
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent coordination to palladium can invert the selectivity of oxidative addition†
Emily K.
Elias‡
 ,
Steven M.
Rehbein‡
and
Sharon R.
Neufeldt
*
,
Steven M.
Rehbein‡
and
Sharon R.
Neufeldt
*
Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, USA. E-mail: sharon.neufeldt@montana.edu
First published on 22nd December 2021
Abstract
Reaction solvent was previously shown to influence the selectivity of Pd/PtBu3-catalyzed Suzuki–Miyaura cross-couplings of chloroaryl triflates. The role of solvents has been hypothesized to relate to their polarity, whereby polar solvents stabilize anionic transition states involving [Pd(PtBu3)(X)]− (X = anionic ligand) and nonpolar solvents do not. However, here we report detailed studies that reveal a more complicated mechanistic picture. In particular, these results suggest that the selectivity change observed in certain solvents is primarily due to solvent coordination to palladium. Polar coordinating and polar noncoordinating solvents lead to dramatically different selectivity. In coordinating solvents, preferential reaction at triflate is likely catalyzed by Pd(PtBu3)(solv), whereas noncoordinating solvents lead to reaction at chloride through monoligated Pd(PtBu3). The role of solvent coordination is supported by stoichiometric oxidative addition experiments, density functional theory (DFT) calculations, and catalytic cross-coupling studies. Additional results suggest that anionic [Pd(PtBu3)(X)]− is also relevant to triflate selectivity in certain scenarios, particularly when halide anions are available in high concentrations.
Oxidative addition is a key elementary step in diverse transformations catalyzed by transition metals.1 For instance, this step is common to traditional cross-coupling reactions, which are among the most widely used methods for small molecule synthesis. During the oxidative addition step of cross-coupling reactions, a low valent metal [usually Pd(0)] inserts into a C–X bond with concomitant oxidation of the metal by two electrons. The “X” group of the C–X bond is commonly a halogen or triflate. Despite a wealth of research into this step,2–5 uncertainties remain about its mechanistic nuances. The mechanistic details are especially pertinent to issues of selectivity that arise when substrates contain more than one potentially reactive C–X bond.6
One of the best-studied examples of divergent selectivity at the oxidative addition step is the case of Pd-catalyzed Suzuki couplings of chloroaryl triflates. In 2000, Fu reported that a combination of Pd(0) and PtBu3 in tetrahydrofuran (THF) effects selective coupling of 1 with o-tolylB(OH)2via C–Cl cleavage, resulting in retention of the triflate substituent in the final product 2a (Scheme 1A).7 In contrast, the use of PCy3 (ref. 7) or most other phosphines8 provides complementary selectivity (product 2b) under similar conditions. The unique selectivity imparted by PtBu3 was later attributed to this ligand's ability to promote a monoligated oxidative addition transition state on account of its bulkiness.5,8 Smaller ligands, on the other hand, favor bisligated palladium, which prefers to react at triflate. The relationship between palladium's ligation state and chemoselectivity has been rationalized by Schoenebeck and Houk through a distortion/interaction analysis.5 In brief, the selectivity preference of PdL2 is dominated by a strong interaction between the electron-rich Pd and the more electrophilic site (C–OTf). On the other hand, PdL is less electron-rich and its selectivity preference mainly relates to minimizing unfavorable distortion energy by reacting at the more easily-distorted C–Cl bond.
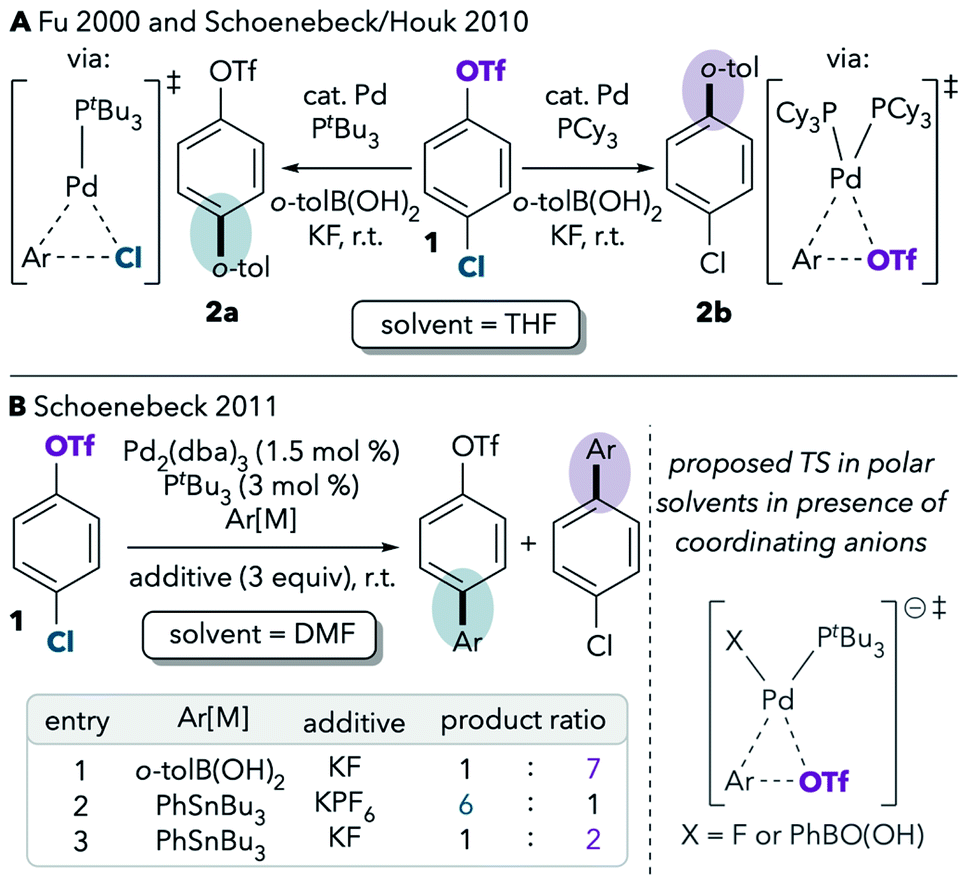 | ||
| Scheme 1 Seminal reports on the effects of (A) ligands and (B) solvents on the selectivity of cross-coupling of a chloroaryl triflate.5,7,9 | ||
Proutiere and Schoenebeck later discovered that replacing THF with dimethylformamide (DMF, Scheme 1B, entry 1) or acetonitrile caused a change in selectivity for the Pd/PtBu3 system.9,10 In these two polar solvents, preferential reaction at triflate was observed, and PtBu3 no longer displayed its unique chloride selectivity. The possibility of solvent coordination to Pd was considered, as bisligated Pd(PtBu3)(solv) would be expected to favor reaction at triflate. However, solvent coordination was ruled out on the basis of two intriguing studies. First, DFT calculations using the functional B3LYP suggested that solvent-coordinated transition states are prohibitively high in free energy (about 16 kcal mol−1 higher than the lowest-energy monoligated transition structure). Second, the same solvent effect was not observed in a Pd/PtBu3-catalyzed base-free Stille coupling in DMF (Scheme 1B, entry 2). Instead, the Stille coupling was reported to favor reaction at chloride despite the use of a polar solvent. This result appears inconsistent with the possibility that solvent coordination induces triflate-selectivity, as coordination of DMF to Pd should be possible in both the Stille and Suzuki conditions, if it happens at all. Instead, it was proposed that the key difference between the Suzuki and Stille conditions was the absence of coordinating anions in the latter (unlike traditional Suzuki couplings, Stille couplings do not necessarily require basic additives such as KF to promote transmetalation). Indeed, when KF or CsF was added to the Stille reaction in DMF, selectivity shifted to favor reaction at triflate (Scheme 1B, entry 3), thereby displaying the same behavior as the Suzuki coupling in this solvent. On the basis of this and the DFT studies, it was proposed that polar solvents induce a switch in chemoselectivity if coordinating anions like fluoride are available by stabilizing anionic bisligated transition structures (Scheme 1B, right).
