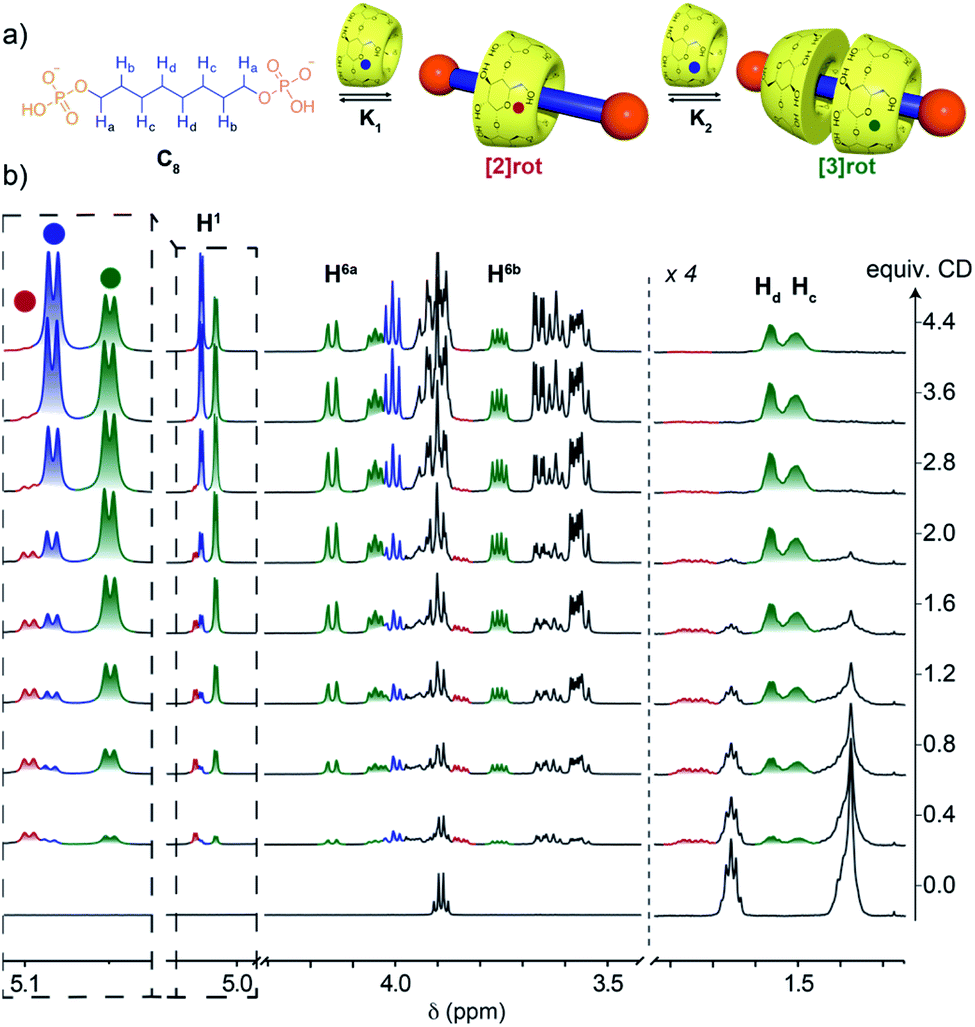 | ||
| Fig. 3 (a) Equilibrium of formation of [2] and [3]pseudorotaxanes with the C8 axle. (b) 1H NMR (600 MHz, D2O, 300 K) titration of the C8 axle (2.8 mM) with CD in acetate buffer at pH = 5 showing the progressive appearance of [2]rot (red), [3]rot (green) and free CD (blue). | ||
The effect of the axle length was evaluated by further increasing the number of carbons between the two phosphate moieties from 8 to 12. In all cases the formation of [2] and [3]pseudorotaxanes was observed upon addition of CD to the axle at pH 5 (see Fig. S2–S5†). The binding constants were determined from 1H NMR and ITC titration experiments and are both in agreement (Table 1). The K1 association constant for the formation of the [2]pseudorotaxane presents an overall increase upon the chain length as expected for an increase of the hydrophobicity of the axle that favors the threading of the CD. K2 values do not follow the same trend with a decrease from C8 to C10 followed by an increase to the maximum value for C12. The thermodynamics of the binding were extracted from the ITC titrations. For all the axles, the binding of the first CD to form a [2]pseudorotaxane presents an enthalpic (ΔH1) contribution that predominates over the entropic one with TΔS1 values close to zero. However, for the formation of [3]pseudorotaxane, the entropic contribution is no longer negligible and its negative value disfavors the binding. This decrease in entropy reflects an overall decrease in the degrees of freedom of the system, associated with conformational restrictions, which is not compensated by the release of water molecules inside the CD cavity. However, the enthalpy ΔH2 values are also significantly more negative than ΔH1, showing a common phenomenon of enthalpy/entropy compensation that results in values in the same order of magnitude between ΔG1 and ΔG2. The formation of a network of hydrogen bonds between the secondary rim of the CD, as well as the possible interaction of the primary rim with the terminal phosphates may justify this strong enthalpic stabilization of the [3]pseudorotaxane.
