DOI:
10.1039/D1SC06315D
(Review Article)
Chem. Sci., 2022,
13, 2202-2217
Redox dyshomeostasis strategy for tumor therapy based on nanomaterials chemistry
Received
15th November 2021
, Accepted 23rd December 2021
First published on 12th January 2022
Abstract
Redox homeostasis, as an innate cellular defense mechanism, not only contributes to malignant transformation and metastasis of tumors, but also seriously restricts reactive oxygen species (ROS)-mediated tumor therapies, such as chemotherapy, radiotherapy, photodynamic therapy (PDT), and chemodynamic therapy (CDT). Therefore, the development of the redox dyshomeostasis (RDH) strategy based on nanomaterials chemistry is of great significance for developing highly efficient tumor therapy. This review will firstly introduce the basic definition and function of cellular redox homeostasis and RDH. Subsequently, the current representative progress of the nanomaterial-based RDH strategy for tumor therapy is evaluated, summarized and discussed. This strategy can be categorized into three groups: (1) regulation of oxidizing species; (2) regulation of reducing species and (3) regulation of both of them. Furthermore, the current limitations and potential future directions for this field will be briefly discussed. We expect that this review could attract positive attention in the chemistry, materials science, and biomedicine fields and further promote their interdisciplinary integration.
 Yelin Wu | Yelin Wu received his PhD degree from East China Normal University. He is now a professor at Tongji University Cancer Center, Shanghai Tenth People's Hospital. His research interests focus on the design and application of nanomaterials for cancer and wound healing. |
 Yanli Li | Yanli Li received her PhD degree in 2021 from the East China Normal University (ECNU), under the supervision of Prof. Wenbo Bu. She is now an associate professor at Guangzhou Medical University. Her research interests focus on the design and synthesis of multifunctional nanomaterials for biomedical applications. |
 Guanglei Lv | Guanglei Lv received his PhD degree from Fudan University under the supervision of Prof. Tao Yi. Then, he worked as a lecturer at Zhejiang Normal University until 2020. He is now working as a postdoctoral researcher in the group of Prof. Wenbo Bu at Fudan University. His current research interests focus on the design and synthesis of organic/inorganic materials and their bio-applications. |
 Wenbo Bu | Wenbo Bu received his PhD degree from Nanjing University of Technology. He is a full professor at Fudan University and an adjunct professor at Chinese Academy of Sciences (SICCAS). His current research interests include the design and synthesis of multifunctional nanomaterials for cancer imaging and therapeutic applications. |
1. Introduction
Cellular redox homeostasis, which refers to the dynamic balance between intracellular oxidizing species and reducing species, plays an important role in maintaining normal physiological processes such as cell growth, metabolism, differentiation, aging and apoptosis.1 Oxidizing species are a class of chemical substances that tend to obtain electrons from reducing agents in redox reactions, including reactive oxygen species (ROS),2 reactive nitrogen species (RNS),3 and so on. A moderate concentration of intercellular oxidizing species such as ROS is required for various biological processes including cell signaling, biosynthetic processes, and host defense.4 However, excessive amounts of ROS can cause oxidative damage to lipids, proteins, and DNA, which leads to oxidative stress.5 Under this persistent intrinsic oxidative stress, reducing species, a class of oxidizing species-scavenging substances, are activated to protect cells from ROS-induced oxidative damage. Therefore, it is crucial to keep cellular redox homeostasis for normal cell survival and growth.
Compared with normal cells, numerous types of cancer cells tend to elevate levels of oxidizing species owing to the genetic instability, increased chronic inflammation, and aberrant metabolism.6 Oxidizing species in turn induce gene mutation and inflammation, leading to the generation of more oxidizing species.7 This cycle ultimately results in persistent oxidative stress. Cancer cells that survive oxidative stress mobilize a set of adaptive mechanisms through enhancing antioxidant capacity.8,9 This adaptive system contributes to malignant transformation, metastasis, and resistance of tumor cells to ROS-mediated tumor therapies such as part of chemotherapeutic drugs, radiotherapy (RT), photodynamic therapy (PDT), and chemodynamic therapy (CDT).4,10–12 Therefore, it is crucial to disrupt tumor redox homeostasis to make tumor cells sensitive to current tumor treatment modalities.
The redox dyshomeostasis (RDH) strategy for tumor therapy is first proposed by our group, aiming to make tumor cells more sensitive to different tumor therapy patterns13 through disrupting the cellular redox homeostasis using nanomaterials (Fig. 1). Unlike traditional chemotherapeutic drugs, nanomaterials have their special characteristics such as passive targeting, drug delivery, and tendency of losing or gaining electrons. Therefore, based on these characteristics, nanomaterials could be designed to modulate the redox homeostasis of tumor cells by delivering and reacting with oxidizing or reducing species, under endogenous stimuli such as low pH and hypoxia, or external stimuli such as light and ultrasound.14 Here, this review summarizes the most recent progress of the nanomaterial-based RDH strategy for novel and efficient tumor therapies, mainly including the following regulations to realize RDH: (1) regulation of oxidizing species; (2) regulation of reducing species, and (3) regulation of both oxidizing species and reducing species.
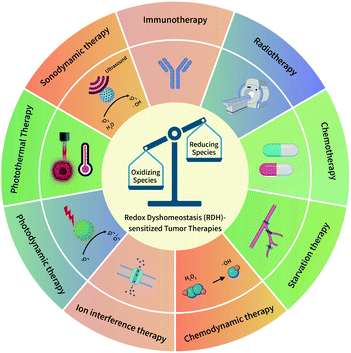 |
| | Fig. 1 A schematic overview of the redox dyshomeostasis (RDH) strategy for tumor therapy. | |
2. Regulation of oxidizing species to realize RDH
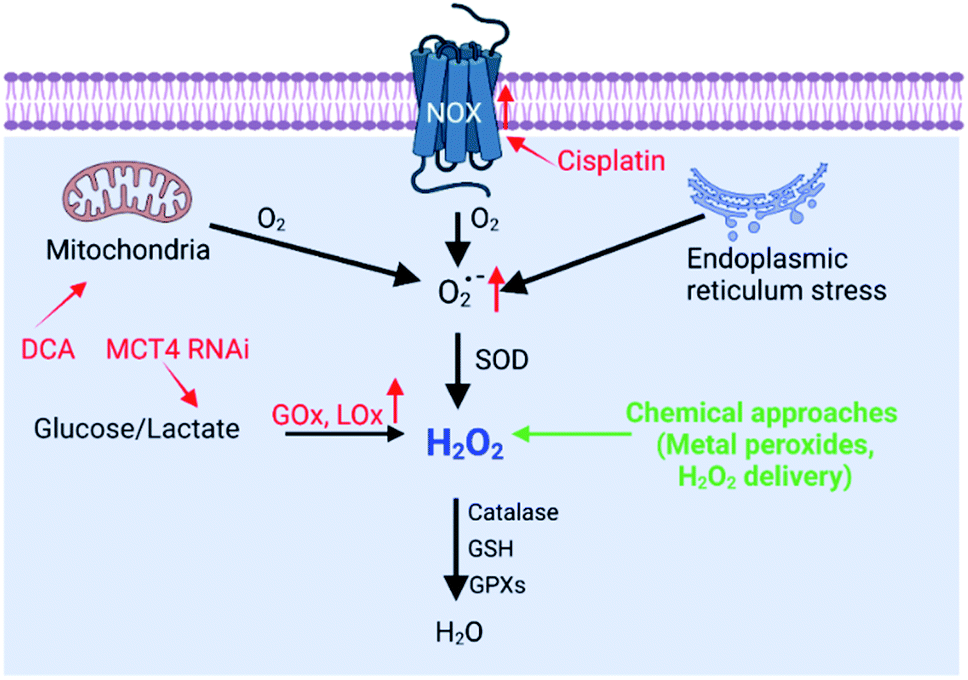
Oxidizing species mainly include ROS and RNS. ROS are oxygen-containing molecules with higher chemical reactivity including superoxide anions (O2˙−), hydrogen peroxide (H2O2), and hydroxyl radicals (˙OH). RNS are nitrogen-containing oxidants, mainly including nitric oxide (NO) and peroxynitrite (ONOO−). Among these oxidizing species, H2O2 is a significantly stable and diffusible form of ROS.9 Furthermore, as an intermediate product of mammalian oxygen metabolism, H2O2 is easily transformed into various active species in cells.6 Therefore, H2O2 is one of the most important oxidizing species, which is capable of being regulated to disrupt redox homeostasis. In the past few years, a series of nanomaterials have been constructed to increase H2O2 production, realizing the RDH strategy to enhance the sensitivity of tumor therapy. On the basis of working principles, increasing the H2O2 level can be realized through two distinctive strategies: (1) biosynthesis approaches; (2) chemical approaches.
2.1 Increasing the H2O2 level in tumor cells via biosynthesis approaches
H2O2 can be generated in tumor cells through biosynthesis approaches.4,6 For example, electron leakage from the mitochondrial respiratory chain or endoplasmic reticulum (ER) stress can promote H2O2 production. In addition, the production of H2O2 is controlled by a series of biosynthesis-related enzymes, such as NADPH oxidase (NOX), glucose oxidase (GOx) and lactate oxidase (LOx) (Fig. 2). Therefore, enzymes, the function of mitochondria and ER, and the substrate (O2 and O2˙−) have been regulated using a wide range of nanomaterials to increase the H2O2 level, realizing RDH to enhance the sensitivity of tumor therapy. H2O2 production is driven by both enzyme-dependent and -independent strategies.
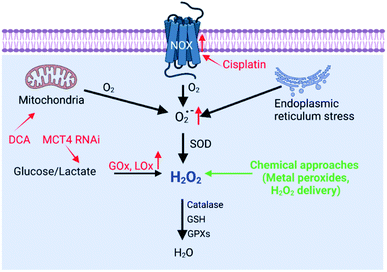 |
| | Fig. 2 Biosynthesis and strategies of increasing H2O2via biosynthesis approaches and chemical approaches (red font represents biosynthesis approaches and green font represents chemical approaches). | |
2.1.1 Enzyme-dependent strategies.
NOX, the main H2O2-generating enzymes in eukaryotic cells, are closely associated with redox homeostasis.15 It has been reported that cisplatin induces the production of ROS via NADPH oxidase activation in human prostate cancer cells.16 Jun Lin et al. constructed self-sacrificing iron-oxide nanoparticles with cisplatin(IV) drug FePt-NP2. As shown in Fig. 3A, once the nanoparticles entered the cells, cisplatin was released to activate NOX, which catalyzed the formation of superoxide and H2O2 from O2. The accumulation of H2O2 disrupted redox homeostasis, leading to RDH in tumor cells. The RDH enhanced Fe2+/Fe3+-induced Fenton reaction resulted in tumor cell death.17 GOx and LOx are the main producers of H2O2 in prokaryotic cells. Among them, GOx can catalyze glucose to generate glucose acid and H2O2, and LOx can catalyze lactic acid to produce H2O2. Therefore, GOx and LOx are promising enzymes to increase the H2O2 level, achieving RDH. Jianlin Shi et al. loaded GOx and ultra-small Fe3O4 into macroporous silica nanoparticles (MSN) to prepare a biodegradable and sequentially functioning nanocatalyst GOx-Fe3O4@DMSN. GOx catalyzed glucose into abundant H2O2, resulting in RDH in tumors, which promoted Fe3O4 NP-induced Fenton-like reactions.18 Similarly, Changfeng Wu constructed a nanoparticle platform by covalent conjugation of GOx to small polymer dots. This nanosystem could be persistently immobilized into a tumor and catalyze glucose oxidation to generate H2O2, leading to RDH, which was beneficial for phototherapy (Fig. 3B).19 Therefore, these examples indicate that RDH in tumor cells enhances the sensitivity of ˙OH induced by the Fenton reaction or phototherapy. Besides GOx, LOx-catalyzed generation of H2O2 has been used to disrupt redox homeostasis. Chengzhong Yu et al. reported a nanoplatform, where LOx and a chemotherapy drug AQ4N were loaded into openwork@dendritic mesoporous silica nanoparticles.20 LOx catalyzed lactate depletion and H2O2 production. The former downregulated VEGF expression and inhibited angiogenesis, while the latter induced RDH promoting AQN4-induced chemotherapy (Fig. 3C).
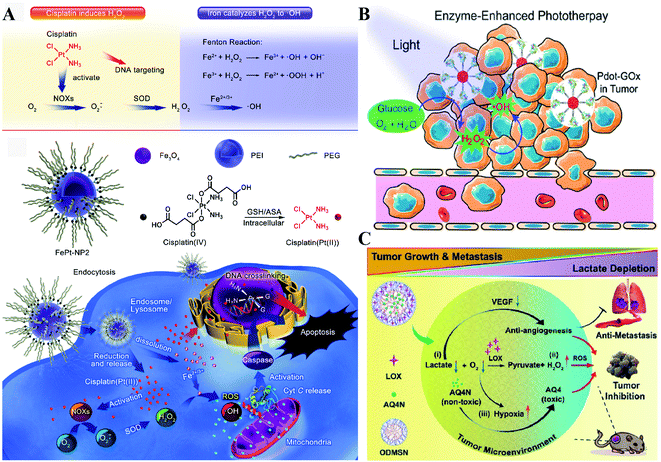 |
| | Fig. 3 Schematic illustrations of enzyme-dependent strategies to realize RDH. (A) Cisplatin from FePt-NP2 activated NOX to produce H2O2 and then induced RDH, which sensitized Fe2+/Fe3+-induced cell death. Adapted from ref. 17 with permission. Copyright 2017 American Chemical Society. (B) GOx from Pdot-GOx catalyzed glucose oxidation to generate H2O2 realizing RDH which enhanced phototherapy. Adapted from ref. 19 with permission. Copyright 2017 American Chemical Society. (C) LOx from ODMSN catalyzed lactate depletion and H2O2 production. The latter induced RDH promoting AQN4-induced chemotherapy. Adapted from ref. 20 with permission. Copyright 2020 Wiley-VCH. | |
2.1.2 Enzyme-independent strategies.
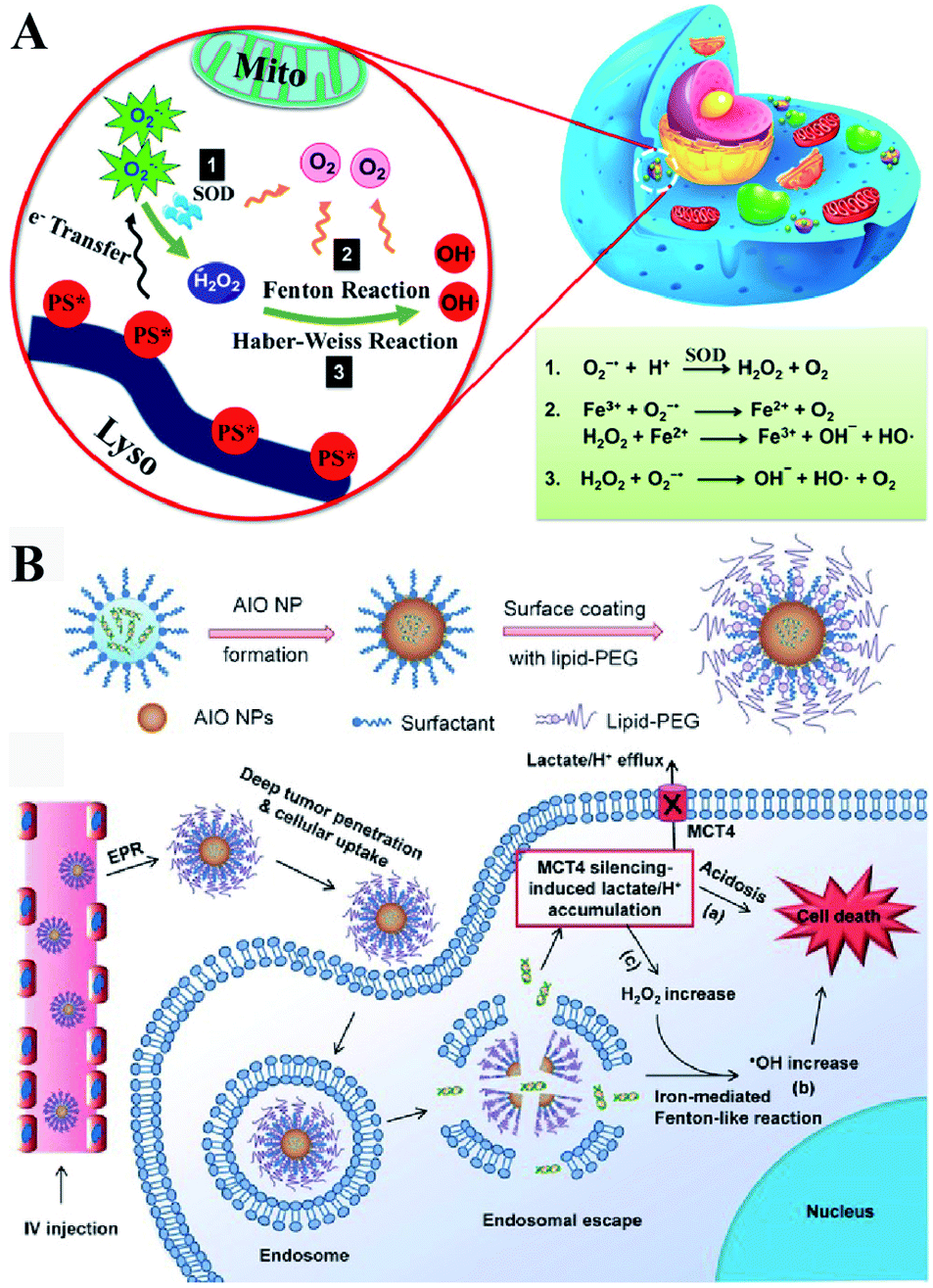
Besides enzyme-regulated H2O2 production, the biosynthesis of H2O2 can be regulated by the increase of substrates and enhanced metabolic processes. O2 and O2˙− are the indispensable substrates for H2O2 biosynthesis. O2˙− can be produced by photosensitizers upon light excitation.21 Xiaojun Peng et al. developed a near-infrared light-triggered molecular superoxide radical generator (ENBS-B).22 ENBS-B can produce a considerable amount of O2˙−, which can be transformed to H2O2 by SOD-mediated reactions. The increased H2O2 induced RDH, promoting Fenton reaction and Haber–Weiss reaction-induced cancer cell apoptosis (Fig. 4A). Similarly, Haifeng Dong et al. synthesized metal–organic framework (MOF) nanosheets (DBBC-UiO), which were composed of 5,15-di(p-benzoato)bacteriochlorin (H2DBBC) as blocks and heavy Hf6(μ3-O)4(μ3-OH)4 clusters as centers.23 This system could produce considerable O2˙− under a hypoxic environment with 750 nm irradiation. The produced O2˙− could not only kill tumor cells but transform to H2O2via SOD-mediated reactions. The robust H2O2 induced the imbalance of redox homeostasis to realize RDH, which ultimately improved the PDT efficacy.
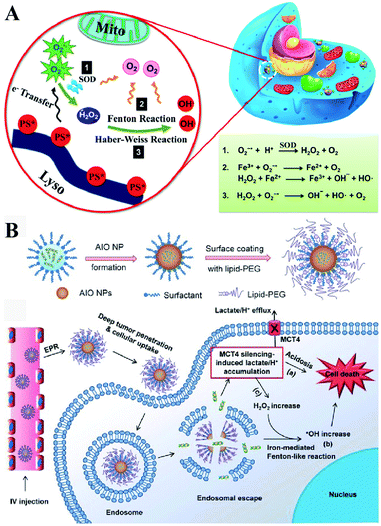 |
| | Fig. 4 Schematic illustrations of enzyme-independent strategies to induce RDH. (A) Near-infrared light triggered ENBS-B to produce a considerable amount of O2˙− which is transformed to H2O2 leading to RDH, promoting Fenton reaction and Haber–Weiss reaction-induced cell apoptosis. Adapted from ref. 22 with permission. Copyright 2018 American Chemical Society. (B) Silencing of MCT4 induced the accumulation of H2O2, which realized RDH, exacerbating Fenton-like reaction-mediated tumor treatment. Adapted from ref. 26 with permission. Copyright 2018 Wiley-VCH. | |
Altering lactic acid and energy metabolism can also trigger the production of H2O2.24 The high rate of glycolysis in the tumor is associated with the excessive generation of lactic acid, which leads to the upregulation of MCT4,25 an efflux of lactate/H+ for maintaining a stable intracellular pH and acidic tumor microenvironment. It has been reported that MCT4 silencing could block the intracellular lactate efflux, resulting in more H2O2 production. Jinjun Shi et al. developed a unique amorphous iron oxide (AIO) MCT4 RNAi NP platform. Silencing of MCT4 induced the accumulation of H2O2, leading to RDH, which exacerbated the Fenton-like reaction for tumor treatment (Fig. 4B).26 In addition, Xinghai Ning et al. used dichloroacetic acid (DCA) to increase mitochondrial aerobic oxidation for the production of H2O2.27 They constructed a liposomal formulation MD@Lip containing DCA and MOF-Fe2+. DCA promoted the production of H2O2 leading to RDH, which sensitized the MOF-Fe2+-associated antitumor strategy.
2.2 Increasing the H2O2 level in tumor cells via chemical approaches
Besides regulation of H2O2 production via biosynthesis strategies, the most straightforward way to generate H2O2 in tumor cells is to directly deliver or produce H2O2 at tumor sites via chemical approaches. Compared with biosynthesis, chemical approaches can rapidly increase the H2O2 content in a short time, leading to RDH. By ingeniously designing functional nanomaterials, H2O2 can be produced in two ways: (1) direct delivery and (2) chemical reactions.
2.2.1 Increasing the H2O2 level in tumor cells by direct delivery.
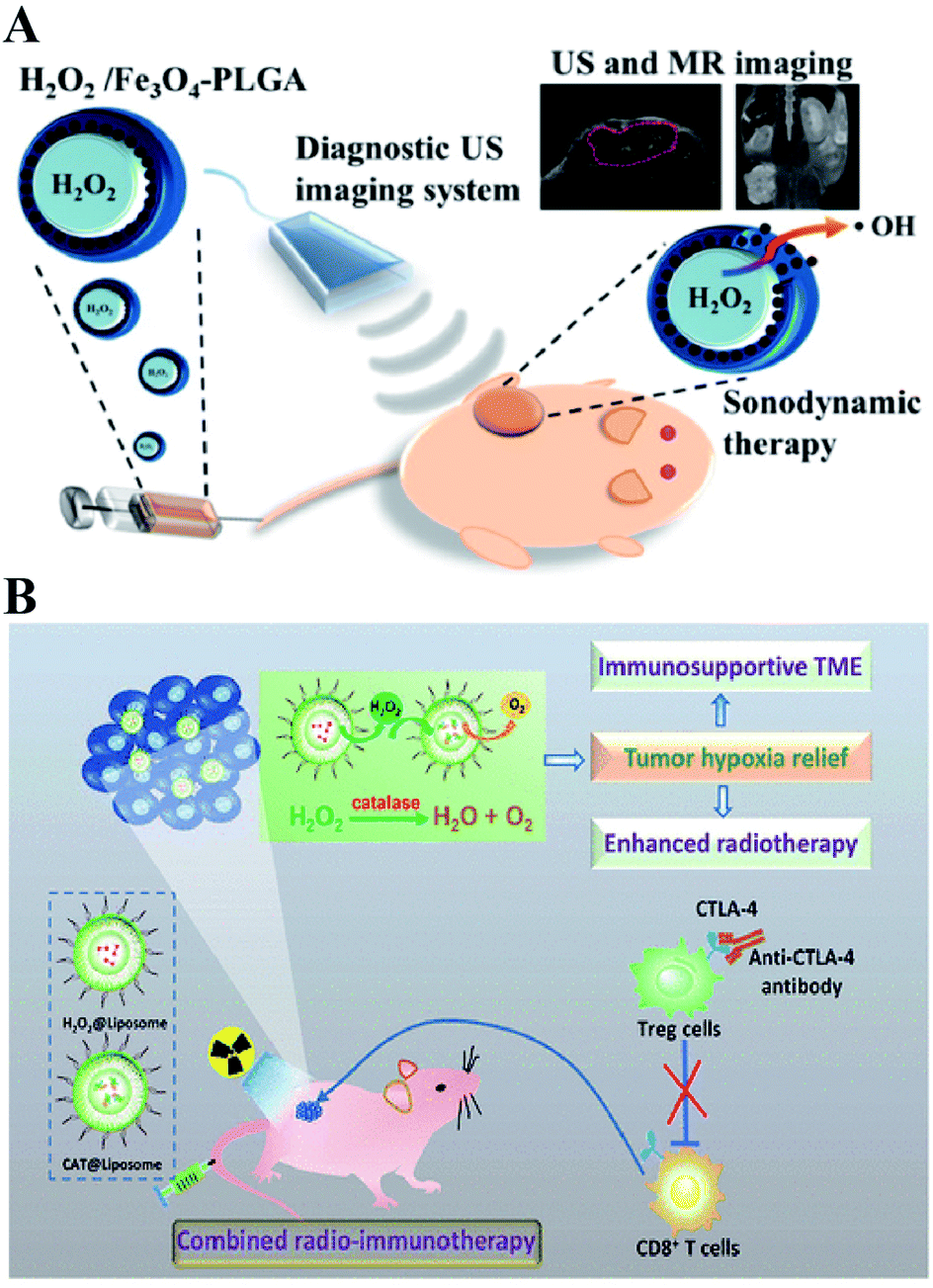
Directly delivering H2O2 into tumor cells is the most straightforward way to increase the H2O2 content, resulting in RDH. Chen-Sheng Yeh et al. encapsulated H2O2 into the Fe3O4-PLGA polymersome.28 On exposure to ultrasound, the polymersome can be easily disrupted to release H2O2, causing RDH, which enhanced the Fe3O4-induced Fenton reaction (Fig. 5A). Similarly, in order to supply enough oxygen for enhanced radio-immunotherapy of cancer, Zhuang Liu et al. separately loaded H2O2 and catalase (CAT) into stealthy liposomes.29 The CAT@liposome was preinjected into tumor sites followed by H2O2@liposome injection. Therefore, the sustainably released H2O2 of H2O2@liposome could be decomposed by the catalase from CAT@liposome, resulting in a long-lasting oxygen release and RDH, which further enhanced the therapeutic effects of radiotherapy and CTLA4 (cytotoxic T lymphocyte-associated antigen 4) antibody-mediated immunotherapy (Fig. 5B).
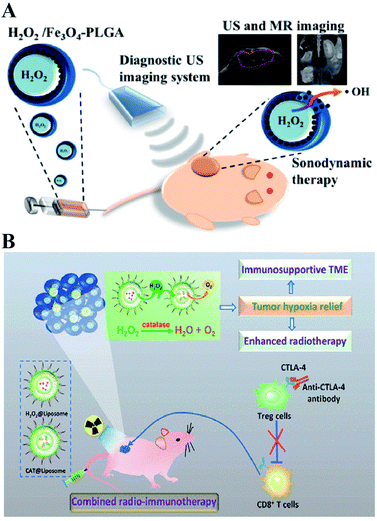 |
| | Fig. 5 Schematic illustrations of increasing the H2O2 level in tumor cells by direct delivery. (A) H2O2/Fe3O4-PLGA polymersome-induced RDH. On exposure to ultrasound, the polymersome can be easily disrupted to release H2O2, causing RDH which enhanced the Fe3O4-induced Fenton reaction. Adapted from ref. 28 with permission. Copyright 2016 American Chemical Society. (B) H2O2@liposome and CAT@liposome-induced RDH. The sustainably released H2O2 of H2O2@liposome could be decomposed by catalase from CAT@liposome, resulting in a long-lasting oxygen release and RDH, which enhanced the radiotherapy and immunotherapy. Adapted from ref. 28 with permission. Copyright 2018 American Chemical Society. | |
2.2.2 Increasing the H2O2 level in tumor cells by chemical reactions.
Direct delivery of H2O2 into tumor sites may have some disadvantages such as poor stability and uncontrollable action. Undoubtedly, producing H2O2in situ is a promising alternative. Metal peroxides are typically composed of metal ions and peroxo groups, which can react with H2O to produce H2O2 under acid conditions.30 Therefore, metal peroxides are capable of specifically responding to the acidic tumor microenvironment to produce a large number of H2O2 in tumor sites, which could disrupt the redox homeostasis of tumors and ultimately enhance the sensitivity of tumor cells to other treatments. For example, our group constructed a biodegradable nanoprodrug of transferrin-modified MgO2 nanosheets. In response to the acidic and low catalase activity of the tumor microenvironment, MgO2 produced abundant H2O2 to disrupt redox homeostasis, ultimately enhancing transferrin-mediated CDT (Fig. 6A).31 Similarly, Xiaoyuan Chen et al. reported a method of fabricating copper peroxide nanodots (CP).32 The CP nanodots decomposed under the acidic environment of endo/lysosomes, allowing the simultaneous release of Fenton catalytic Cu2+ and H2O2. The increase of H2O2 induced RDH, sensitizing Cu2+-induced CDT. Altering the metal ion content balance could induce a series of intracellular responses, even cell death. This approach has been used to treat tumors and was defined as ion interference therapy (IIT).33 For example, calcium ion (Ca2+) accumulation is a cause of damage or cell death in various cell types. With this in mind, we constructed pH-sensitive sodium-hyaluronate-modified calcium peroxide nanoparticles (SH-CaO2 NPs) to produce abnormal H2O2 accumulation and calcium overload.33 The increased level of H2O2 disturbed cellular redox homeostasis and enhanced calcium overload-induced cell death (Fig. 6B). Taken together, these strategies combined RDH with CDT and IIT, opening a new door for further material design and corresponding tumor treatment.
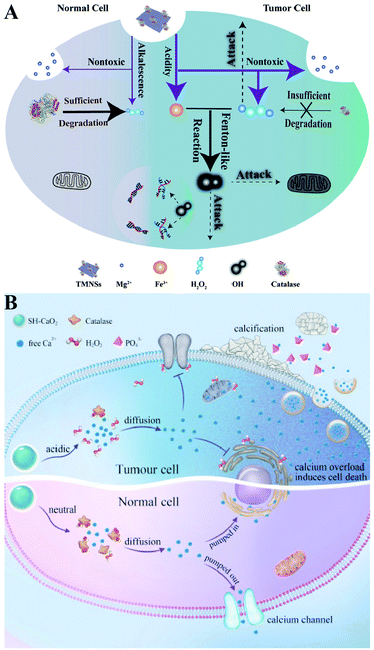 |
| | Fig. 6 Schematic illustrations of increasing the H2O2 level by chemical reactions. (A) MgO2-induced RDH. MgO2 nanosheets produced abundant H2O2 to disrupt redox homeostasis enhancing transferrin-mediated CDT. Adapted from ref. 31 with permission. Copyright 2019 Wiley-VCH. (B) SH-CaO2-induced RDH. SH-CaO2 produced abundant H2O2 to disrupt redox homeostasis enhancing Ca2+-mediated ion interference therapy (IIT). Adapted from ref. 33 with permission. Copyright 2019 Elsevier. | |
2.3 Increasing other oxidizing species to realize RDH
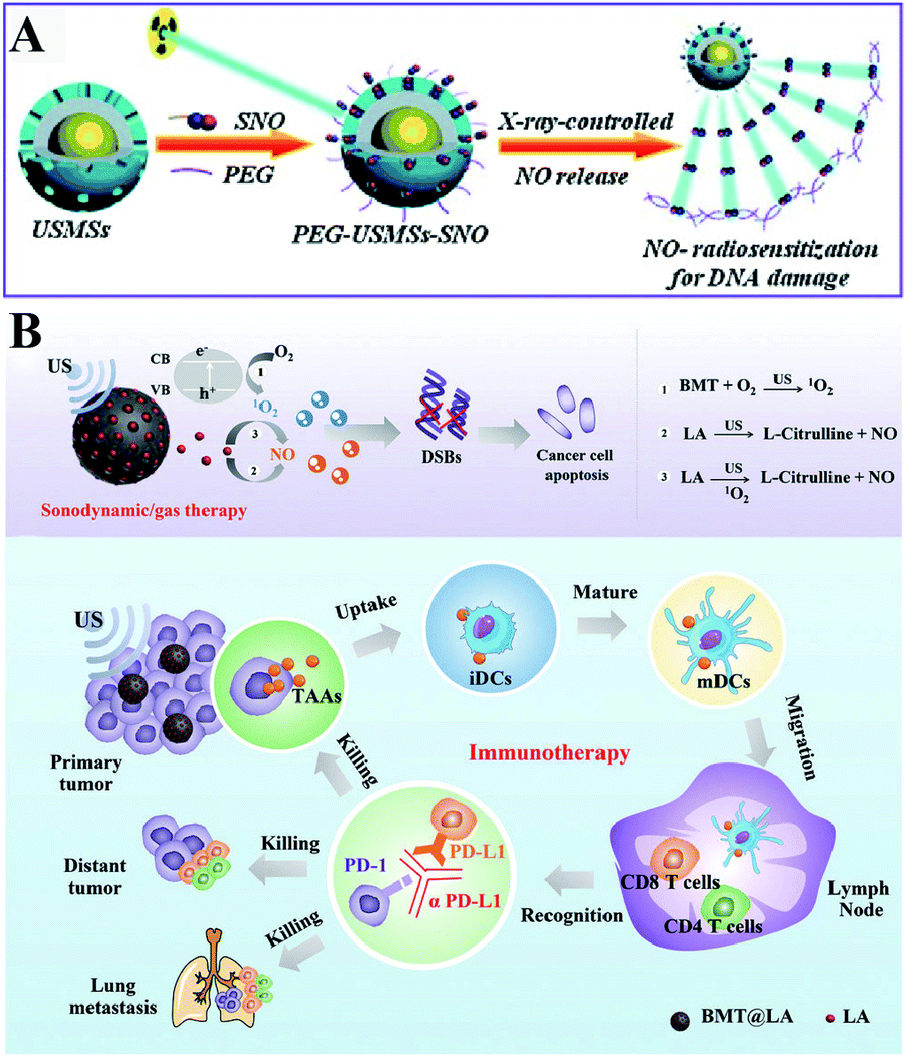
Besides ROS, RNS including NO and ONOO− are another important oxidizing species. A few studies have been designed to produce RNS modulating the redox homeostasis of tumor cells. For example, our group constructed an intelligent X-ray-controlled NO-releasing upconversion nano-theranostic system PEG-USMSx-SNO. The increase of NO could induce RDH consequently sensitizing radiotherapy (Fig. 7A).34 In order to achieve a better antitumor effect and inhibit tumor metastasis, Jun Lin et al. fabricated a multifunctional nanovaccine based on L-arginine (LA)-loaded black mesoporous titania (BMT). By using ultrasound, BMT and LA were triggered to produce NO which not only induced RDH but also induced cell apoptosis through DNA double-strand breaks. All of these eventually enhanced PD-L1 antibody-mediated immunotherapy, further inhibiting metastatic tumors (Fig. 7B).35
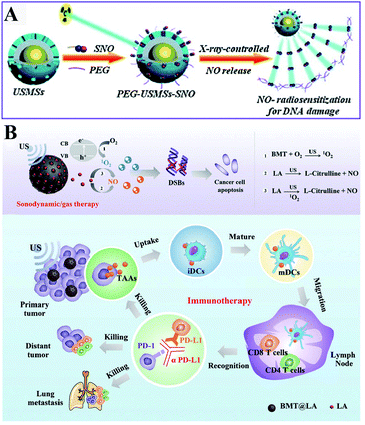 |
| | Fig. 7 Schematic illustrations of NO-induced RDH. (A) X-ray-controlled NO release induced RDH, sensitizing X-ray-mediated radiotherapy. Adapted from ref. 34 with permission. Copyright 2015 Wiley-VCH. (B) BMT and LA were triggered by ultrasound to produce NO which induces RDH, sensitizing PD-L1 antibody-mediated immunotherapy. Adapted from ref. 35 with permission. Copyright 2021 Wiley-VCH. | |
3. Regulation of reducing species to realize RDH
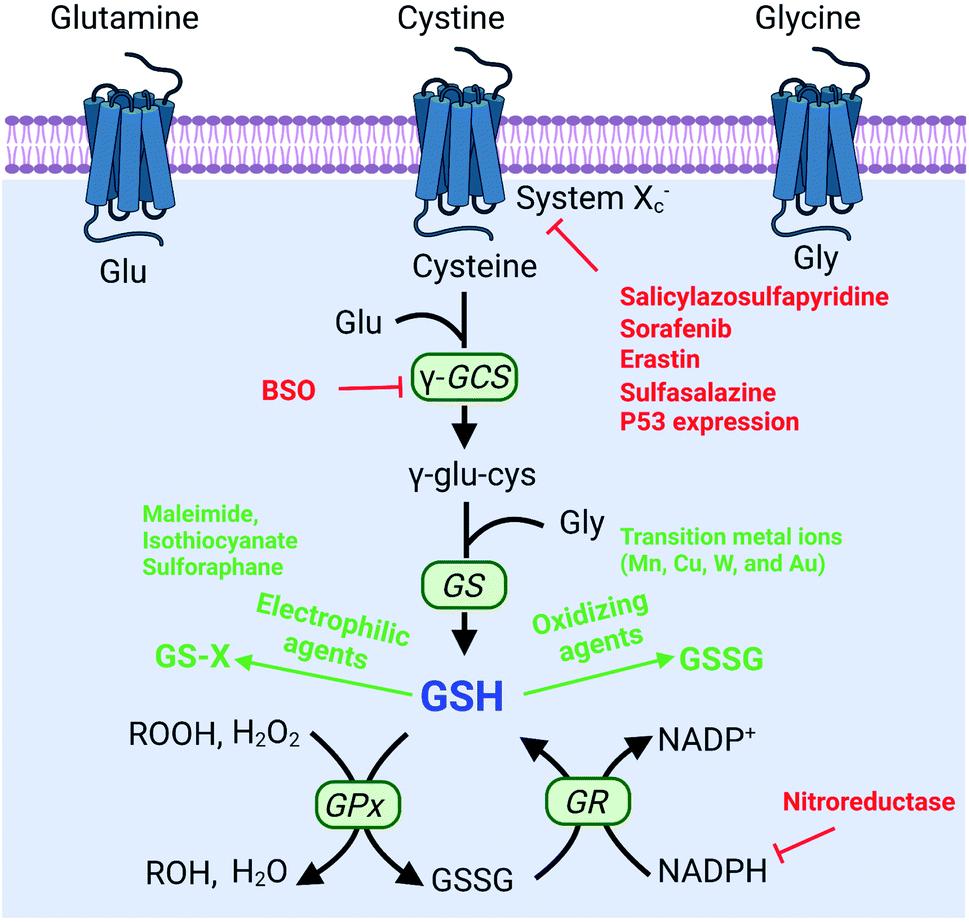
To protect tumor cells from damage by oxidative stress, the powerful antioxidant defense system of cells maintains the balance of intracellular redox by producing reducing species.36 Reducing species, as another part of redox homeostasis, essentially transfer electrons to oxidizing species, which would decrease the sensitivity of tumor cells to ROS-medicated tumor therapy. Therefore, inhibiting the production of reducing species could effectively break the redox balance and also promote the efficacy of tumors.37–41 It is well established that glutathione (GSH) is the primary reducing species for intracellular ROS scavenging, and its presence significantly reduces intracellular ROS levels and weakens the effect of tumor therapy. Therefore, GSH is one of the most important reducing species capable of being regulated to disrupt redox homeostasis. Thus, how to reduce GSH to break the redox homeostasis of tumor cells is one of the cutting-edge research directions of the RDH strategy. In the past few years, numerous nanomaterials have been constructed to decrease GSH production,42,43 adopting the RDH strategy to enhance the sensitivity of tumor therapy. Based on working principles, inhibition of GSH can be divided into two distinctive strategies: (1) suppressing GSH biosynthesis; (2) chemical approaches (Fig. 8).
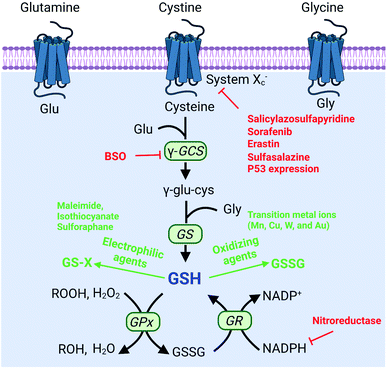 |
| | Fig. 8 Biosynthesis pathways of GSH and strategies of decreasing the GSH level via biosynthesis approaches and chemical approaches (red font represents biosynthesis approaches and green font represents chemical approaches). | |
3.1 Decreasing the GSH level of tumor cells to realize RDH via suppressing GSH biosynthesis
The biosynthesis of GSH consists of the production of GSH and the recycling of GSH. The process of GSH production includes the formation of γ-glutamylcysteine from glutamate and cysteine, and the formation of GSH from γ-glutamylcysteine and glycine.44 In this system, cysteine is an indispensable precursor for GSH. γ-glutamine cysteine synthase (γ-GCS) and GSH synthetase (GS) are two rate-limiting enzymes for the biosynthesis of GSH. After GSH production, GSH acts as an antioxidant to reduce hydrogen peroxide and lipid peroxide through GSH peroxidase (GPx)-catalyzed reactions. In this process, GSH is oxidized to oxidized-glutathione (GSSG). In turn, GSSG is reduced back to GSH by GSSG reductase (GR) at the expense of NADPH, thereby forming a redox cycle. Therefore, the biosynthesis level of GSH is dependent on synthesis-related enzymes (γ-GCS or GS), substances (cysteine, glutamate and glycine) and the recycling level (NADPH) (Fig. 8). Thus, with this knowledge, we conclude that nanomaterials have the potential to suppress GSH biosynthesis. The suppression of GSH is also driven by enzyme-dependent and -independent strategies.
3.1.1 Enzyme-dependent strategies.
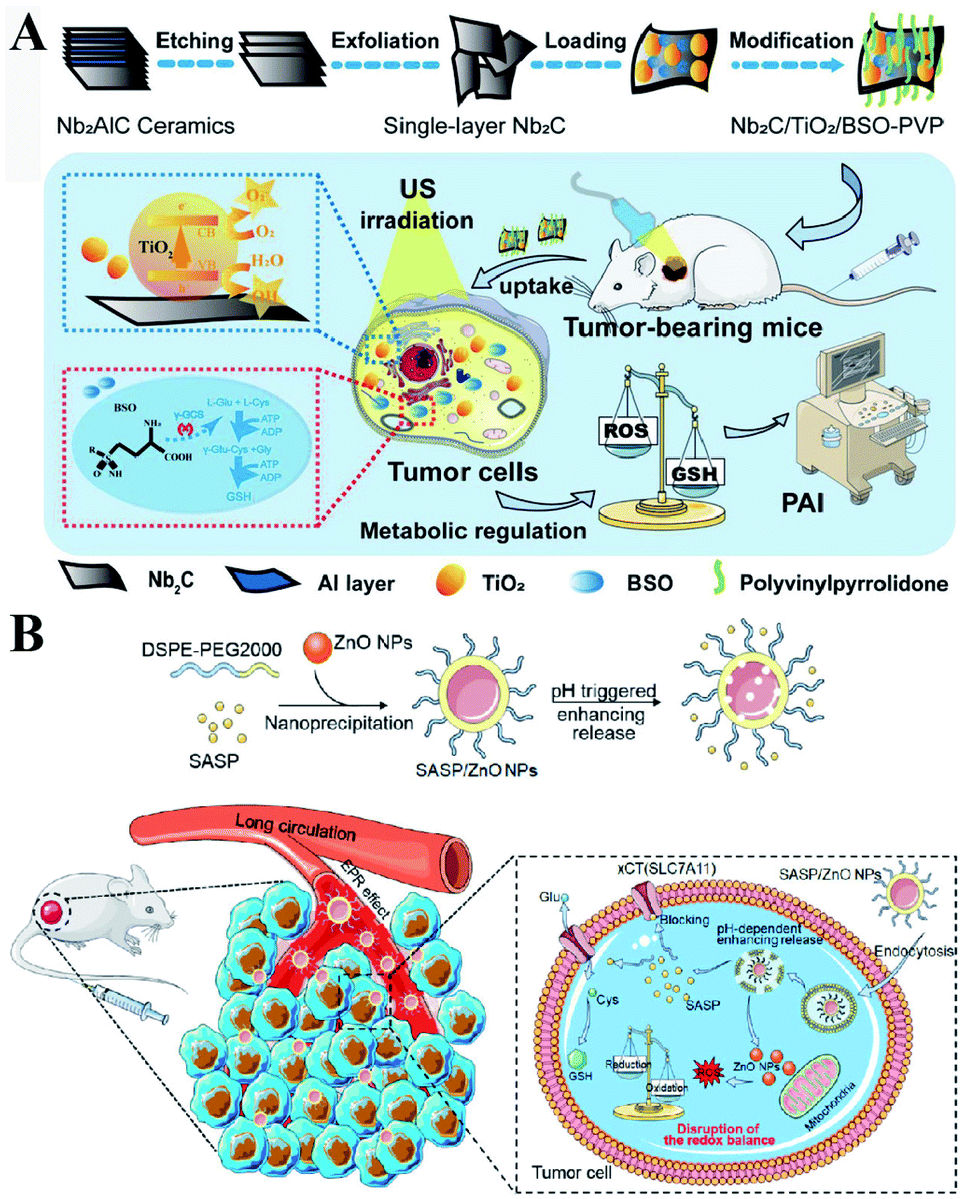
γ-GCS and GS are two important enzymes for the biosynthesis of GSH. However, compared with the overexpression of GS, the overexpression of γ-GCS could elevate higher levels of GSH. Therefore, γ-GCS is used as a more popular target for the regulation of GSH synthesis.45,46 The γ-GCS inhibitor L-buthionine-(S,R)-sulfoximine (BSO) is the most widely used chemical compound for the inhibition of GSH biosynthesis.47 Xiaolin Wang et al. developed near-infrared photothermal liposomal nano-antagonists (PLANs) containing a photosensitizer indocyanine (ICG) and BSO. BSO inhibited GSH synthesis to disrupt redox homeostasis, amplifying ICG-mediated cancer PDT.48 Similarly, Kun Zhang et al. constructed a tumor metabolism-engineered composite nanoplatform Nb2C/TiO2/BSO-PVP. Nb2C was used to load TiO2 sonosensitizers and BSO. The loaded BSO can block GSH synthesis to break redox homeostasis, which can maximally increase SDT (Fig. 9A).49
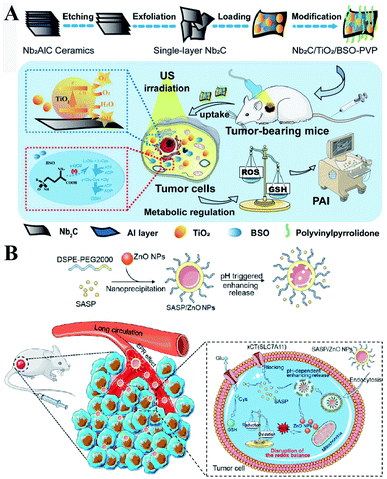 |
| | Fig. 9 (A) Schematic illustrations of the synthesis process and function of the Nb2C/TiO2/BSO-PVP. Adapted from ref. 49 with permission. Copyright 2020 Wiley-VCH. (B) Design and application of SASP/ZnO NPs for cancer therapy. Adapted from ref. 53 with permission. Copyright 2019 American Chemical Society. | |
3.1.2 Enzyme-independent strategies.
In addition to enzyme-dependent strategies, the biosynthesis of GSH can be regulated by substrate availability and GSH recycling. Cysteine is the indispensable substrate for GSH biosynthesis.50–52 However, cysteine is unstable in the cytosol of cells and easily oxidized to cystine. Therefore, uptake of stable cystine that is reduced to cysteine in cell cytosol is another rate-limiting step in GSH biosynthesis. Several transporters, such as cystine/glutamate transporter System XC−(SLC7A11 and SLC3A2), P-glycoprotein (P-gp, ABCB1), and breast cancer resistance protein (BCRP, ABCG2),14 have been confirmed to be involved in the control of cysteine concentration. Among them, System XC− and its inhibitors (salicylazosulfapyridine,53 sorafenib,54 erastin,55 and sulfasalazine56) have been broadly used to inhibit GSH biosynthesis. Ruijie Chen et al. reported a nanocomposite to inhibit the GSH synthesis, where ZnO nanoparticles (NPs) were used as a carrier, loaded with salicylazosulfapyridine (SASP) and stabilized with DSPE-PEG to form ultra-small NPs (SASP/ZnO NPs).53 SASP was used to inhibit the transport function of SLC7A11, resulting in the decrease of cysteine for GSH synthesis. As a consequence, treatment of tumor cells with SASP/ZnO NPs resulted in a disruption of redox balance in tumor cells, enhancing ZnO-induced cell death (Fig. 9B). Similarly, sorafenib (SFB) could block the System XC− transport system to inhibit the biosynthesis of GSH.57,58 With this in mind, Fanzhu Li et al. constructed targeted SFB loaded manganese-doped silica nanoparticles (FaPEG-MnMSN@SFB), which were capable of destroying the intracellular redox homeostasis by the consumption of GSH and the inhibition of GSH synthesis.54 This strategy ultimately enhanced ROS-dependent ferroptosis. Apart from the XC− inhibitor, endogenous protein expression can also regulate SLC7A11 expression. It has been reported that the famous tumor suppressor gene P53 can inhibit cystine uptake by repressing the expression of SLC7A11.59 Xianzheng Zhang et al. used a metal–organic network (MON) to encapsulate P53 plasmid (MON-P53). Once MON-P53 was internalized by cancer cells, the expression of P53 can inhibit the expression of SLC7A11 and then decrease the transport of cysteine for GSH synthesis. The decrease of GSH promoted RDH in tumor cells, increasing the sensitivity of cells to ferroptosis-mediated cell death.60
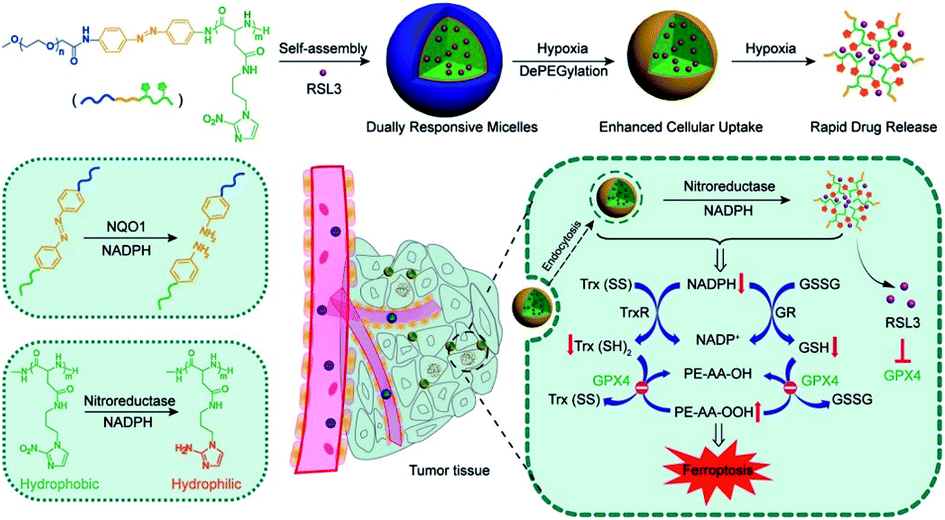
In the process of GSH recycling, GSSG can be in turn reduced back to GSH under the catalysis of NADPH and the GSH reductase (GR), which forms a redox cycle. Therefore, decreasing the level of NADPH may inhibit GSH production by breaking the redox cycle.43 Yanjun Zhao et al. constructed a polymer containing hydrophilic poly(ethyleneglycol) (PEG) that was linked by azobenzene with a nitroimidazole-conjugated polypeptide.61 The nitroimidazole moiety was reduced by the overexpressed nitroreductase with reduced NADPH as the cofactor, resulting in transient depletion of NADPH. The lack of NADP impaired both the glutathione and thioredoxin redox cycle, leading to diminished intracellular glutathione and thioredoxin. As a result, tumor redox homeostasis was destroyed, which enhanced the sensitivity of RSL3-induced ferroptosis (Fig. 10). This RDH strategy via blocking NADPH to decrease GSH is still less studied. Besides azobenzene, etomoxir, a carnitine palmitoyltransferase 1 inhibitor, can also decrease NADPH levels, thereby reducing the GSH content by inhibiting fatty acid oxidation.62 Etomoxir has not been used for the construction of nanomaterials for RDH.
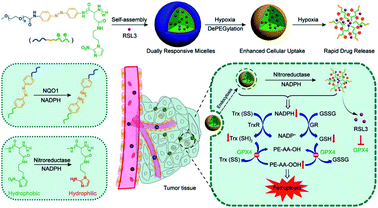 |
| | Fig. 10 Schematic illustrations of NADPH inhibition-mediated RDH. NADPH depletion diminished intracellular glutathione and thioredoxin, which destroyed tumor redox homeostasis enhancing the sensitivity of RSL3-induced ferroptosis. Reprinted from ref. 61 with permission. Copyright 2020 American Chemical Society. | |
3.2 Decreasing the GSH level of tumor cells to realize RDH via chemical approaches
Decreasing GSH via biosynthesis approaches is an effective and specific strategy, but the underlying biological mechanisms and possible side effects require further study. Using chemical approaches to deplete GSH involves introducing some substances that can react with GSH. Compared with the suppression of GSH biosynthesis, chemical approaches are a more straightforward way. However, the function is temporary and short-lived, as tumor cells may increase the GSH level through alternative pathways. Currently, chemical approaches can be divided into two categories: (1) consumption of GSH by oxidizing agents, and (2) consumption of GSH by electrophiles.
3.2.1 Consumption of GSH by oxidizing agents.
Converting GSH to GSSG using oxidizing agents is a rapid method to consume GSH. Oxidizing agents include transition metal (Mn, Cu, W, Mo, and Au) ions. Numerous research groups made extensive effort in this area. Weihong Tan et al. reported a smart chlorine 6 (Ce6)–manganese dioxide (MnO2) nano drug delivery system.63 MnO2 consumed GSH to induce RDH, thereby sensitizing photosensitizer Ce6-induced PDT effects (Fig. 11A). Mn2+ could not only consume GSH but also display Fenton-like properties.64–66 Based on this, Xiaoyuan Chen et al. constructed MnO2-based mesoporous silica nanoparticles (MS@MnO2 NP).67 Once taken up by cancer cells, the MnO2 shell was used to oxidize GSH to accumulate H2O2, which led to RDH, ultimately enhancing Mn2+-mediated CDT. Compared with Mn ions, Cu+ displays higher efficacy of CDT.68–70 Our group developed self-assembled copper-amino acid mercaptide nanoparticles (Cu-Cys NPs).71 Cu2+ in Cu-Cys NPs was first reduced by local GSH to generate Cu+ and simultaneously realize GSH depletion, leading to RDH. Subsequently, this state of the cells was more sensitive to the cytotoxicity of Cu+-catalyzed toxic ˙OH (CDT). These cascade reactions can perfectly achieve “two birds with one stone” in cancer treatment (Fig. 11B). In addition, Yuliang Zhao et al. found that oxidative W(VI) also had the capacity to react with intracellular GSH in cancer cells.72 They developed Gd-containing polyoxometalate-conjugated chitosan (GdW10@CS nanosphere) as a radio-sensitization system. The GdW10@CS nanospheres were able to promote the depletion of intracellular GSH by synergy W6+-triggered GSH oxidation. As a result, the as-synthesized GdW10@CS nanospheres disrupted the redox homeostasis of tumor cells, thereby facilitating the efficiency of radiotherapy (Fig. 11C). To achieve more specific radiotherapy, they further explored bismuth heteropolytungstate (BiP5W30) nanoclusters as radiosensitizers.73 In this system, BiP5W30 had the capability of depleting GSH via redox reaction owing to its unique electron structure and multi-electron properties. GSH consumption induced RDH, sensitizing high-Z elements like Bi and W-enhanced radiotherapy. In recent years, it has been widely reported that gold (Au) as a radiosensitizer can exhaust GSH by the formation of an Au–S bond, showing great promise in preclinical studies.74 Wu et al. prepared histidine-capped gold nanoclusters (Au NCs@His) for GSH depletion and RDH, thereby sensitizing Au-mediated cancer radiotherapy.75
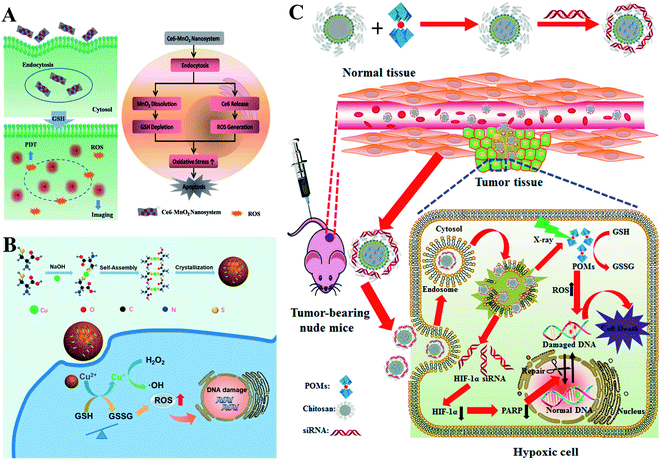 |
| | Fig. 11 Schematic illustrations of GSH depletion by oxidizing agents. (A) MnO2 of the Ce6–MnO2 nanosystem consumed GSH to induce RDH, thereby sensitizing photosensitizer Ce6-induced PDT effects. Adapted from ref. 63 with permission. Copyright 2016 Wiley-VCH. (B) Schematic illustrations of Cu-induced GSH depletion. Cu2+ in Cu-Cys NPs consumed GSH to induce RDH and simultaneously generate Cu+. This RDH state of the cells was more sensitive to the cytotoxicity of Cu+-mediated CDT. Adapted from ref. 71 with permission. Copyright 2019 American Chemical Society. (C) W6+ of GdW10@CS triggered GSH oxidation leading to RDH, thereby facilitating the efficiency of radiotherapy. Adapted from ref. 72 with permission. Copyright 2017 American Chemical Society. | |
3.2.2 Consumption of GSH by electrophiles.
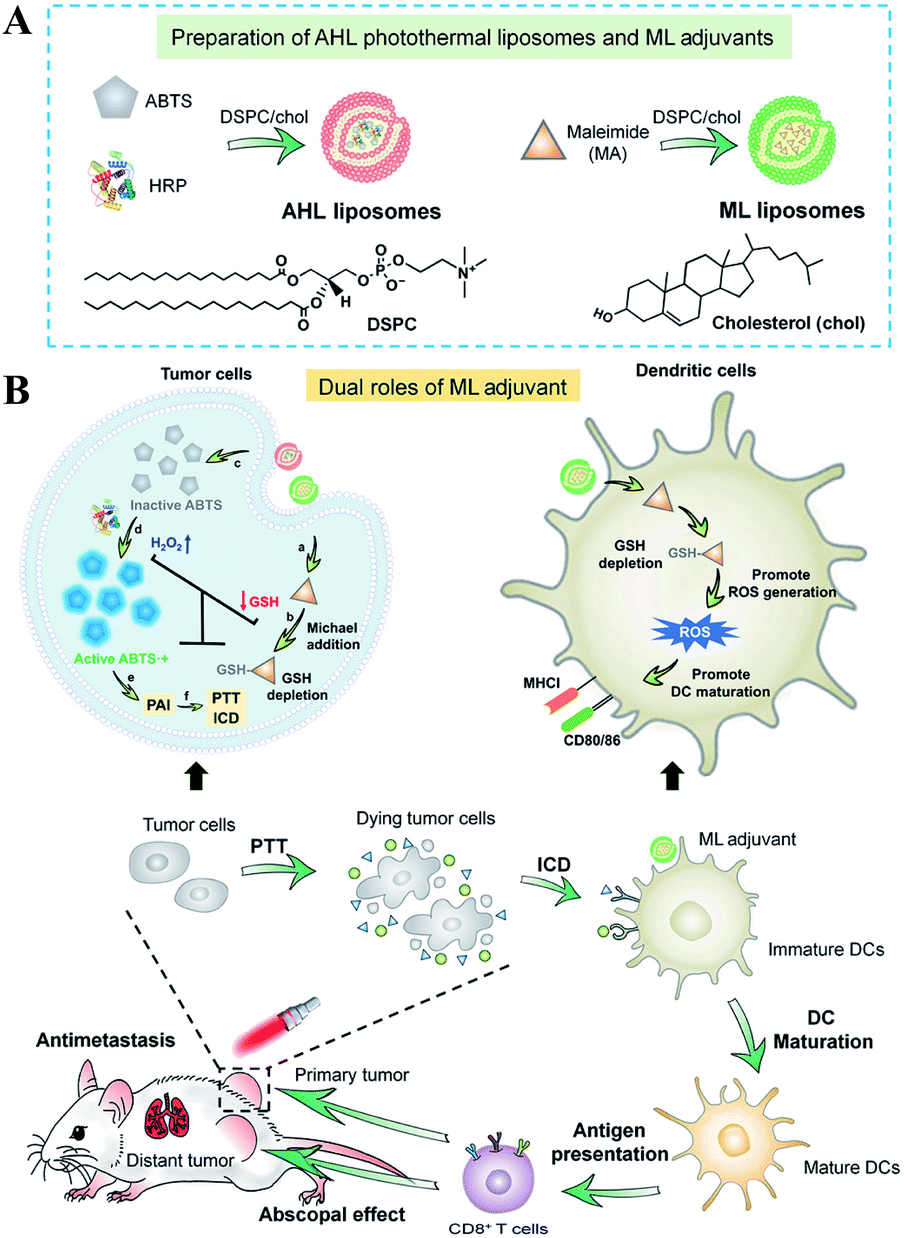
Besides the use of oxidizing agents to deplete GSH, many electrophilic reagents that can directly react with thiol groups are also used to consume GSH, leading to the RDH of tumors. Compared with oxidizing agents, electrophilic reagents may non-selectively react with nucleophiles, leading to undesirable toxic effects. Therefore, moderately active electrophilic such as maleimide, isothiocyanate and sulforaphane were used to consume GSH. For example, Minjie Sun et al. developed maleimide liposome (ML) adjuvants to deplete GSH for augmenting the photothermal immunotherapy of breast cancer.76 The maleimide group of ML can irreversibly react with thiols of GSH by Michael addition. The depletion of GSH caused RDH, enhancing photothermal immunotherapy of breast cancer through promoting immunogenic cell death (ICD) and dendritic cell maturation (Fig. 12). In addition, Xiangliang Yang et al. employed a natural compound, β-phenylethyl isothiocyanate (PEITC), found in cruciferous vegetables, to deplete intracellular GSH through the formation of the conjugate of PEITC with GSH.77 They combined ICG-loaded hydroxyethylstarch-oleic acid conjugate (HES-OA) nanoparticles with PEITC for potent PDT. GSH depletion by PEITC led to the imbalance of redox homeostasis in tumor cells and significantly enhanced ICG-based PDT. Furthermore, Guangjun Nie et al. utilized another naturally occurring compound, sulforaphane (SFN), stemming from broccoli, to deplete GSH via directly forming the GSH–SFN complex, which could be exported outside cells and result in disrupting tumor redox homeostasis.78 Based on this, the authors constructed a codelivery system to simultaneously encapsulate two drugs, anticancer agent cisplatin (cis-dichlorodiammineplatinum(II), CDDP) and SFN. The use of SFN greatly improved the therapeutic efficacy of platinum (Pt)-based chemotherapy.
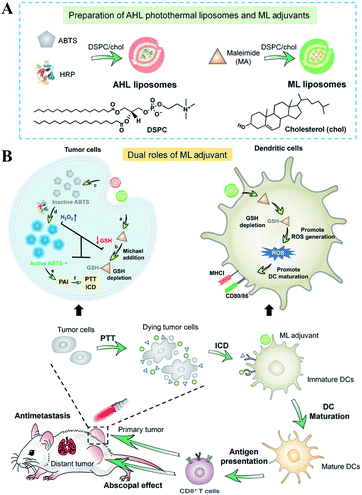 |
| | Fig. 12 Schematic illustrations of the design (A) and application (B) of the AHL photothermal liposomes and ML adjuvants. Maleimide induced the depletion of GSH causing RDH, which enhanced the photothermal immunotherapy of breast cancer through promoting immunogenic cell death (ICD). Adapted from ref. 76 with permission. Copyright 2020 American Association for the Advancement of Science. | |
3.3 Decreasing other reducing species to realize RDH
Besides GSH, the thioredoxin/thioredoxin reductase (Trx/TrxR) system in cancer is another important antioxidant defense system.79 TrxR, which is the major disulfide reductase, can reduce the disulfide of oxidized Trx (Trx-S2) to a dithiol (Trx-(SH)2). It has been reported that Au nanoclusters could effectively inhibit TrxR in the tumor cell cytoplasm. Xueyun Gao et al. constructed spatiotemporal controllable liposomal nanocomposites, co-loaded with Au nanoclusters and photosensitizer Chlorine 6 (Ce6).80 Au nanoclusters effectively inhibited TrxR to induce RDH, which enhanced the Ce6-mediated PDT efficacy. Similarly, Xianzheng Zhang et al. reported a porous MOF of Zirconium-metalloporphyrin PCN-22 (PCN), loaded with a TrxR inhibitor alkaloid piperlongumine (PL).81 Inside tumor cells, the released PL could effectively inhibit TrxR-mediated H2O2 elimination to disturb cellular redox homeostasis, improving the PDT efficacy (Fig. 13).
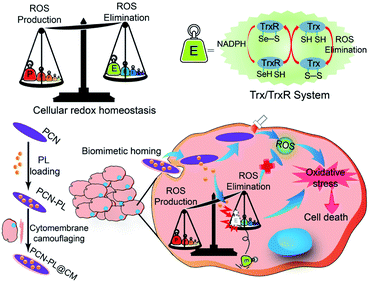 |
| | Fig. 13 Schematic illustration of interfering with the TrxR pathway in cancer cells for improved PDT. The released PL from PCN could effectively inhibit TrxR-mediated H2O2 elimination to disturb cellular redox homeostasis, improving the MOF-mediated PDT efficacy. Adapted from ref. 81 with permission. Copyright 2019 Elsevier. | |
4. Regulation of both reducing and oxidizing species to realize RDH
In addition to single regulation of oxidizing species or reducing species in cells, simultaneous regulation of both oxidizing species and reducing species to realize RDH has also been widely developed. Compared with single regulation, simultaneous regulation of both of them may induce a faster and higher degree of RDH. Therefore, this “Two-way regulation” strategy has been widely used in various cancer therapeutic regimens including PDT, SDT, CDT, RT, starvation therapy, chemotherapy, immunotherapy, and ferroptosis. Currently, this section is divided into two groups: (1) two-way regulated RDH combined with monotherapy, and (2) two-way regulated RDH combined with combinational therapies.
4.1 Two-way regulated-RDH combined with monotherapy
To enhance the PDT efficiency, Xianzheng Zhang et al. constructed SRF@Fe(III)TA nanoparticles in which Fe3+ and tannic acid (TA) spontaneously formed a network-like corona onto sorafenib (SRF) nanocores.82 TA was arranged to chemically reduce Fe3+ to Fe2+, inducing a Fenton-like reaction to produce cytotoxic oxidizing species ˙OH. Meanwhile, SRF can deactivate glutathione peroxidase 4 (GPX4) to block cellular antioxidant defense. Both of the above processes disrupted the redox homeostasis of tumor cells, enhancing photosensitizer methylene blue (MB)-induced PDT (Fig. 14A). To promote the SDT efficiency, Zhuang Liu et al. utilized CaCO3 nanoparticles as the template to obtain pH-dissociable hollow metal–organic coordination nanostructures, where meso-tetra-(4-carboxyphenyl)porphine (TCPP) and ferric ions were chosen as the sonosensitizer and metallic center, respectively.83 Upon endocytosis by tumor cells, the nanocomplex could fast dissociate to release Ca2+ and BSO. The released Ca2+ remarkably elevated intracellular Ca2+ concentrations and resulted in mitochondria damage, which then induced severe ROS production and subsequently synergized with BSO-mediated GSH depletion to cause RDH. This strategy ultimately enhanced TCPP-mediated SDT (Fig. 14B). For starvation therapy,84,85 Xiue Jiang et al. presented a GOx-loaded nanozyme IrOx-GOx, which can covert H2O2 at tumor tissues to O2, which could further react with glucose to produce H2O2 by using GOx.86 IrOx not only can generate O2 and ˙OH by its gluconic acid-unlocked oxidase and peroxidase-like activities, but also can consume GSH through its self-cyclic valence alteration of Ir(IV) and Ir(III). Consequently, introduction of this as-prepared nanozyme into tumor cells could cause RDH, enhancing GOx-induced starvation therapy (Fig. 15A). For immunotherapy87–92 Jun Lin et al. reported an artificial cascade enzyme–drug conjugate (GHZD NCs), in which GOx, ferric protoporphyrin (Hemin), and dihydroartemisinin (DHA) were automatically localized into zeolitic imidazolate framework-8 (ZIF-8) scaffolds via a one-pot reaction. GOx catalyzed glucose to produce H2O2.93 Moreover, hemin (Fe3+) acted as not only peroxidase producing ˙OH, but also glutathione peroxidase consuming antioxidant GSH, resulting in the destruction of redox homeostasis. Consequently, Fe3+ would be converted to Fe2+, which sequentially triggered the drug DHA to burst C-centered free radicals. All the above processes amplified the immunogenic cell death effect and combated immunosuppressive tumors (Fig. 15B).
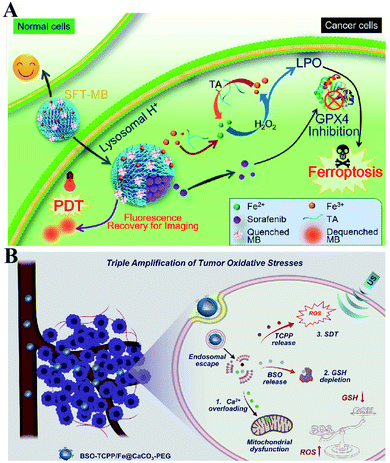 |
| | Fig. 14 Schematic illustration of two-way regulated-RDH combined with (A) PDT and (B) SDT. (A) SRF@Fe(III)TA nanoparticles for RDH-sensitized PDT. TA is arranged to induce a Fenton-like reaction to produce cytotoxic ˙OH. Meanwhile, SRF can deactivateGPX4 to block cellular antioxidant defense. Adapted from ref. 82 with permission. Copyright 2018 American Chemical Society. (B) BSO-TCPP/Fe@CaCO3 nanoparticles for RDH-sensitized SDT. Ca2+-overloading-induced ROS generation and BSO-induced GSH depletion led to RDH, enhancing TCPP-mediated SDT. Adapted from ref. 83 with permission. Copyright 2020 Elsevier. | |
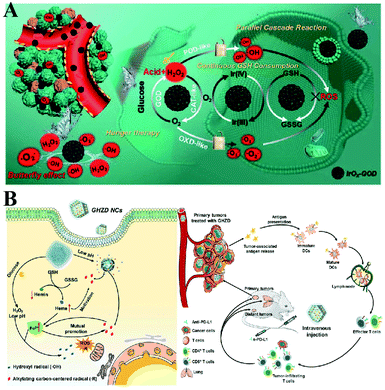 |
| | Fig. 15 (A) Schematic illustration of two-way regulation leading to RDH, sensitizing GOx-induced starvation therapy. Adapted from ref. 86 with permission. Copyright 2020 Wiley-VCH. (B) Two-way regulation led to RDH, sensitizing PD-L1-mediated immunotherapy. Adapted from ref. 93 with permission. Copyright 2020 Wiley-VCH. | |
Two-way regulated-RDH strategies have been widely developed to enhance the CDT efficiency.94 Our group synthesized FeCysPW@ZIF-82@CAT Dz nanoparticles13 in which the shell ZIF-82 can be degraded into Zn2+ as a cofactor for catalase DNAzyme (CAT Dz) mediated CAT silencing and the electrophilic ligand for GSH depletion. This two-way regulation could induce RDH, sensitizing tumor cells to FeCysPW-induced CDT (Fig. 16A). To further enhance the efficacy of CDT, our group constructed DMON@Fe0/AT nanoparticles.95 On one hand, S–S bond-rich dendritic mesoporous organic silica (DMON) could decrease the content of GSH in tumor cells. On the other hand, the catalase inhibitor (3-amino-1,2,4-triazole (AT)) could suppress the activity of catalase for H2O2 accumulation. DMON@Fe0/AT can effectively inhibit the expression of ferroportin 1 to disrupt the cellular iron metabolism system, leading to the retention of iron in the cytoplasm for CDT. With DMON-mediated GSH-depletion and AT-mediated H2O2 production, DMON@Fe0/AT would dramatically induce RDH in tumor cells to enhance the CDT efficacy (Fig. 16B). This RDH-sensitized CDT strategy was also performed by other groups. For example, Xiaogang Qu et al. designed a nanosystem PZIF67-AT by modifying 3-AT and PEG on zeolitic imidazole framework-67 (ZIF-67) nanoparticles that were formed by bridging 2-methylimidazole (2-mim) and cobalt ions with a sodalite zeolite-type topology.96 This system not only displayed SOD-like activity capable of converting O2˙− to H2O2, but also could deplete GSH via catalase inhibitor AT, which led to an accumulation of H2O2. The above processes would ultimately cause RDH and intensify the Fenton-like reaction-based CDT. Furthermore, Guanbin Song et al. constructed β-lapachone (Lapa)-loaded iron oxide nanocarriers (Fe3O4-HSA@Lapa).97 The release of Lapa selectively increased the generation of H2O2via NADPH:NQO1 catalysis. Meanwhile, Lapa severely consumed intracellular NADPH, leading to the inhibition of GSSG to GSH catalyzed by glutathione reductase. Therefore, Lapa not only supplied sufficient H2O2 but also inhibited GSH levels, causing RDH. Ultimately, this strategy boosted the efficiency of CDT.
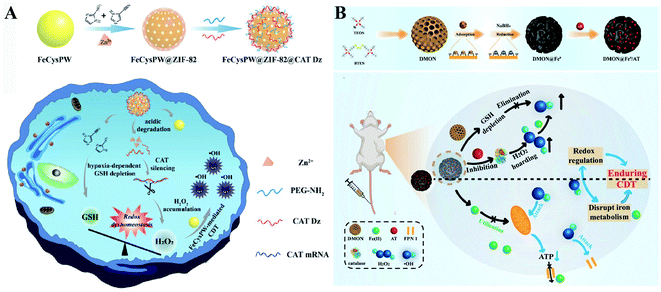 |
| | Fig. 16 Schematic illustration of two-way regulated-RDH strategies to enhance the CDT efficiency. (A) The design and multiple functions of FeCysPW@ZIF-82@CAT Dz nanoparticles. The shell ZIF-82 can be degraded into Zn2+ as a cofactor for CAT Dz mediated CAT silencing and the electrophilic ligand for GSH depletion, leading to RDH, sensitizing FeCysPW-induced CDT. Adapted from ref. 13 with permission. Copyright 2020 Wiley-VCH. (B) The design and application of DMON@Fe0/AT. DMON-mediated GSH-depletion and AT-mediated H2O2 production induced RDH in tumor cells sensitizing CDT efficacy. Adapted from ref. 95 with permission. Copyright 2021 Wiley-VCH. | |
4.2 Two-way regulated-RDH combined with combinational therapies
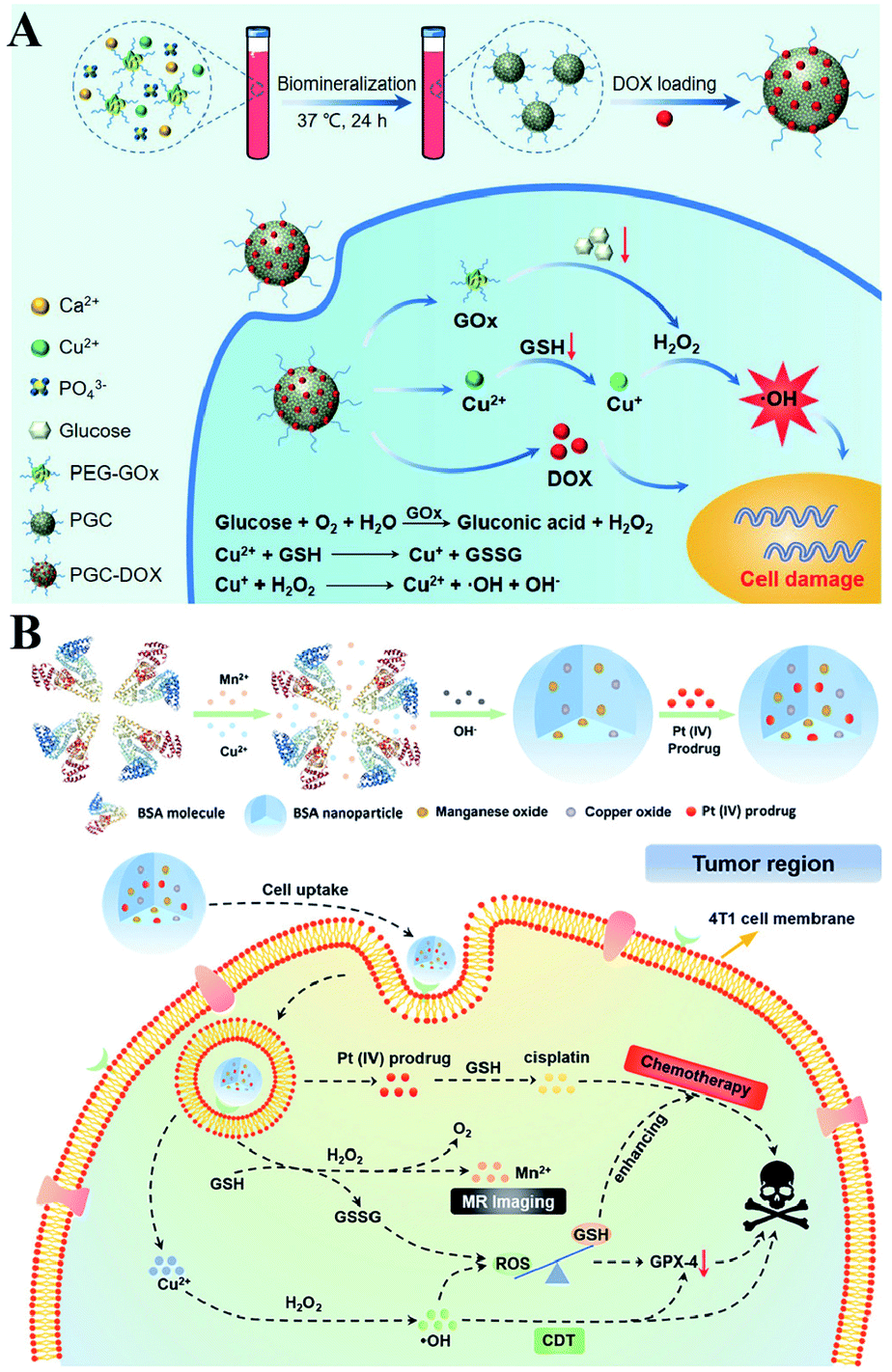
It is well established that monotherapy is less effective than combinational therapies. Therefore, the two-way regulated-RDH could be used to further strengthen the combinational therapy, which has received tremendous attention. Peng Huang et al. used PEG-modified GOx as a template to construct biodegradable copper-doped calcium phosphate nanoparticles, followed by the loading of doxorubicin (DOX), named PGC-DOX. GOx could effectively catalyze intracellular glucose to produce H2O2.98 Meanwhile, the released Cu2+ ions could react with GSH and induce GSH depletion. Cu2+ could simultaneously be reduced to Fenton agent Cu+ ions. The above processes would ultimately perturb redox homeostasis, resulting in enhanced Cu+-induced CDT and DOX-mediated chemotherapy (Fig. 17A). Similarly, Hangrong Chen et al. constructed a novel albumin-based multifunctional nanoagent, in which CuO and MnOxin situ grew inside the bovine serum albumin (BSA) through a facile biomineralization process, followed by the conjugation of the Pt(IV) prodrug to obtain the final nanoagent.99 In this system, copper species can catalyze H2O2 to produce ˙OH by a Fenton-like reaction, while MnOx can react with GSH, leading to the GSH depletion. ˙OH accumulation and GSH depletion gave rise to a disruption of redox homeostasis, which promoted better chemotherapy and CDT efficacies (Fig. 17B).
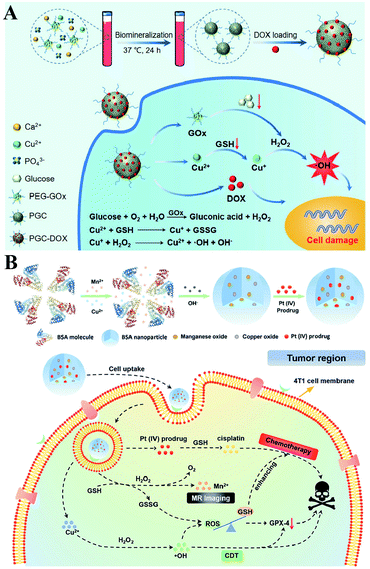 |
| | Fig. 17 Schematic illustrations of the two-way regulation combining chemotherapy and CDT. (A) Synthetic process and function of PGC-DOX. GOx can effectively catalyze intracellular glucose to generate H2O2. Meanwhile, the released Cu2+ ions could react with GSH and induce GSH depletion. This two-way regulation led to RDH sensitizing Cu+-induced CDT and DOX-mediated chemotherapy. Adapted from ref. 98 with permission. Copyright 2021 Wiley-VCH. (B) Synthetic process and function of CMBP NPs. Copper species can catalyze H2O2 to produce ˙OH by a Fenton-like reaction, while MnOx can react with GSH, leading to the GSH depletion. This two-way regulation gave rise to the disruption of redox homeostasis, which promoted better chemotherapy and CDT efficacies. Adapted from ref. 99 with permission. Copyright 2021 Wiley-VCH. | |
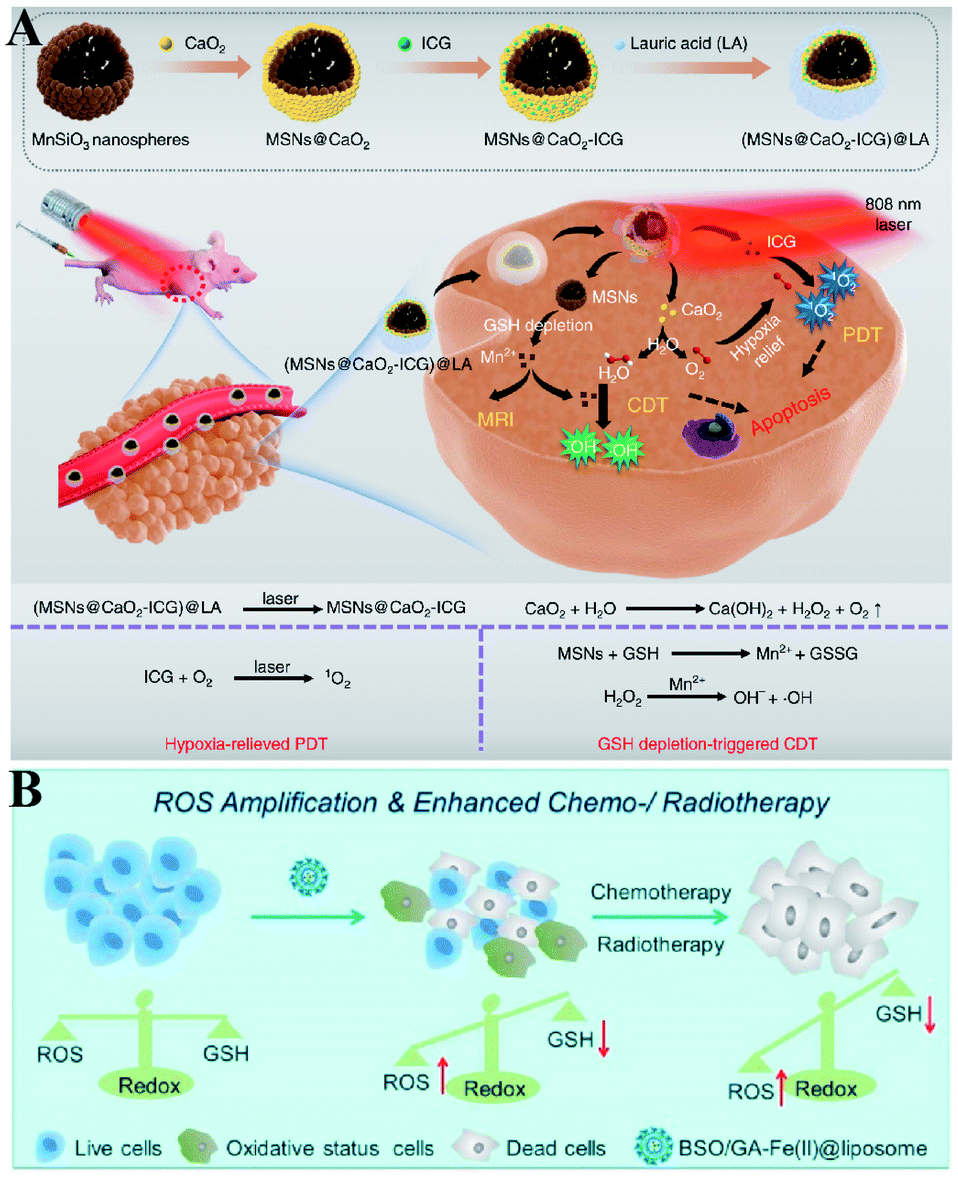
Furthermore, Haifeng Dong et al. reported an H2O2/O2 self-supplying nanoagent (MSNs@CaO2-ICG)@LA.100 Under laser irradiation, ICG simultaneously generated singlet oxygen and emitted heat to melt the phase-change material lauric acid (LA). The exposed CaO2 reacted with water to rapidly generate H2O2 and O2. The interaction between MSNs and GSH led to the release of Mn2+ and GSH depletion. Thus, this system could accumulate H2O2 and eliminate GSH to destroy redox homeostasis, which enhanced the efficacies of PDT and CDT (Fig. 18A). To further promote the efficiency of SDT-induced tumor therapy, Zhuang Liu et al. synthesized new nanomaterials to combine RDH with chemotherapy and radiotherapy. Gallic acid-ferrous (GA-Fe(II)) and BSO were co-encapsulated within liposome (BSO/GA-Fe(II)@liposome).101 GA-Fe(II) nanocomplexes were used as the catalyst of Fenton reaction to enable persistent conversion of H2O2 to highly cytotoxic hydroxyl radicals (˙OH), leading to the increase of oxidizing species. Meanwhile, the released BSO could inhibit the biosynthesis of GSH, resulting in the decrease of reducing species. Thus, GA-Fe(II)-mediated ˙OH production and BSO-mediated GSH depletion would dramatically enhance RDH in tumor cells, which then resulted in remarkably improved therapeutic efficacies of concurrently applied chemotherapy or radiotherapy (Fig. 18B).
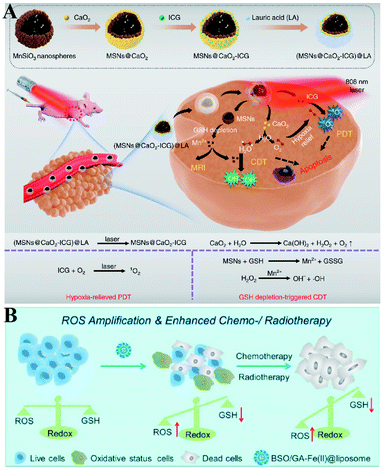 |
| | Fig. 18 Two-way regulated-RDH combined with combinational therapies. (A) Schematic illustrations of the synthetic process and function of (MSNs@CaO2-ICG)@LA NPs. This system can induce redox homeostasis, which enhanced the efficacies of PDT and CDT. Adapted from ref. 100 with permission. Copyright 2020 Nature Publishing Group. (B) Schematic illustrations of the function of BSO/GA-Fe(II)@liposome. GA-Fe(II)-mediated ˙OH production and BSO-mediated GSH depletion dramatically induced RDH in tumor cells, which then resulted in remarkably improved therapeutic efficacies of concurrently applied chemotherapy or RT. Adapted from ref. 101 with permission. Copyright 2019 American Chemical Society. | |
5. Conclusions and perspectives
This review summarizes the current progress of RDH for tumor therapy. This strategy focuses on the use of nanomaterials to disturb redox homeostasis by regulating intracellular oxidizing states, reducing states or both of them at the same time, which increases the sensitivity of tumor cells to various therapeutic modalities. Although nanomaterial-mediated RDH has been widely applied in the research of tumor therapy, many important scientific problems still need to be considered before it is ready for clinical transformation.
First, it is critically vital to deeply explore cellular biological mechanisms involved in redox homeostasis, which is beneficial to further optimize the RDH strategy at the genetic and molecular levels. Second, redox homeostasis is ubiquitous in various cells.102,103 Thus, it is necessary to make tumor cells more specific and sensitive for the RDH strategy, avoiding potential toxic and side effects on normal tissues and organs. Finally, the most important issue is to improve the efficacy of RDH strategy-mediated tumor treatment. At present, most of the tumor treatments are based on ROS-mediated oxidative stress to kill tumor cells. In view of the resistance of tumor cells to ROS-mediated therapy, some new tumor therapeutic modalities could be combined with RDH. On the one hand, there is an urgent need to explore novel active species other than ROS/RNS, such as alkyl radicals (R˙)104,105 or chlorine radicals (˙Cl),106 which may exhibit higher efficacy in tumor treatment. On the other hand, reductive stress attracts increasing attention,107 which could break through the bottleneck of existing oxidative stress-mediated tumor treatment.
In summary, the RDH strategy, as a research hotspot in the field of tumor therapy, has attracted increasing attention of worldwide researchers, despite the existence of some unresolved issues. Under the guidance of the RDH strategy, researchers are still exploring various novel nanomaterials for accurate regulation of redox homeostasis to enhance tumor therapy efficacy. We expect that the development of the RDH strategy will not only provide valuable insights for the design of various functional nanomaterials, but also bring more breakthroughs in the treatment of cancer or other diseases in the future.
Author contributions
Y. L. Wu and Y. L. Li contributed equally to this work. All authors participated in this review work. Y. L. Wu, Y. L. Li and W. B. Bu conceived the structure of this review. Y. L. Wu, Y. L. Li and G. L. Lv prepared the draft. G. L. Lv and W. B. Bu reviewed and refined the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would greatly acknowledge the financial support by the National Funds for Distinguished Young Scientists (Grant No. 51725202), the Key Project of Shanghai Science and Technology Commission (Grant No. 19JC1412000), and the National Natural Science Foundation of China (Grant No. 51872094, 82172091).
References
- P. H. G. M. Willems, R. Rossignol, C. E. J. Dieteren, M. P. Murphy and W. J. H. Koopman, Cell Metab., 2015, 22, 207–218 CrossRef CAS PubMed.
- G. S. Shadel and T. L. Horvath, Cell, 2015, 163, 560–569 CrossRef CAS.
- N. T. Moldogazieva, I. M. Mokhosoev, N. B. Feldman and S. V. Lutsenko, Free Radical Res., 2018, 52, 507–543 CrossRef CAS PubMed.
- A. Ghoneum, A. Y. Abdulfattah, B. O. Warren, J. Shu and N. Said, Int. J. Mol. Sci., 2020, 21, 3100 CrossRef CAS PubMed.
- D. Samanta and G. L. Semenza, Redox Biol., 2017, 13, 331–335 CrossRef CAS.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- D. S. A. Simpson and P. L. Oliver, Antioxidants, 2020, 9, 743 CrossRef CAS PubMed.
- E. Panieri and M. M. Santoro, Cell Death Dis., 2016, 7, e2253 CrossRef CAS PubMed.
- B. Marengo, M. Nitti, A. L. Furfaro, R. Colla, C. D. Ciucis, U. M. Marinari, M. A. Pronzato, N. Traverso and C. Domenicotti, Oxid. Med. Cell. Longevity, 2016, 2016, 6235641 Search PubMed.
- D. H. Hyun, Cancers, 2020, 12, 1822 CrossRef CAS PubMed.
- T. C. Jorgenson, W. Zhong and T. D. Oberley, Cancer Res., 2013, 73, 6118–6123 CrossRef CAS.
- C. L. Grek and K. D. Tew, Curr. Opin. Pharmacol., 2010, 10, 362–368 CrossRef CAS PubMed.
- Y. Li, P. Zhao, T. Gong, H. Wang, X. Jiang, H. Cheng, Y. Liu, Y. Wu and W. Bu, Angew. Chem., Int. Ed., 2020, 59, 22537–22543 CrossRef CAS PubMed.
- L. Kou, X. Jiang, H. Huang, X. Lin, Y. Zhang, Q. Yao and R. Chen, Asian J. Pharm. Sci., 2020, 15, 145–157 CrossRef PubMed.
- R. P. Brandes, N. Weissmann and K. Schroeder, Free Radical Biol. Med., 2014, 76, 208–226 CrossRef CAS PubMed.
- T. Itoh, R. Terazawa, K. Kojima, K. Nakane, T. Deguchi, M. Ando, Y. Tsukamasa, M. Ito and Y. Nozawa, Free Radical Res., 2011, 45, 1033–1039 CrossRef CAS PubMed.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- K. Chang, Z. Liu, X. Fang, H. Chen, X. Men, Y. Yuan, K. Sun, X. Zhang, Z. Yuan and C. Wu, Nano Lett., 2017, 17, 4323–4329 CrossRef CAS PubMed.
- J. Tang, A. K. Meka, S. Theivendran, Y. Wang, Y. Yang, H. Song, J. Fu, W. Ban, Z. Gu, C. Lei, S. Li and C. Yu, Angew. Chem., Int. Ed., 2020, 59, 22054–22062 CrossRef CAS PubMed.
- Z. Zhuang, J. Dai, M. Yu, J. Li, P. Shen, R. Hu, X. Lou, Z. Zhao and B. Z. Tang, Chem. Sci., 2020, 11, 3405–3417 RSC.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- K. Zhang, Z. Yu, X. Meng, W. Zhao, Z. Shi, Z. Yang, H. Dong and X. Zhang, Adv. Sci., 2019, 6, 1900530 CrossRef PubMed.
- S. K. Parks, J. Chiche and J. Pouyssegur, Nat. Rev. Cancer, 2013, 13, 611–623 CrossRef CAS PubMed.
- V. L. Payen, E. Mina, V. F. Van Hee, P. E. Porporato and P. Sonveaux, Mol. Metabol., 2020, 33, 48–66 CrossRef CAS PubMed.
- Y. Liu, X. Ji, W. W. L. Tong, D. Askhatova, T. Yang, H. Cheng, Y. Wang and J. Shi, Angew. Chem., Int. Ed., 2018, 57, 1510–1513 CrossRef CAS PubMed.
- L. Sun, Y. Xu, Y. Gao, X. Huang, S. Feng, J. Chen, X. Wang, L. Guo, M. Li, X. Meng, J. Zhang, J. Ge, X. An, D. Ding, Y. Luo, Y. Zhang, Q. Jiang and X. Ning, Small, 2019, 15, e1901156 CrossRef PubMed.
- W.-P. Li, C.-H. Su, Y.-C. Chang, Y.-J. Lin and C.-S. Yeh, ACS Nano, 2016, 10, 2017–2027 CrossRef CAS PubMed.
- X. Song, J. Xu, C. Liang, Y. Chao, Q. Jin, C. Wang, M. Chen and Z. Liu, Nano Lett., 2018, 18, 6360–6368 CrossRef CAS PubMed.
- M. Zhang, B. Shen, R. Song, H. Wang, B. Lv, X. Meng, Y. Liu, Y. Liu, X. Zheng, W. Su, C. Zuo and W. Bu, Mater. Horiz., 2019, 6, 1034–1040 RSC.
- Z. Tang, Y. Liu, D. Ni, J. Zhou, M. Zhang, P. Zhao, B. Lv, H. Wang, D. Jin and W. Bu, Adv. Mater., 2020, 32, e1904011 CrossRef PubMed.
- L. S. Lin, T. Huang, J. Song, X. Y. Ou, Z. Wang, H. Deng, R. Tian, Y. Liu, J. F. Wang, Y. Liu, G. Yu, Z. Zhou, S. Wang, G. Niu, H. H. Yang and X. Chen, J. Am. Chem. Soc., 2019, 141, 9937–9945 CrossRef CAS PubMed.
- M. Zhang, R. Song, Y. Liu, Z. Yi, X. Meng, J. Zhang, Z. Tang, Z. Yao, Y. Liu, X. Liu and W. Bu, Chem, 2019, 5, 2171–2182 CAS.
- W. Fan, W. Bu, Z. Zhang, B. Shen, H. Zhang, Q. He, D. Ni, Z. Cui, K. Zhao, J. Bu, J. Du, J. Liu and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 14026–14030 CrossRef CAS PubMed.
- M. Wang, Z. Hou, S. Liu, S. Liang, B. Ding, Y. Zhao, M. Chang, G. Han, A. A. A. Kheraif and J. Lin, Small, 2021, 17, e2005728 CrossRef PubMed.
- B. Poljsak, D. Suput and I. Milisav, Oxid. Med. Cell. Longevity, 2013, 2013, 956792 Search PubMed.
- A. Glasauer and N. S. Chandel, Biochem. Pharmacol., 2014, 92, 90–101 CrossRef CAS PubMed.
- L. He, T. He, S. Farrar, L. Ji, T. Liu and X. Ma, Cell. Physiol. Biochem., 2017, 44, 532–553 CrossRef PubMed.
- C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS PubMed.
- A. V. Snezhkina, A. V. Kudryavtseva, O. L. Kardymon, M. V. Savvateeva, N. V. Melnikova, G. S. Krasnov and A. A. Dmitriev, Oxid. Med. Cell. Longevity, 2019, 2019, 1–16 CrossRef PubMed.
- C. Zhang, W. Bu, D. Ni, C. Zuo, C. Cheng, Q. Li, L. Zhang, Z. Wang and J. Shi, J. Am. Chem. Soc., 2016, 138, 8156–8164 CrossRef CAS PubMed.
- B. Niu, K. Liao, Y. Zhou, T. Wen, G. Quan, X. Pan and C. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed.
- Y. Xiong, C. Xiao, Z. Li and X. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC.
- S. C. Lu, Biochim. Biophys. Acta, 2013, 1830, 3143–3153 CrossRef CAS PubMed.
- S. J. Kim, H. G. Kim, B. C. Kim, K. Kim, E. H. Park and C. J. Lim, J. Microbiol., 2004, 42, 233–238 CAS.
- S.-O. Kang and M.-K. Kwak, J. Microbiol. Biotechnol., 2021, 31, 79–91 CrossRef CAS PubMed.
- I. S. Harris, A. E. Treloar, S. Inoue, M. Sasaki, C. Gorrini, K. C. Lee, K. Y. Yung, D. Brenner, C. B. Knobbe-Thomsen, M. A. Cox, A. Elia, T. Berger, D. W. Cescon, A. Adeoye, A. Bruestle, S. D. Molyneux, J. M. Mason, W. Y. Li, K. Yamamoto, A. Wakeham, H. K. Berman, R. Khokha, S. J. Done, T. J. Kavanagh, C.-W. Lam and T. W. Mak, Cancer Cell, 2015, 27, 211–222 CrossRef CAS PubMed.
- H. Sun, M. Feng, S. Chen, R. Wang, Y. Luo, B. Yin, J. Li and X. Wang, J. Mater. Chem. B, 2020, 8, 7149–7159 RSC.
- X. Guan, H. H. Yin, X. H. Xu, G. Xu, Y. Zhang, B. G. Zhou, W. W. Yue, C. Liu, L. P. Sun, H. X. Xu and K. Zhang, Adv. Funct. Mater., 2020, 30, 2000326 CrossRef CAS.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS PubMed.
- L. Kou, R. Sun, S. Xiao, Y. Zheng, Z. Chen, A. Cai, H. Zheng, Q. Yao, V. Ganapathy and R. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 26722–26730 CrossRef CAS PubMed.
- H. Tang, C. Li, Y. Zhang, H. Zheng, Y. Cheng, J. Zhu, X. Chen, Z. Zhu, J. G. Piao and F. Li, Theranostics, 2020, 10, 9865–9887 CrossRef CAS PubMed.
- L. Wang, Y. Liu, T. Du, H. Yang, L. Lei, M. Guo, H. F. Ding, J. Zhang, H. Wang, X. Chen and C. Yan, Cell Death Differ., 2020, 27, 662–675 CrossRef CAS PubMed.
- L. Sleire, B. S. Skeie, I. A. Netland, H. E. Forde, E. Dodoo, F. Selheim, L. Leiss, J. I. Heggdal, P. H. Pedersen, J. Wang and P. O. Enger, Oncogene, 2015, 34, 5951–5959 CrossRef CAS PubMed.
- E. Lachaier, C. Louandre, C. Godin, Z. Saidak, M. Baert, M. Diouf, B. Chauffert and A. Galmiche, Anticancer Res., 2014, 34, 6417–6422 CAS.
- Y. S. Chang, J. Adnane, P. A. Trail, J. Levy, A. Henderson, D. Xue, E. Bortolon, M. Ichetovkin, C. Chen, A. McNabola, D. Wilkie, C. A. Carter, I. C. A. Taylor, M. Lynch and S. Wilhelm, Cancer Chemother. Pharmacol., 2007, 59, 561–574 CrossRef CAS PubMed.
- L. Jiang, N. Kon, T. Li, S. J. Wang, T. Su, H. Hibshoosh, R. Baer and W. Gu, Nature, 2015, 520, 57–62 CrossRef CAS PubMed.
- D. W. Zheng, Q. Lei, J. Y. Zhu, J. X. Fan, C. X. Li, C. Li, Z. Xu, S. X. Cheng and X. Z. Zhang, Nano Lett., 2017, 17, 284–291 CrossRef CAS PubMed.
- X. Guo, F. Liu, J. Deng, P. Dai, Y. Qin, Z. Li, B. Wang, A. Fan, Z. Wang and Y. Zhao, ACS Nano, 2020, 14, 14715–14730 CrossRef PubMed.
- L. S. Pike, A. L. Smift, N. J. Croteau, D. A. Ferrick and M. Wu, Biochim. Biophys. Acta, 2011, 1807, 726–734 CrossRef CAS PubMed.
- H. Fan, G. Yan, Z. Zhao, X. Hu, W. Zhang, H. Liu, X. Fu, T. Fu, X. B. Zhang and W. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477–5482 CrossRef CAS PubMed.
- J. Ou, H. Tian, J. Wu, J. Gao, J. Jiang, K. Liu, S. Wang, F. Wang, F. Tong, Y. Ye, L. Liu, B. Chen, X. Ma, X. Chen, F. Peng and Y. Tu, ACS Appl. Mater. Interfaces, 2021, 13, 38050–38060 CrossRef CAS PubMed.
- L.-S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H.-H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- H. He, Q. Yang, H. Li, S. Meng, Z. Xu, X. Chen, Z. Sun, B. Jiang and C. Li, Microchim. Acta, 2021, 188, 141 CrossRef CAS PubMed.
- L.-S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H.-H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- C. Liu, D. Wang, S. Zhang, Y. Cheng, F. Yang, Y. Xing, T. Xu, H. Dong and X. Zhang, ACS Nano, 2019, 13, 4267–4277 CrossRef CAS PubMed.
- M. Chang, M. Wang, M. Wang, M. Shu, B. Ding, C. Li, M. Pang, S. Cui, Z. Hou and J. Lin, Adv. Mater., 2019, 31, 1905271 CrossRef CAS PubMed.
- L.-H. Fu, Y. Wan, C. Qi, J. He, C. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2006892 CrossRef CAS PubMed.
- B. Ma, S. Wang, F. Liu, S. Zhang, J. Duan, Z. Li, Y. Kong, Y. Sang, H. Liu, W. Bu and L. Li, J. Am. Chem. Soc., 2019, 141, 849–857 CrossRef CAS PubMed.
- Y. Yong, C. Zhang, Z. Gu, J. Du, Z. Guo, X. Dong, J. Xie, G. Zhang, X. Liu and Y. Zhao, ACS Nano, 2017, 11, 7164–7176 CrossRef CAS PubMed.
- R. Zhou, H. Wang, Y. Yang, C. Zhang, X. Dong, J. Du, L. Yan, G. Zhang, Z. Gu and Y. Zhao, Biomaterials, 2019, 189, 11–22 CrossRef CAS PubMed.
- S. Her, D. A. Jaffray and C. Allen, Adv. Drug Delivery Rev., 2017, 109, 84–101 CrossRef CAS PubMed.
- X. Zhang, X. Chen, Y.-W. Jiang, N. Ma, L.-Y. Xia, X. Cheng, H.-R. Jia, P. Liu, N. Gu, Z. Chen and F.-G. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 10601–10606 CrossRef CAS PubMed.
- Z. Zhou, H. Wu, R. Yang, A. Xu, Q. Zhang, J. Dong, C. Qian and M. Sun, Sci. Adv., 2020, 6, eabc4373 CrossRef CAS PubMed.
- H. Hu, J. Chen, H. Yang, X. Huang, H. Wu, Y. Wu, F. Li, Y. Yi, C. Xiao, Y. Li, Y. Tang, Z. Li, B. Zhang and X. Yang, Nanoscale, 2019, 11, 6384–6393 RSC.
- Y. Xu, X. Han, Y. Li, H. Min, X. Zhao, Y. Zhang, Y. Qi, J. Shi, S. Qi, Y. Bao and G. Nie, ACS Nano, 2019, 13, 13445–13455 CrossRef CAS PubMed.
- J. Zhang, X. Li, X. Han, R. Liu and J. Fang, Trends Pharmacol. Sci., 2017, 38, 794–808 CrossRef CAS PubMed.
- F. Gao, W. Zheng, L. Gao, P. Cai, R. Liu, Y. Wang, Q. Yuan, Y. Zhao and X. Gao, Adv. Healthcare Mater., 2017, 6, 1601453 CrossRef PubMed.
- Q. Cheng, W. Yu, J. Ye, M. Liu, W. Liu, C. Zhang, C. Zhang, J. Feng and X. Z. Zhang, Biomaterials, 2019, 224, 119500 CrossRef CAS PubMed.
- T. Liu, W. Liu, M. Zhang, W. Yu, F. Gao, C. Li, S. B. Wang, J. Feng and X. Z. Zhang, ACS Nano, 2018, 12, 12181–12192 CrossRef CAS PubMed.
- Z. Dong, L. Feng, Y. Hao, Q. Li, M. Chen, Z. Yang, H. Zhao and Z. Liu, Chem, 2020, 6, 1391–1407 CAS.
- C. Wang, J. Yang, C. Dong and S. Shi, Adv. Ther., 2020, 3, 2000110 CrossRef CAS.
- M. Wang, D. Wang, Q. Chen, C. Li, Z. Li and J. Lin, Small, 2019, 15, 1903895 CrossRef CAS PubMed.
- W. Zhen, Y. Liu, W. Wang, M. Zhang, W. Hu, X. Jia, C. Wang and X. Jiang, Angew. Chem., Int. Ed., 2020, 59, 9491–9497 CrossRef CAS PubMed.
- J. Li, Y. Luo and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 12682–12705 CrossRef CAS PubMed.
- L. A. Emens, Clin. Cancer Res., 2018, 24, 511–520 CrossRef CAS PubMed.
- K. Esfahani, L. Roudaia, N. Buhlaiga, S. V. Del Rincon, N. Papneja and W. H. Miller Jr, Curr. Oncol., 2020, 27, S87–S97 CrossRef CAS PubMed.
- L. A. Emens, P. A. Ascierto, P. K. Darcy, S. Demaria, A. M. M. Eggermont, W. L. Redmond, B. Seliger and F. M. Marincola, Eur. J. Cancer, 2017, 81, 116–129 CrossRef CAS PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- Y. Zhao, X. Xiao, M. Zou, B. Ding, H. Xiao, M. Wang, F. Jiang, Z. Cheng, P. Ma and J. Lin, Adv. Mater., 2021, 33, e2006363 CrossRef PubMed.
- Z. Tang, P. Zhao, H. Wang, Y. Liu and W. Bu, Chem. Rev., 2021, 121, 1981–2019 CrossRef CAS PubMed.
- Y. Liu, S. Zhai, X. Jiang, Y. Liu, K. Wang, C. Wang, M. Zhang, X. Liu and W. Bu, Adv. Funct. Mater., 2021, 31, 2010390 CrossRef CAS.
- Y. Sang, F. Cao, W. Li, L. Zhang, Y. You, Q. Deng, K. Dong, J. Ren and X. Qu, J. Am. Chem. Soc., 2020, 142, 5177–5183 CrossRef CAS PubMed.
- Q. Chen, J. Zhou, Z. Chen, Q. Luo, J. Xu and G. Song, ACS Appl. Mater. Interfaces, 2019, 11, 30551–30565 CrossRef CAS PubMed.
- L. H. Fu, Y. Wan, C. Qi, J. He, C. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, e2006892 CrossRef PubMed.
- K. Liang, H. Sun, Z. Yang, H. Yu, J. Shen, X. Wang and H. Chen, Adv. Funct. Mater., 2021, 31, 2100355 CrossRef CAS.
- C. Liu, Y. Cao, Y. Cheng, D. Wang, T. Xu, L. Su, X. Zhang and H. Dong, Nat. Commun., 2020, 11, 1735 CrossRef CAS PubMed.
- Z. Dong, L. Feng, Y. Chao, Y. Hao, M. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS PubMed.
- J. Jezek, K. F. Cooper and R. Strich, Antioxidants, 2018, 7, 13 CrossRef PubMed.
- E.-M. Hanschmann, J. R. Godoy, C. Berndt, C. Hudemann and C. H. Lillig, Antioxid. Redox Signaling, 2013, 19, 1539–1605 CrossRef CAS PubMed.
- X.-Q. Wang, F. Gao and X.-Z. Zhang, Angew. Chem., Int. Ed., 2017, 56, 9029–9033 CrossRef CAS PubMed.
- S. Wu, X. Liu, J. Ren and X. Qu, Small, 2019, 15, 1904870 CrossRef CAS PubMed.
- R. Song, H. Wang, M. Zhang, Y. Liu, X. Meng, S. Zhai, C. Wang, T. Gong, Y. Wu, X. Jiang and W. Bu, Angew. Chem., Int. Ed., 2020, 59, 21032–21040 CrossRef CAS PubMed.
- K.-S. Chun, D.-H. Kim and Y.-J. Surh, Cells, 2021, 10, 758 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yanli
Li†
c,
Guanglei
Lv
*b and
Wenbo
Bu
ab,
Yanli
Li†
c,
Guanglei
Lv
*b and
Wenbo
Bu