However, our recent extended solvent effect studies produced confounding results.11 In a Pd/PtBu3-catalyzed Suzuki cross-coupling of chloroaryl triflate 1, we observed no correlation between solvent polarity and chemoselectivity (Scheme 2). Although some polar solvents such as MeCN, DMF, and dimethylsulfoxide (DMSO) favor reaction at triflate, a number of other polar solvents provide the same results as nonpolar solvents by favoring reaction at chloride. For example, cross-coupling primarily takes place through C–Cl cleavage when the reaction is conducted in highly polar solvents like methanol, water, acetone, and propylene carbonate. In fact, the only solvents that promote reaction at triflate are ones that are commonly thought of as “coordinating” in the context of late transition metal chemistry.12 These are solvents containing nitrogen, sulfur, or electron-rich oxygen lone pairs (nitriles, DMSO, and amides). The observed solvent effects were upheld for a variety of chloroaryl triflates and aryl boronic acids.11
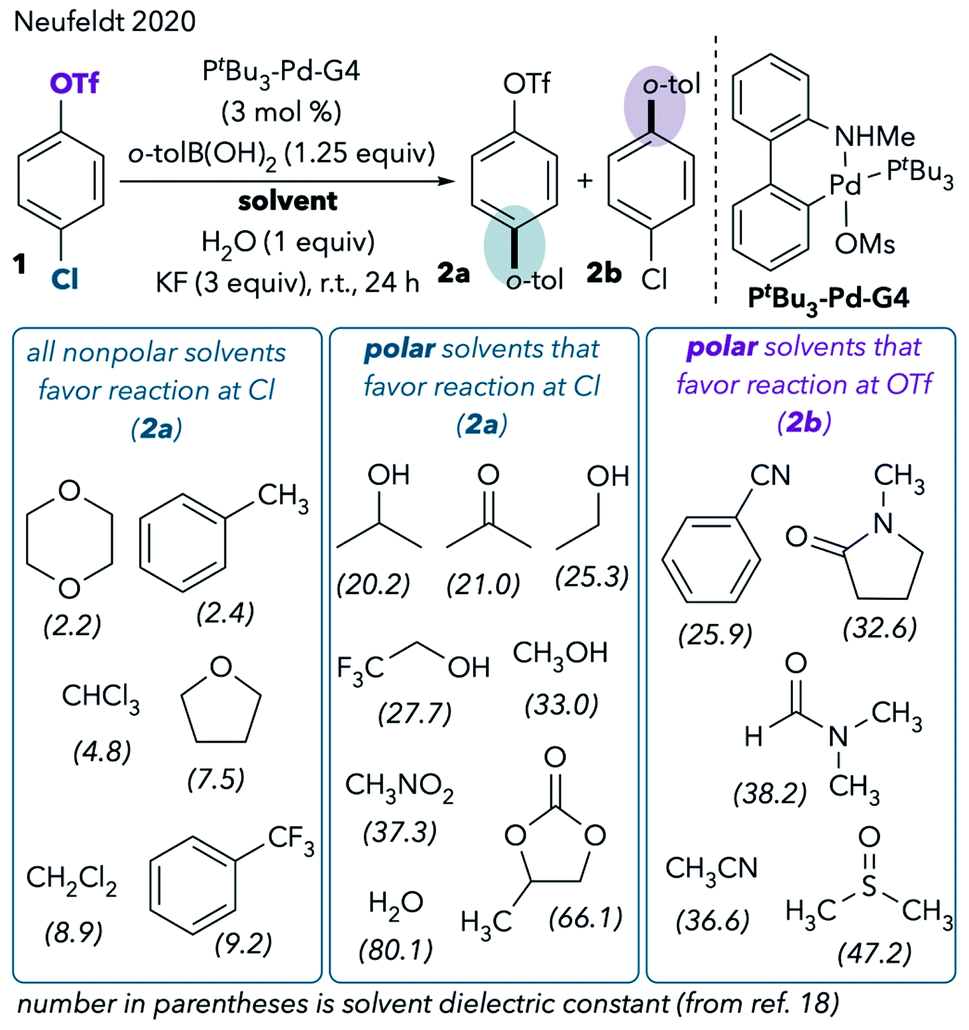 | ||
| Scheme 2 Expanded solvent effect studies in the Pd/PtBu3-catalyzed Suzuki coupling.11 | ||
We have sought to reconcile these observations with the earlier evidence9 against solvent coordination. Herein we report detailed mechanistic studies indicating that coordinating solvents alone are sufficient to induce the observed selectivity switch. In solvents like DMF and MeCN, stoichiometric oxidative addition is favored at C–OTf even in the absence of anionic additives. The apparent contradiction between our observations and the previously-reported DFT calculations and base-free Stille couplings is reconciled by a reevaluation of those studies. In particular, when dispersion is considered in DFT calculations, neutral solvent-coordinated transition structures involving Pd(PtBu3)(solv) become energetically feasible. Furthermore, we find that the selectivity analysis in the Stille couplings is convoluted by low yields, the formation of side products, and temperature effects. When these factors are disentangled, the Stille coupling in DMF displays selectivity similar to the Suzuki coupling in the same coordinating solvent. In light of these new results, anionic bisligated [Pd(PtBu3)(X)]− does not appear to be the dominant active catalyst in nonpolar or polar solvents unless special measures are taken to increase the concentration of free halide, such as adding tetraalkylammonium halide salts or crown ethers.
Results and discussion
Based on the observed solvent trends in Scheme 2, we hypothesized that selectivity depends on the solvent's ability to coordinate to palladium during oxidative addition. In this scenario, oxidative addition of triflate in solvents such as DMF would not necessarily involve anionic [Pd(PtBu3)(X)]−, but rather would involve neutral solvent-bound palladium such as Pd(PtBu3)(DMF). The experiments described below support this hypothesis. We also considered the hypothesis that selectivity could relate to the solvent's ability to solubilize the base (KF). In solvents in which KF is more soluble, a higher concentration of anions [F−, HO−, or ArBO(OH)−] would be available to promote formation of anionic palladium. However, we found no correlation between KF solubility and selectivity (see ESI† for details on this and other alternative hypotheses).Effect of benzonitriles on selectivity
Nitriles, including acetonitrile and benzonitrile, were observed to promote reaction at triflate (Scheme 2). Because nitriles are common σ-donor ligands for Pd, we were interested in whether the use of nitriles as substoichiometric additives rather than solvents would still provoke a change in selectivity. To answer this question, the Suzuki reaction was conducted under our standard optimized conditions with THF as solvent in the presence or absence of 0.5 equiv. of benzonitrile (Table 1). This small amount of benzonitrile, corresponding to <3% of the solvent volume, should have a minimal effect on the polarity of the reaction medium. Our optimized conditions use Buchwald's air-stable PtBu3-Pd-G4 precatalyst (see Scheme 2) for convenience of handling. Importantly, the use of the Buchwald precatalyst does not impact selectivity when compared to Fu's original system using Pd2(dba)3 (see ref. 11 and ESI†).7,13 One equivalent of water was routinely added to the Suzuki couplings because this leads to better reproducibility. Small amounts of water are needed to promote transmetallation, and our lab is located in a particularly dry climate where adventitious water can be negligible. However, the addition of 1 equiv. of water does not impact observed selectivities (see ESI†). All results reported herein are the average of at least 2 trials (see ESI† for results of individual replicates). The estimated uncertainty in total mass balance for each catalytic trial is about ±3% due to measurement and instrument error (see ESI†).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | PhCN (equiv.) | 1 (%) | 2a (%) | 2b (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2b :2b |
| a GC yields calibrated against undecane as the internal standard. Average of at least three trials. ≤4% yield of the diarylated product was detected in all trials. | ||||||
| 1 | THF | 0 | 20 | 74 | <1 | >74:1 |
| 2 | THF | 0.5 | 39 | 55 | 4 | 14:1 |
| 3 | PhCN | (Solvent) | 27 | 7 | 53 | 1:8 |
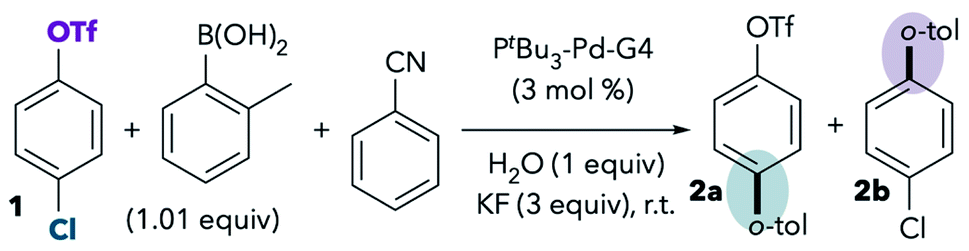
Consistent with the hypothesis that benzonitrile can coordinate to palladium and influence selectivity, even a substoichiometric amount of PhCN (0.5 equiv.) in THF leads to erosion of chloride-selectivity when compared to the reaction without PhCN. Although the reaction gives >70:1 selectivity for 2a in the absence of PhCN (Table 1, entry 1), the addition of 0.5 equiv. of PhCN leads to a lower selectivity of 14:1 (2a:2b, entry 2). For comparison, the use of PhCN as solvent provides a complete reversal of selectivity, affording 2a and 2b in a ratio of 1:8 (entry 3). Analysis of other ratios of PhCN:THF reveals that selectivity inverts when PhCN comprises 10–25% of the total solvent volume, and that small amounts of PhCN lead to lower conversion than observed in either neat THF or PhCN (see ESI† for details and discussion).