Hydrophobicity-controlled inclusion is characterized by an exothermic binding and a positive entropy ΔS variation due to the release of solvating water molecules. Previous studies on the binding of amphiphiles with CD have shown that for α-CD the binding constants are higher than those observed for the β-CD complex, and the entropy change is negative.13 The algebraic value of the entropy change is determined by two competing factors: (i) the positive release of water molecules from the alkyl chain and the cavity of the CD and (ii) the negative formation of the inclusion complex that causes a decrease in the accessible chain conformations. The values we have determined thus tend to indicate that the formation of the pseudorotaxanes is a favorable process of mostly enthalpic origin, and unfavorable from the entropic point of view, especially for the [3]pseudorotaxane. CD threading has been described on various aliphatic axles in the literature,14 and shows similar behaviors with a main enthalpic contribution to the formation of inclusion complexes.
The effect of the first binding event on the second one was further investigated by calculating the cooperativity factor α = 4K2/K1 (Table 1). A positive cooperativity (α > 1) indicating a favorable binding of the second CD to form the [3]pseudorotaxane was observed for C8 and C12 axles while an inhibition (α < 1) was clearly obtained for the C10 axle. These values indicate a specific behavior for C10 where the formation of the [3]pseudorotaxane is disfavored.
Kinetics of threading at basic pH
The effect of the protonation state of the phosphate groups on the threading process was also investigated. At pH = 10 when the two phosphate groups are doubly deprotonated, the threading becomes very slow and its kinetics can be easily monitored by 1H and 31P NMR. Starting from a 1:1 mixture between CD and the C6 axle a progressive disappearance of the signal of the free CD and axle and the appearance of new set of signals corresponding to the [2]pseudorotaxane were observed over time (see Fig. S8†). The formation of a [2]pseudorotaxane was also confirmed by 31P{1H} NMR with the appearance of two singlets (at 3.51 and 3.59 ppm) and the decrease of the initial singlet (at 3.92 ppm) indicating the desymmetrization of the axle due to the threading of the CD. The equilibrium was reached after ca. 4 hours in sharp contrast with the experiment at pH = 5 where the threading was almost instantaneous. The energy barrier for the CD threading is increased at basic pH probably due to the increased solvation sphere of the doubly anionic phosphate end groups.
The threading studies at pH = 10 on axles with more than 6 carbons were performed with two equivalents of CDs to favor the formation of a [3]pseudorotaxane. NMR monitoring showed the progressive formation of [2]rot and [3]rot with characteristic signals in the region of anomeric protons of the CD, in the same line as for the titrations at pH = 5 (see Fig. S9–S13†). From the integration of the NMR signals the proportion of the different species can be determined and fitted to access kinetic constants. For C8 to C12 axles, [2]rot was first formed and reached a maximum after ca. one day and was then slowly converted into [3]pseudorotaxane over several weeks (see Fig. S14†). The model rate constant k1 (Table 2) shows a 10-fold increase for the formation of [2]pseudorotaxane with axle C6 rather than with C8–C12 axles. The latter are all of the same order of magnitude, however small variations occur. From C8 to C12 axles, the formation rate constants k1 and k2 present a regular increase with the length of the axle that can be related to the increase in hydrophobicity of the axle with its length. Notably, the rate constant for the threading of the first CD (k1) is one order of magnitude higher than the one for the second (k2). While the threading of the first CD can occur from both sides and in both orientations, the second CD has to thread with the secondary rim facing the one of the CD on the axle to form a head to head dimer leading to reduction in possibilities and a significant decrease in the rate constant. The binding constants associated with both equilibria and the cooperativity factors were determined from the kinetic constants (Table 2). The set of constants are similar to the one obtained at pH = 5 indicating that the change in pH drastically slows the kinetics of threading without significantly affecting the thermodynamic stability of the [2] and [3]pseudorotaxanes. The sequential formation of [2] and [3]pseudorotaxanes associated with slow threading and even slower dethreading at basic pH is particularly interesting to isolate and study the conformation of the pseudo-rotaxanes in solution and in the solid state. Such kinetic stability under basic conditions thus enables rotaxane-like features for the alkyldiphosphate [3]pseudorotaxanes in terms of stability and interlocking effect. These findings highlight the elusive nature of pseudorotaxanes,15 where depending on the external conditions the phosphate groups enable fast equilibrium or become almost like real stoppers.
