
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salicylate metal-binding isosteres as fragments for metalloenzyme inhibition†
Moritz K.
Jackl
,
Hyeonglim
Seo
 ,
Johannes
Karges
,
Mark
Kalaj
and
Seth M.
Cohen
*
,
Johannes
Karges
,
Mark
Kalaj
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: scohen@ucsd.edu
First published on 21st January 2022
Abstract
Metalloenzyme inhibitors typically share a common need to possess a metal-binding pharmacophore (MBP) for binding the active site metal ions. However, MBPs can suffer from physicochemical liabilities, impeding the pharmacological properties and drug-likeliness of inhibitors. To circumvent this, problematic features of the MBP can be identified and exchanged with isosteric replacements. Herein, the carboxylic and hydroxyl group of the salicylic acid MBP were replaced and a total of 27 salicylate metal-binding isosteres (MBIs) synthesized. Of these 27 MBIs, at least 12 represent previously unreported compounds, and the metal-binding abilities of >20 of the MBIs have not been previously reported. These salicylate MBIs were examined for their metal-binding features in model complexes, physicochemical properties, and biological activity. It was observed that salicylate MBIs can demonstrate a range of attractive physicochemical properties and bind to the metal in a variety of expected and unexpected binding modes. The biological activity of these novel MBIs was evaluated by measuring inhibition against two Zn2+-dependent metalloenzymes, human glyoxalase 1 (GLO1) and matrix metalloproteinase 3 (MMP-3), as well as a dinuclear Mn2+-dependent metalloenzyme, influenza H1N1 N-terminal endonuclease (PAN). It was observed that salicylate MBIs could maintain or improve enzyme inhibition and selectivity. To probe salicylate MBIs as fragments for fragment-based drug discovery (FBDD), an MBI that showed good inhibitory activity against GLO1 was derivatized and a rudimentary structure–activity relationship was developed. The resulting elaborated fragments showed GLO1 inhibition with low micromolar activity.
Introduction
Metalloenzymes have been found to play important roles in the propagation of various diseases, ranging from microbial infections to cancer, which makes them an important class of targets for drug development.1 Fragment-based drug discovery (FBDD)2–5 strategies have been successfully used to develop potent inhibitors against metalloenzymes. Starting from a metal-binding pharmacophore (MBP), fragment growth strategies have been applied to identify hits from which lead-like compounds with good potency and selectivity can be obtained.6–8Unfortunately, many MBPs are highly polar molecules that contain functional groups that can suffer from pharmacological liabilities, such as poor membrane permeability, challenging solubility, or toxicity. An attractive approach to circumvent these problems is to install isosteric replacements. Problematic functional groups can be exchanged with alternative, isosteric groups with improved properties while maintaining similar biological activity. This strategy is frequently applied in modern medicinal chemistry and drug development and it has been demonstrated that isosteric replacement can improve solubility, lipophilicity, cell permeability, and reduce toxicity. Furthermore, isosteres can be used to produce novel compositions of matter for intellectual property purposes.9–11
Recently, isosteric replacement has been applied to MBPs for metalloenzyme inhibition. In a proof-of-concept study, the carboxylic acid moiety of picolinic acid was exchanged for known carboxylic acid isosteres to obtain a library of metal-binding isosteres (MBIs). These picolinic acid MBIs showed a broad range of physicochemical properties while maintaining similar metal-binding properties and biological activity as the parent MBP. This demonstrated the potential value of the MBI concept to allow for modulation of properties in the development of more drug-like metalloenzyme inhibitors by FBDD.12,13
Herein, MBIs of salicylic acid, a known metal binding ligand and metalloenzyme inhibitor, were prepared.14 A library of 27 salicylate MBIs was designed, synthesized, and studied for metal-binding and physicochemical properties. Furthermore, their inhibitory activity against two Zn2+-dependent metalloenzymes – human glyoxalase I (GLO1) and matrix metalloproteinase-3 (MMP-3) – and a dinuclear Mn2+-dependent metalloenzyme – influenza H1N1 N-terminal endonuclease (PAN) – was investigated. A selected salicylate MBI, which showed good inhibitory activity against GLO1, was computationally docked into the enzyme active site and a rudimentary SAR study was performed. It was demonstrated that salicylate MBIs not only cover a wide range of physicochemical properties and chemical space but also showcase various metal-binding modes, opening new avenues for metalloenzyme inhibitor development. It was observed that salicylate MBIs not only maintain, but also can surpass the parent salicylic acid MBP in terms of metalloenzyme inhibition and selectivity in select cases. Finally, it was shown that salicylate MBI fragments are easy to derivatize, which makes them strong candidates for lead development and rational drug design.
Results and discussion
MBI library design and synthesis
Salicylic acid 1 is known to bind to various metal ions and salicylic acids have been reported as metalloenzyme inhibitors.14 Two sites of the molecule – the hydroxyl group and the carboxylic acid group – are generally involved in metal binding. Consequently, these functional groups were the target of isosteric replacement (Fig. 1).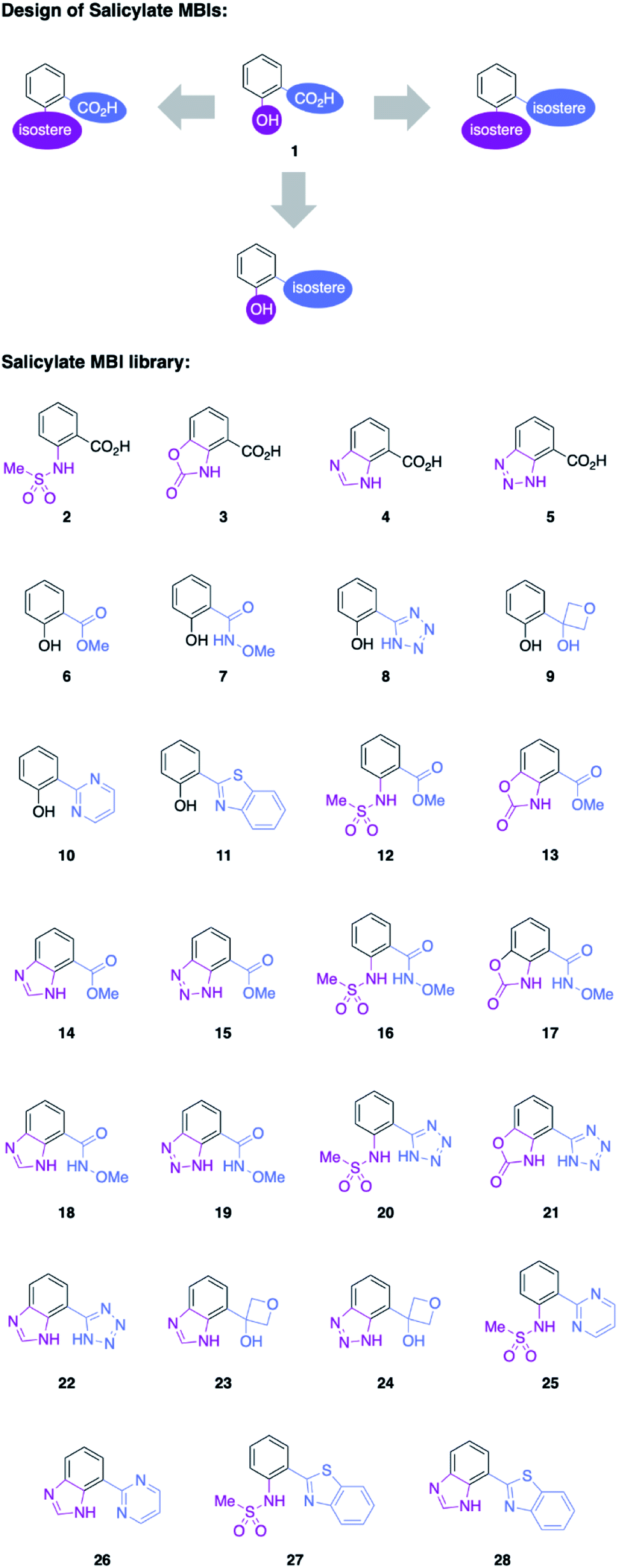 | ||
| Fig. 1 Top: Salicylic acid (1) and conceptual approach to salicylic acid metal-binding isosteres (MBIs). Bottom: MBIs of salicylic acid. The hydroxyl group (purple) and the carboxylic acid group (blue) were replaced with isosteres to obtain 27 new MBIs. | ||
Similar to a previous study on picolinic acid MBIs, the carboxylic acid moiety was substituted by a hydroxamic acid ester, tetrazole, and oxetane.13 These isosteric replacements were assumed to bind to the metal in their deprotonated, anionic form. Furthermore, methyl ester, pyrimidine, and benzothiazole were identified as neutral metal coordinators and potential carboxylic acid isosteres. To date, hydroxyl group isosteres have not been widely reported in the context of MBIs. Inspired by the literature on classical, non-metal-binding isosteres, several functional groups, including sulfonamides, carbamates, imidazoles, and triazoles were selected as isostere replacements for the hydroxyl group.9–11
MBIs with one or both of these isosteric replacements were designed and synthesized, resulting in a library of 27 MBIs (Fig. 1). Salicylate MBIs 2, 4, and 6 were purchased from commercial suppliers, while all other MBIs were prepared in a few steps from widely available starting materials using established synthetic methods (see ESI†). Esters and hydroxamic acid esters were derived from carboxylic acids by simple functional group interconversions, whereas tetrazoles were prepared from nitriles by 1,3-dipolar cycloaddition. Oxetanes were accessible from halides by lithiation/addition to 3-oxetanone, pyrimidines and benzothiazoles were synthesized using cross-coupling approaches. Sulfonamides were prepared by reaction of anilines with methylsulfonyl chloride, carbamates by annulation of amino alcohols with 1,1′-carbonyldiimidazole (CDI), and benzotriazoles by a diazotization/ring-closing protocol. Importantly, 12 out of 27 MBIs (compounds 16–19, 21–28) are new, unreported molecules. This indicates that by simply applying the principle of isosters to salicylic acid, new chemical space and new compositions of matter can be readily obtained.
Synthesis and characterization of model complexes
To evaluate the structural features of metal binding by the salicylate MBIs, [(TpPh,Me)Zn(MBI)] (TpPh,Me = hydrotris(3,5-phenylmethylpyrazolyl)borate) model complexes were prepared and characterized by X-ray crystallography (Fig. 2). The [(TpPh,Me)Zn(MBI)] complex models the tris(histidine) active site of Zn2+-dependent metalloenzymes (e.g., matrix metalloproteinases, MMPs) and has been widely used to evaluate the structural characteristics of MBPs and MBIs as potential metalloenzyme inhibitors.15–17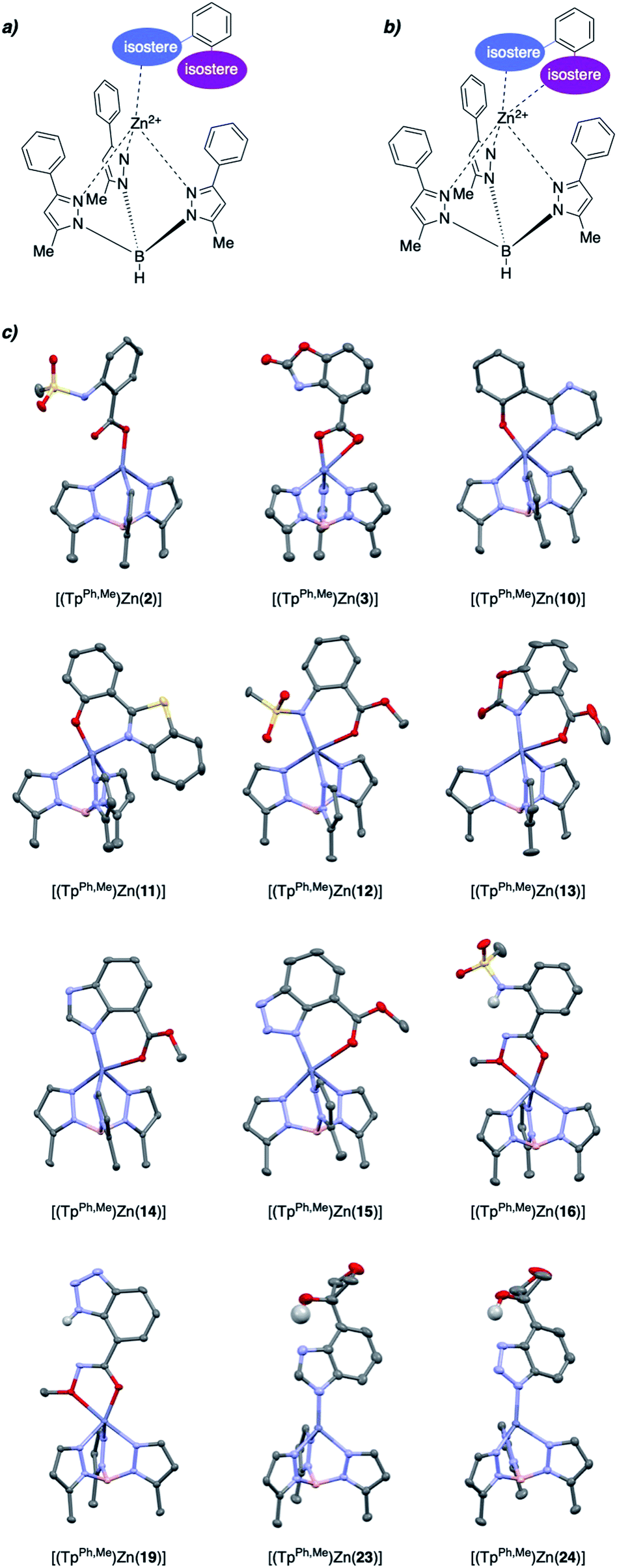 | ||
| Fig. 2 (a) Monodentate and (b) bidentate binding modes for MBIs. (c) Crystal structures of [(TpPh,Me)Zn(MBI)] model complexes. Hydrogen atoms and phenyl groups on TpPh,Me were omitted for clarity except for hydrogen atoms involved in intramolecular hydrogen bonds. Color code: carbon = gray, nitrogen = blue, oxygen = red, sulfur = yellow, boron = pink, zinc = dark blue. | ||
Various binding modes were observed with different salicylate MBIs (Fig. 2). While MBIs 10–15 formed 6-membered chelate rings engaging both hydroxyl and carboxylic acid groups (or isosteres) MBIs 2, 3, 16, 19, 23, and 24 bound the Zn2+ ion by only one isostere donor in either a monodentate or bidentate fashion.
Both monodentate and bidentate coordination in [(TpPh,Me)Zn(MBP)] complexes has been observed for salicylate ligands18 and it was hypothesized that the differences in binding behaviour observed here can be explained, in part, by charge neutrality. To obtain an overall neutral [(TpPh,Me)Zn(MBI)] complex, monoanionic MBIs are required, as dianionic MBIs will produce charged species. However, some functional groups, such as carboxylic acids, alcohols, or sulfonamides must be deprotonated (e.g., form anionic ligands) to effectively bind to the metal centre. MBIs that combine two ionizable groups, such as 2, 3, 16, 19, 23, and 24, consequently can only bind as 6-membered chelates when double deprotonated (i.e., dianionic), which disfavours charge neutrality. It should be noted that this is a limitation of the [(TpPh,Me)Zn(MBI)] model complex and does not necessarily correlate within the context of a metalloenzyme active site. Indeed, several protein crystal structures of dianionic MBPs (e.g., catechol ligands) bound to active site metal ions in metalloenzymes have been reported.19–21
An interesting observation was made for hydroxamic ester MBIs 16 and 19 (Fig. 2). These MBIs bind to the Zn2+ ion via the deprotonated hydroxamic ester moiety forming 5-membered chelates. The deprotonated hydroxamic ester is present in the enol form and presumably stabilized by intramolecular hydrogen bonding from the sulfonamide and benzotriazole groups, respectively. Considering the widespread use of hydroxamic acids as MBPs in metalloenzyme inhibitors,22 this observation could inspire the use of hydroxamic esters for new avenues in the development of MBIs and metalloenzyme inhibitors. For example, the hydroxamic ester moiety could be easily elaborated by applying more complex O-substituted hydroxylamines. This would enable additional interactions with enzyme pockets near the metal centre, which cannot be readily achieved with conventional hydroxamic acid MBPs.
Internal hydrogen bonds were also observed for oxetane MBIs 23 and 24 (Fig. 2). These MBIs are bound to the metal with a deprotonated nitrogen atom of the imidazole or triazole ring. Another nitrogen of the heteroaromatic moiety engages in internal hydrogen bonding with the aliphatic hydroxyl group adjacent to the oxetane. Apart from stabilization, this also rigidifies the molecules and could enable secondary interactions of the oxetane moiety with an enzyme pocket. Overall, this rich variations in metal binding modes among the 27 novel MBIs, serves to highlight the fact that only five of these compounds have previously characterized metal complexes (compounds 4, 6, 8, 11, 14), further emphasizing their utility as novel moieties for metal coordination.
Physicochemical analysis
In addition to gaining an understanding about the metal coordinating properties of the salicylate MBIs by using [(TpPh,Me)Zn(MBI)] model complexes, the physicochemical properties of the MBIs were also studied. The acidity (pKa) and lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) were measured using potentiometric methods and the lipophilicity at physiological pH (logD7.4) was calculated from these two values (Table 1).23–25
P) were measured using potentiometric methods and the lipophilicity at physiological pH (logD7.4) was calculated from these two values (Table 1).23–25
| Compound | pKa | logP |
logD7.4 |
|---|---|---|---|
| 1 | 2.82, >12 | 2.18 | −2.40 |
| 2 | 3.04, 10.8 | 1.39 | −2.97 |
| 3 | 3.65, 10.29 | 1.28 | −2.47 |
| 4 | 3.04, 5.49, 12.79 | −1.40 | −2.09 |
| 5 | 3.49, 9.29 | 1.38 | −2.37 |
| 6 | 9.8 | 2.19 | 2.19 |
| 7 | 7.23, 12 | 0.85 | 0.49 |
| 8 | 4.63, 10.92 | 1.54 | −0.46 |
| 9 | 9.38 | 0.43 | 0.42 |
| 12 | 8.86 | 1.51 | 1.49 |
| 13 | 8.78 | 1.24 | 1.22 |
| 14 | 4.51, 12.32 | 1.73 | 1.73 |
| 15 | 7.96 | 1.31 | 1.21 |
| 16 | 7.15, 11.31 | 0.12 | −0.33 |
| 17 | 7.58, 12.51 | 0.40 | 0.18 |
| 18 | 4.23, 8.40, >12 | 0.05 | 0.01 |
| 19 | 6.99, 9.93 | 0.75 | 0.22 |
| 20 | 3.62, 9.52 | 1.18 | −0.43 |
Overall, a wide range of acidities and lipophilicities was observed among the MBIs. Except for MBI 4 (Table 1, entry 4), which is an ampholyte and presumably exist as the zwitterion, all logP values were in the range of 0–3. MBIs 2–5 (Table 1, entries 2–5) were rather acidic (pKa,1 ∼3) and are deprotonated at physiological pH, which explains the high polarity of these MBIs at this pH (logD7.4 approx. −2). When the carboxylic moiety was exchanged for a less acidic isostere, the lipophilicity at physiological pH increased significantly. Interestingly, this was also observed for the tetrazole MBIs – despite the rather low pKa of this functional group (pKa,1 ∼4), lipophilicities at physiological pH were in the range logD7.4 ∼0. This highlights that by careful selection and combination of isosteric groups, MBIs with tailor-made physicochemical properties can be designed.
Unfortunately, the physicochemical properties of benzothiazole MBIs (11, 27, 28) could not be measured with the applied method due to the poor aqueous solubility of these compounds. Literature data of structurally related compounds (e.g., 4-(benzo[d]thiazol-2-yl)phenol, logP = 1.86, determined by a HPLC based method) suggests higher lipophilicity compared to other salicylate MBIs. This was confirmed by computationally calculated physicochemical data (Tables S2 and S3†). The calculated logP and logD7.4 values of MBIs 11 and 28 were the highest of the entire MBI library, which potentially makes these MBIs interesting candidates for central nervous system applications.26–28
MBI screening
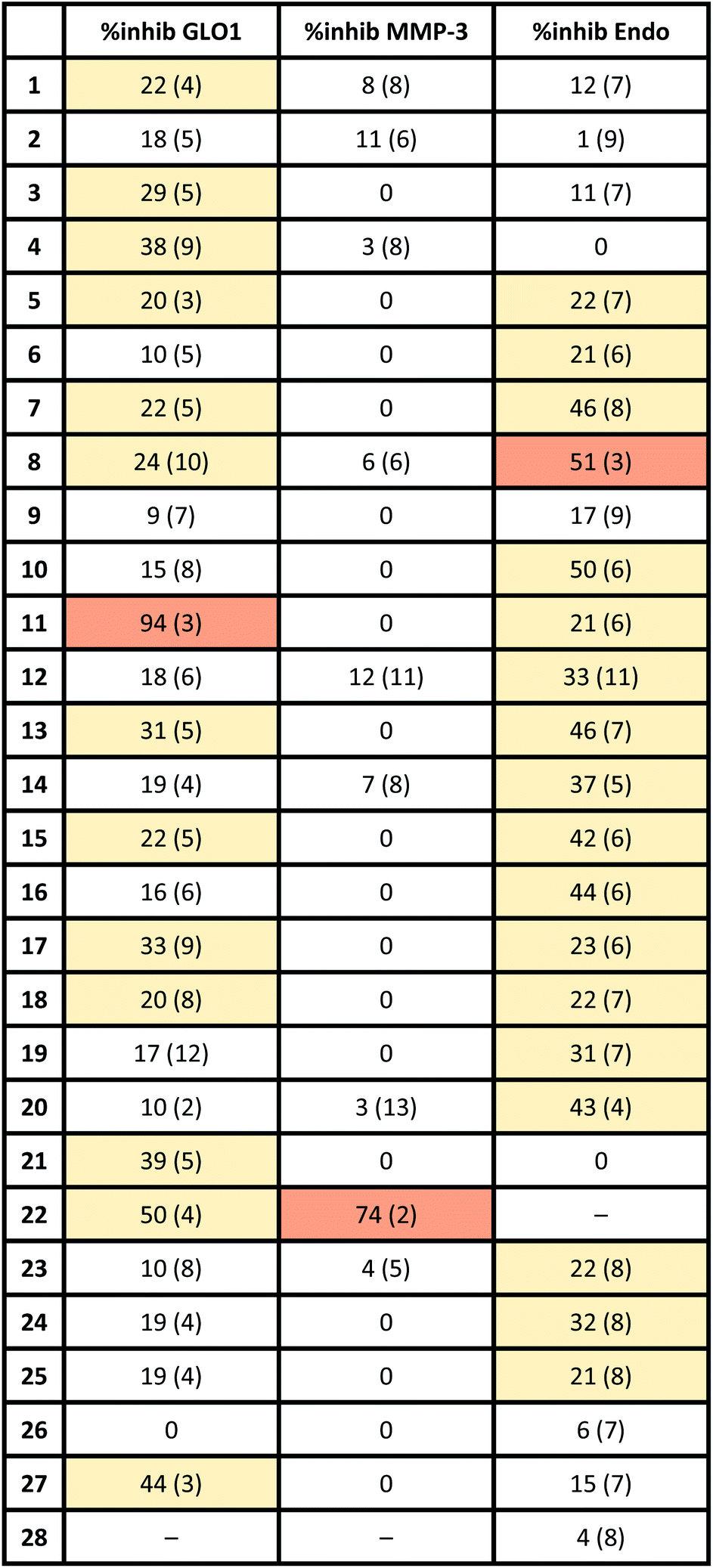
To evaluate if the salicylate MBIs could serve as starting points for future FBDD campaigns, the MBI library was screened for inhibitory activity against human glyoxalase I (GLO1) at fragment concentrations of 100 μM (Table 2). GLO1 is a Zn2+-dependent metalloenzyme and is associated with the propagation of several diseases.29–31 The catalytic Zn2+ is coordinated to four amino acids (Gln33, Glu99, Glu172, His126) and is in proximity to two hydrophobic pockets.1,32 Previous studies (unpublished) found salicylic acid 1 to be a weak GLO1 inhibitor (IC50 >50 μM). This result was confirmed in our screening and around half of the salicylate MBIs showed similar or better inhibitory activity compared to salicylic acid. MBIs 11, 22, and 27 inhibited GLO1 significantly better, with 22 showing 50% inhibition and 11 almost complete inhibition at 100 μM. The good inhibitory activity of 11 compared to the other salicylate MBIs could be rationalized by hydrophobic (and potentially π–π) interactions of the benzothiazole moiety with the hydrophobic pockets in proximity to the catalytic Zn2+.8,33 This indicates that isosteric replacements in MBPs could not only maintain, but also improve enzyme binding for metalloenzyme inhibition.
|
To probe the selectivity of the salicylate MBIs, the library was screened against matrix metalloproteinase-3 (MMP-3) at fragment concentrations of 100 μM (Table 2). MMP-3 is another Zn2+-dependent metalloenzyme that is inhibited by a variety of MBPs – however, salicylic acid 1 is not a known MMP-3 inhibitor.1,14,34,35 Except for MBI 22, which showed 74% inhibitory activity against MMP-3, the salicylate MBIs showed only very weak or no inhibition at all. These results indicate that salicylate MBIs could serve as selective fragments for the development of more elaborate metalloenzyme inhibitors, even against enzymes that contain common active site metal ions (e.g., Zn2+).
Finally, the salicylate MBI library was screened against influenza H1N1 N-terminal PA endonuclease (PAN), a dinuclear Mn2+-dependent metalloenzyme.38 PAN plays a major role in the viral lifecycle and is highly conserved among various influenza strains, which makes it an interesting target for the development of influenza antivirals.1,7 While the inhibitory activity of salicylic acid 1 was rather low at a concentration of 100 μM, more than half of the MBIs showed some significant inhibition (Table 2), with MBIs 8 and 10 showing inhibition ≥50%. This highlights that salicylate MBIs are not limited to Zn2+-dependent metalloenzymes and could potentially serve as starting points for inhibitor development against a variety of metalloenzyme targets.
Computational docking and SAR
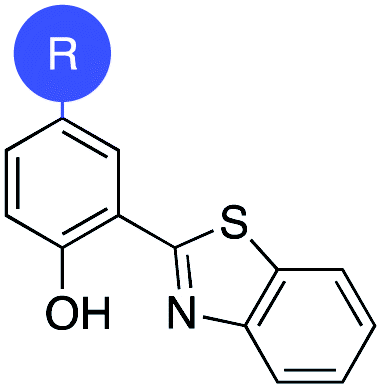
To rationalize the high inhibitory activity of 11 against GLO1 and to probe salicylate MBIs as starting points for lead development, computational docking studies were performed. A computational model was built using a reported GLO1 crystal structure (PDB: 3VW9, see ESI† for details).33 The docking study confirmed the initial hypothesis – 11 coordinates to the catalytic Zn2+ with the benzothiazole nitrogen and the deprotonated hydroxyl group, thereby directing the aromatic moieties towards the hydrophobic pockets of the GLO1 active site (Fig. 3a). The first hydrophobic pocket, also referred to as the glutathione (GSH) binding pocket, consists of Phe67, Leu69, Met157, and Phe162. A second hydrophobic pocket consists of Cys60, Phe62, Ile88, Met179, Leu182, and Met183 residues.1,8,32 The benzothiazole moiety of 11 is predicted to occupy a large part of the GSH binding pocket, possibly engaging in hydrophobic or π–π interactions with Phe67, Leu69, and Phe162. However, the second hydrophobic pocket is only partially occupied by 11. To better fill this pocket, a phenyl group was attached to 11 in the para position relative to the hydroxy group and this new fragment 11a was docked into the GLO1 active site (Fig. 3b and c, S2 and Table S4†). Compound 11a fills the hydrophobic active site almost completely with the phenyl group possibly engaging in hydrophobic or π–π interactions with Phe62, Leu182, or Cys60. Consequently, it was expected that 11a should also show improved inhibitory activity compared to 11. To validate the computation predictions, 11a and six other para substituted derivatives of 11 were synthesized and tested for their inhibitory activity against GLO1 (Table 3). All compounds were obtained in just a few synthetic steps using well established methods (see ESI†). Aryl substituted derivatives 11a–11c and 11g were prepared by Suzuki cross-coupling from aryl boronic acids, while saturated N-heterocycle substituted derivatives 11d–11f by Buchwald–Hartwig coupling with cyclic amines. This highlights that salicylate MBIs are easy to synthesize and derivatize, making them ideal candidates for FBDD and lead development. It was found that aryl substituted derivatives 11a–11c showed significantly better inhibition of GLO1 compared to 11 (Table 3). The IC50 of 11 was 19.6 ± 4.7 μM whereas the IC50 of 11c was determined to be 3.3 ± 0.7 μM, which confirms the docking and virtual fragment growth results. Exchanging the phenyl ring for smaller aromatic moieties did not improve inhibitory activity. The IC50 of furane 11g was similar to phenyl derivatives 11a–11c. The inhibitory activity of piperidine 11d and morpholine 11e was significantly worse than 11. Interestingly, the derivative substituted with the smaller pyrrolidine 11f showed similarly good inhibition as the aryl substituted derivatives. This suggests that ring-size and shape has a larger effect on GLO1 inhibition for saturated N-heterocycles than for the aryl substituted derivatives of 11. The inhibitory activity of compounds 11a–c and 11f–g is comparable to the known micromolar inhibitor Myricetin,36 which gave an IC50 of 4.8 ± 1.3 μM under identical assay conditions (Fig. S13†).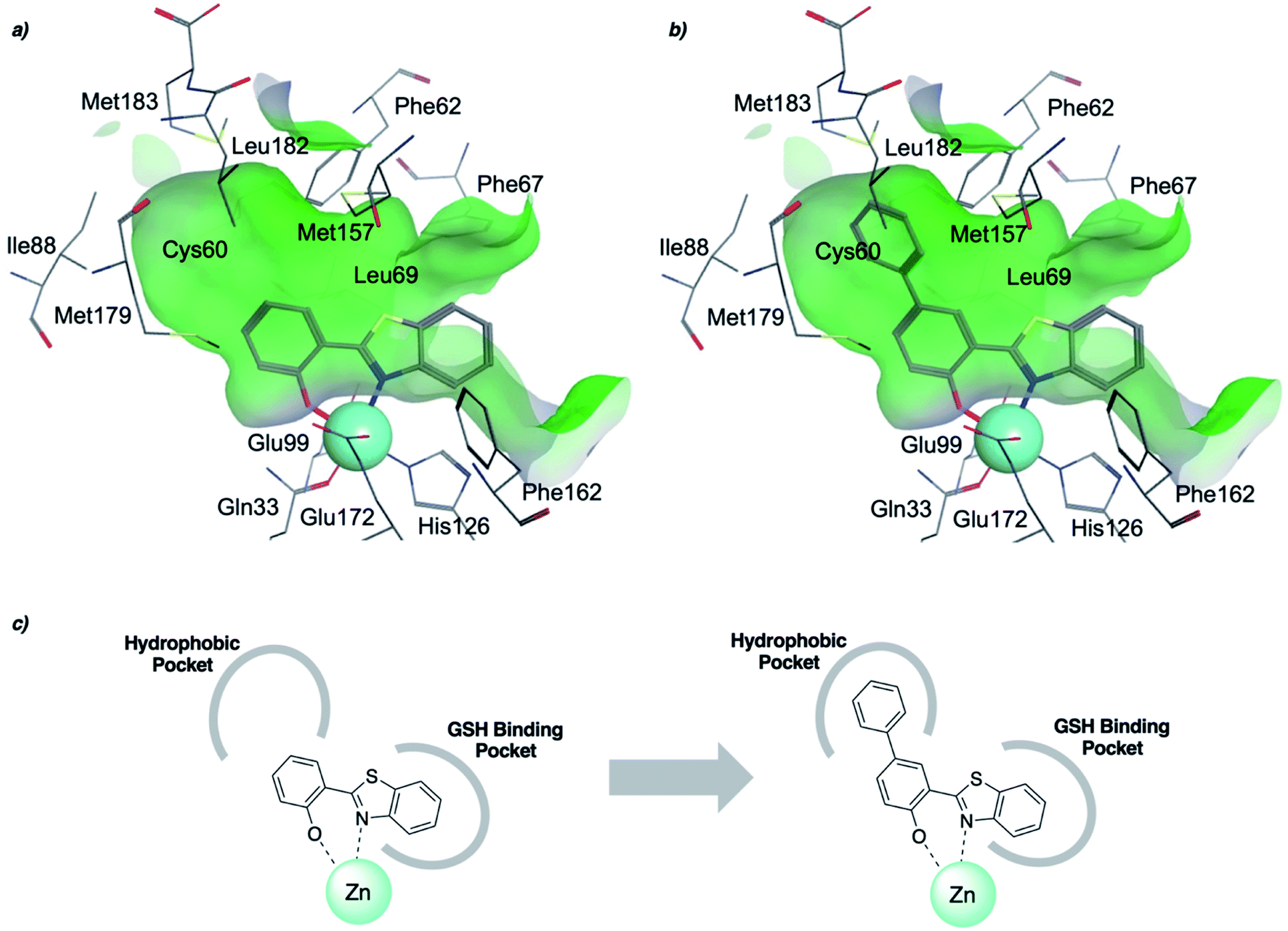 | ||
| Fig. 3 (a) Computational docking of 11 and (b) 11a in the GLO1 active site (PDB: 3VW9). The docking was performed using GOLD and visualized using MOE. Zn2+ is represented as a turquoise sphere, active site surface is colored in green with hydrophilic areas in blue. Inhibitor and key amino acid residues are drawn as sticks with carbon atoms in gray, oxygen in red, nitrogen in blue, and sulfur in yellow. (c) Schematic representation of fragment growth and inhibitor enzyme interactions. | ||
|
|
||
|---|---|---|
| Compound | R | IC50 [μM] |
| 11 | H | 19.6 ± 4.7 |
| 11a |
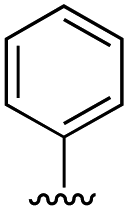
|
5.0 ± 1.5 |
| 11b |
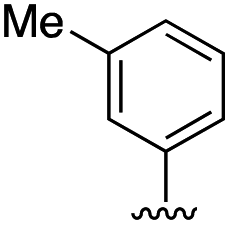
|
6.8 ± 1.4 |
| 11c |
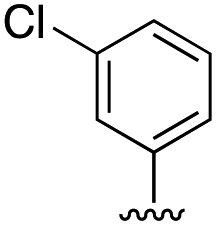
|
3.3 ± 0.7 |
| 11d |
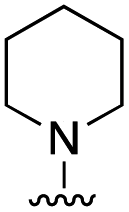
|
>20 |
| 11e |
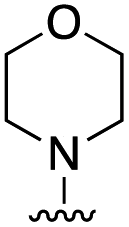
|
>50 |
| 11f |
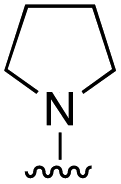
|
4.4 ± 1.8 |
| 11g |
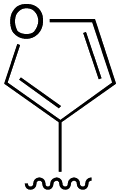
|
6.9 ± 2.8 |
Conclusions
Salicylate MBIs are easy to synthesize and allowed for rapid preparation of 27 compounds, of which nearly half were wholly new compositions of matter. They show a variety of metal binding modes in a [(TpPh,Me)Zn(MBI)] model system. Indeed the majority (>20) of the MBIs generated had no previously described coordination chemistry. Unexpected binding modes were observed for salicylate MBIs capable of forming intramolecular hydrogen bonds, which will open new avenues in the development of metalloenzyme inhibitors. The physicochemical properties of salicylate MBIs are tuneable with a wide range of acidities and lipophilicities achieved. Importantly, the lipophilicities of most salicylate MBIs tested are in a desirable range for FBDD. The screening of their inhibitory activity against three metalloenzyme targets revealed that isosteric replacements in MBPs cannot only maintain, but potentially improve inhibitory activity and that salicylate MBIs can inhibit various metalloenzyme targets containing different metals. Supported by computational docking studies and rudimentary SAR, the enzyme binding of a selected salicylate MBI was investigated and the inhibitory activity improved. This demonstrates that salicylate MBIs are an attractive starting point for FBDD and lead development. In conclusion, salicylate MBIs are an interesting new class of metalloenzyme binding fragments with beneficial physicochemical properties and will be useful for the development of new metalloenzyme targeting inhibitors with more drug-like properties.Experimental
General information
All solvents and reagents, unless otherwise noted, were obtained from commercial sources and used without further purification. MBIs 2, 4, 6, and 11 were purchased from Combi-Blocks Inc. and used without further purification. All reactions, unless otherwise stated, were performed under a nitrogen atmosphere. Silica chromatography was performed using a CombiFlash Rf Teledyne ISCO system using hexane, ethyl acetate, methylene chloride, or methanol as eluents. C18 reverse-phase chromatography was performed using the same instrument using 0.1% formic acid in methanol, acetonitrile, or water as eluent. Separations were monitored by mass spectrometry via a Teledyne ISCO RF+ PurIon ESI-MS detector with 1 Da resolution. 1H- and 13C-NMR spectra were obtained on a Varian (400 MHz) spectrometer or Jeol (500 MHz) spectrometer or a VX (500 MHz) equipped with an XSens cold probe (Varian) spectrometer in the Department of Chemistry and Biochemistry at U.C. San Diego. Standard resolution MS was performed either at U.C. San Diego Molecular Mass Spectrometry Facility or on the aforementioned Teledyne ISCO RF + PurIon MS. High resolution mass spectrometry (HRMS) analysis was performed using an Agilent 6230 accurate-mass liquid chromatography time-of-flight mass spectrometry LC-TOFMS located at the U.C. San Diego Molecular Mass Spectrometry Facility.General synthesis and characterization of model complexes
[(TpPh,Me)ZnOH] was prepared according to literature known procedures.37 [(TpPh,Me)ZnOH] (50 mg, 0.09 mmol) was dissolved in 15 mL of CH2Cl2 in a 50 mL round-bottom flask. The MBI (0.09 mmol, 1 equiv.) in 10 mL of MeOH was added, and the reaction mixture was stirred overnight under a nitrogen atmosphere. The resulting mixture was evaporated to dryness and subsequently dissolved in a minimal amount (∼1 mL) of benzene. The solution was filtered using a syringe filter to remove any undissolved solids. The resulting complex in benzene was recrystallized using vapor diffusion with pentane. Crystals typically formed within a few days.Physicochemical properties analysis
Physicochemical properties were determined using a Sirius T3 instrument.23–25 All titrations, both pKa and logP, were performed in 0.15 M KCl with 0.5 M HCl and KOH. The pKa of a compound was determined by analysing each MBI sample in triplicate using potentiometric titrations. Experiments were typically performed over a pH range of 2.0–12.0. Standard deviations were derived from fitting all three replicate experiments. For water insoluble compounds methanol was added and the obtained apparent pKa (psKa) was extrapolated to the aqueous pKa by the Yasuda–Shedlovsky procedure. logP was determined via potentiometric titrations in the presence of varying ratios of octanol and water. The presence of octanol shifts the pKa of ionizable species, and based on the shifts, a logP can be determined. Measurements for logP determination were typically performed over a pH range of 2.0–12.0. Three experiments with varying ratios of water:octanol was performed, allowing for a standard deviation to be determined from the fitting of all measurements. MBI sample sizes were ∼0.5 mg for both pKa and logP measurements.
GLO1 assay
Recombinant human glyoxalase I (GLO1) was purchased from R&D Systems (catalog #4959-GL). Assays were carried out in 0.1 M sodium phosphate, pH 7.0 buffer, utilizing 96-well clear UV plates (Corning UV Transparent Microplates, catalog #3635). A fresh solution of GSH (100 mM) and methylglyoxal (MG) (100 mM) was prepared in Millipore grade water. The substrate for the assay was prepared by adding 0.43 mL of GSH and 0.43 mL of MG to 15.14 mL of buffer. The substrate mixture was vortexed vigorously for 45 s and then allowed to sit at room temperature for 15 min. The initial well volume was 50 μL containing GLO1 (40 ng) and inhibitor. This protein and inhibitor mixture was incubated for 15–20 min prior to addition of the substrate. The substrate (150 μL) was then added to the wells yielding a maximum amount of 5% DMSO per well. The enzyme activity was measured utilizing a Biotek Synergy HT or H4 plate reader by measuring absorbance at 240 nm every 1 min for a duration of 8 min. The rate of absorbance increase was compared for samples versus controls containing no inhibitor (set at 100% activity). The absorbance reading for background wells containing DMSO, buffer, and substrate (no enzyme or inhibitor) was subtracted from the experimental wells. A positive control (1-hydroxy-4,6-diphenylpyridin-2(1H)-one, 50 μM)33 showed complete inhibition under the assay conditions described above. Dose-response curves were generated, analysed and fitted to obtain IC50 values (see ESI†). Myricetin,36 a known micromolar inhibitor, gave an IC50 value of 4.8 ± 1.3 μM under these assay conditions.MMP-3 assay
Human recombinant MMP-3 catalytic domain was purchased from ENZO Life Sciences (catalog # BML-SE109-9090). Assays were carried out in clear Costar 96-well, half-area, flat-bottom assay plates (catalog # 80-2404). Each well contained a total volume of 100 μL including buffer (50 mM MES, 10 mM CaCl2, 0.05% Brij-35, 1 mM DNTB, pH 6.0), MMP-3 (2 U), and the fragment solution. After a 30 min incubation period at 37 °C, the reaction was initiated by the addition of 10 μL of chromogenic MMP-3 substrate (100 μM final concentration, Ac-Pro-Leu-Gly-[2-mercap-to-4-methyl-pentanoyl]-Leu-Gly-OC2H5, ENZO Life Sciences, catalog #BML-P125-0005). Absorbance was monitored at 412 nm using a Bio-Tek ELx 808 colorimetric plate reader, and measurements were recorded every minute for 10 min. The rate of absorbance increase was compared for samples versus negative controls (no inhibitor, arbitrarily set as 100% activity). A positive control (NNGH, 1.3 μM) showed complete inhibition under the assay conditions described above.PAN assay
Endonuclease was expressed and purified as previously reported.38 The activity assays were carried in black Costar 96-well plates with a total volume of 100 μL per well. Assay buffer consisted of 20 mM Tris pH 8.0, 150 mM NaCl, 2 mM MnCl2, 2 mM MgCl2, 10 mM 2-mercaptoethanol, and 0.2% Triton-X100. Endo was included at a final concentration of 25 nM, and MBIs were added from a 50 mM DMSO stock to a final concentration of 100 μM. A fluorescent ssDNA-oligo substrate [([6-FAM]AATCGCAGGC-AGCACTC[TAM]), FAM = 6-carboxyfluorescein, TAM = tetrame-thylrhodamine] was used to monitor enzymatic activity. After addition of substrate, fluorescence (λex = 490 nm, λem = 520 nm) was monitored over 45 min at 39 s intervals at 37 °C. Negative control wells contained no inhibitor and were set to 100% activity. A reported pyridinone-based inhibitor was included as a positive control for inhibition.38 Percent inhibition was determined by comparing the activity of wells containing MBIs to the activity of those without.Author contributions
M. K. J. and S. M. C. outlined the project, interpreted results, and wrote the manuscript. M. K. J. and S. M. C. designed the MBI library, M. K. J. planned and executed the synthesis of all compounds. M. K. J. and H. S. performed the enzyme activity assays and interpreted the results. H. S., M. K., and M. K. J. performed X-ray crystallographic measurements and edited the data. J. K. performed computational docking studies.Conflicts of interest
S.M.C. is a cofounder of and has an equity interest in Cleave Therapeutics, Forge Therapeutics, and Blacksmith Medicines, companies that may potentially benefit from the research results. S.M.C. also serves on the Scientific Advisory Board for Blacksmith Medicines and serves on the Scientific Advisory Board and receives compensation from Forge Therapeutics. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies.Acknowledgements
This work was supported by the National Institute of Health (R01 AI149444). Financial support for M. K. J. was provided by the Swiss National Science Foundation (SNSF) (Early Postdoc Mobility Fellowship – Project No. P2EZP2_195505). We thank Dr Milan Gembicky, and Dr Jake Bailey (U.C. San Diego) for assistance with crystallo-graphic data collection and structure determination. We also thank Dr Yongxuan Su for mass spectrometry sample analysis at the Molecular Mass Spectrometry Facility at U.C. San Diego. This research was supported in part by W. M. Keck Foundation through computing resources at the W. M. Keck Laboratory for Integrated Biology. Finally, we thank Ryjul Stokes, Conor O'Herin, and Alysia Kohlbrand (U.C. San Diego) for helpful discussions.Notes and references
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2019, 119, 1323–1455 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- M. Congreve, G. Chessari, D. Tisi and A. J. Woodhead, J. Med. Chem., 2008, 51, 3661–3680 CrossRef CAS PubMed.
- P. J. Hajduk, J. Med. Chem., 2006, 49, 6972–6976 CrossRef CAS PubMed.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS PubMed.
- A. Y. Chen, P. W. Thomas, A. C. Stewart, A. Bergstrom, Z. Cheng, C. Miller, C. R. Bethel, S. H. Marshall, C. V. Credille, C. L. Riley, R. C. Page, R. A. Bonomo, M. W. Crowder, D. L. Tierney, W. Fast and S. M. Cohen, J. Med. Chem., 2017, 60, 7267–7283 CrossRef CAS PubMed.
- C. V. Credille, C. N. Morrison, R. W. Stokes, B. L. Dick, Y. Feng, J. Sun, Y. Chen and S. M. Cohen, J. Med. Chem., 2019, 62, 9438–9449 CrossRef CAS PubMed.
- C. Perez, A. M. Barkley-Levenson, B. L. Dick, P. F. Glatt, Y. Martinez, D. Siegel, J. D. Momper, A. A. Palmer and S. M. Cohen, J. Med. Chem., 2019, 62, 1609–1625 CrossRef CAS PubMed.
- P. Lassalas, B. Gay, C. Lasfargeas, M. J. James, V. Tran, K. G. Vijayendran, K. R. Brunden, M. C. Kozlowski, C. J. Thomas, A. B. Smith, D. M. Huryn and C. Ballatore, J. Med. Chem., 2016, 59, 3183–3203 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- G. A. Patani and E. J. Lavoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- B. L. Dick, A. Patel and S. M. Cohen, Chem. Sci., 2020, 11, 6907–6914 RSC.
- B. L. Dick and S. M. Cohen, Inorg. Chem., 2018, 57, 9538–9543 CrossRef CAS PubMed.
- J. A. Jacobsen, J. L. Fullagar, M. T. Miller and S. M. Cohen, J. Med. Chem., 2011, 54, 591–602 CrossRef CAS PubMed.
- D. T. Puerta and S. M. Cohen, Inorg. Chem., 2003, 42, 3423–3430 CrossRef CAS PubMed.
- G. Parkin, Chem. Commun., 2000, 1971–1985 RSC.
- H. Vahrenkamp, Acc. Chem. Res., 1999, 32, 589–596 CrossRef CAS.
- F. E. Jacobsen and S. M. Cohen, Inorg. Chem., 2004, 43, 3038–3047 CrossRef CAS PubMed.
- K. Rutherford, I. Le Trong, R. E. Stenkamp and W. W. Parson, J. Mol. Biol., 2008, 380, 120–130 CrossRef CAS PubMed.
- M. Kawatani, H. Okumura, K. Honda, N. Kanoh, M. Muroi, N. Dohmae, M. Takami, M. Kitagawa, Y. Futamura, M. Imoto and H. Osada, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11691–11696 CrossRef CAS PubMed.
- E. Kowalinski, C. Zubieta, A. Wolkerstorfer, O. H. J. Szolar, R. W. H. Ruigrok and S. Cusack, PLoS Pathog., 2012, 8, e1002831 CrossRef CAS PubMed.
- M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735–2776 CrossRef CAS PubMed.
- D. Schönherr, U. Wollatz, D. Haznar-Garbacz, U. Hanke, K. J. Box, R. Taylor, R. Ruiz, S. Beato, D. Becker and W. Weitschies, Eur. J. Pharm. Biopharm., 2015, 92, 155–170 CrossRef PubMed.
- K. Y. Tam and K. Takács-Novák, Anal. Chim. Acta, 2001, 434, 157–167 CrossRef CAS.
- B. Slater, A. McCormack, A. Avdeef and J. E. A. Comer, J. Pharm. Sci., 1994, 83, 1280–1283 CrossRef CAS PubMed.
- Y. Wang, C. A. Mathis, G.-F. Huang, M. L. Debnath, D. P. Holt, L. Shao and W. E. Klunk, J. Mol. Neurosci., 2003, 20, 255–260 CrossRef PubMed.
- W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergström, I. Savitcheva, G.-F. Huang, S. Estrada, B. Ausén, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Långström, Ann. Neurol., 2004, 55, 306–319 CrossRef CAS PubMed.
- Y. Huang, H.-J. Cho, N. Bandara, L. Sun, D. Tran, B. E. Rogers and L. M. Mirica, Chem. Sci., 2020, 11, 7789–7799 RSC.
- H. Sakamoto, T. Mashima, A. Kizaki, S. Dan, Y. Hashimoto, M. Naito and T. Tsuruo, Blood, 2000, 95, 3214–3218 CrossRef CAS PubMed.
- A. Hutschenreuther, M. Bigl, N. Hemdan, T. Debebe, F. Gaunitz and G. Birkenmeier, Int. J. Mol. Sci., 2016, 17, 2133 CrossRef PubMed.
- I. Hovatta, R. S. Tennant, R. Helton, R. A. Marr, O. Singer, J. M. Redwine, J. A. Ellison, E. E. Schadt, I. M. Verma, D. J. Lockhart and C. Barlow, Nature, 2005, 438, 662–666 CrossRef CAS PubMed.
- A. D. Cameron, B. Olin, M. Ridderström, B. Mannervik and T. A. Jones, EMBO J., 1997, 16, 3386–3395 CrossRef CAS PubMed.
- T. Chiba, J. Ohwada, H. Sakamoto, T. Kobayashi, T. A. Fukami, M. Irie, T. Miura, K. Ohara and H. Koyano, Bioorg. Med. Chem. Lett., 2012, 22, 7486–7489 CrossRef CAS PubMed.
- G. Murphy, J. Biol. Chem., 2017, 292, 7708–7717 CrossRef CAS PubMed.
- P. J. Hajduk, S. B. Shuker, D. G. Nettesheim, R. Craig, D. J. Augeri, D. Betebenner, D. H. Albert, Y. Guo, R. P. Meadows, L. Xu, M. Michaelides, S. K. Davidsen and S. W. Fesik, J. Med. Chem., 2002, 45, 5628–5639 CrossRef CAS PubMed.
- R. Takasawa, S. Takahashi, K. Saeki, S. Sunaga, A. Yoshimori and S.-I. Tanuma, Bioorg. Med. Chem., 2008, 16, 3969–3975 CrossRef CAS PubMed.
- D. T. Puerta and S. M. Cohen, Inorg. Chem., 2002, 41, 5075–5082 CrossRef CAS PubMed.
- C. V. Credille, Y. Chen and S. M. Cohen, J. Med. Chem., 2016, 59, 6444–6454 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, characterization data and spectra; details about computational docking studies; dose-response curves; crystallographic data for all [(TpPh,Me)Zn(MBI)] complexes. CCDC 2117767–2117778. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06011b |
| This journal is © The Royal Society of Chemistry 2022 |