We next repeated this experiment using a series of substituted benzonitriles, inspired by a similar experiment performed by Watson and coworkers.14 This study led to a linear free energy relationship plot that revealed a positive correlation between Hammett σ+ values and preference for reaction at chloride (Fig. 1).15 Benzonitriles bearing more electron-donating groups in the para position are expected to be better σ-donors (better ligands) for palladium. Indeed, more reaction at triflate is observed with more electron-rich benzonitriles, consistent with the hypothesis that σ-donation of the nitrile to palladium during oxidative addition promotes reaction at triflate. However, even the most electron-deficient benzonitriles lead to more reaction at triflate compared to the results in the absence of benzonitrile additives (horizontal dashed line in Fig. 1).
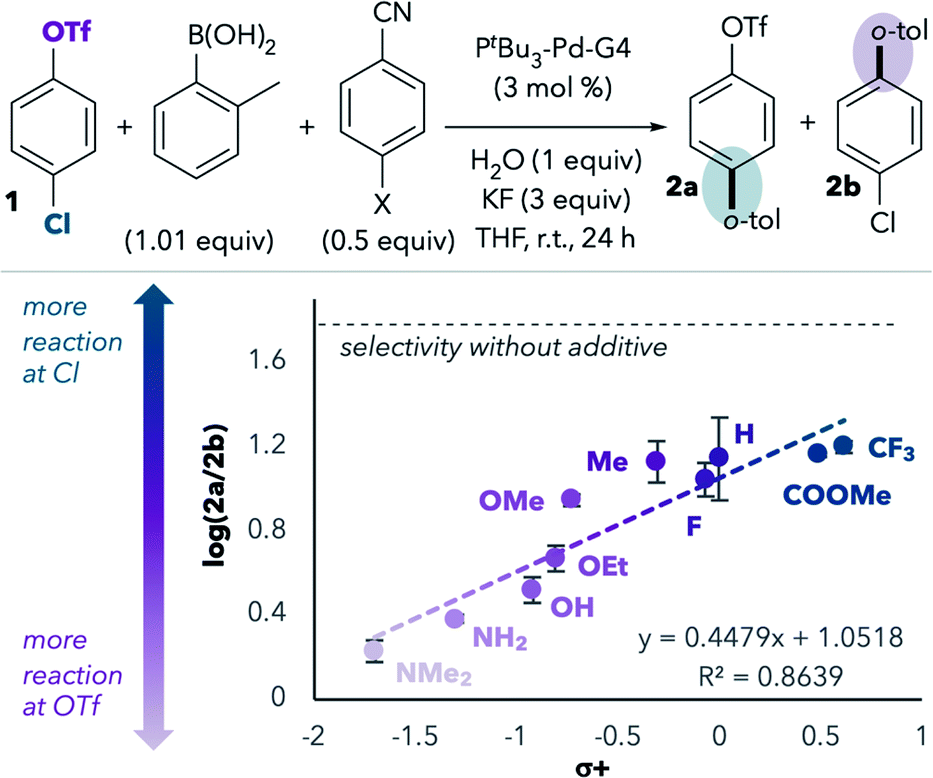 | ||
| Fig. 1 Hammett-type plot shows a correlation between benzonitrile donor ability and increasing reaction at triflate. Each data point represents the average of three trials, and the error bars represent standard deviation. | ||
Solvent variants with modified coordinating ability
Like benzonitrile, acetonitrile also promotes reaction at triflate. We hypothesized that a more electron-deficient derivative of acetonitrile would coordinate more poorly to palladium, and thereby display less of a preference for triflate selectivity than acetonitrile itself. Indeed, the use of fluoroacetonitrile leads to poor selectivity (2a:2b = 1:2; Table 2, entry 2), whereas MeCN strongly favors reaction at triflate (entry 1). This observation is consistent with weaker coordination of fluoroacetonitrile to palladium, resulting in a lower concentration of Pd(PtBu3)(solv) and less reaction at triflate.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | ε | 2a (%) | 2b (%) |
2a:2b |
| a GC yields calibrated against undecane as the internal standard. Average of two trials. ≤3% yield of the diarylated product was detected in all trials. b Dielectric constants from ref. 18 (entries 1–4) and ref. 19 (entry 2). | |||||
| 1 | CH3CN | 36.6 | 2 | 77 | 1:39 |
| 2 | CH2FCN | ∼36 | 4 | 10 | 1:2 |
| 3 |
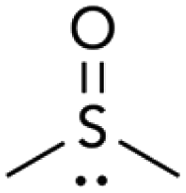
|
47.2 | 1 | 45 | 1:45 |
| 4 |
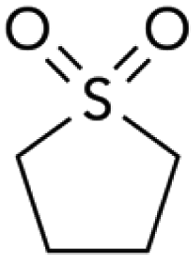
|
42.2 | 44 | 11 | 4:1 |
We also compared DMSO to sulfolane. DMSO, a highly polar sulfoxide, can coordinate to soft metals through sulfur's lone electron pair.16 The use of DMSO as solvent strongly favors reaction at triflate (Table 2, entry 3). Sulfolane has a similar polarity to DMSO, but because it is a sulfone (no lone pair on sulfur) it cannot coordinate to metals in the same manner as DMSO. Consistent with coordinating ability being a critical solvent feature, the use of sulfolane as solvent provides the opposite selectivity as DMSO (entry 4).17
Stoichiometric oxidative addition studies
The observed solvent trends in the Suzuki coupling support the hypothesis that solvent coordination to Pd during oxidative addition can lead to a switch in chemoselectivity. To eliminate confounding factors, we next sought to study the oxidative addition step in isolation. We conducted NMR studies on the reaction of Pd(PtBu3) with competing aryl chloride and aryl triflate electrophiles, 3a and 3b.20 These fluorinated substrates were chosen so that their consumption could be monitored by 19F NMR. The putative Pd0 complex Pd(PtBu3) was generated in situ from a 1:1 mixture of Pd(COD)(CH2TMS)2 and PtBu3.21,22 C6D6 was added to the reactions to facilitate locking the NMR samples.
The reaction in the noncoordinating solvents toluene, THF, acetone, sulfolane, and propylene carbonate leads to consumption of 3a (Table 3, entries 1–5, 8 and 9), and the known complex (PtBu3)Pd(2-CF3C6H4)(Cl) is detected by 19F and 31P NMR in most of these solvents.23 In contrast, nearly all of the aryl triflate 3b remains unreacted. This result is consistent with the high selectivity for C–Cl activation observed in the catalytic Suzuki coupling of 1 in these solvents. The lower reactivity in toluene at room temperature (entry 1) is ascribed to the formation of Pd(PtBu3)2, which can be detected by 31P NMR and has been shown previously to react sluggishly with aryl chlorides.2a,b
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Recovereda (%) | Reacted | |
| 3a | 3b |
3a:3b |
||
| a 19F NMR yields calibrated against C6H5F as an internal standard. Average of two trials. b Heated to 80 °C for 2 h. c PC = propylene carbonate. d Pd(COD)(CH2TMS)2 was omitted from the reaction mixture. e 3a was omitted from the reaction mixture (n.a. = not applicable). | ||||
| 1 | Toluene | 89 | ≥99 | ≥11:1 |
| 2b | Toluene | 52 | 98 | 24:1 |
| 3 | THF | 55 | 97 | 15:1 |
| 4 | Acetone | 71 | 98 | 15:1 |
| 5 | MeOH | 70 | ≥99 | ≥30:1 |
| 6 | MeCN | 97 | 1 | 1:33 |
| 7 | DMF | 78 | 44 | 1:3 |
| 8 | Sulfolane | 86 | 95 | 3:1 |
| 9 | PCc | 83 | 94 | 3:1 |
| 10d | MeCN | ≥99 | ≥99 | — |
| 11d | DMF | ≥99 | ≥99 | — |
| 12e | MeCN | n.a. | 3 | — |
| 13e | DMF | n.a. | 28 | — |
In contrast to the results in non-coordinating solvents, the use of MeCN or DMF leads to preferential consumption of 3bvia C–OTf activation. In MeCN, 3b is nearly fully consumed after 6 h at room temperature (entry 6), with concomitant formation of palladium black,24–26 an intractable mixture of trifluoromethyl-containing by-products, and a new 19F signal at −77.5 ppm assigned to “free” triflate anion by comparison to the chemical shift of NBu4OTf in the same solvent mixture. Similar results are observed in DMF (entry 7), albeit with lower conversion and lower selectivity compared to MeCN.27
Interestingly, the 31P NMR spectrum of the reaction in DMF reveals a signal at ∼75 ppm that is not detected in any of the other solvents. This signal is also observed when 3a is omitted from the reaction mixture (entry 13). Monitoring the reaction over time reveals that a signal at ∼64 ppm is also present at early time points, but is completely replaced by the ∼75 ppm signal within 3 h (see ESI†). The ∼64 ppm signal can also be detected at 6 h when the reaction is run at 0 °C (see ESI†). We hypothesize that these two signals, which are unique to the reaction in DMF, represent DMF-coordinated species such as [ArPd(PtBu3)(DMF)2]OTf, resulting from oxidative addition of the triflate. Analogous signals are not observed in MeCN, despite its strong coordinating ability. MeCN-bound oxidative addition adducts may decompose more readily due to the smaller sterics of MeCN. For example, NMR and GCMS data indicate that one decomposition pathway under these conditions involves Heck coupling with COD (see ESI†), and coordination of COD to Pd(II) may be less favored in the presence of bulkier DMF ligands compared to MeCN ligands.
Importantly, palladium-free control studies demonstrate that the substrates do not decompose in the absence of Pd (entries 10 and 11). Furthermore, the presence of 3a is not necessary for reaction of 3b to take place in coordinating solvents, suggesting that formation of an anionic species like [Pd(PtBu3)(Cl)]−via initial C–Cl activation of 3a, is not a prerequisite for C–OTf activation in these experiments (entries 12 and 13).28
The results of these stoichiometric studies provide strong evidence that coordinating solvents alone—in the absence of anionic ligands for Pd—can promote a switch in the chemoselectivity of oxidative addition compared to the use of noncoordinating solvents. Based on the known preference for bisligated Pd to react at triflate over chloride, the most likely explanation for the observed selectivity in coordinating solvents is involvement of Pd(PtBu3)(solv) during oxidative addition. We next turned to computational studies to further evaluate the feasibility of solvent-bound species.
DFT studies
In the earlier report by Proutiere and Schoenebeck, solvent coordination to Pd during oxidative addition at C–OTf was tentatively ruled out, in part on the basis of the high energy values that were calculated for such transition structures.9 The prior DFT results had indicated that solvent-coordinated transition structures involving Pd(PtBu3)(solv) were prohibitively high-energy compared to monoligated transition structures with Pd(PtBu3). However, those calculations were conducted with a DFT method (B3LYP)33 that does not take dispersion into account. Schoenebeck and others have since demonstrated that, in general, the computed energies of crowded structures are significantly lower when dispersion is considered.34,35Therefore, we reevaluated the energetics of solvent coordination to Pd using methods that include dispersion. Fig. 2 compares the results of calculations performed with B3LYP (no dispersion) and with MN15L36 (accounts for dispersion) using the CPCM solvation model for DMF. When dispersion is not considered (Fig. 2A), the DMF-coordinated transition structure for oxidative addition at triflate (TS5b-dmf) is about 16 kcal mol−1 higher in energy than the lowest-energy monoligated transition structure (TS5a), suggesting that TS5b-dmf is unfeasible. However, use of MN15L leads to very different results: TS5b-dmf is now predicted to be favored over TS5a (Fig. 2B).37 At minimum, these results demonstrate that solvent coordination should not be ruled out on the basis of DFT calculations. Instead, solvent coordination to palladium is likely to be energetically feasible during oxidative addition, especially under experimental conditions in which solvent is in large excess relative to Pd and substrate.
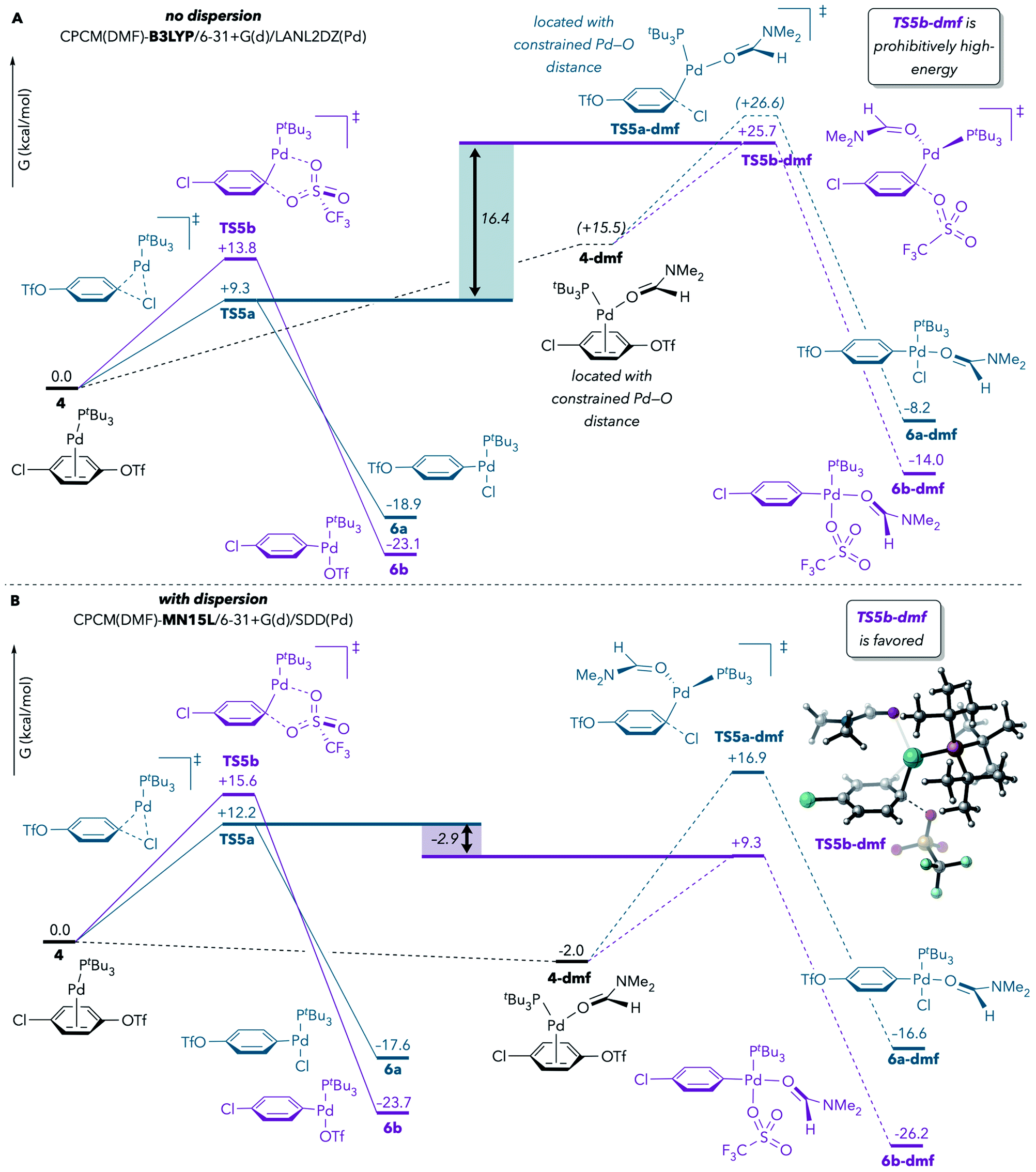 | ||