| Axle | 104k1 (M−1 s−1) | 107k−1 (s−1) | 104k2 (M−1 s−1) | 107k−2 (s−1) | log(K1) | log(K2) | α |
|---|---|---|---|---|---|---|---|
| C6 | 760 ± 5 | 770 ± 10 | — | — | 3.0 | — | — |
| C8 | 78 ± 4 | 11 ± 2 | 2.1 ± 0.1 | 0.25 ± 0.1 | 3.9 | 3.9 | 4.5 |
| C9 | 105 ± 22 | 37 ± 2 | 2.0 ± 0.1 | 2.5 ± 0.2 | 3.4 | 2.9 | 0.79 |
| C10 | 89 ± 4 | 19 ± 3 | 3.2 ± 0.5 | 14 ± 3 | 3.7 | 2.4 | 0.19 |
| C11 | 76 ± 4 | 3.2 ± 0.5 | 8.4 ± 0.8 | 18 ± 4 | 4.4 | 2.7 | 0.15 |
| C12 | 185 ± 12 | 6.4 ± 2.9 | 10.7 ± 0.8 | 3.8 ± 0.9 | 4.5 |
3.4 | 0.39 |
Conformational studies in the solid state
Single crystals suitable for X-ray diffraction of C8, C9, C10 and C12 [3]pseudorotaxanes were obtained by slow diffusion of DMF into a solution of [3]pseudorotaxane in water with K2CO3 (Fig. 4). The [3]pseudorotaxane with the C8 axle crystalized in the tetragonal P42212 space group with a unit cell (a = b = 23.43 Å; c = 24.28 Å; α = β = γ = 90°) and presented the expected head to head conformation for the CD dimers. The average distance between the oxygen atoms of the secondary rim of the two CDs of 2.82 Å is consistent with that of other structures of CD [3]rotaxanes16 and enables hydrogen bonding between the two CDs. The alkyl chain adopts a fully trans linear conformation with an average C–C–C angle of 121° and an average dihedral angle of 179.01°. The distance between the two phosphorous atoms of 14.07 Å enables close contacts between the terminal oxygen atoms of the phosphate groups and the hydroxyl groups of the primary rim of the CD. Indeed the O–O distances of 2.59 and 2.70 Å are consistent with the formation of stabilizing hydrogen bonds between the primary alcohol rim and the phosphate groups. Five of the six primary hydroxyl groups are involved in this hydrogen bonding that results in a closed rim conformation with the hydroxyl pointing toward the cavity (see Fig. S1†). Such an optimal fit between the head to head CD cavity and the C8 axle probably explains the strong cooperativity and stability of the [3]pseudorotaxane observed in solution. The [3]pseudorotaxane with the C9 axle was crystallized and also presented an interaction between both phosphate groups and the primary rim of the head to head CD dimer. This induces a P–P distance of 14.40 Å and a 5% compression of the chain compared to its fully extended conformation (15.22 Å). However, due to the important disorder on the inner part of the axle observed in the crystallographic structure, we will not further comment on its conformation.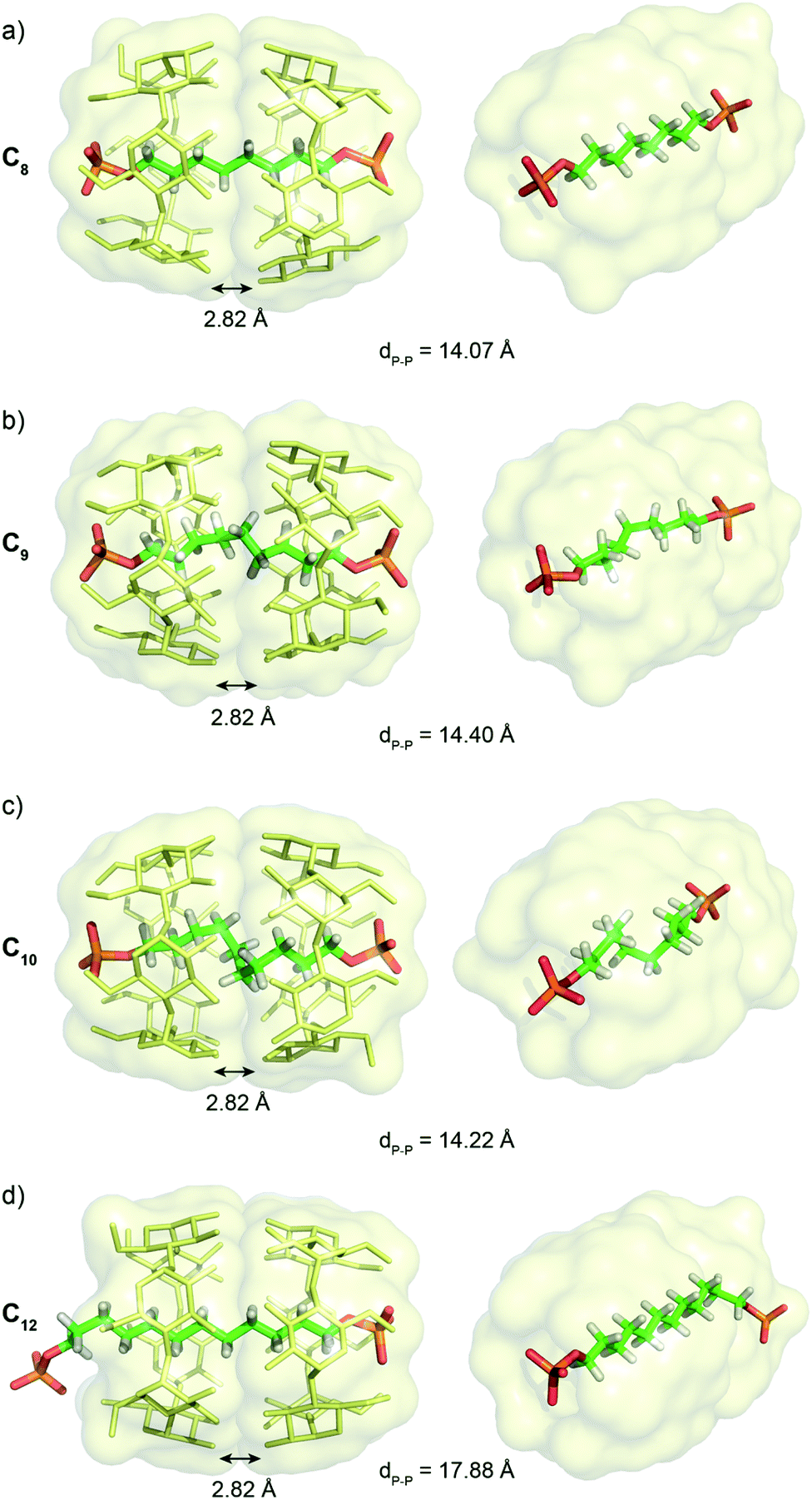 | ||
| Fig. 4 Crystallographic structures of [3]pseudorotaxanes with (a) C8, (b) C9, (c) C10 and (d) C12 axles showing 5 gauche conformations for the C10 axle resulting in a compression of the alkyl chain. Hydrogen atoms, except on the axle, and counter anions have been omitted for clarity. | ||
For the [3]pseudorotaxane with the C10 axle, the CDs adopt a similar dimeric structure with an average distance of 2.82 Å between the oxygen atoms of their secondary rims and close contact between the terminal oxygens of the phosphate groups with the primary rim (average O–O distance of 2.65 Å). This results in a similar P–P distance of 14.20 Å that forces the compression of the central alkyl chain, as the fully elongated conformation of the axle would impose a P–P distance of 16.53 Å. The resulting 15% compression is obtained by five gauche conformations from C4 to C7 with successive dihedral angles along the chain of 175.64, 65.13, 52.15, 51.58, 50.89, 64.37 and 171.27°. The stabilizing supramolecular interactions between the CD dimer and the terminal phosphate groups compensate for the energy cost necessary to twist the alkyl chain. Such chain compression, if also present in solution, might explain the strong negative cooperativity for the formation of this [3]pseudorotaxane observed in the titration experiments. Remarkably, only one helicity of the chain is present in the crystal structure as the pseudorotaxane crystalizes in the non-centrosymmetric P43212 space group. The intrinsic chirality of the cyclodextrin head to head dimer favors the M helicity of the compressed chain. Such stereoselective folding was observed in self-assembled achiral capsules only by using a chiral and optically enriched guest.17 This underlines the potential interest of the head to head CD [3]rotaxane architecture. Finally, with the C12 axle, only one phosphate group interacts with the primary rim of the CD dimers and the alkyl chain adopts an extended conformation with the second phosphate group interacting intermolecularly with hydroxyl groups of the secondary rim on another CD dimer in the crystal packing (see Fig. S1†). Thus, the C12 axle is too long to be compressed inside the α-CD dimeric structure and protrudes from the CD dimer. This threading disrupts the hydrogen bond network on the primary rim with all the C–H6 pointing toward the alkyl chain and the hydroxyl outward of the cavity.
Thus, the crystallographic structures clarify the binding constants obtained in solution, with the best fit for the C8 axle to maximize the interactions of the primary rim with the two terminal phosphate groups and to enable a linear all-trans conformation for the alkyl chain. This can explain the strong positive cooperativity to form a [3]pseudorotaxane with this axle. Such interactions between the phosphate groups and CD primary rim enable a compression of the alkyl chain with the C10 axle that should be responsible for the negative cooperativity for the [3]pseudorotaxane in this case.
Conformational studies in solution
2D NMR experiments were performed in order to evaluate the chain conformation of the rotaxanes in solution. For the C8 axle, NOE correlations are observed only between hydrogen atoms on carbon Ci and Ci+2 (i.e. Ha/Hc and Hb/Hd) as expected for an elongated zig-zag conformation of the chain (see Fig. 5 and S15†). HOESY 1H/31P experiments showed correlations between the phosphorous atoms on the axle and the H6 and H5 protons of the CD, indicating a close proximity between the primary rim of both CDs and the terminal phosphate groups as in the crystallographic structure. Upon increasing the axle length, additional Ci/Ci+3 and Ci/Ci+4 such as Ha/Hd/e and Hb/He/f correlations are observed for C9, C10 and C11 axles. Such NOE correlations are indicative of a compressed chain with gauche conformations occurring in solution as already observed in the crystallographic structure of C10[3]rot. In addition, the spatial proximity between both terminal phosphate groups and the primary rim of the CDs is confirmed for C9 and C10 axles by the heteronuclear NOE correlations between P and H6 and H5 protons.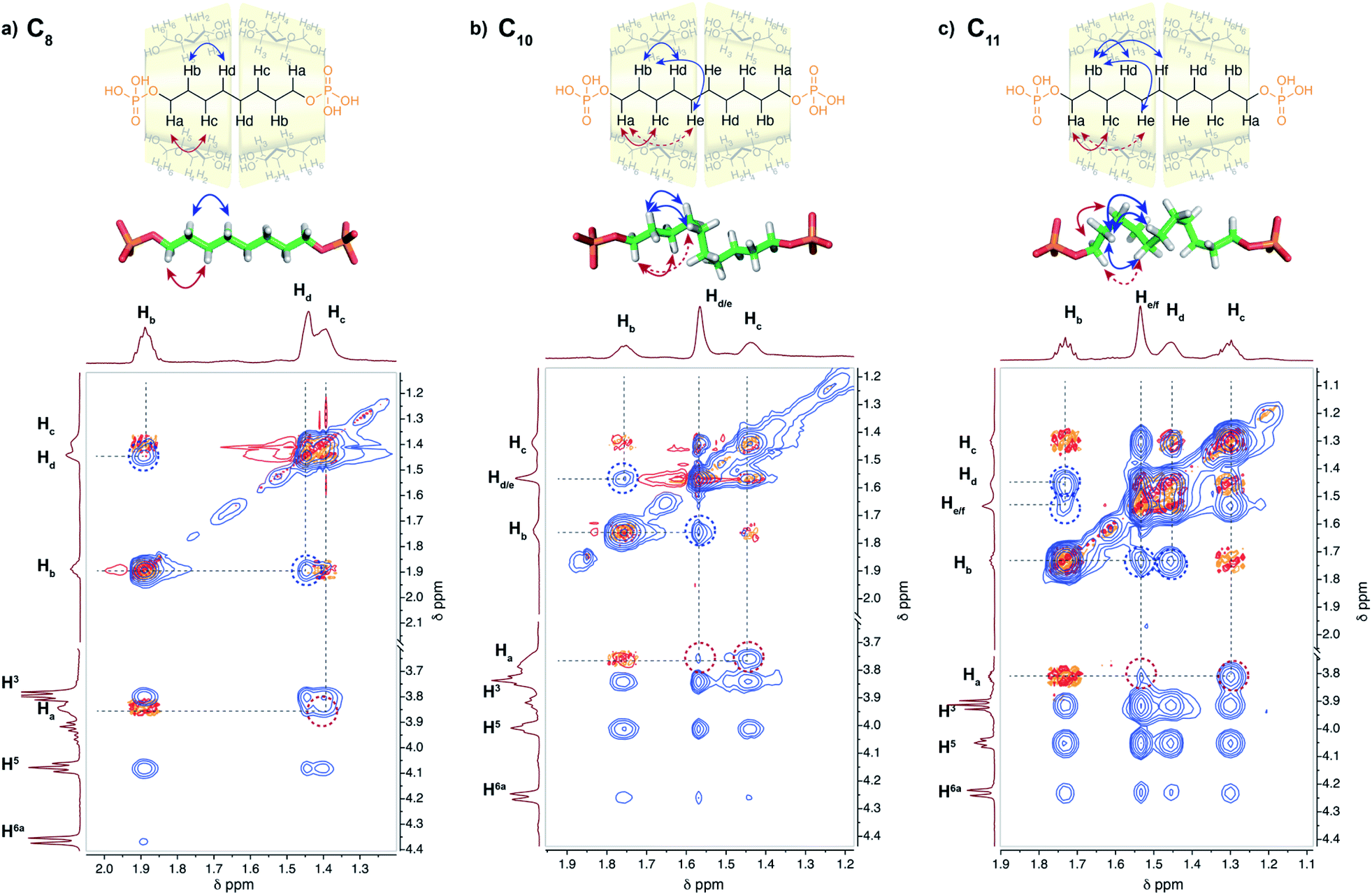 | ||
| Fig. 5 Stack between the COSY (red) and NOESY (blue) NMR spectra (600 MHz, D2O, pH = 10) of [3]pseudorotaxane with (a) C8, (b) C10, and (c) C11 axles and optimized geometries of axles in the corresponding [3]rot. Long range NOE correlations Ha–Hd/e and Hb–Hd/e and Ha–He/f and Hb–He/f for C10 and C11 axles, respectively, are observed in agreement with the conformation of the axles. | ||
Molecular modeling was performed with ORCA18 at the B3LYP-D3 level and def2-SV(P) basis set for the [3]pseudorotaxanes with C8–C11 axles (see Fig. S28†). The DFT geometry optimizations using a dispersion corrected hybrid functional showed a progressive compression of the alkyl chain caused by the hydrogen bonding interactions between the terminal phosphate groups and the hydroxyl groups of the primary rim of the CDs and the head-to-head CD dimer that maintains P–P distances between 13.8 and 14.0 Å. In addition, the optimized coiled conformation of the axles present H–H distances in agreement with the observed NOE correlations (highlighted in Fig. 5 and S15†).
In contrast, the C12 axle presents NOE correlations only up to 2 carbons and no HOESY correlation between the P and the CD protons (see Fig. S15 and S25†). This is characteristic of an elongated chain and the absence of compression with a fast shuttling of the CD dimer along the axle. Thus the CD [3]pseudorotaxanes enable a remarkable compression of alkyl chains from 9 up to 11 carbons in solution, thanks to the hydrogen bonding networks between the secondary rims of the two CDs that enable a dimeric structure and the interactions between the phosphate end groups and the primary rims of the CDs. Such a compression mechanism is conceptually different from the one observed with self-assembled capsules that exist in the absence of a guest. Here the presence of the guest is necessary to form the interlocked molecule in a three-component self-assembly process. The compression of the alkyl chain results from a delicate balance between destabilizing gauche conformations and stabilizing hydrophobic effect and hydrogen bonding between the CD head to head assembly and with the terminal groups of the axles.
Conclusion
In conclusion, we have demonstrated the formation of [2] and [3]pseudorotaxanes between α-CD and alkyldiphosphate axles depending on their lengths. The protonation state of the phosphate groups allows the control of the kinetics of threading which is slowed down at basic pH when the phosphate groups are doubly deprotonated. Such pH-dependent control enabled the preparation of [3]pseudorotaxanes at acidic pH and their trapping and isolation by switching to basic conditions. Studies of the chain conformations were then possible in aqueous solution by NMR. While the C8 axle presents a fully extended conformation in the [3]pseudorotaxane, the CD dimer induces an unexpected compression of the alkyl chains from C9 to C11 axles. A combination of the hydrophobic effect and hydrogen bonding interactions between the phosphate end groups and the primary rim hydroxyl groups of the CD dimer stabilizes the compressed conformation of the chain. While alkyl chain compression has been observed in molecular capsules, such an effect has not been reported in solution for pseudorotaxane architectures. Such a compression mechanism is significantly different from aliphatic guests inside self-assembled capsules that maximize their packing coefficient without preferred orientation inside the cavity. Here, the head to head CD dimer, only obtained by the three-component self-assembly process forming the [3]pseudorotaxane, allows specific interactions between both terminal groups of the axle and CD's primary rims and thus imposes the orientation of the chain and its end to end distance like in a clamp. Extension by one carbon to a C12 axle results in an extended conformation of the chain and its protrusion from the CD dimer, highlighting the delicate balance of interactions needed to stabilize high energy conformations. Our approach thus demonstrates the interest of interlocked architectures to stabilize high energy conformations with fine control.Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI files,† and also from the corresponding authors upon reasonable request.Author contributions
J. S, H. V. carried out the experimental work, analysis and interpretation of the results. L. B. performed the ITC measurements and their analysis. L.-M. C. resolved the crystallographic structures. M. S. supervised the project and participated to the writing and editing of the manuscript. G. V. conceived and designed the experiments, supervised the project, supported the analysis and interpretation of the results and wrote the original draft. B. H. conceived and supervised the project, secured the funding, supported the analysis and interpretation of the results, participated to the writing of the manuscript. All authors discussed the results and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work benefited from the support of the French National Research Agency (ANR) for the project Rotaximage (16-CE09-0018) and the LabEx MiChem programme under reference ANR-11-IDEX-0004-02.Notes and references
- (a) D. Ajami and J. Rebek, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed; (b) J. J. Rebek, Chem. Commun., 2007, 2777–2789 RSC.
- Y. Yu, J.-M. Yang and J. Rebek Jr, Chem, 2020, 6, 1265–1274 CAS.
- (a) G. Zanotti, G. Scapin, P. Spadon, J. H. Veerkamp and J. C. Sacchettini, J. Biol. Chem., 1992, 267, 18541–18550 CrossRef CAS PubMed; (b) G. K. Balendiran, F. Schnütgen, G. Scapin, T. Börchers, N. Xhong, K. Lim, R. Godbout, F. Spener and J. C. Sacchettini, J. Biol. Chem., 2000, 275, 27045–27054 CrossRef CAS PubMed; (c) G. W. Han, J. Y. Lee, H. K. Song, C. Chang, K. Min, J. Moon, D. H. Shin, M. L. Kopka, M. R. Sawaya, H. S. Yuan, T. D. Kim, J. Choe, D. Lim, H. J. Moon and S. W. Suh, J. Mol. Biol., 2001, 308, 263–278 CrossRef CAS PubMed.
- (a) L. Trembleau and J. Rebek, Science, 2003, 301, 1219–1220 CrossRef CAS PubMed; (b) S. Mosca, D. Ajami and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11181 CrossRef CAS PubMed.
- (a) A. Scarso, L. Trembleau and J. Rebek, Angew. Chem., Int. Ed., 2003, 42, 5499–5502 CrossRef CAS PubMed; (b) A. Scarso, L. Trembleau and J. Rebek, J. Am. Chem. Soc., 2004, 126, 13512–13518 CrossRef CAS PubMed; (c) D. Ajami and J. Rebek, Nat. Chem., 2009, 1, 87–90 CrossRef CAS PubMed; (d) A. Asadi, D. Ajami and J. Rebek, J. Am. Chem. Soc., 2011, 133, 10682–10684 CrossRef CAS PubMed; (e) D. Ajami and J. Rebek, J. Am. Chem. Soc., 2006, 128, 15038–15039 CrossRef CAS PubMed.
- (a) C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2006, 128, 16498–16499 CrossRef CAS PubMed; (b) C. L. D. Gibb and B. C. Gibb, Chem. Commun., 2007, 1635–1637 RSC; (c) S. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, J. Am. Chem. Soc., 2013, 135, 4314–4324 CrossRef CAS PubMed; (d) H. Gan and B. C. Gibb, Chem. Commun., 2012, 48, 1656–1658 RSC; (e) S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709–3716 RSC.
- (a) D. Dumitrescu, Y.-M. Legrand, E. Petit, A. van der Lee and M. Barboiu, Chem. Sci., 2015, 6, 2079–2086 RSC; (b) D. Dumitrescu, Y.-M. Legrand, E. Petit, A. van der Lee and M. Barboiu, Chem. Commun., 2014, 50, 14086–14088 RSC; (c) W.-X. Feng, A. van der Lee, Y.-M. Legrand, E. Petit, D. Dumitrescu, C.-Y. Su and M. Barboiu, Org. Lett., 2016, 18, 5556–5559 CrossRef CAS PubMed; (d) D. G. Dumitrescu, W.-X. Feng, Y.-M. Legrand, E. Petit, A. van der Lee and M. Barboiu, Eur. J. Org. Chem., 2017, 2017, 3282–3287 CrossRef CAS.
- (a) Y. Akae, Y. Koyama, S. Kuwata and T. Takata, Chem.–Eur. J., 2014, 20, 17132–17136 CrossRef CAS PubMed; (b) Y. Akae, H. Okamura, Y. Koyama, T. Arai and T. Takata, Org. Lett., 2012, 14, 2226–2229 CrossRef CAS PubMed.
- (a) S. Makedonopoulou and I. M. Mavridis, Carbohydr. Res., 2001, 335, 213–220 CrossRef CAS PubMed; (b) S. Makedonopoulou and I. M. Mavridis, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 322–331 CrossRef PubMed.
- T. Bojinova, H. Gornitzka, N. Lauth-de Viguerie and I. Rico-Lattes, Carbohydr. Res., 2003, 338, 781–785 CrossRef CAS PubMed.
- (a) E. Jeamet, J. Septavaux, A. Héloin, M. Donnier-Maréchal, M. Dumartin, B. Ourri, P. Mandal, I. Huc, E. Bignon, E. Dumont, C. Morell, J.-P. Francoia, F. Perret, L. Vial and J. Leclaire, Chem. Sci., 2019, 10, 277–283 RSC; (b) S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855–8864 CrossRef CAS PubMed.
- (a) J. W. Fredy, J. Scelle, A. Guenet, E. Morel, S. Adam de Beaumais, M. Ménand, V. Marvaud, C. S. Bonnet, E. Tóth, M. Sollogoub, G. Vives and B. Hasenknopf, Chem.–Eur. J., 2014, 20, 10915–10920 CrossRef CAS PubMed; (b) J. W. Fredy, J. Scelle, G. Ramniceanu, B.-T. Doan, C. S. Bonnet, É. Tóth, M. Ménand, M. Sollogoub, G. Vives and B. Hasenknopf, Org. Lett., 2017, 19, 1136–1139 CrossRef CAS PubMed; (c) S. Cherraben, J. Scelle, B. Hasenknopf, G. Vives and M. Sollogoub, Org. Lett., 2021, 23, 7938–7942 CrossRef CAS PubMed.
- (a) M. Bastos, L. E. Briggner, I. Shehatta and I. Wadsö, J. Chem. Thermodyn., 1990, 22, 1181–1190 CrossRef CAS; (b) A. J. M. Valente and O. Söderman, Adv. Colloid Interface Sci., 2014, 205, 156–176 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- (a) P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS; (b) J. Groppi, L. Casimiro, M. Canton, S. Corra, M. Jafari-Nasab, G. Tabacchi, L. Cavallo, M. Baroncini, S. Silvi, E. Fois and A. Credi, Angew. Chem., Int. Ed., 2020, 59, 14825–14834 CrossRef CAS PubMed.
- Y. Akae, Y. Koyama, H. Sogawa, Y. Hayashi, S. Kawauchi, S. Kuwata and T. Takata, Chem.–Eur. J., 2016, 22, 5335–5341 CrossRef CAS PubMed.
- C. Siering, J. Toräng, H. Kruse, S. Grimme and S. R. Waldvogel, Chem. Commun., 2010, 46, 1625–1627 RSC.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2114960–2114963. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05697b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |