
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bond activated glycosylation under mild conditions†
Ke
Xiao
a,
Yongxin
Hu
a,
Yongyong
Wan
a,
XinXin
Li
a,
Qin
Nie
a,
Hao
Yan
a,
Liming
Wang
*a,
Jinxi
Liao
a,
Deyong
Liu
a,
Yuanhong
Tu
a,
Jiansong
Sun
 *a,
Jeroen D. C.
Codée
b and
Qingju
Zhang
*ac
*a,
Jeroen D. C.
Codée
b and
Qingju
Zhang
*ac
aNational Research Centre for Carbohydrate Synthesis, Jiangxi Normal University, 99 Ziyang Avenue, Nanchang 330022, China. E-mail: zhangq@jxnu.edu.cn; jssun@jxnu.edu.cn; wang-lm@jxnu.edu.cn
bLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
cKey Laboratory of Functional Small Molecule, Ministry of Education, Jiangxi Normal University, 99 Ziyang Avenue, Nanchang 330022, China
First published on 15th December 2021
Abstract
Herein, we report a new glycosylation system for the highly efficient and stereoselective formation of glycosidic bonds using glycosyl N-phenyl trifluoroacetimidate (PTFAI) donors and a charged thiourea hydrogen-bond-donor catalyst. The glycosylation protocol features broad substrate scope, controllable stereoselectivity, good to excellent yields and exceptionally mild catalysis conditions. Benefitting from the mild reaction conditions, this new hydrogen bond-mediated glycosylation system in combination with a hydrogen bond-mediated aglycon delivery system provides a reliable method for the synthesis of challenging phenolic glycosides. In addition, a chemoselective glycosylation procedure was developed using different imidate donors (trichloroacetimidates, N-phenyl trifluoroacetimidates, N-4-nitrophenyl trifluoroacetimidates, benzoxazolyl imidates and 6-nitro-benzothiazolyl imidates) and it was applied for a trisaccharide synthesis through a novel one-pot single catalyst strategy.
Introduction
Oligosaccharides and glycoconjugates play critical roles in an array of biological processes1 and they have been widely explored for drug and vaccine development.2 The key step in oligosaccharide and glycoconjugate synthesis is the glycosidic bond formation. In the past few decades, various glycosylation methods have been developed, including Brønsted acid or Lewis acid promoted glycosylation,3,4 base promoted glycosylation,5 gold-catalyzed glycosylation,6 and recently emerged organo-catalyst effected glycosylation.7 (Thio)ureas are the most well explored organo-catalysts, which generally act as hydrogen-bond donors and exhibit many important advantages over widely applied Brønsted acid or Lewis acid catalysts, such as shelf stability and excellent functional group tolerance. In 2013, Schmidt and co-workers reported a novel glycosylation system based on the activation and SN2-type substitution of trichloroacetimidate (TCAI) donors under the cooperative catalysis of phosphorous acid and Schreiner's thiourea.8 In 2016, Ye and coworkers realized stereoselective glycosylation with glycosyl chlorides using Schreiner's thiourea in the presence of K2CO3.9 Toshima and Takemoto reported that arylthioureas can act as organo photoacids and co-catalysts for the activation of TCAI-donors through halogen bond activation (Fig. 1A).10 Recently, inspired by glycosyltransferase-catalyzed glycosylation, Jacobsen and co-workers developed a novel type of chiral macrocyclic bis-thiourea for the activation of glycosyl chloride and phosphate donors with various glycoside acceptors, achieving stereoselective SN2-type glycosylation (Fig. 1A).11–13 Moreover, Galan and coworkers showed that 2-deoxyglycosides and 2-amino-2-deoxyglycosides can be assembled through the activation of glycal and 2-nitroglycal donors with an organothiourea catalyst.7,14 Loh and coworkers described a stereoselective glycosylation method using a unique constrained cyclopropyl ketone glycosyl donor, which could be activated with a hydrogen-bond donor thiourea catalyst, the reactivity of which was enhanced by cationic charge in the activator.15 | ||
| Fig. 1 Representative (thio)urea-catalyzed glycosylation (A) and the proposed hydrogen bond activated glycosylation in this study (B). | ||
Despite these important advancements in organocatalyzed glycosylation, several limitations remain, including the requirement of acidic co-catalyst species, or the necessity to scavenge acidic species generated in the reaction, as well as poor stereoselectivity. Herein, we describe a novel and generally applicable hydrogen bond activated glycosylation protocol that can proceed under mild conditions for the assembly of various O-, C-, S- and N-glycosides. We envisioned that glycosyl imidate-type donors (the H-bond acceptor) can be activated with organo-(thio)urea catalysts (the H-bond donor) under mild catalysis conditions and that the released secondary amide leaving group is neither acidic nor nucleophilic enough to compete with the acceptor toward the activated donor (Fig. 1B).3c,d Depending on the structure of the activator system, the hydrogen bond activation may generate oxocarbenium ion-type species,15 which can be harnessed to achieve stereoselective glycosylation procedures.3
Results and discussion
Perbenzyl-D-glucopyranosyl 6-nitro-2-benzothiazole 1a16 was first evaluated as a donor for our proposed hydrogen bond activated glycosylation system since this imidate-type donor does not generate an acidic side product,3c,d and the nitro group may function as a hydrogen bond acceptor which may help the activation. As shown in Table 1, a total of 10 hydrogen bond donor catalysts C1–C10 were prepared including mono-(thio)urea C1–C3, N,N′-diarylsquaramide C4,17 and bis-(thio)ureas C5–C9 inspired by Jacobsen's work,11–13 and the charge-enhanced H-bond donor thiourea-catalyst C10.18 In initial studies, glucosyl acceptor 2a was chosen as a model acceptor and the reactions were conducted in acetonitrile at 50 °C (Table 1, entries 1–11). The screening of the thiourea, urea, and squaramide catalysts C1–C9 revealed poor reactivity of these catalysts with bis-(thio)urea C6 and C7 giving the highest yields (entries 7 and 8). The optimization of H-bonding catalysts without increasing the acidity of the catalysts is very challenging. The Kass group reported that the charge-enhanced H-bond donor thiourea-catalyst C10 showed much higher catalytic efficiency than Schreiner's thiourea, despite both of them having similar acidity.18 The condensation of 1a and 2a under the activation of Kass' catalyst C10 proceeded smoothly and yielded desired glycosylation product 3a in 72% yield (entry 11). Switching the solvent to toluene or dichloroethane further increased the yield, to give 3a in 87% and 81% yield, respectively (entries 12 and 13). Next, the effects of temperature and reaction time were examined. Reducing the temperature from 50 °C to 30 °C and shortening the reaction time from 36 hours to 16 hours resulted in a decrease in yield (52%, entry 14). In order to further optimize the glycosylation system, other imidate donors, including perbenzyl 1-O-(N-4-nitro-phenyl-trifluoroacetimidoyl)-D-glucopyranoside 1b and perbenzyl 1-O-(N-phenyl-trifluoroacetimidoyl)-D-glucopyranoside 1c (Table 1), were investigated next. To our delight, donor 1b reacted with acceptor 2a in the presence of Kass catalyst C10 at 30 °C producing disaccharide 3a in 80% yield in 16 hours (entry 15). Perbenzyl 1-O-(N-phenyl-trifluoroacetimidoyl)-D-glucopyranoside donor 1c, without the nitro substituent on the leaving group, afforded the desired product with an even higher yield (86%) under the same reaction conditions (entry 16). The enhanced reactivity of 1c over 1b may be due to the electron withdrawing effect of the nitro substituent on the leaving group of donor 1b that decreases the electron density of the nitrogen of the glycosyl imidate, which acts as the actual H-bond acceptor.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Donor | Catalyst | Solvent | Temp. | Time | Yield |
| 1 | 1a | No | CH3CN | 50 °C | 36 h | <5% |
| 2 | 1a | C1 (0.1 eq.) | CH3CN | 50 °C | 36 h | 12% |
| 3 | 1a | C2 (0.1 eq.) | CH3CN | 50 °C | 36 h | 17% |
| 4 | 1a | C3 (0.1 eq.) | CH3CN | 50 °C | 36 h | 5% |
| 5 | 1a | C4 (0.1 eq.) | CH3CN | 50 °C | 36 h | 10% |
| 6 | 1a | C5 (0.1 eq.) | CH3CN | 50 °C | 36 h | 15% |
| 7 | 1a | C6 (0.1 eq.) | CH3CN | 50 °C | 36 h | 24% |
| 8 | 1a | C7 (0.1 eq.) | CH3CN | 50 °C | 36 h | 20% |
| 9 | 1a | C8 (0.1 eq.) | CH3CN | 50 °C | 36 h | 10% |
| 10 | 1a | C9 (0.1 eq.) | CH3CN | 50 °C | 36 h | 16% |
| 11 | 1a | C10 (0.1 eq.) | CH3CN | 50 °C | 36 h | 72% |
| 12 | 1a | C10 (0.1 eq.) | Toluene | 50 °C | 36 h | 87% |
| 13 | 1a | C10 (0.1 eq.) | DCE | 50 °C | 36 h | 81% |
| 14 | 1a | C10 (0.1 eq.) | DCM | 30 °C | 16 h | 52% |
| 15 | 1b | C10 (0.1 eq.) | DCM | 30 °C | 16 h | 80% |
| 16 | 1c | C10 (0.1 eq.) | DCM | 30 °C | 16 h | 86% |
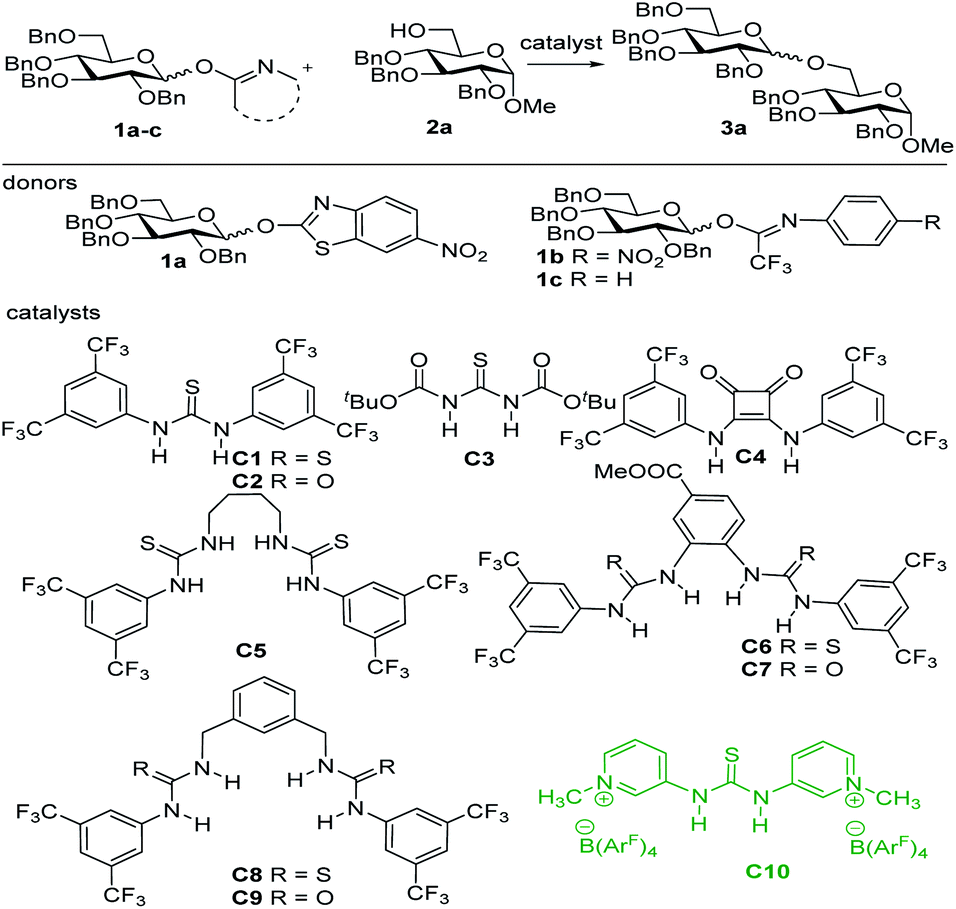
With an optimized glycosylation protocol in hand, the scope and general application of this activation system were further explored using different donors and acceptors (Table 2). We were pleased to find that primary alcohol 2a, secondary alcohol 2b, and tertiary alcohol 1-adamantanol 2c all coupled smoothly with glucosyl donor 1c, providing disaccharides 3a, 3b and compound 3c in 88–93% yields (entries 1–3). The reactions can be readily scaled up and disaccharide 3a could be produced on a gram scale in 89% yield. Notably, the hydrogen bond mediated glycosylation system works very well for acid labile nucleophiles. The steviol aglycon 2d, ergocalciferol 2e and perillyl alcohol 2f were glycosylated with 1c or 1d, yielding the coupled glycosides 3d–3f in 61–86% yield (entries 4–6). Therefore, this mild hydrogen bond activation system provides a powerful tool for the direct glycosylation of acid labile acceptors, enabling the first chemical synthesis of the extremely acid labile glycosylated ergocalciferol 3e. To show the general applicability of the mild glycosylation system, a wide array of glycosyl donors 1d–1i including D-galacto-, D-manno-, L-rhamno-, L-fuco-, D-xylopyranosyl donors, and a D-ribofuranosyl donor were coupled successfully with various acceptors to produce the corresponding glycosides 3g–3n in excellent yields (77–99%, entries 7–14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Temperature | Product | Yielda |
α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :βb :βb |
|
a The reaction yields depended on isolated products.
b The anomeric ratios were determined by 1H NMR analysis or separation.
c 0.15 equivalent C10 was used; N1 product: 59% (α:β 1:4.7); N2 product: (37% 1:3).
d 0.2 equivalent C10 was used.
|
||||||
| 1 | 1c | 2a | 27 °C | 3a | 93% | 1:1 |
| 2 | 1c | 2b | 30 °C | 3b | 88% | 3:1 |
| 3 | 1c | 2c | 30 °C | 3c | 92% | 1.2:1 |
| 4 | 1c | 2d | 30 °C | 3d | 81% | 1:1.4 |
| 5 | 1c | 2e | 3 °C | 3e | 61% | 1.2:1 |
| 6 | 1d | 2f | 27 °C | 3f | 86% | 1:1 |
| 7 | 1d | 2g | 30 °C | 3g | 85% | 2.2:1 |
| 8 | 1d | 2h | 30 °C | 3h | 97% | 1:1.5 |
| 9 | 1e | 2a | 30 °C | 3i | 91% | 1.6:1 |
| 10 | 1f | 2a | 19 °C | 3j | 99% | 1:1 |
| 11 | 1f | 2i | 19 °C | 3k | 88% | 1.5:1 |
| 12 | 1g | 2a | 25 °C | 3l | 77% | 1:1.4 |
| 13 | 1h | 2a | 27 °C | 3m | 93% | 1:1.5 |
| 14 | 1i | 2a | 27 °C | 3n | 88% | 1:1 |
| 15 | 1c | 2j | 30 °C | 3o | 85% | 1:1 |
| 16 | 1c | 2k | 30 °C | 3p | 84% | 2.2:1 |
| 17 | 1e | 2l | 25 °C | 3q | 75% | 2.5:1 |
| 18 | 1c | 2m | 50 °C | 3r | 66% | 1:1.3 |
| 19 | 1c | 2n | 30 °C | 3s | 67% | 2:1 |
| 20 | 1c | 2o | 50 °C | 3t | 81% | 2:1 |
| 21 | 1c | 2p | 30 °C | 3u | 98% | 1.3:1 |
| 22 | 1d | 2q | 30 °C | 3v | 94% | 1.5:1 |
| 23 | 1c | 2r | 30 °C | 3w | 96%c | |
| 24 | 1i | 2s | 40 °C | 3x | 62%d | 1.5:1 |

Furthermore, both thioglycoside acceptor 2j and o-(p-methoxyphenylethynyl)phenyl glycoside acceptor 2k reacted smoothly with imidate donor 1c under C10-mediated activation to yield 3o and 3p, indicating that this activation system could be applied for glycosylation between glycosyl donors and acceptors bearing orthogonal leaving groups (entries 15 and 16). Notably, the very acid labile glycal acceptor 2l could be successfully condensed with glycosyl donor 1e under the mild glycosylation conditions to provide disaccharide 3q (entry 17). Encouraged by these results in O-glycoside synthesis, the assembly of C-glycoside, S-glycosides and N-glycosides in the activation system was investigated. 1,3,5-Trimethoxybenzene 2m, thiophenol 2p and L-cysteine acceptors 2n–2o were glycosylated successfully with 1c to afford C-glycoside 3r and S-glycosides 3s–3u in good yield (66–98%), respectively (entries 18–21). In addition, the coupling of N,4-dimethyl benzenesulfonamide 2q with donor 1d gave N-glycosyl sulfonamide 3v in 94% yield (entry 22). This result shows that the mild activation system can be adopted for the synthesis of glycosyl sulfonamides, which have been investigated as cytostatic agents.19 Benzotriazole 2r and purine 2s could be glycosylated with donor 1c and ribofuranosyl imidate 1i to give the desired N-glycoside 3w in 96% yield and nucleoside 3x in 62% yield, respectively (entries 23 and 24).
We then focussed our attention on the development of stereoselective glycosylation using the hydrogen bond activated glycosylation system (Scheme 1). The activation of the imidate donors with catalyst C10 likely leads to the generation of an oxocarbenium ion intermediate with a weakly coordinating tetrakis(3,5-bis(trifluoromethyl)phenyl)borate counterion (for more details of initial studies into the reaction mechanism see the ESI†). To control the stereoselectivity of SN1-type glycosylation,20 different strategies were explored including neighboring group participation, conformational constriction, additive-controlled glycosylation and hydrogen bond mediated aglycon delivery. Neighboring group participation is the most widely used strategy for the construction of 1,2-trans-glycosidic bonds.20b,c,21 However, when perbenzoyl-1-O-(N-phenyl-trifluoroacetimidoyl)-D-glucopyranoside donor 4a was used to glycosylate acceptor 2a, disaccharide 5a was obtained with relatively moderate stereoselectivity. The formation of the undesired α-anomer may originate from the orthoester intermediate under the mild catalysis conditions. Therefore, the bulky trimethylbenzoyl (TMBz) group,22 a more potent neighboring group, was evaluated, resulting in the formation of the desired products 5b–5f in good yield (77–95%) with excellent stereoselectivity (see Scheme 1A). Several conformationally restricted donor systems have been introduced over the years to enable the stereoselective construction of glycosidic linkages.23 We tested a silylidene protected galactosyl donor 4e to generate the desired products 5g–5i, which were isolated in good yield and as single anomers (see Scheme 1B). Much effort has recently been directed towards the in situ generation of reactive species through the use of nucleophilic additives or reactivity modulators to allow for stereoselective glycosylation reactions. Especially the use of DMF has found wide application in the construction of 1,2-cis-glucosides.24 We successfully combined this strategy with the new glycosylation system to produce 1,2-cis-glucosides (5j–5k) and 1,2-cis-galactosides (3g and 5l) in good yield and excellent stereoselectivity (see Scheme 1C). Hydrogen bond-mediated aglycon delivery (HAD) was introduced by Demchenko and co-workers to direct the acceptor to the desired face of the donor glycoside. This approach has now been applied for the stereoselective construction of many different glycosidic linkages.25 We investigated the combination of this strategy with our glycosylation system for the selective formation of β-glucosides, β-galactosides and β-rhamnosides (see Scheme 1D). The glucosyl donor with the directing picoloyl group (Pico) on the C-6 position delivered the desired β-product 5m (81%, β:α = 7.5:1). The use of the directing picoloyl group on the C-4 position of a galactosyl donor led to the desired β-product 5n in 83% yield with good stereoselectivity (β:α = 11:1). Even the very challenging β-rhamnoglycosidic bond could be forged using the C-3-picoloyl group and disaccharide 5o was generated in 87% yield with acceptable stereoselectivity (β:α = 3:1).
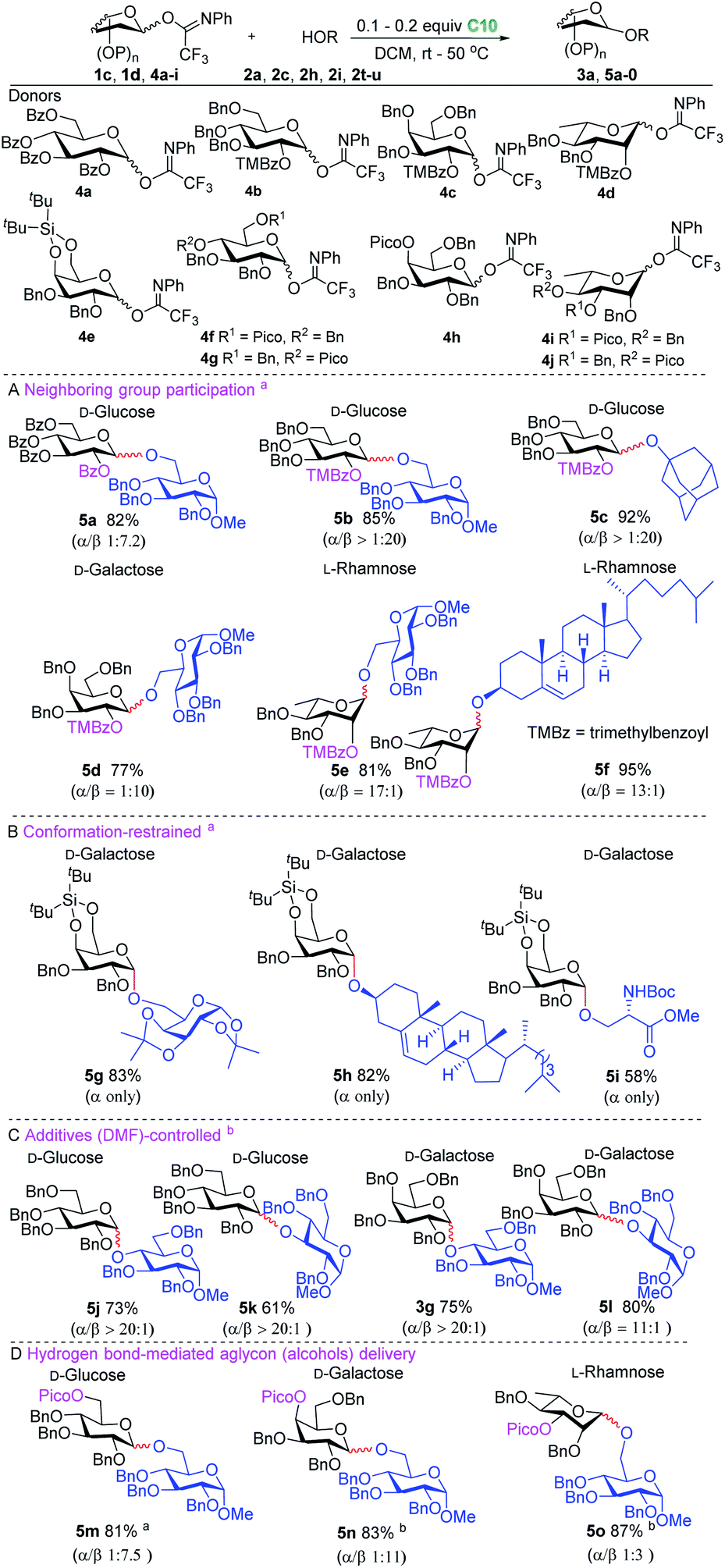 | ||
| Scheme 1 Stereoselective glycosylation using the hydrogen bond mediated glycosylation system combined with different stereoselective glycosylation strategies ((A) neighboring group participation; (B) conformation-restrained; (C) additive-controlled; (D) hydrogen bond-mediated aglycon delivery). Highlighted in blue are nucleophilic acceptors and in red are glycosidic bonds cleaved and formed. a0.1 equivalent C10 was used; b0.2 equivalent C10 was used. The reaction yields depended on isolated products; the anomeric ratios were determined by 1H NMR analysis or separation of the anomers by chromatography. | ||
Having successfully combined our hydrogen bond-mediated activation with the HAD system, we realized that this strategy could provide access to the effective synthesis of phenolic glycosides, which have been shown to display diverse biological activities and pharmaceutical potential.26 Phenolic O-glycosylation is challenging due to the poor nucleophilicity of phenols (especially those bearing electron-withdrawing groups) as well as the Fries-type rearrangement of O-aryl glycosides to C-glycosides of phenols, bearing electron-donating groups, under acidic reaction conditions.
We envisioned that the relatively low pKa of phenols leads to a stronger hydrogen bond with the directing picoloyl group on the glycosyl donor, which not only increases the phenol acceptors' nucleophilicity but also increases the stereoselectivity. As shown in Scheme 2, the glucosyl donor with a directing picoloyl group at the C-6 position was coupled with 4-methoxyphenol to produce the desired phenolic glycoside 7a in 94% yield with good stereoselectivity (β:α = 8:1). A wide range of glycosyl donors with a Pico-directing group were next glycosylated with electron-rich and electron-poor phenols to afford the O-aryl glycosides 7b–7i in excellent yield with good to excellent stereoselectivity. The challenging β-O-aryl rhamnosides 7f–7h were obtained in high yield with excellent stereoselectivity (82–98%, α:β ≥ 1:10). The increase in the stereoselectivity of this system with respect to the O-alkyl β-rhamnosylation (towards 5o, Scheme 1D) supports the notion that the stronger H-bond leads to more effective delivery of the aglycon. Encouraged by these results, we investigated the glycosylation of the very electron-poor 3,4-dicarbomethoxyphenol, generating the desired product 7j and 7k in 85% and quantitative yield, respectively, with good stereoselectivity (β:α = 10:1 and α:β > 20/1). To benchmark our procedure, some experiments under ‘classic’ TfOH-catalyzed conditions were performed (indicated in red in Scheme 2). These experiments showed that these reactions proceeded with lower yield and/or poorer stereoselectivity (7a: 94%, α:β = 1:8 vs. 67%, α:β = 1:3; 7c: 70%, α:β > 20:1 vs. 50%, α:β > 20:1; 7d: 92%, α:β = 1:7 vs. 82%, α:β = 1:3.5; 7j: quantitative yield, α:β = 1:10 vs. 81%, α:β = 1:5.5; 7k: 85%, α:β > 20:1 vs. 53%, α:β > 20:1). Likely, TfOH can protonate the Pico group, which not only prevents the formation of a hydrogen bond with the acceptor, resulting in poorer stereoselectivity, but also decreases the donor's reactivity resulting in a lower yield.
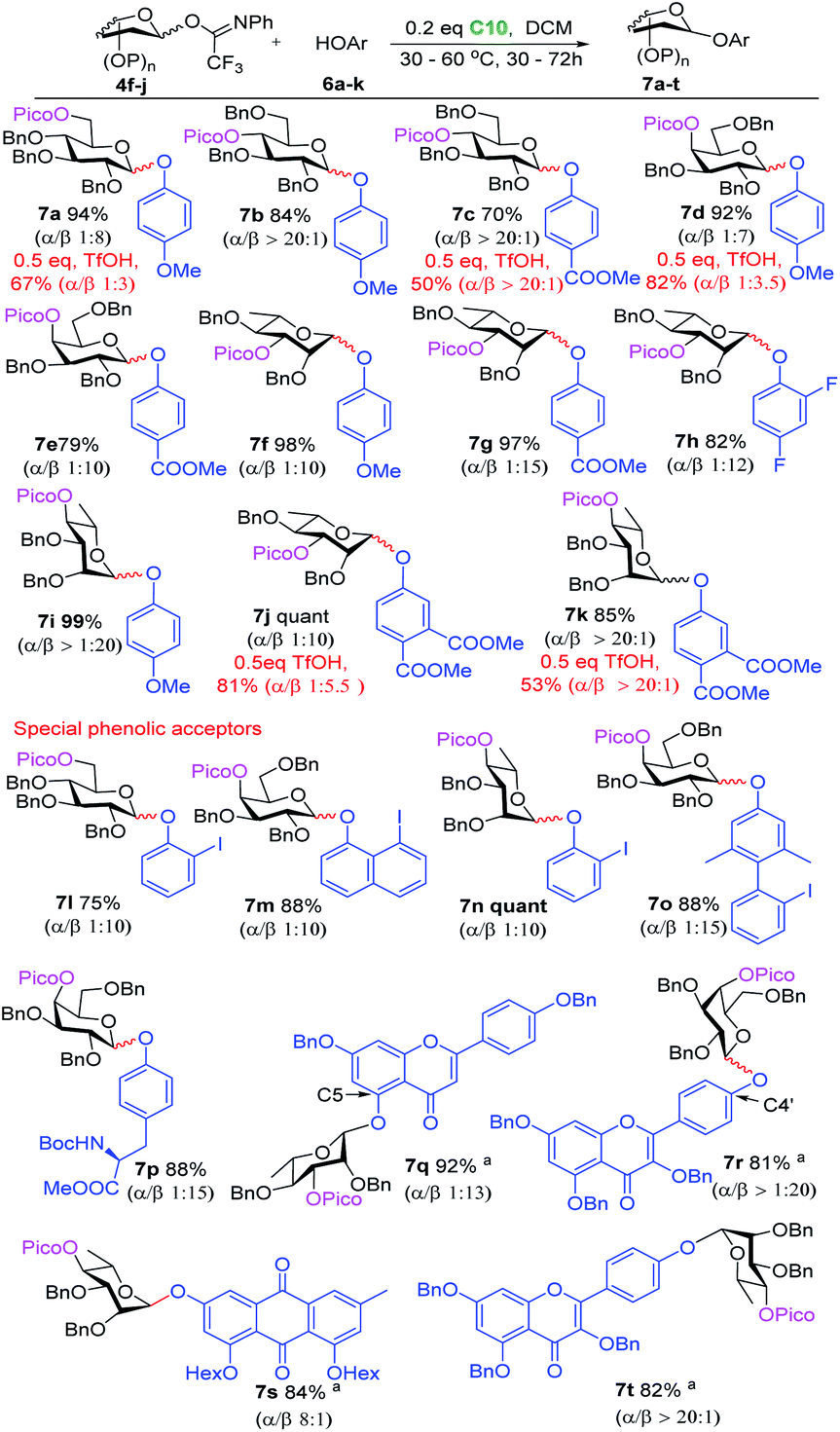 | ||
| Scheme 2 Phenolic glycosylation substrate scope. The nucleophilic acceptors are highlighted in blue and glycosidic bonds cleaved and formed are highlighted in red. a0.3 equivalent C10 was used. The reaction yields depended on isolated products; the anomeric ratios were determined by 1H NMR analysis or separation of the anomers by chromatography. | ||
We then explored more elaborate phenolic acceptors. 2-Iodophenol, 8-iodo-1-naphthol and 2′-iodo-2,6-dimethyl-[1,1′-biphenyl]-4-ol were glycosylated with different Pico-bearing glycosyl donors to give the desired products 7l–7o in 75–100% yield with excellent stereoselectivity (>10:1). The former can be easily transferred into an o-(p-methoxyphenylethynyl)phenyl glycoside (MPEP donor),4e while the latter can provide a 3,5-dimethyl-4-(2′-phenylethynylphenyl)phenyl glycoside (EPP donor).4b Boc-protected L-tyrosine could be coupled with galactosyl donor 4h to afford glycosyl amino acid 7p in 88% yield with excellent stereoselectivity (β:α = 15:1). The mild glycosylation conditions ensured that the acid-labile Boc-group remained unscathed. Flavonoid O-glycosides possess a wide spectrum of biological activities, showing antimicrobial, anticancer, and radical-scavenging potential.26 The extraordinary low reactivity of the flavonoid phenolic OHs makes the formation of glycosidic linkages with these alcohols very challenging.27 Gratifyingly, 4′,7-di-O-benzyl-apigenin and 3,5,7-tri-O-benzyl-kaempferol were glycosylated uneventfully to give the desired 7q in 92% yield (β:α = 13:1) and 7r in 81% yield with excellent stereoselectivity (β:α > 20:1). Finally, the utility of our glycosylation protocol was successfully demonstrated in the efficient synthesis of two fully protected natural phenolic glycosides: emodin-3-O-α-L-rhamnopyranoside and kaempferol-4′-O-α-L-rhamnopyranoside, which show interesting antioxidant, anti-inflammatory and antitumor properties and which are important components of medicinal herbs.28 Thus, 1,8-di-O-n-hexanoyl-emodin was coupled with rhamnosyl donor 4j to produce the phenolic glycoside 7s in 84% yield with good stereoselectivity (α:β = 8:1). Similarly, tri-O-benzyl kaempferol was glycosylated with this donor to afford the phenolic glycoside 7t in excellent yield with stereoselectivity (82%, α:β > 20:1).
Having established the utility of our protocol in the assembly of these targets, we realized that the difference in the reactivity of the different imidate-type donors (see Table 1) and the mildness of the developed activation procedure might enable the development of a chemoselective glycosylation procedure using different glycosyl imidates. The conception of chemoselective glycosylation procedures has streamlined oligosaccharide synthesis and found application in the assembly of a plethora of biologically relevant oligosaccharides. However, the use of glycosyl imidate acceptors in chemoselective glycosylation sequences has only been reported once due to the high reactivity of these glycosides.29 We first sought to establish the relative reactivity of different types of imidates under our hydrogen bond mediated activation conditions. Therefore, the perbenzyl glucose N-phenyl trifluoroacetimidate donor 1c was used to compete with the corresponding nitro-benzothiazolyl imidate 1a, N-4-nitro-phenyl trifluoroacetimidate 1b, trichloroacetimidate 8a, and benzoxazolyl imidate 8b (Scheme 3A–D). Thus, Kass' catalyst C10 (0.1 equiv.) was added to a mixture of N-phenyl-trifluoroacetimidoyl 1c, perbenzyl 6-nitro-2-benzothiazolyl glucopyranoside 1a (1.0 equiv. each) and sugar alcohol 2a (1.0 equiv.) in CH2Cl2 at 0 °C. The reaction mixture was stirred for 10 hours, during which time it was gradually warmed to room temperature. It was observed that the N-phenyl-trifluoroacetimidate donor 1c was completely consumed, leading to the disaccharide product 3a in 72% isolated yield, while the 6-nitro-benzothiazolyl imidate donor 1a could be recovered in 81%. In a similar competitive reaction of N-phenyl-trifluoroacetimidate 1c and N-(4-nitro-phenyl)-trifluoroacetimidate 1b, the former was fully activated, leading to coupled disaccharide 3a in 95% yield, while the latter remained largely intact (82% recovery) (Scheme 3A). The competitive reaction between 1c and trichloroacetimidate 8a revealed that the latter was more reactive under the H-bond activation conditions, although the difference in reactivity was relatively small and N-phenyl trifluoroacetimidate donor 1c could be recovered in 40% along with 12% of donor 8a. Benzoxazolyl imidate donor 8b was shown to be most reactive according to the competitive reaction with 1c, and the latter could be recovered in 50% yield (Scheme 3B). By adjusting the protecting groups of the donors, a larger difference in relative reactivity could be created. As shown in Scheme 3C, in the competitive reactions of the trimethyl benzoyl protected N-phenyl trifluoroacetimidate donor 4b with 8a or 8b, the latter donors were completely consumed, while donor 4b could be recovered in 84% and 85% yield respectively. Similar competitive reactions using TMSOTf catalysis provided both disaccharides 3a and 5b, while none of the donors could be recovered (Scheme 3D). Thus, H-bond activation appears to be superior to other well-established approaches, for the selective activation of different imidate donors. On the basis of these findings, the one-pot assembly of trisaccharide 10 was explored (Scheme 3E). Initially, 2-O-benzoyl-3,4,6-tri-O-benzyl mannosyl trichloroacetimidate 8c and mannosyl N-phenyl trifluoroacetimidate acceptor 9 were treated with 15 mol% of Kass' catalyst C10 for 17 h. Subsequently, acceptor 2a was added to couple with the newly formed disaccharide, affording trisaccharide 10 in 36% yield. This yield could be further improved through the use of the more reactive benzoxazolyl imidate donor 8d delivering the trisaccharide 10 in 45% yield. The main reason for the moderate overall yield was the fact that acceptor 9 was not completely consumed in the first step coupling. This one-pot reaction sequence illustrates the potential of C10 in the chemoselective activation of different imidate donors. In future one-pot assemblies, fine tuning of donors may be required because of the generally high reactivity of imidate type glycosides.
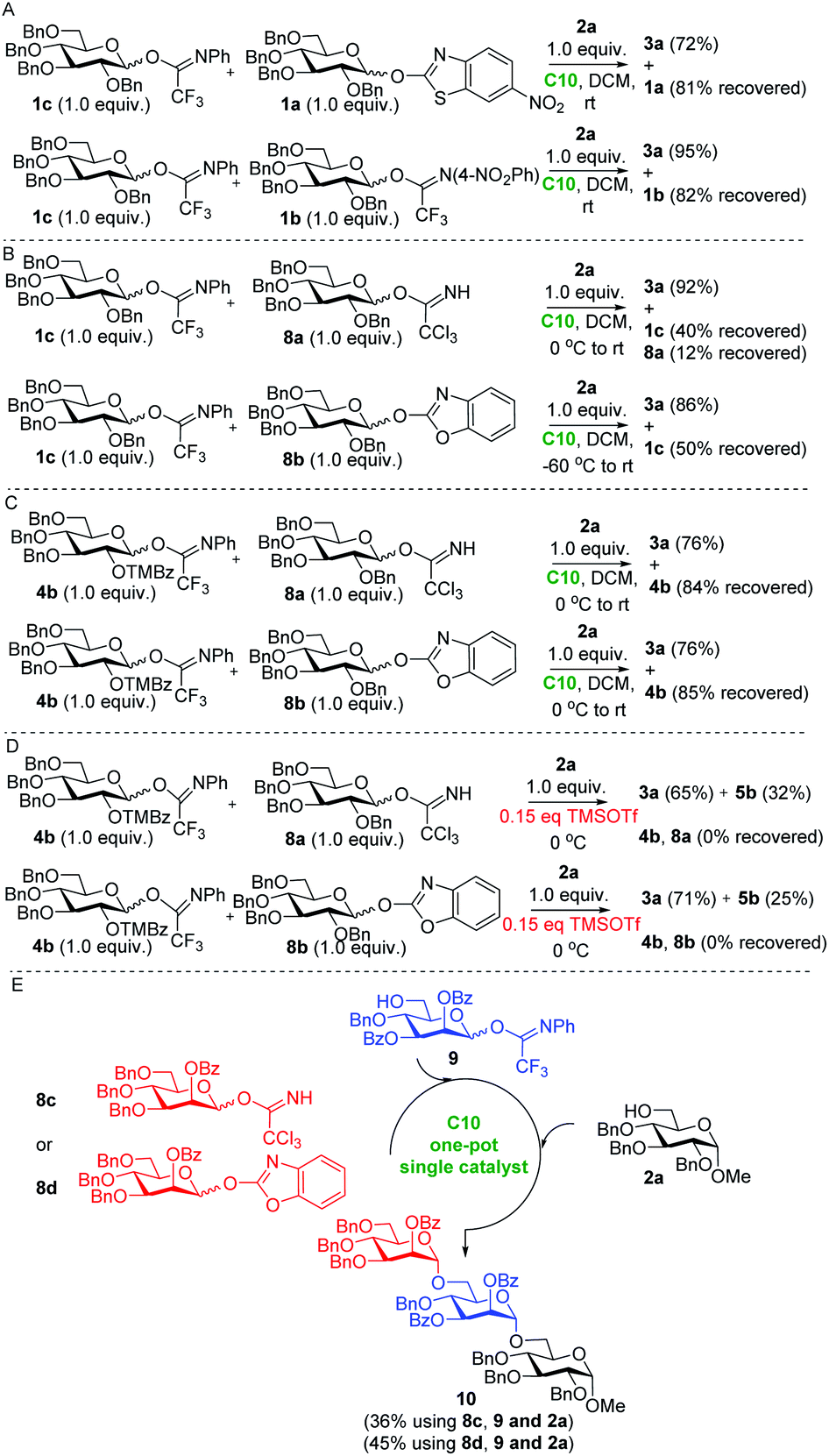 | ||
| Scheme 3 (A–D) Comparison of the different imidate-type donors' reactivity under the action of organocatalyst C10; (E) one-pot single catalyst synthesis of trisaccharide 10. | ||
Conclusions
A novel glycosylation system using glycosyl imidate donors and a charge-enhanced thiourea hydrogen-bond-donor catalyst has been developed for the highly efficient and stereoselective formation of glycosidic bonds. This hydrogen bond-mediated glycosylation system operates under mild conditions and offers broad reaction scope, allowing the effective synthesis of O-, C-, S- and N-glycosides. Taking advantage of the mild reaction conditions, the combination of the new hydrogen bond-mediated glycosylation system with hydrogen bond-mediated aglycon delivery provides a reliable method for the synthesis of phenolic glycosides. The directing picoloyl group on the glycosyl donors can form a hydrogen bond with the phenol alcohol, increasing the acceptors' nucleophilicity and effectively guiding the nucleophile to the same face of the donor to which the Pico group is attached. Benefitting from the mild reaction conditions, glycosyl N-phenyl trifluoroacetimidate donors can be used in conjunction with more reactive imidate-type donors, such as trichloroacetimidates and benzoxazolyl imidates, opening new avenues for chemoselective, one-pot glycosylation strategies. With these promising features, we foresee many applications of this new glycosylation protocol in the assembly of biologically relevant oligosaccharides and glycoconjugates. The method will prove exceptionally useful in the construction of target structures that are highly acid labile.Author contributions
Ke Xiao did most of the experiments; Yongxin Hu did part of work on hydrogen bond-mediated aglycon delivery; Yongyong Wan work on synthesis of the catalyst; Xinxin li and Qin Nie did part of work on phenolic glycosylation; Hao Yan did some work on optimization the glycosylation conditions. Liming Wang did experiments on additives-controlled glycosylation and results analysis/discussion. Jinxi Liao, Deyong Liu and Yuanhong Tu did NMR on mechanism study. Jiansong Sun and Qingju Zhang: funding acquisition, project administration, supervision, writing – review and editing. Jeroen Codee provided many suggestions on this project and organization of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21977039, 21877055, 21762024, 22007039 and 21867012).Notes and references
- (a) C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed; (b) T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- (a) R. Y. Kuo, K. Qian, S. L. Morris-Natschke and K. H. Lee, Nat. Prod. Rep., 2009, 26, 1321–1344 RSC; (b) M. K. Kharel, P. Pahari, M. D. Shepherd, N. Tibrewal, S. E. Nybo, K. A. Shaaban and J. Rohr, Nat. Prod. Rep., 2012, 29, 264–325 RSC; (c) V. K. Tiwaria, R. C. Mishrab, A. Sharmac and R. P. Tripathi, Mini-Rev. Med. Chem., 2012, 12, 1497–1519 CrossRef PubMed; (d) T. Buskas, P. Thompson and G. J. Boons, Chem. Commun., 2009, 5335–5349 RSC; (e) D. Zhu and B. Yu, Chin. J. Chem., 2018, 36, 681–691 CrossRef CAS; (f) F. Micoli, L. Del Bino, R. Alfini, F. Carboni, M. R. Romano and R. Adamo, Expert Rev. Vaccines, 2019, 18, 881–895 CrossRef CAS PubMed.
- (a) K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503–1531 CrossRef CAS; (b) J. D. Codee, R. E. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769–782 RSC; (c) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed; (d) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC; (e) P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS PubMed; (f) M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS PubMed; (g) P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- (a) Y. Zu, C. Cai, J. Sheng, L. Cheng, Y. Feng, S. Zhang, Q. Zhang and Y. Chai, Org. Lett., 2019, 21, 8270–8274 CrossRef CAS PubMed; (b) Z. Hu, Y. Tang and B. Yu, J. Am. Chem. Soc., 2019, 141, 4806–4810 CrossRef CAS PubMed; (c) P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405 CrossRef CAS PubMed; (d) H. Y. Wang, C. J. Simmons, S. A. Blaszczyk, P. G. Balzer, R. Luo, X. Duan and W. Tang, Angew. Chem., Int. Ed., 2017, 56, 15698–15702 CrossRef CAS PubMed; (e) Y. Hu, K. Yu, L. L. Shi, L. Liu, J. J. Sui, D. Y. Liu, B. Xiong and J. S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS PubMed; (f) L. Meng, P. Wu, J. Fang, Y. Xiao, X. Xiao, G. Tu, X. Ma, S. Teng, J. Zeng and Q. Wan, J. Am. Chem. Soc., 2019, 141, 11775–11780 CrossRef CAS PubMed; (g) K. Le Mai Hoang, J. X. He, G. Bati, M. B. Chan-Park and X. W. Liu, Nat. Commun., 2017, 8, 1146 CrossRef PubMed; (h) S. S. Nigudkar, K. J. Stine and A. V. Demchenko, J. Am. Chem. Soc., 2014, 136, 921–923 CrossRef CAS PubMed; (i) D.-K. Liu, D.-C. Xiong, X. Wu, Q. Li and X.-S. Ye, Org. Chem. Front., 2019, 6, 2756–2759 RSC.
- (a) T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef CAS PubMed; (b) M. H. Zhuo, D. J. Wilbur, E. E. Kwan and C. S. Bennett, J. Am. Chem. Soc., 2019, 141, 16743–16754 CrossRef CAS PubMed; (c) J. P. Issa and C. S. Bennett, J. Am. Chem. Soc., 2014, 136, 5740–5744 CrossRef CAS PubMed; (d) F. Yu, J. Li, P. M. DeMent, Y. J. Tu, H. B. Schlegel and H. M. Nguyen, Angew. Chem., Int. Ed., 2019, 58, 6957–6961 CrossRef CAS PubMed.
- (a) B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed; (b) X. Li, C. Li, R. Liu, J. Wang, Z. Wang, Y. Chen and Y. Yang, Org. Lett., 2019, 21, 9693–9698 CrossRef CAS PubMed; (c) W. Li and B. Yu, Chem. Soc. Rev., 2018, 47, 7954–7984 RSC.
- R. Williams and M. C. Galan, Eur. J. Org. Chem., 2017, 2017, 6247–6264 CrossRef CAS.
- Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 10089–10092 CrossRef CAS PubMed.
- L. Sun, X. Wu, D. C. Xiong and X. S. Ye, Angew. Chem., Int. Ed., 2016, 55, 8041–8044 CrossRef CAS PubMed.
- (a) T. Kimura, T. Eto, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 3190–3193 CrossRef CAS PubMed; (b) M. Shaw, Y. Kumar, R. Thakur and A. Kumar, Beilstein J. Org. Chem., 2017, 13, 2385–2395 CrossRef CAS PubMed; (c) A. Dubey, R. Sangwan and P. K. Mandal, Catal. Commun., 2019, 125, 123–129 CrossRef CAS; (d) Y. Kobayashi, Y. Nakatsuji, S. Li, S. Tsuzuki and Y. Takemoto, Angew. Chem., Int. Ed., 2018, 57, 3646–3650 CrossRef CAS PubMed.
- (a) Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162–166 CrossRef CAS PubMed; (b) Q. Li, S. M. Levi and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 11865–11872 CrossRef CAS PubMed.
- S. M. Levi, Q. Li, A. R. Rotheli and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 35–39 CrossRef CAS PubMed.
- A. B. Mayfield, J. B. Metternich, A. H. Trotta and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 4061–4069 CrossRef CAS PubMed.
- G. A. Bradshaw, A. C. Colgan, N. P. Allen, I. Pongener, M. B. Boland, Y. Ortin and E. M. McGarrigle, Chem. Sci., 2019, 10, 508–514 RSC.
- C. Xu and C. C. J. Loh, Nat. Commun., 2018, 9, 4057 CrossRef PubMed.
- T. Hashihayata, H. Mandai and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2004, 77, 169–178 CrossRef CAS.
- A. Rostami, A. Colin, X. Y. Li, M. G. Chudzinski, A. J. Lough and M. S. Taylor, J. Org. Chem., 2010, 75, 3983–3992 CrossRef CAS PubMed.
- Y. Fan and S. R. Kass, Org. Lett., 2016, 18, 188–191 CrossRef CAS PubMed.
- R. Crespo, M. G. de Bravo, P. A. Colinas and R. D. Bravo, Bioorg. Med. Chem. Lett., 2010, 20, 6469–6471 CrossRef CAS PubMed.
- (a) R. A. Jeanneret, S. E. Johnson and M. C. Galan, J. Org. Chem., 2020, 85, 15801–15826 CrossRef CAS PubMed; (b) K. Sasaki and K. Tohda, Tetrahedron Lett., 2018, 59, 496–503 CrossRef CAS; (c) Y. Kobayashi and Y. Takemoto, Tetrahedron, 2020, 76, 131328 CrossRef CAS; (d) A. Khanam, A. Tiwari and P. K. Mandal, Carbohydr. Res., 2020, 495, 108045 CrossRef CAS PubMed; (e) S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC; (f) C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed; (g) H. Yao, M. D. Vu and X. W. Liu, Carbohydr. Res., 2019, 473, 72–81 CrossRef CAS PubMed; (h) J. Ling and C. S. Bennett, Asian J. Org. Chem., 2019, 8, 802–813 CrossRef CAS PubMed; (i) J. Zeng, Y. Xu, H. Wang, L. Meng and Q. Wan, Sci. China: Chem., 2017, 60, 1162–1179 CrossRef CAS; (j) W. L. Leng, H. Yao, J. X. He and X. W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed.
- H. Liu, T. Hansen, S. Y. Zhou, G. E. Wen, X. X. Liu, Q. J. Zhang, J. D. C. Codee, R. R. Schmidt and J. S. Sun, Org. Lett., 2019, 21, 8713–8717 CrossRef CAS PubMed.
- R. J. Williams, N. W. McGill, J. M. White and S. J. Williams, J. Carbohydr. Chem., 2010, 29, 236–263 CrossRef CAS.
- (a) D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed; (b) A. Imamura, N. Matsuzawa, S. Sakai, T. Udagawa, S. Nakashima, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2016, 81, 9086–9104 CrossRef CAS PubMed; (c) L. J. van den Bos, R. E. J. N. Litjens, R. J. B. H. N. van den Berg, H. S. Overkleeft and G. A. van der Marel, Org. Lett., 2005, 7, 2007–2010 CrossRef CAS PubMed; (d) S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666–10667 CrossRef CAS PubMed; (e) X. Zhu, S. Kawatkar, Y. Rao and G.-J. Boons, J. Am. Chem. Soc., 2006, 128, 11948–11957 CrossRef CAS PubMed; (f) Y. Geng, Q. Qin and X.-S. Ye, J. Org. Chem., 2012, 77, 5255–5270 CrossRef CAS PubMed.
- (a) S. R. Lu, Y. H. Lai, J. H. Chen, C. Y. Liu and K. K. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed; (b) L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
- (a) J. P. Yasomanee and A. V. Demchenko, J. Am. Chem. Soc., 2012, 134, 20097–20102 CrossRef CAS PubMed; (b) Y.-F. Wu and Y.-F. Tsai, Org. Lett., 2017, 19, 4171–4174 CrossRef CAS PubMed; (c) M. Teruaki, H. Takashi and M. Hiroki, Chem. Lett., 2003, 32, 340–341 CrossRef; (d) M. P. Mannino and A. V. Demchenko, Chem.–Eur. J., 2020, 26, 2938–2946 CrossRef CAS PubMed; (e) Y. Xu, H.-C. Bin, F. Su and J.-S. Yang, Tetrahedron Lett., 2017, 58, 1548–1552 CrossRef CAS; (f) D.-M. Liu, H.-L. Wang, J.-C. Lei, X.-Y. Zhou and J.-S. Yang, Eur. J. Org. Chem., 2020, 2020, 575–585 CrossRef CAS.
- (a) S. P. B. Ovenden, M. Cobbe, R. Kissell, G. W. Birrell, M. Chavchich and M. D. Edstein, J. Nat. Prod., 2011, 74, 74–78 CrossRef CAS PubMed; (b) J. K. MacLeod, H. B. Rasmussen and A. C. Willis, J. Nat. Prod., 1997, 60, 620–622 CrossRef CAS PubMed; (c) J. B. Harborne and C. A. Williams, Nat. Prod. Rep., 2001, 18, 310–333 RSC; (d) N. C. Veitch and R. J. Grayer, Nat. Prod. Rep., 2011, 28, 1626–1695 RSC; (e) M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266–1281 CrossRef CAS PubMed.
- J.-X. Liao, N.-L. Fan, H. Liu, Y.-H. Tu and J.-S. Sun, Org. Biomol. Chem., 2016, 14, 1221–1225 RSC.
- (a) G. Song, H. Liu, W. Zhang, M. Geng and Y. Li, Bioorg. Med. Chem., 2010, 18, 5183–5193 CrossRef CAS PubMed; (b) J. Alsarraf, P. Gormand, J. Legault, M. Mihoub and A. Pichette, Tetrahedron Lett., 2020, 61, 151706 CrossRef CAS; (c) N. Y. Lee, W. K. Khoo, M. A. Adnan, T. P. Mahalingam, A. R. Fernandez and K. Jeevaratnam, J. Pharm. Pharmacol., 2016, 68, 953–969 CrossRef CAS PubMed; (d) F. Shaheen, L. Ali, S. Ali, N. Erdemoglu and B. Sener, Chem. Nat. Compd., 2009, 45, 346–349 CrossRef CAS; (e) A. Ibrahim, S. I. Khalifa, I. Khafagi, D. T. Youssef, S. Khan, M. Mesbah and I. Khan, Chem. Pharm. Bull., 2008, 56, 1253–1258 CrossRef CAS PubMed.
- M. Adinolfi, A. Iadonisi and A. Ravidà, Synlett, 2006, 583–586 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05772c |
| This journal is © The Royal Society of Chemistry 2022 |