| Fig. 2 Effect of dispersion on the stability of DMF-coordinated oxidative addition transition structures. Calculations were performed (A) without dispersion or (B) with dispersion. Free energy values are corrected for concentration (DMF:Pd = 765:1) and are reported after applying Cramer and Truhlar's29 and Head-Gordon's30 quasi-harmonic approximations to vibrational entropy and enthalpy, respectively.31,32 | ||
Stille cross-couplings
Although the Pd/PtBu3-catalyzed Suzuki coupling of 1 in DMF favors reaction at triflate, the analogous base-free Stille coupling was reported to favor reaction at chloride in this solvent (see Scheme 1B, entry 2). This result is perplexing, because Suzuki and Stille couplings are thought to proceed through the same oxidative addition step, and our stoichiometric NMR studies indicate that oxidative addition of triflate is preferred in DMF even in the absence of anionic additives.To understand this contradiction, we examined the Stille coupling between 1 and PhSnMe3 in DMF (Scheme 3).38,39 Poor conversion is observed at room temperature,40 but 7b is slightly favored over 7a. Methylated products 7e and 7f were also detected in the reaction mixture.41 Overall, the ratio of chloride:triflate activation under these conditions is approximately 4:7 [(7a + 7e):(7b + 7f)], which is qualitatively consistent with the triflate selectivity observed in the Suzuki coupling and in the stoichiometric oxidative addition studies. These results suggest that there may not be a discrepancy between the behavior of Pd/PtBu3 in the catalytic Suzuki vs. Stille couplings in DMF. However, the low conversion in the Stille coupling calls into question the accuracy of selectivity analysis for at least two reasons. First, measurement errors may be introduced when integrating small product peaks on a GC chromatogram or when isolating small quantities of materials. More importantly, the low conversion suggests a very sluggish rate of cross-coupling. If cross-coupling is slow, it is possible for undesired processes like catalyst decomposition to compete with coupling, which could lead to different catalyst speciation compared to the much faster Suzuki coupling system.42
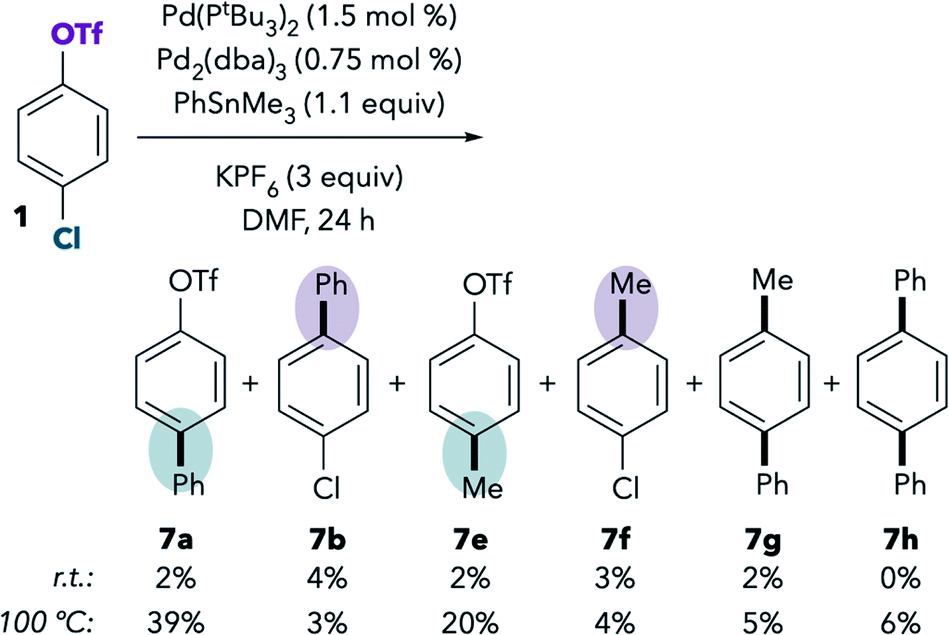 | ||
| Scheme 3 Base-free stille coupling of 1 Catalyzed by Pd/PtBu3. GC yields calibrated against dodecane as an internal standard. Results are the average of two trials. Leftover 1: 83% at r.t., 4% at 100 °C. | ||
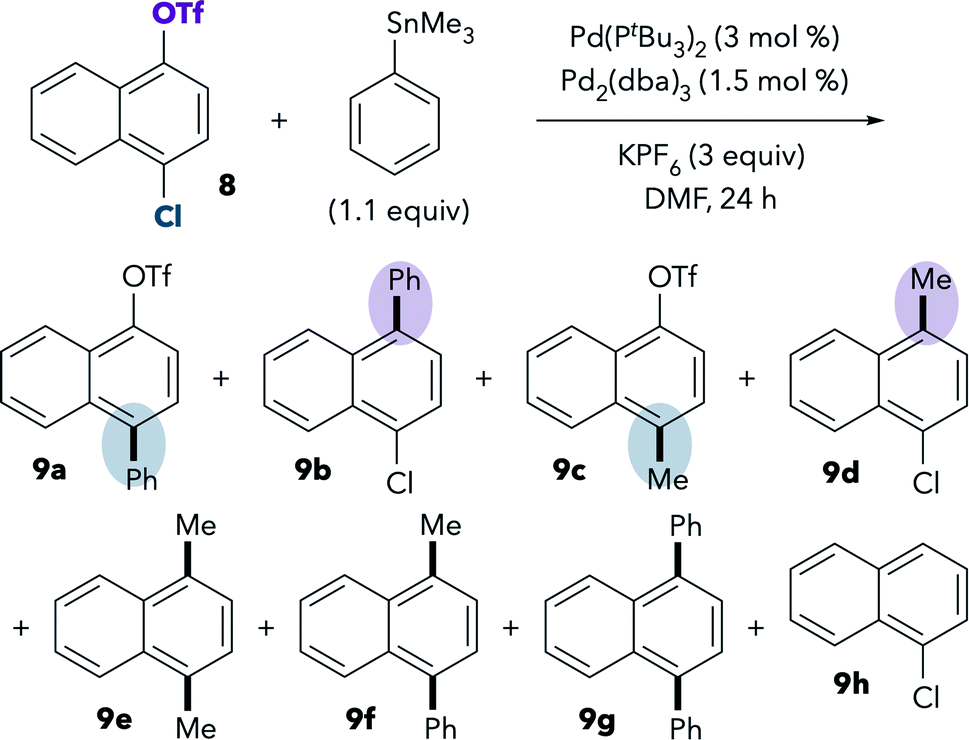
In an effort to increase the conversion of the Stille coupling, the reaction was heated to 100 °C. Slightly higher yields were observed at this temperature, and, interestingly, reaction at chloride becomes preferred. The ratio of chloride:triflate activation under these conditions is approximately 59:7 [(7a + 7e):(7b + 7f)]. The chloride preference under these conditions is consistent with the prior report.9 However, further examination suggests that the selectivity at 100 °C should not be compared to the selectivity at room temperature. We observe an analogous change in selectivity upon heating the Suzuki coupling of 1 in DMF. At 100 °C, selectivity is eroded when using Buchwald's PtBu3-Pd-G4 catalyst (Table 4, compare entries 1 and 2). Furthermore, selectivity actually inverts at 100 °C when using a mixture of Pd2(dba)3 and Pd(PtBu3)2 (entries 3 and 4).43 High temperatures are expected to disfavor bisligated Pd (i.e., reaction at triflate) for entropic reasons. However, the observation that precatalyst influences the selectivity at 100 °C indicates that the increased formation of 2a may reflect more than simply an entropic penalty. We hypothesize that the palladium catalyst is transformed at high temperature into a species that displays different selectivity than the active catalyst at room temperature.44,45 As such, a high-temperature Stille coupling is not comparable to a room-temperature Suzuki coupling or to the room-temperature oxidative addition studies. For these reasons, and in order to achieve a faster rate for the room temperature Stille coupling, we next examined the use of a more reactive substrate, 8.46
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Temp (°C) | 1 (%) | 2a (%) | 2b (%) | Ratio 2a:2b |
| a GC yields calibrated against undecane as an internal standard. Results are the average of at least two trials. ≤2% yield of the diarylated product was detected in all trials. | ||||||
| 1 | A | r.t. | 23 | 10 | 60 | 1:6.0 |
| 2 | A | 100 | 2 | 21 | 30 | 1:1.4 |
| 3 | B | r.t. | 17 | 11 | 67 | 1:6.1 |
| 4 | B | 100 | 7 | 33 | 23 | 1.4:1 |
The room-temperature Stille coupling between PhSnMe3 and chloronaphthol derivative 8 proceeds to higher conversion compared to the reaction of 1 (Table 5, entry 1). After 24 h, only 24% of 8 remained unreacted, and numerous products were detected by GCMS. The two major products are 9b and 9d, resulting from triflate activation. Overall, the ratio of chloride:triflate activation is ∼4:52 [(9a + 9c):(9b + 9d + 9h)]. Again, these results show that the Stille coupling displays the same oxidative addition preference as the Suzuki coupling in DMF. Consistent with earlier observations, more reaction at chloride is observed upon heating (entry 2). At 100 °C, the ratio of chloride:triflate activation is ∼46:17 [(9a + 9c):(9b + 9d + 9h)]. Similar selectivity trends are observed when using PhSnnBu3 instead of PhSnMe3 (see ESI†).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Temp | 8 | 9a | 9b | 9c | 9d | 9e | 9f | 9g | 9h |
| a GC yields calibrated against undecane as an internal standard. Results are the average of two trials. No other products derived from 8 could be identified by GCMS. | ||||||||||
| 1 | r.t. | 24 | 1 | 9 | 3 | 42 | 1 | 1 | 1 | 1 |
| 2 | 100 °C | 1 | 13 | 4 | 33 | 12 | 1 | 8 | 9 | 1 |
Taken together, our studies indicate that the selectivity of oxidative addition is similar under stoichiometric, catalytic Suzuki, and base-free Stille conditions at room temperature in DMF. This conclusion is consistent with the hypothesis that DMF coordinates to palladium and promotes C–OTf oxidative addition at a bisligated Pd(PtBu3)(DMF) complex.
Role of anions on selectivity
The evidence presented thus far strongly suggests that coordinating solvents can promote oxidative addition at triflate through solvent-ligated transition states. As such, it is not necessary to invoke anionic [Pd(PtBu3)(X)]− to explain the selectivity of Suzuki couplings of chloroaryl triflates in coordinating solvents. However, oxidative addition of PhOTf at [PdL(X)]− in the presence of quaternary ammonium chloride and bromide salts has been described by Hartwig [L = (P(o-tolyl)3)].47 Oxidative addition at anionic palladium complexes bearing electron-deficient phosphines or in the absence of phosphines or other strong ancillary ligands has also been proposed.48 Furthermore, DFT calculations suggest that if [Pd(PtBu3)(X)]− participates in oxidative addition, it would preferentially react at triflate as seen for other PdL2 complexes.5,9,11 We speculated that transition states involving [Pd(PtBu3)(X)]− might be primarily responsible for triflate selectivity under some conditions.To evaluate this possibility, we subjected substrate 1 to Suzuki coupling conditions in THF in the presence of a variety of anionic additives (Table 6). As previously reported, reaction at chloride is strongly favored in the absence of extra additives, despite the use of KF (a source of fluoride anion) as a base (entry 1). This suggests that bisligated palladium is not the active catalyst under these conditions. However, addition of 18-crown-6, a chelator for K+, induces a reversal of selectivity (entry 2). Alternatively, replacing potassium with a noncoordinating tetrabutylammonium cation also promotes reaction at triflate, albeit with significant substrate decomposition (entry 3). These results suggest that increased availability of free fluoride facilitates reaction at triflate through a mechanism involving [Pd(PtBu3)(X)]−. A similar effect is observed with added tetrabutylammonium salts of Cl− and Br− (entries 4 and 5). Because chloride and bromide are not basic, their effect on selectivity is unlikely to result from an increase in concentration of hydroxide (from water) or deprotonated boronic acid. Instead, it is reasonable to conclude that these salts promote reaction at triflate through formation of [Pd(PtBu3)(Cl)]− or [Pd(PtBu3)(Br)]−. The quaternary ammonium salt of a non-coordinating anion (NBu4OTf) does not have the same effect as the halide salts (entry 6), suggesting that the selectivity change with NBu4F, NBu4Cl, and NBu4Br is not due to a medium effect. In the polar noncoordinating solvents iPrOH and propylene carbonate, the addition of 18-crown-6 also leads to a reversal of selectivity with an increase in the total yield of 2b (entries 7–10). However, poor mass balance is observed, likely due to an enhanced rate of triflate hydrolysis under the more basic conditions. The preexisting triflate selectivity appears to be enhanced in DMF upon the addition of 18-crown-6 (compare entries 11 and 12), although the overall quantity of 2b produced is lower and the poor mass balance complicates interpretation of the selectivity. Taken together, the results in Table 6 show that higher concentrations of “naked” anions promote reaction at triflate. This observation is consistent with the involvement of [Pd(PtBu3)(X)]− under specific conditions that provide free anions in high concentration.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Additive (equiv.) | 1 (%) | 2a (%) | 2b (%) | Ratio 2a:2b |
| a GC yields calibrated against undecane as the internal standard. Average at least of two runs. ≤2% yield of the diarylated product was detected in all trials except entry 2 (8% yield of diarylated product). PC = propylene carbonate. b KF was omitted from the reaction mixture. | ||||||
| 1 | THF | — | 20 | 74 | <1 | >74:1 |
| 2 | THF | 18-Crown-6 (3) | 0 | 1 | 76 | 1:38 |
| 3b | THF | NBu4F (3) | <1 | <1 | 13 | <1:13 |
| 4 | THF | NBu4Cl (1) | 20 | 4 | 69 | 1:17 |
| 5 | THF | NBu4Br (1) | 3 | 2 | 91 | 1:46 |
| 6 | THF | NBu4OTf (1) | 26 | 70 | 3 | 23:1 |
| 7 | PC | — | 15 | 71 | 5 | 14:1 |
| 8 | PC | 18-Crown-6 (3) | 5 | 1 | 11 | 1:11 |
| 7 | iPrOH | — | 2 | 87 | 1 | 87:1 |
| 8 | iPrOH | 18-Crown-6 (3) | 0 | 0 | 3 | <1:3 |
| 11 | DMF | — | 20 | 10 | 61 | 1:6 |
| 12 | DMF | 18-Crown-6 (3) | 0 | 0 | 47 | <1:47 |
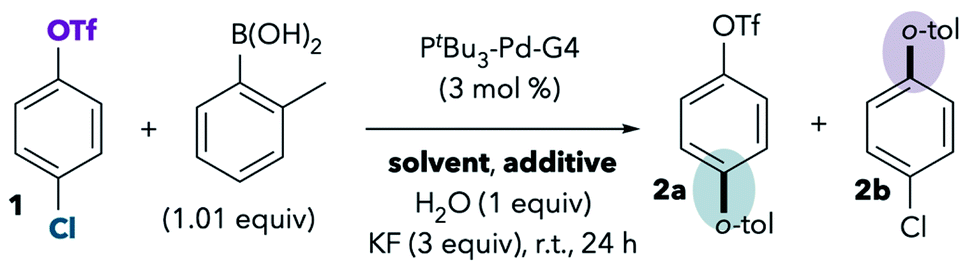
Conclusions
A series of studies were conducted to understand the observed solvent effects in the Pd/PtBu3-catalyzed Suzuki coupling of chloroaryl triflates. The results provide strong evidence for solvent coordination to Pd during triflate-selective cross-coupling. A summary of the key results is as follows: the use of coordinating solvents including nitriles, amides, and sulfoxides leads to Suzuki coupling through C–OTf bond cleavage. Conversely, nonpolar solvents as well as polar solvents that are poorly coordinating to palladium, such as propylene carbonate, alcohols, and sulfolane, favor reaction at chloride. Addition of substoichiometric amounts of benzonitrile derivatives to the Suzuki reaction in THF leads to erosion of chloride selectivity, and a correlation is observed between benzonitrile donating ability and increased reaction at triflate. The solvent-dependent selectivity observed in the stoichiometric reaction between Pd/PtBu3 and a competing aryl chloride and triflate mirrors that of the catalytic Suzuki coupling. DFT calculations reveal that dispersion dramatically lowers the predicted barrier to oxidative addition at Pd(PtBu3)(solv). Although previous results suggested that the Pd/PtBu3-catalyzed Stille coupling in a coordinating solvent leads to different selectivity than the corresponding Suzuki coupling, we find that the selectivity of these two reactions is approximately identical after controlling for convoluting factors such as low yields, formation of side products, and catalyst decomposition.To our knowledge, this work represents the first compelling evidence for involvement of a solvent ligand during Ar–X oxidative addition at homogeneous Pd in the presence of a phosphine.49 Taken together, the studies reported herein are consistent with the following mechanistic paradigm: in the presence of PtBu3, preferential reaction of chloride proceeds through an oxidative addition transition state involving monoligated PdL (L = PtBu3), as previously established. This selectivity for chloride is observed in non-coordinating solvents in the absence of a high concentration of “naked” halide ions. In coordinating solvents, however, palladium prefers to react at triflate through a transition state involving PdL(solv). Anionic bisligated [PdL(X)]−, where X = halide, also reacts preferentially at triflate. However, this anionic species does not appear to be the dominant active catalyst, even in polar solvents, unless extra measures are taken to increase the concentration of free halide. Such measures include the addition of a crown ether to chelate potassium from KF or the addition of quaternary ammonium halide salts.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
EKE: data curation, formal analysis, investigation, methodology, visualization, writing – original draft; SMR: data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft; SRN: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, visualization, writing – original draft; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
These studies are based upon work supported by NSF CAREER (CHE-1848090) and a Cottrell Scholar Award from the Research Corporation for Science Advancement (#26808). Calculations were performed on Comet and Expanse at the San Diego Supercomputer Center and on Bridges-2 at the Pittsburgh Supercomputing Center through XSEDE (CHE-170089), which is supported by NSF (ACI-1548562). Support for MSU's NMR Center and the 400 MHz NMR spectrometer used in this research has been provided by the National Science Foundation (Grant No. NSF-MRI:CHE-2018388) and MSU's office of the Vice President for Research and Economic Development, and support for the 500 MHz NMR spectrometer was provided by the NSF (NSF-MRI:DBI-1532078) and the Murdock Charitable Trust Foundation (2015066:MNL). We are grateful to Emily Entz, Oliver Jackson, Nate Larson, Jérome Lépeule, and John Russell for evaluating reproducibility, to Emma Baumgardner for assistance in the laboratory, and to Professor Thomas Livinghouse for helpful conversations.Notes and references
- (a) J. A. Labinger, Organometallics, 2015, 34, 4784 CrossRef CAS; (b) J. J. Hartwig, in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- Selected experimental mechanistic studies with implications for oxidative addition at Pd(0) ligated by bulky monodentate phosphines: (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232 CrossRef CAS PubMed; (b) J. P. Stambuli, C. D. Incarvito, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 1184 CrossRef CAS PubMed; (c) E. Galardon, S. Rhamdeehul, J. M. Brown, A. Cowley, K. K. Hii and A. Jutand, Angew. Chem., Int. Ed., 2002, 41, 1760 CrossRef CAS; (d) F. Barrios Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141 CrossRef CAS PubMed.
- Selected computational studies: (a) H. M. Senn and T. Ziegler, Organometallics, 2004, 26, 758 Search PubMed; (b) K. C. Lam, T. B. Marder and Z. Lin, Organometallics, 2007, 26, 758 CrossRef CAS; (c) M. Besora and F. Maseras, Dalton Trans., 2019, 48, 16242 RSC; (d) J. Joy, T. Stuvyer and S. Shaik, J. Am. Chem. Soc., 2020, 142, 3836 CrossRef CAS PubMed.
- Selected reviews: (a) C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254 CrossRef CAS; (b) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692 RSC; (c) M. García-Melchor, A. A. C. Braga, A. Lledós, G. Ujaque and F. Masaras, Acc. Chem. Res., 2013, 46, 2626 CrossRef PubMed; (d) K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014, 43, 6609 RSC; (e) D. J. Jones, M. Lautens and G. P. McGlacken, Nat. Catal., 2019, 2, 843 CrossRef CAS; (f) M. C. D'Alterio, É. Casals-Cruańas, N. V. Tzouras, G. Talarico, S. P. Nolan and A. Poater, Chem.–Eur. J., 2021, 27, 13481 CrossRef PubMed.
- F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496 CrossRef CAS PubMed.
- Selected reviews: (a) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef CAS PubMed; (b) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40 RSC; (c) E. K. Reeves, E. D. Entz and S. R. Neufeldt, Chem.–Eur. J., 2021, 27, 6161 CrossRef PubMed; (d) M. Cao and H. Xie, Chin. Chem. Lett., 2021, 32, 319 CrossRef CAS; (e) V. Palani, M. A. Perea and R. Sarpong, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1c00513.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610 CrossRef CAS PubMed.
- F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192 CrossRef CAS PubMed.
- F. Proutiere and F. Schoenebeck, Synlett, 2012, 23, 645 CrossRef CAS.
- E. K. Reeves, O. R. Bauman, G. B. Mitchem and S. R. Neufeldt, Isr. J. Chem., 2020, 60, 406 CrossRef CAS PubMed.
- To our knowledge, none of the published solvent coordinative ability scales closely match the observed trends. However, none of these scales have been developed specifically for soft, late transition metals like palladium. For a review, see: S. Alvarez, Chem.–Eur. J., 2020, 26, 4350 CrossRef CAS PubMed.
- Anecdotally, we have sometimes encountered worse reproducibility with Pd2(dba)3, likely due to challenges with controlling its purity: S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- Q. Zhou, H. D. Srinivas, S. Zhang and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 11989 CrossRef CAS PubMed.
- The correlation to σ+ is a little stronger than to σ, see ESI† for details.
- For examples, see: (a) T. Diao, P. White, I. Guzei and S. S. Stahl, Inorg. Chem., 2012, 51, 11898 CrossRef CAS PubMed; (b) F. Schroeter, J. Soellner and T. Strassner, ACS Catal., 2017, 7, 3004 CrossRef CAS.
- We also examined the use of tetrahydrothiophene (THT), a sulfur analog of THF with a similar dielectric constant. However, as a softer atom, sulfur is generally a better ligand for palladium compared to oxygen. Consistent with a coordination hypothesis, THT and THF promote opposite chemoselectivities, but the reaction in THT gives extremely poor conversion (∼2% yield of 2b, the only product observed). The low yield in THT may be due to its very strong coordinating ability, leading to catalyst poisoning (see ESI†).
- C. Wohlfarth, in CRC Handbook of Chemistry and Physics, ed. J. Rumble Jr, D. Lide and T. Bruno, CRC Press, Boca Raton, FL, 2020, Permittivity (Dielectric Constant) of Liquids Search PubMed.
- K. Suzuki, M. Shinya, Y. Ono, T. Matsumoto, N. Nanbu, M. Takehara, M. Ue and Y. Sasaki, Electrochemistry, 2007, 75, 611 CrossRef CAS.
- An intermolecular competition between 3a and 3b under the catalytic Suzuki coupling conditions yields similar selectivities as observed under stoichiometric conditions (see ESI†). Compared to 3a/3b, the para substituted analogues of these substrates give similar results in catalytic and stoichiometric studies, but with increased decomposition (see ESI†).
- The reaction of Pd(COD)(CH2TMS)2 with ArI + PtBu3 in toluene at room temperature has been shown to yield the oxidative addition adduct (PtBu3)Pd(Ar)(I): T. L. Andersen, S. D. Friis, H. Audrain, P. Nordeman, G. Antoni and T. Skrydstrup, J. Am. Chem. Soc., 2015, 137, 1548 CrossRef CAS PubMed.
- For other examples of the use of Pd(COD)(CH2SiR3)2 as a precursor to LnPd(Ar)(X) oxidative addition complexes (n = 1 or 2, X = halide); see: (a) B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552 CrossRef CAS PubMed; (b) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. Garcia-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (c) A. G. Sergeev, T. Schultz, C. Torborg, A. Spannenberg, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 7595 CrossRef CAS PubMed; (d) P. Larini, C. E. Kefalidis, R. Jazzar, A. Renaudat, E. Clot and O. Baudoin, Chem.–Eur. J., 2012, 18, 1932 CrossRef CAS PubMed; (e) P. J. Milner, Y. Yang and S. L. Buchwald, Organometallics, 2015, 34, 4775 CrossRef CAS PubMed.
- This complex has been previously reported as a dimer in the solid state and a monomer in solution: see ref. 2d.
- A. L. Casado, P. Espinet and A. M. Gallego, J. Am. Chem. Soc., 2000, 122, 11771 CrossRef CAS.
- We observe formation of Pd black during the stoichiometric reactions in all solvents, typically after about 30–60 minutes or longer. This can partly be attributed to decomposition of the Pd triflate oxidative addition adduct (see ref. 2b). Although stable palladium complexes ArPd(PPh3)2(OTf) and ArPd(PPh3)2(THF) have been previously reported (see ref. 24), we see no evidence of inner-sphere Pd(OTf) in any solvents nor evidence for THF-stabilized cationic palladium complexes under our conditions with PtBu3.
- The route to formation of Pd black involves clusters and nanoparticles, and the selectivity of such intermediate multinuclear species during oxidative addition is a topic of ongoing study in our group.
- The 31P NMR spectra for the DMF reactions in entries 7 and 13 display a unique signal that may correspond to a solvent-bound cationic oxidative addition product from the reaction of 3b (see ESI†).
- We also considered, and tentatively ruled out, the possibility that autocatalysis mediated by PtBu3·HCl is primarily responsible for the triflate selectivity observed in coordinating solvents (see ESI†). Autocatalysis has previously been reported in the oxidative addition of PhBr to Pd(PtBu3)2: F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 5842 CrossRef CAS PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556 CrossRef CAS PubMed.
- Y.-P. Li, J. Gomes, S. M. Sharada, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2015, 119, 1840 CrossRef CAS.
- 3D image was prepared using CYLview 2.0, ed. C. Y. Legault, Université de Sherbrooke, 2020, http://www.cylview.org Search PubMed.
- All thermodynamic quantities were computed with the GoodVibes code (298.15 K): G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data, F1000Research, 2020, 9, 291 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 38, 3098 CrossRef PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- E. Lyngvi, I. A. Sanhueza and F. Schoenebeck, Organometallics, 2015, 34, 805 CrossRef CAS.
- Examples: (a) Y. Zhao and D. G. Truhlar, Org. Lett., 2007, 9, 1967 CrossRef CAS PubMed; (b) D. Benitez, E. Tkatchouk and W. A. Goddard III, Chem. Commun., 2008, 6194 RSC; (c) C. L. McMullin, J. Jover, J. N. Harvey and N. Fey, Dalton Trans., 2010, 39, 10833 RSC; (d) M. S. G. Ahlquist and P.-O. Norrby, Angew. Chem., Int. Ed., 2011, 50, 11794 CrossRef CAS PubMed; (e) S. O. N. Lill, P. Ryberg, T. Rein, E. Bennström and P.-O. Norrby, Chem.–Eur. J., 2012, 18, 1640 CrossRef PubMed; (f) C. McMullin, N. Fey and J. N. Harvey, Dalton Trans., 2014, 43, 13545 RSC; (g) J. Zhu, Y. Liang, L. Wang, Z.-B. Zheng, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2014, 136, 6900 CrossRef CAS PubMed; (h) M. Xie and W. Lu, RSC Adv., 2018, 8, 2240 RSC; (i) L. V. Hooker and S. R. Neufeldt, Tetrahedron, 2018, 74, 6717 CrossRef CAS PubMed.
- H. S. Yu, X. He and D. G. Truhlar, J. Chem. Theory Comput., 2016, 12, 1280 CrossRef CAS PubMed.
- Results are similar with LANL2DZ instead of SDD for Pd, see ESI†.
- The room-temperature reaction of 1 with PhSnnBu3 is low-yielding. See the ESI† for full details of our analysis of this cross-coupling.
- The attempted Stille coupling of 1 with PhSnMe3 in other coordinating solvents (MeCN and DMSO) gives only trace yields of 7a and 7b at room temperature.
- The poor conversion in the absence of a base is consistent with a previous report under slightly different conditions: A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343 CrossRef CAS PubMed.
- Aryl group transfer is usually favored over alkyl group trans-fer from tin in Stille couplings, but, in at least one example, methylation was observed as the major product from a Stille coupling using PhSnMe3: E. Gomez-Bengoa and A. M. Echavarren, J. Org. Chem., 1991, 56, 3497 CrossRef CAS.
- Replacing KPF6 with KF in the room temperature reaction in Scheme 3 leads to 18% yield of 7a and 45% of 7b, with 33% of 1 remaining after 24 h. The clear preference for 7b under these conditions is consistent with the prior report (ref. 9). We hypothesize that fluoride accelerates the slow transmetallation step, allowing the desired cross-coupling to outcompete catalyst speciation changes (decomposition). For an example of the accelerating effect of fluoride on a Pd/PtBu3-catalyzed Stille cross-coupling of aryl chlorides, see ref. 40.
- Other precatalyst systems give similar results to system B. See ESI† for details.
- For example, palladium clusters and monomeric Pd have been found to promote different selectivity in cross-coupling reactions of 2,4-dibromopyridine: N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682 CrossRef CAS PubMed.
- Attempts to recycle the catalyst after using it in a 100 °C Suzuki coupling in DMF provides results that are consistent with catalyst decomposition to a species that preferentially reacts at chloride in DMF (see ESI†).
- M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 194 CrossRef CAS.
- (a) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed; (b) A. H. M. De Vries, F. J. Parlevliet, L. Schmieder-van de Vondervoort, J. H. Mommers, H. J. W. Henderickx, M. A. M. Walet and J. G. De Vries, Adv. Synth. Catal., 2002, 344, 996 CrossRef CAS; (c) L. J. Gooßen, D. Koley, H. L. Hermann and W. Thiel, J. Am. Chem. Soc., 2005, 127, 11102 CrossRef PubMed; (d) S. Kozuch, C. Amatore, A. Jutand and S. Shaik, Organometallics, 2005, 24, 2319 CrossRef CAS; (e) M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066 CrossRef CAS; (f) M. Ahlquist and P.-O. Norrby, Organometallics, 2007, 26, 550 CrossRef CAS; (g) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 79 CrossRef CAS; (h) F. Schroeter, J. Soellner and T. Strassner, ACS Catal., 2017, 7, 3004 CrossRef CAS.
- For examples of other solvent effects on Pd-catalyzed cross-couplings, see: J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details; NMR spectra; calculated energies; cartesian coordinates of minimum-energy calculated structures. See DOI: 10.1039/d1sc05862b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |