
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of an ambient stable pyreno[4,5-b]pyrrole monoanion and pyreno[4,5-b:9,10-b′]dipyrrole dianion: from serendipity to design†
Sharvan
Kumar
*a,
Kohshi
Yoshida‡
a,
Yusuke
Hattori‡
a,
Tomohiro
Higashino
 a,
Hiroshi
Imahori
ab and
Shu
Seki
*a
a,
Hiroshi
Imahori
ab and
Shu
Seki
*a
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: seki@moleng.kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 4th January 2022
Abstract
The stability of singly or multiply negatively charged π-conjugated organic compounds is greatly influenced by their electronic delocalization. Herein, we report a strategic methodology for isolation of a mysterious compound. The isolated compounds, a pyreno[4,5-b]pyrrole monoanion and pyreno[4,5-b:9,10-b′]dipyrrole dianion, were highly stable under ambient conditions due to high delocalization of the negative charge over multiple electron deficient C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups and pyrene π-scaffolds and allowed purification by column chromatography. To our knowledge, this is the first report on TCNE type reductive condensation of malononitrile involving pyrene di- and tetraone and formation of pyrenopyrrole. All compounds were characterized by spectroscopic methods and X-ray crystallography. A UV-vis spectroscopic study shows an intense low energy absorption band with a large absorption coefficient (ε).
N groups and pyrene π-scaffolds and allowed purification by column chromatography. To our knowledge, this is the first report on TCNE type reductive condensation of malononitrile involving pyrene di- and tetraone and formation of pyrenopyrrole. All compounds were characterized by spectroscopic methods and X-ray crystallography. A UV-vis spectroscopic study shows an intense low energy absorption band with a large absorption coefficient (ε).
Introduction
Extended π-conjugated negatively charged organic materials continue to attract considerable attention due to their fundamental importance in understanding the π-electron delocalization within a system and fascinating optoelectronic properties.1–3 Such π-conjugated charged materials can be useful as electrode active materials in batteries,4 organic conductors5,6 and solar cells.7 The strong visible absorbance of these materials provides access to high energy photoexcited electron-transfer reactions and they can also be employed as organic dyes.8,9 Although singly charged π-conjugated organic molecules often show significant stability under ambient conditions, doubly or multiply charged π-conjugated organic molecular materials are intrinsically unstable even in condensed phases due to their high reactivity towards several oxidants, protic solvents, and chlorinated hydrocarbons.9–12 Therefore, novel strategic designs that help in the isolation of such highly electron-rich materials with satisfactory stability and good environment compatibility are needed in view of their feasibility in high density electron doping of molecular materials.Transition metal promoted reductive condensation of tetracyanoethylene (TCNE) yielded an unexpected stable pyrrolenine monoanion, a precursor for a series of pyrrolizinato metal complexes (Scheme 1a).13–15 These metal complexes show low energy intense electronic transitions around 660 nm, similar to metal phthalocyanines (MPs).16 This condensation reaction is considered as the missing element in the preparation of π-conjugated macrocycles starting with polycyano compounds.15 While cis-1,2-dicyanoethylene and 1,2-dicyanobenzene form metal tetraazaporphyrins and metal phthalocyanines through cyclotetramerization under appropriate conditions, TCNE follows cyclodimerization.15 They could also be useful as dyes,17 and in biological applications.16 However, despite the extraordinary optoelectronic properties of the resultant molecules, this reaction was limited to the TCNE scaffold and only pyrrolenine monoanion derivatives have been reported so far. Moreover, this reaction involves hazards and costly transition metals.
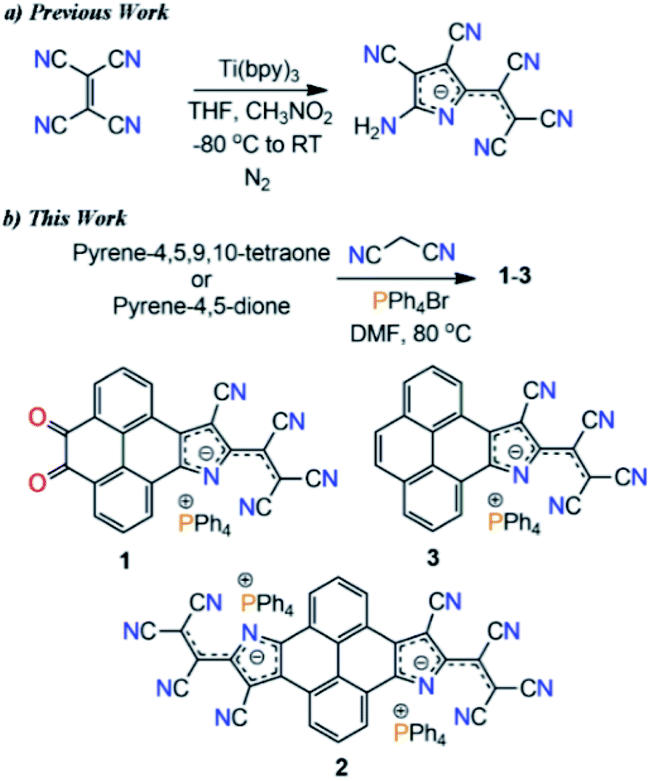 | ||
| Scheme 1 (a) Previously reported synthetic scheme, and (b) metal-free synthesis via reductive Knoevenagel condensation in this work. | ||
Pyrene and its derivatives have witnessed significant advancements in the areas of organic electronics,18–20 sensing,21,22 bio-imaging,23–25 and photocatalysis26,27 due to their unique photophysical and electrochemical properties. It is well established that the optical and electrical properties of a system can be regulated by π-extension over the planar structures in neutral states with small reorganization in charged states, hence their relevant performance on application. Although there are several reports on the extension of pyrene π-systems,28,29 heterocyclic fused pyrene systems are limited,30–32 and only in a few cases have pyrenopyrrole systems been reported.33–35 At the same time, there is no report on a successful synthetic route to pyreno[4,5-b:9, 10-b′]dipyrrole. Moreover, there are only a few reports on singly or multiply charged pyrene anions.36–38
We have been interested in the preparation of π-conjugated electron deficient/rich molecules to yield stable radical ions as platforms with electron conductive pathways and/or magnetism via spin localization.39–41 In this context, we envisaged that tetracyanoquinodimethane (TCNQ) type electron deficient compounds could be obtained through the Knoevenagel condensation reaction of pyrene-4,5,9,10-tetraone with malononitrile (ESI Scheme S1†). In addition, a report on tetracyano-4,5-pyrenoquinodimethanes42 encouraged us to prepare our designed molecules by the condensation reactions of polycyano compounds.
Experimental
General
All the starting materials were sourced either from Sigma-Aldrich, TCI or Wako (Japan). All the chemicals were used as received. Thin layer chromatography (TLC) was carried out on aluminium plates coated with silica gel mixed with a fluorescent indicator sourced from Merck, Germany. NMR (1H and 13C) spectra were recorded on a JEOL JNM-AL 400 MHz FT-NMR spectrometer in CH2Cl2 with TMS as an internal standard. 31P NMR spectra were recorded on a JEOL JNM-ECZ500R/S1 500 MHz FT-NMR spectrometer in CH2Cl2. Spin multiplicities are reported as a singlet (s), doublet (d), triplet (t), and quartet (q) with coupling constants (J) given in Hz, or a multiplet (m). ESI-HRMS mass spectrometry data were obtained using a Thermo Fisher Scientific made Dionex Ultimate 3000 UHPLC system.UV-vis, photoluminescence, and FT-IR spectroscopy
UV-vis and photoluminescence spectra were recorded on JASCO V-570 and HITACHI F-2700 spectrophotometers, respectively. All UV-Vis measurements were performed in a quartz cuvette with a 10.0 mm optical pathlength. UV-grade dichloromethane was used for the spectroscopic experiments. Fourier transform infrared (FT-IR) spectra were recorded on a VERTEX 70 spectrometer. Neat dry powders were directly analysed. A blank scan was run to cut out the air effects before analysis.Cyclic and Differential Pulse Voltammetry (CV/DPV)
CV and DPV were carried out using a computer controlled potentiostat (CHI 612E) and a standard three-electrode arrangement that consisted of platinum as a working and an auxiliary electrode and Ag/AgCl as a reference electrode. All the electrochemical measurements were carried out in Ar-purged CH2Cl2 with n-Bu4NBF4 as the supporting electrolyte. The scan rate for the measurements was typically 200 mV s−1. DPV was carried out keeping the peak amplitude at 50 mV, pulse width at 0.05 s, pulse period at 0.5 s and increment E at 4 mV.X-ray crystallography
Crystals of 1, 2, and 3 were grown in hexane:DCM solution by a slow evaporation method at ∼10 °C in a refrigerator. The reported dataset was collected by mounting the crystal with parabar oil on the loop at 153 K. The X-ray data were collected on a Rigaku Saturn724+ (4 × 4 bin mode) diffractometer using Mo-Kα radiation (λ = 0.71073 Å), generated from the micro-focus sealed tube using φ and ω-scans of 0.5° steps at 153 K. Cell determination, data collection and data reduction were performed with the help of CrysAlisPro 1.171.39.46e (Rigaku OD, 2018) software. Structure solution and refinement were performed using SHELXS-2014 (Sheldrick, 2014). Refinement of coordinates and anisotropic thermal parameters of non-hydrogen atoms was carried out by the full-matrix least-squares method. The hydrogen atoms were generated with idealized geometries and refined isotropically using a riding model.Results and discussion
Toward the synthesis of the target molecules, we followed the reported protocols of the Knoevenagel condensation reaction involving the TiCl4 catalyst, giving a dark blue colored, highly polar, non-isolable, sticky compound. Next, we performed a metal-free reaction of pyrene-4,5,9,10-tetraone and malononitrile in DMF. Again, a highly polar blue colored compound was formed. Meanwhile, upon reaction with 2.0 eq. of malononitrile a highly polar purple color compound was obtained. With the anticipation that this might be forming some ionic material, PPh4Br salt was added into a reaction mixture of 2.0 eq. malononitrile and pyrene-4,5,9,10-tetraone in DMF. Surprisingly, a nonpolar column chromatographically separable purple color compound was formed. Mass-spectrometry analysis suggested the formation of a substituted pyreno[4,5-dioxo-9,10-b]pyrrole monoanion, 1 (Scheme 1b). We found that malononitrile followed the TCNE reductive condensation pathway with pyrene di- and tetraone. To our knowledge, this is the first report on the metal-free Knoevenagel condensation and cyclization reaction.Thereafter, with our best optimized reaction conditions, we prepared 1, pyreno[4,5-b:9,10-b′]dipyrrole dianion (2), and pyreno[4,5-b]pyrrole monoanion (3) with good yields (Scheme 2). All these anions were highly stable and allowed purification via column chromatography. This unprecedented stability of these anions is because of the delocalization of the negative charge over electron deficient tricyanoethanide and cyano groups.13–15 These anions showed a wide range of solubility from non-polar CHCl3 to highly polar DMSO with great stability, and were characterized via spectroscopic methods as well as by X-ray crystallography. On the basis of the Knoevenagel condensation and TCNE cyclodimerization reaction mechanism, we also proposed the plausible synthetic pathway of this reaction (ESI Scheme S2†).
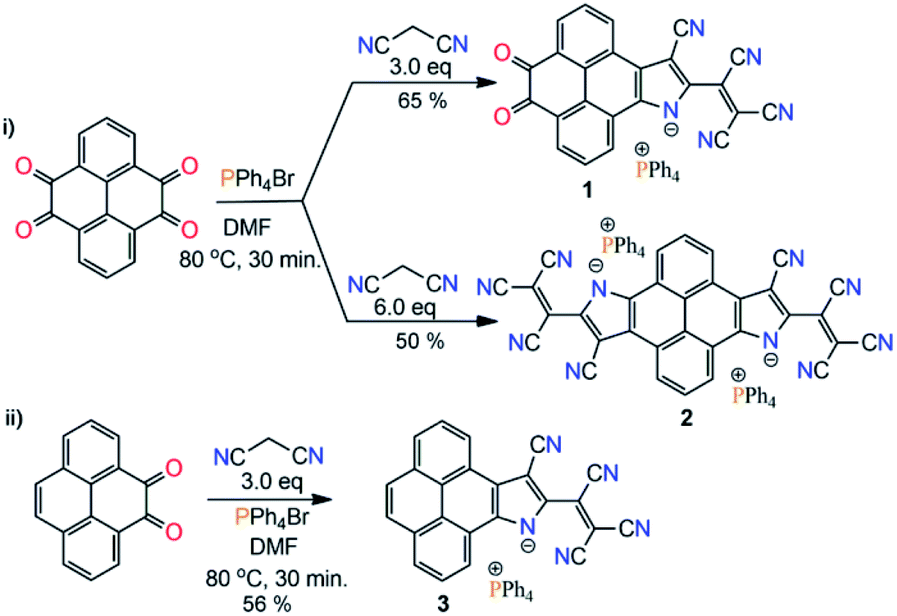 | ||
| Scheme 2 Synthetic scheme of 1–3. | ||
X-ray suitable single crystals of 1, 2, and 3 were grown from a hexane–DCM solution at ∼10 °C.§ X-ray diffraction analysis unequivocally confirms the formation of the pyreno[4,5-b]pyrrole monoanion and pyreno[4,5-b:9,10-b′]dipyrrole dianion and its ion pairing with the PPh4+ counter-cation (Fig. 1 and S1, ESI†). The structural parameters and crystallographic details are given in Tables S1 and S2.† The CN bond length in 1 ranged between 1.146 and 1.160 Å, and in 2 it was between 1.147 and 1.192 Å, while in 3 it ranged between 1.141 and 1.149 Å. Such bond lengthening in the malononitrile CN is observed in the dimalononitrile NDI dianion;43 however, the CN bond lengthening in dianion 2 (1.192 Å) is ∼0.03 Å longer than the reported CN bond lengthening for an NDI-based dianion and tetracyanoquinodimethane (TCNQ) dianion.44 The carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond lengths in 1 ranged between 1.217 and 1.228 Å which is similar to the CO bond length in pyrenedione.45 Moreover, the bond length alterations (BLAs) in the pyrrole part is similar to what is reported for the pyrrolenine monoanion. DFT optimized structures of these anionic compounds also show similar BLAs (ESI Table S1 and Fig. S2†).
O) bond lengths in 1 ranged between 1.217 and 1.228 Å which is similar to the CO bond length in pyrenedione.45 Moreover, the bond length alterations (BLAs) in the pyrrole part is similar to what is reported for the pyrrolenine monoanion. DFT optimized structures of these anionic compounds also show similar BLAs (ESI Table S1 and Fig. S2†).
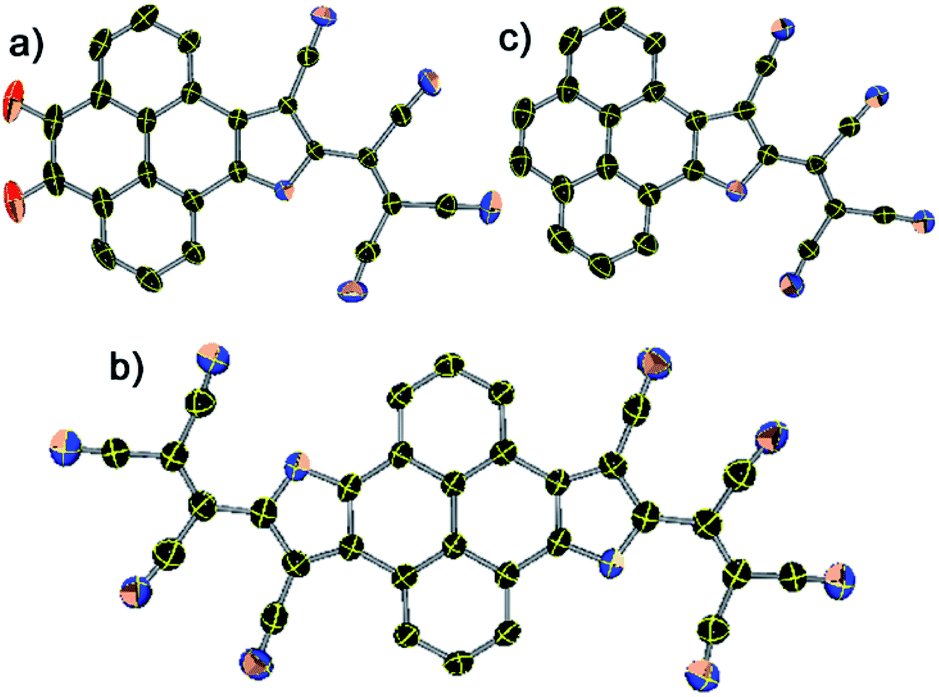 | ||
| Fig. 1 Crystal structure of (a) 1, (b) 2, and (c) 3 (ORTEP representation). Thermal ellipsoids are shown at 50% probability. In all structures, H atoms and counter-cation PPh4+ have been removed for clarity. | ||
The pyrene scaffold forms a strong face-to-face π-stacking interaction with a distance ranging from 3.135 to 3.388 Å in 1, while 3 shows weak face-to-face π-stacking interaction with a distance ranging between 3.417 and 4.240 Å (ESI Fig. S3†). On the other hand, 2 forms strong off-set π-stacking in such a manner that two CN groups directly face the pyrene-π scaffold with a distance of 3.352–3.678 Å (ESI Fig. S4†). Importantly, CN group forms multiple CN⋯H–C H-bonding with the PPh4+ counter cation having N⋯H contacts between 2.514 and 2.733 Å for 1, 2.526–2.858 Å for 2, and 2.563–2.730 Å for 3, respectively (ESI Fig. S5†). These ionic moieties are believed to be stabilized significantly by the multiple H-bonding in the single pyrene scaffold, via an infinite network of the molecules.
UV-vis spectroscopy of 1, 2, and 3 exhibited intense absorption maxima in the low energy region (<2.5 eV) accompanied by a vibronic progression of multiple low intensity high energy absorption peaks (Fig. 2). The transition patterns of these molecules are similar to those of the reported pyrrolenine monoanion and their complexes, and accordingly the main low energy intense absorption was due to the π–π* transition.16,17 Compound 1 absorbs at 549 and 327 nm with a molar extinction coefficient (ε) of 5.7 × 104 dm3 mol−1 cm−1, while 3 shows main absorptions at 596, 557, 403, and 380 nm with ε = 3.8 × 104 dm3 mol−1 cm−1. On the other hand, 2 shows the highest ε (7.1 × 104 dm3 mol−1 cm−1) and lowest energy transition at 640 nm with a clear shoulder at 592 nm along with minor peaks at 472, 353, and 339 nm. The intense visible transition with high ε suggests the feasibility of the compounds as dye materials. All the compounds were, however, non-emissive upon excitation at the corresponding electronic transition maxima: they are likely cases of ion-pairing molecular systems with multiple energy dissipating pathways via charge transfer processes.46,47 Only compound 1 exhibited extremely weak photoluminescence at 645 nm under excitation at 550 nm (ESI Fig. S8†).
 | ||
| Fig. 2 UV-vis absorption of 1 (red line), 2 (blue line), and 3 (black line) in CH2Cl2 at 1 × 10−5 mol dm−3 (solution pictures were taken at 2 × 10−5 mol dm−3). | ||
The redox properties of these anions were evaluated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) analyses using 0.1 M tetrabutylammonium tetrafluoroborate (n-Bu4NBF4) as a supporting electrolyte, Ag/AgCl as a reference electrode, and platinum as a working and a counter electrode (Fig. 3 and S6, ESI†). The CV demonstrated two reversible reduction waves for 1 (−1.01 and −1.37 V vs. Fc/Fc+) and 2 (−1.25 and −1.36 V vs. Fc/Fc+), corresponding to a sequential two-step electron transfer process. On the other hand, 3 shows one reversible reduction wave at −1.27 V vs. Fc/Fc+ corresponding to a one-electron transfer process. This corresponds to a LUMO level of −3.79 eV for 1, −3.55 eV for 2, and −3.53 eV for 3, on the basis of EHOMO = 4.8 eV for Fc against vacuum.48 It is noteworthy that despite being negatively charged, these anions did not show any clear oxidation voltammograms (ESI Fig. S7†). Moreover, their LUMO level is still significantly low and comparable with the LUMO level of unsubstituted NDI (∼3.67–3.94 eV).49,50 The important optoelectronic and redox property data are summarized in Table 1.
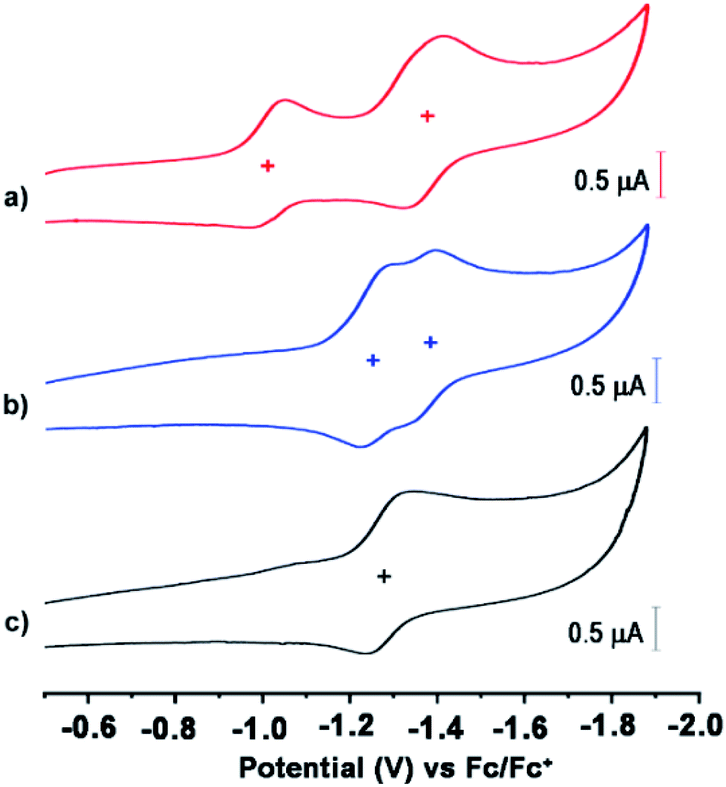 | ||
| Fig. 3 Cyclic voltammetry of (a) 1, (b) 2, and (c) 3. Conditions: 1 × 10−4 mol dm−3 in CH2Cl2; reference electrode, Ag/AgCl; working and auxiliary electrodes, Pt with 0.1 mol dm−3n-Bu4NBF4 and ferrocene/ferrocenium (Fc/Fc+); 298 K. | ||
| Mol. | Potential (V) vs. Fc/Fc+ | LUMO (eV) | Exp. λabsmax [nm] (ε, dm3 mol−1 cm−1) | Exp. λemmax [nm] | |
|---|---|---|---|---|---|
| E Red1 | E Red2 | ||||
| a N.D.: not detected. | |||||
| 1 | −1.01 | −1.37 | −3.79 | 549 (57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) 000) |
645 |
| 2 | −1.25 | −1.36 | −3.55 | 640 (71000) |
N.D. |
| 3 | −1.27 | — | −3.53 | 596 (38000) |
N.D. |
Further, the electron affinity and the importance of electronic delocalization for the extraordinary stability of 1–3 were confirmed by density functional theory (DFT) calculations (Fig. 4). The LUMO energy level was found to be at −3.35 eV for 1, −2.96 eV for 2, and −3.06 eV for 3. The calculated HOMO level was found to be −5.82 eV, −5.18 eV, and −5.39 eV for 1, 2, and 3, respectively. The complete delocalization of the HOMO over the entire moiety in 1–3 anions confirms the delocalization of the extra electrons on the entire π-conjugated skeleton. In addition, the HOMO orbitals are oriented in a similar fashion to the BLAs realized by single crystals. Moreover, the significant electron affinity due to the presence of a flanged electron deficient TCNE type group and supramolecular H-bonding suggests that the electronic delocalization, supramolecular interactions, and electron deficient groups inclusively make these anionic species highly stable.
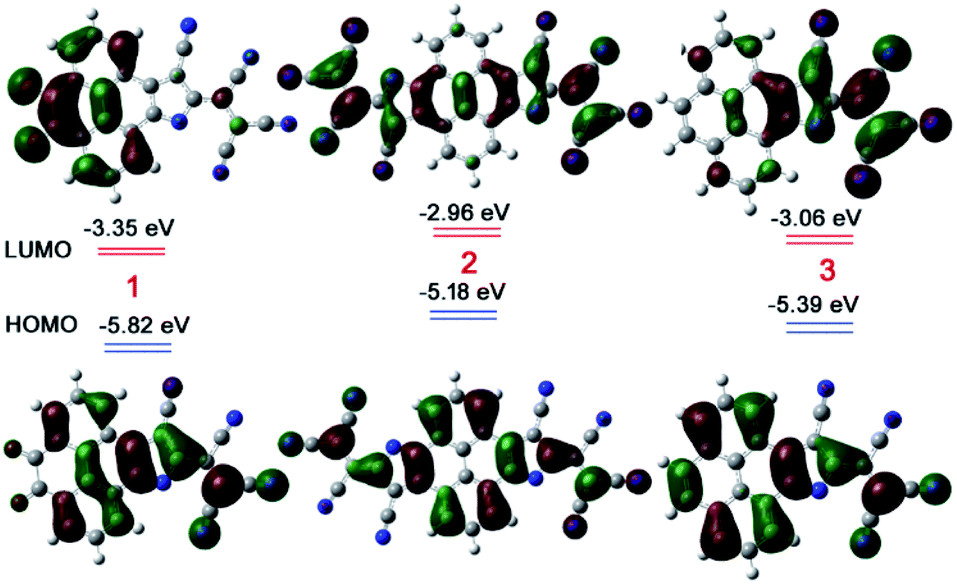 | ||
| Fig. 4 Frontier molecular orbital diagrams of 1, 2, and 3 (in red colour) with the DFT/B3LYP 6-311++G(d,p) basis set and IEFPCM model in CH2Cl2. | ||
Conclusions
In conclusion, we demonstrated the isolation of a pyrene-fused in situ pyrrole mono-anion and dipyrrole dianion. These compounds show extraordinary stability under ambient conditions. We observed a metal-catalyst-free TCNE type reductive condensation reaction involving malononitrile and pyrene di- and tetraones for the first time. These compounds show very high ε. Crystallographic studied revealed strong π–π stacking between pyrene scaffolds. Finally, we are hopeful that this catalyst-free Knoevenagel type condensation and cyclization reaction will provide some new insight into the condensation reaction mechanism.Data availability
All the data are given in ESI.†Author contributions
S. Kumar and S. Seki conceived and designed this work. S. Kumar, K. Yoshida, and Y. Hattori prepared the target compounds. S. Kumar and S. Seki performed electrochemical and spectroscopic measurements. T. Higashino and H. Imahori obtained the single-crystal X-ray diffraction structures. S. Kumar and S. Seki co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research (No. 19F19374, 18H03918, 19KK0134, 20H05867, 20H05862, 20H05831, 20H05832, and 20H05841) from the Japan Society for the Promotion of Science (JSPS). S. K. is thankful for a fellowship (ID No. P19374) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- M. Rabinovitz, I. Willner and A. Minsky, Acc. Chem. Res., 1983, 16, 298–304 CrossRef CAS.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- Y. Cohen, J. Klein and M. Rabinovitz, J. Am. Chem. Soc., 1988, 110, 4634–4640 CrossRef CAS.
- A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed.
- J.-i. Yamada, H. Akutsu, H. Nishikawa and K. Kikuchi, Chem. Rev., 2004, 104, 5057–5084 CrossRef CAS PubMed.
- C. Rovira, Chem. Rev., 2004, 104, 5289–5318 CrossRef CAS PubMed.
- J. E. Coughlin, Z. B. Henson, G. C. Welch and G. C. Bazan, Acc. Chem. Res., 2014, 47, 257–270 CrossRef CAS PubMed.
- M. A. Fox, Chem. Rev., 1979, 79, 253–273 CrossRef CAS.
- M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. A, 2011, 115, 2047–2056 CrossRef CAS PubMed.
- N. L. Holy, Chem. Rev., 1974, 74, 243–277 CrossRef CAS.
- H. Oshima, A. Fukazawa, T. Sasamori and S. Yamguchi, Angew. Chem., Int. Ed., 2015, 54, 7636–7639 CrossRef CAS PubMed.
- S. Kumar, J. Shukla, K. Mandal, Y. Kumar, R. Prakash, P. Ram and P. Mukhopadhyay, Chem. Sci., 2019, 10, 6482–6493 RSC.
- G. Dessy, V. Fares, A. Flamini and A. M. Giuliani, Angew. Chem., Int. Ed. Engl., 1985, 24, 426–427 CrossRef.
- M. Bonamico, V. Fares, A. Flamini, P. Imperatori and N. Poli, Angew. Chem., Int. Ed. Engl., 1989, 28, 1049–1050 CrossRef.
- V. Fares, A. Flamini and N. Poli, J. Am. Chem. Soc., 1995, 117, 11580–11581 CrossRef CAS.
- S. Nakamura, A. Flamini, V. Fares and M. Adachi, J. Phys. Chem., 1992, 96, 8351–8356 CrossRef CAS.
- M. Bonamico, V. Fares, A. Flamini, A. M. Giuliani and P. Imperatori, J. Chem. Soc., Perkin Trans. 2, 1988, 1447–1454 RSC.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- W. L. Jia, T. McCormick, Q. D. Liu, H. Fukutani, M. Motala, R. Y. Wang, Y. Tao and S. N. Wang, J. Mater. Chem., 2004, 14, 3344–3350 RSC.
- Y. Wang, H. M. Wang, Y. Q. Liu, C. A. Di, Y. M. Sun, W. P. Wu, G. Yu, D. Q. Zhang and D. B. Zhu, J. Am. Chem. Soc., 2006, 128, 13058–13059 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- Y. Wu, J. Wang, F. Zeng, S. Huang, J. Huang, H. Xie, C. Yu and S. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 1511–1519 CrossRef CAS PubMed.
- H. J. Lee and B. H. Kim, ACS Appl. Bio Mater., 2021, 4, 1668–1676 CrossRef CAS PubMed.
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef PubMed.
- L. Wang, W. Li, J. Lu, Y.-X. Zhao, G. Fan, J.-P. Zhang and H. Wang, J. Phys. Chem. C, 2013, 117, 26811–26820 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- M. Brasholz, Angew. Chem., Int. Ed., 2017, 56, 10280–10281 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051–6056 CrossRef CAS PubMed.
- B. Gao, M. Wang, Y. Cheng, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2008, 130, 8297–8306 CrossRef CAS PubMed.
- P. Demerseman, J. Einhorn, J.-F. Gourvest and R. J. Royer, J. Heterocycl. Chem., 1985, 22, 39–43 CrossRef CAS.
- R. Pratap, Y. Tominaga, M. L. Lee and R. N. Castle, J. Heterocycl. Chem., 1981, 18, 973–975 CrossRef CAS.
- J. Xiao, B. Yang, J. I. Wong, Y. Liu, F. Wei, K. J. Tan, X. Teng, Y. Wu, L. Huang, C. Kloc, F. Boey, J. Ma, H. Zhang, H. Y. Yang and Q. Zhang, Org. Lett., 2011, 13, 3004–3007 CrossRef CAS PubMed.
- S. Selvi, S.-C. Pu, Y.-M. Cheng, J.-M. Fang and P.-T. Chou, J. Org. Chem., 2004, 69, 6674–6678 CrossRef CAS PubMed.
- C.-I. Lin, S. Selvi, J.-M. Fang, P.-T. Chou, C.-H. Lai and Y.-M. Cheng, J. Org. Chem., 2007, 72, 3537–3542 CrossRef CAS PubMed.
- J. Lee and J. Park, Org. Lett., 2015, 17, 3960–3963 CrossRef CAS PubMed.
- V. Rozenshtein, G. Zilber, M. Rabinovitz and H. Levanon, J. Am. Chem. Soc., 1993, 115, 5193–5203 CrossRef CAS.
- A. Minsky, A. Y. Meyer and M. Rabinovitz, J. Am. Chem. Soc., 1982, 104, 2475–2482 CrossRef CAS.
- C. Tintel, J. Cornelisse and J. Lugtenburg, Recl. Trav. Chim. Pays-Bas, 1983, 102, 231–235 CrossRef CAS.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- K. Tajima, K. Matsuo, H. Yamada, S. Seki, N. Fukui and H. Shinokubo, Angew. Chem., Int. Ed., 2021, 60, 14060–14067 CrossRef CAS PubMed.
- A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035–19042 CrossRef CAS PubMed.
- R. García, S. More, M. Melle-Franco and A. Mateo-Alonso, Org. Lett., 2014, 16, 6096–6099 CrossRef PubMed.
- S. K. Keshri, S. Kumar, K. Mandal and P. Mukhopadhyay, Chem.–Eur. J., 2017, 23, 11802–11809 CrossRef CAS PubMed.
- T. A. Hudson and R. Robson, Cryst. Growth Des., 2009, 9, 1658–1662 CrossRef CAS.
- Z. Wang, V. Enkelmann, F. Negri and K. Müllen, Angew. Chem., Int. Ed., 2004, 43, 1972–1975 CrossRef CAS PubMed.
- B. Qiao, B. E. Hirsch, S. Lee, M. Pink, C.-H. Chen, B. W. Laursen and A. H. Flood, J. Am. Chem. Soc., 2017, 139, 6226–6233 CrossRef CAS PubMed.
- H. Maeda, Chem, 2020, 6, 1847–1860 CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- S. Kumar, J. Shukla, Y. Kumar and P. Mukhopadhyay, Org. Chem. Front., 2018, 5, 2254–2276 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Theoretical and experimental details; synthesis details; crystal and optimized structure of the compounds; DPV of the compounds; table of crystallographic data; EPR, FT-IR, ESI-HRMS, and NMR spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06070h |
| ‡ These authors contributed equally to this work. |
§ Crystallographic data for 1: C24H6N5O2·C24H20P, Mr = 735.71, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a = 9.287(4), b = 14.079(6), c = 14.828(6) Å, α = 99.420(5), β = 100.745(6), γ = 104.380(5)°, V = 1799.7(13) Å3, Z = 2, ρcalcd = 1.358 g cm−3, R1 = 0.0570 [I > 2σ(I)], wR2 = 0.1352 (all data), GOF = 1.025, CCDC 2116983. Crystallographic data for 2: C32H6N10·2(C24H20P)·CH2Cl2, Mr = 1294.13, monoclinic, P21/c(No. 14), a = 7.529(2), b = 30.222(9), c = 13.985(4) Å, β = 92.770(4)°, V = 3178.5(16) Å3, Z = 2, ρcalcd = 1.352 g cm−3, R1 = 0.0889 [I > 2σ(I)], wR2 = 0.2670 (all data), GOF = 1.020, CCDC 2116984. Crystallographic data for 3: C48H16N10·2(C24H20P), Mr = 1411.44, triclinic, P(No. 2), a = 11.934(2), b = 13.207(2), c = 14.026(3) Å, α = 117.428(8), β = 103.381(10), γ = 100.452(16)°, V = 1799.6(6) Å3, Z = 1, ρcalcd = 1.302 g cm−3, R1 = 0.0595 [I > 2σ(I)], wR2 = 0.1568 (all data), GOF = 1.031, CCDC 2116985. (No. 2), a = 9.287(4), b = 14.079(6), c = 14.828(6) Å, α = 99.420(5), β = 100.745(6), γ = 104.380(5)°, V = 1799.7(13) Å3, Z = 2, ρcalcd = 1.358 g cm−3, R1 = 0.0570 [I > 2σ(I)], wR2 = 0.1352 (all data), GOF = 1.025, CCDC 2116983. Crystallographic data for 2: C32H6N10·2(C24H20P)·CH2Cl2, Mr = 1294.13, monoclinic, P21/c(No. 14), a = 7.529(2), b = 30.222(9), c = 13.985(4) Å, β = 92.770(4)°, V = 3178.5(16) Å3, Z = 2, ρcalcd = 1.352 g cm−3, R1 = 0.0889 [I > 2σ(I)], wR2 = 0.2670 (all data), GOF = 1.020, CCDC 2116984. Crystallographic data for 3: C48H16N10·2(C24H20P), Mr = 1411.44, triclinic, P(No. 2), a = 11.934(2), b = 13.207(2), c = 14.026(3) Å, α = 117.428(8), β = 103.381(10), γ = 100.452(16)°, V = 1799.6(6) Å3, Z = 1, ρcalcd = 1.302 g cm−3, R1 = 0.0595 [I > 2σ(I)], wR2 = 0.1568 (all data), GOF = 1.031, CCDC 2116985. |
| This journal is © The Royal Society of Chemistry 2022 |