
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoretentive and regioselective selenium-catalyzed intermolecular propargylic C–H amination of alkynes†
T. Parker
Maloney
,
Alexander F.
Dohoda
,
Alec C.
Zhu
and
Forrest E.
Michael
 *
*
University of Washington, Department of Chemistry, Box 351700, Seattle, Washington 98195-1700, USA. E-mail: michael@chem.washington.edu
First published on 27th January 2022
Abstract
Herein we report an intermolecular propargylic C–H amination of alkynes. This reaction is operationally convenient and requires no transition metal catalysts or additives. Terminal, silyl, and internal alkynes bearing a wide range of functional groups can be aminated in high yields. The regioselectivity of amination for unsymmetrical internal alkynes is strongly influenced by substitution pattern (tertiary > secondary > primary) and by relatively remote heteroatomic substituents. We demonstrate that amination of alkynes bearing α-stereocenters occurs with retention of configuration at the newly-formed C–N bond. Competition experiments between alkynes, kinetic isotope effects, and DFT calculations are performed to confirm the mechanistic hypothesis that initial ene reaction of a selenium bis(imide) species is the rate- and product-determining step. This ene reaction has a transition state that results in substantial partial positive charge development at the carbon atom closer to the amination position. Inductive and/or hyperconjugative stabilization or destabilization of this positive charge explains the observed regioselectivities.
Introduction
Selective functionalization of C–H bonds in organic molecules has proven to be an important strategy for synthesis of new compounds.1–7 The many different types of C–H bonds present in typical organic structures provides both a critical challenge as well as a prime opportunity for the development of new synthetic methods in this area. The large number of new methods for selective C–H functionalization highlights the great potential of this class of transformations. Identifying and addressing unsolved problems in C–H activation selectivity will be critical for continued progress in this area.The C–H amination of alkynes is an appealing application of this strategy due to the availability of alkyne starting materials8,9 and the importance of the propargylamine products as versatile synthetic intermediates.10 Though propargylamines are commonly constructed by the addition of alkynyl nucleophiles to imines, a direct C–H amination strategy would allow the C–N bond to be generated from a simple alkyne starting material by taking advantage of the native reactivity of the propargylic C–H bond (BDE ∼85 kcal mol−1)11 without the need for pre-oxidation at that center. Given the diversity of successful methods for C–H bond functionalization in the allylic position of alkenes,12–23 this analogous functionalization of alkynes might be expected to be a common approach. However, propargylic C–H amination has received relatively little attention (Scheme 1). Though several examples of intramolecular C–H amination of alkynes using metal nitrenoids have been reported,24–31 the need for an appropriately tethered nucleophile fundamentally limits their substrate scope and generality. In contrast, intermolecular propargylic C–H aminations are surprisingly rare. Isolated examples of radical amination of propargyl positions rely on the presence of additional activating groups and/or are severely limited in scope.32–34 Zhang and Lebel have reported enantioselective intermolecular C–H amination reactions that tolerate alkynes, but with only a handful of substrates.35–37
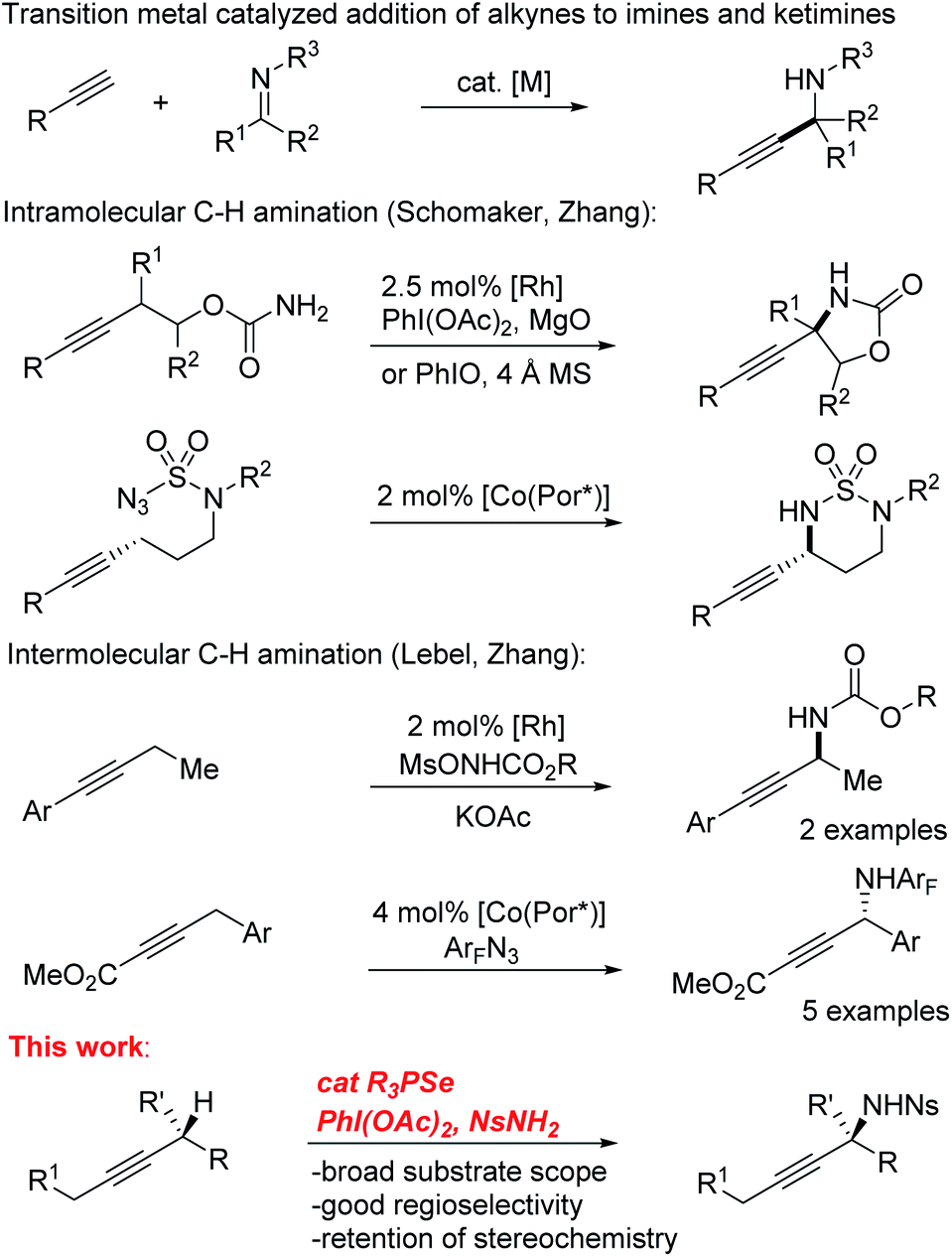 | ||
| Scheme 1 Common routes to propargylamines. | ||
The dearth of such reports may reflect a preference for nitrenoids to react directly with the π system of the triple bond, which is also a common problem for alkene C–H amination reactions.12,13,38 Similarly, allylic amination approaches based on the formation of π-allyl transition metal complexes are not easily extended to alkynes because of the much greater strain inherent in a π-propargyl complex and the difficulty controlling the regioselectivity of attack.39–41 Furthermore, with so few examples of intermolecular propargylic amination, little is known about the factors controlling the regioselectivity of activation in unsymmetrical substrates.
Recently, we developed a practical catalytic system for the allylic amination of alkenes using phosphine selenides as catalysts in place of transition metals.42 In Sharpless' original report describing the allylic amination of alkenes using stoichiometric selenium bis(imide), he reported the amination of just two alkyne substrates, though the yields were low (23–51%).43 We imagined that we could adapt our previously reported catalytic alkene amination system to develop the first general intermolecular propargylic C–H amination reaction. Herein we report the successful development of a selenium-catalyzed intermolecular propargylic C–H amination reaction with broad substrate scope. Critically, we observed that the regioselectivity of amination of internal alkynes could be strongly directed using simple inductively electron-withdrawing and donating groups from as far away as the beta position. As a result of the unique mechanism of action of the selenium catalyst, full retention of stereochemistry is observed when alkynes with alpha stereocenters are employed. We report reactivity studies and mechanistic experiments that explain the factors that control this remarkable selectivity.
We started by subjecting a test alkyne (4-phenyl-1-butyne, 1) to our standard conditions for the allylic amination of alkenes, i.e. using PhI(OAc)2 and NsNH2 in the presence of catalytic SePCy3. This reaction proceeded to give an acceptable yield of the product 2a (74%), but we found that a change of solvent from CH2Cl2 to dioxane generally gave higher yields (85%). As we observed in the corresponding amination of alkenes, commercial Se powder alone failed to catalyze the reaction, highlighting the critical role that the ligand plays in this reaction. However, premixing 15 mol% Se with PCy3 gave nearly identical yields as using isolated SePCy3 (80 vs. 85%), providing a convenient one-pot procedure for generating the catalyst in situ if so desired.
We then tested a range of sulfonamide and sulfamate nitrogen sources under these conditions (Scheme 2). Though all afforded some of the desired product, we found that NsNH2 gave the highest yields of the propargylamine product 2a, followed by TcesNH2 (2b). These two nitrogen sources are especially convenient because they bear protecting groups that are easily handled and removed under mild conditions. These protecting groups had also proved to be the highest yielding in the allylic amination, though we do not find the same simple trend of increasing yield with more electron-poor sulfonamides as we did in our allylic amination.42
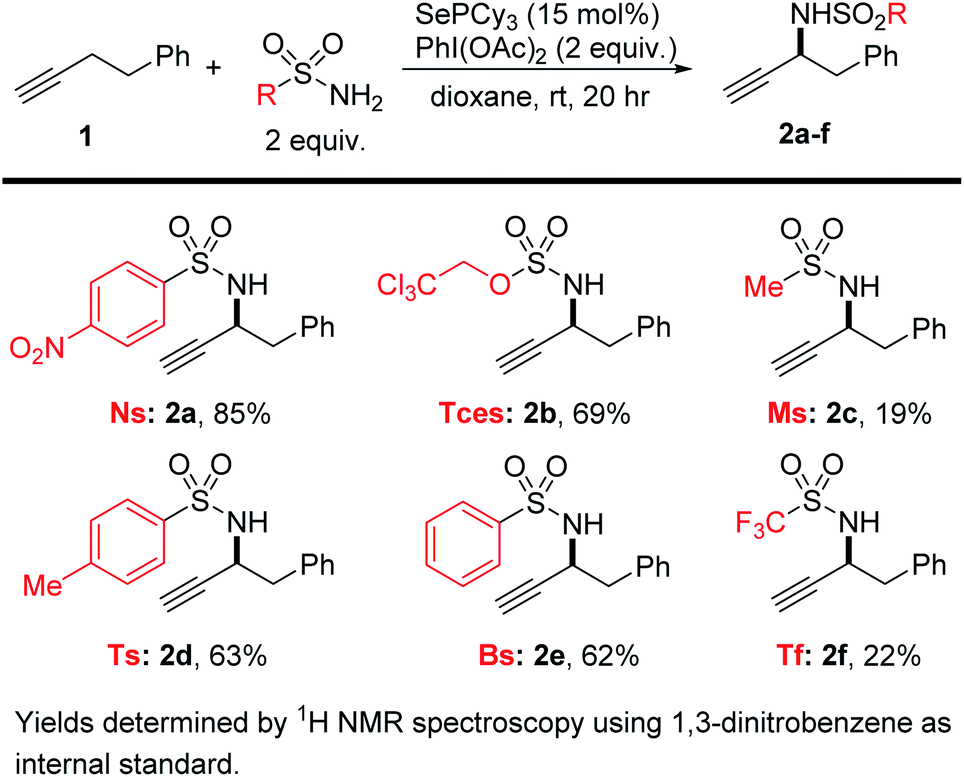 | ||
| Scheme 2 Sulfonamide/sulfamate screen. | ||
We tested a variety of terminal and silyl alkynes (Scheme 3) under our optimized amination conditions. We found that alkynes bearing a wide range of functional groups, including ethers, esters, nitriles, protected alcohols and amines, and alkyl and aryl halides were aminated in high yields. It was especially noteworthy that free carboxylic acids (4s), ketones (4h), and even oxidation-sensitive aldehydes (4i) were also tolerated. Primary (4j,k), secondary, and tertiary (4l) C–H bonds could all be effectively aminated using this procedure. A variety of 1,2- and 1,3-aminoalcohol and diamine derivatives (4d–i,m,p–r,u) can be conveniently prepared from the corresponding simple alkynes, without the need for intramolecular delivery of the nitrogen group, allowing flexibility in the choice of protecting groups on oxygen. The scale of the reaction could be increased to produce 1 gram of product 4a without significant reduction in yield.
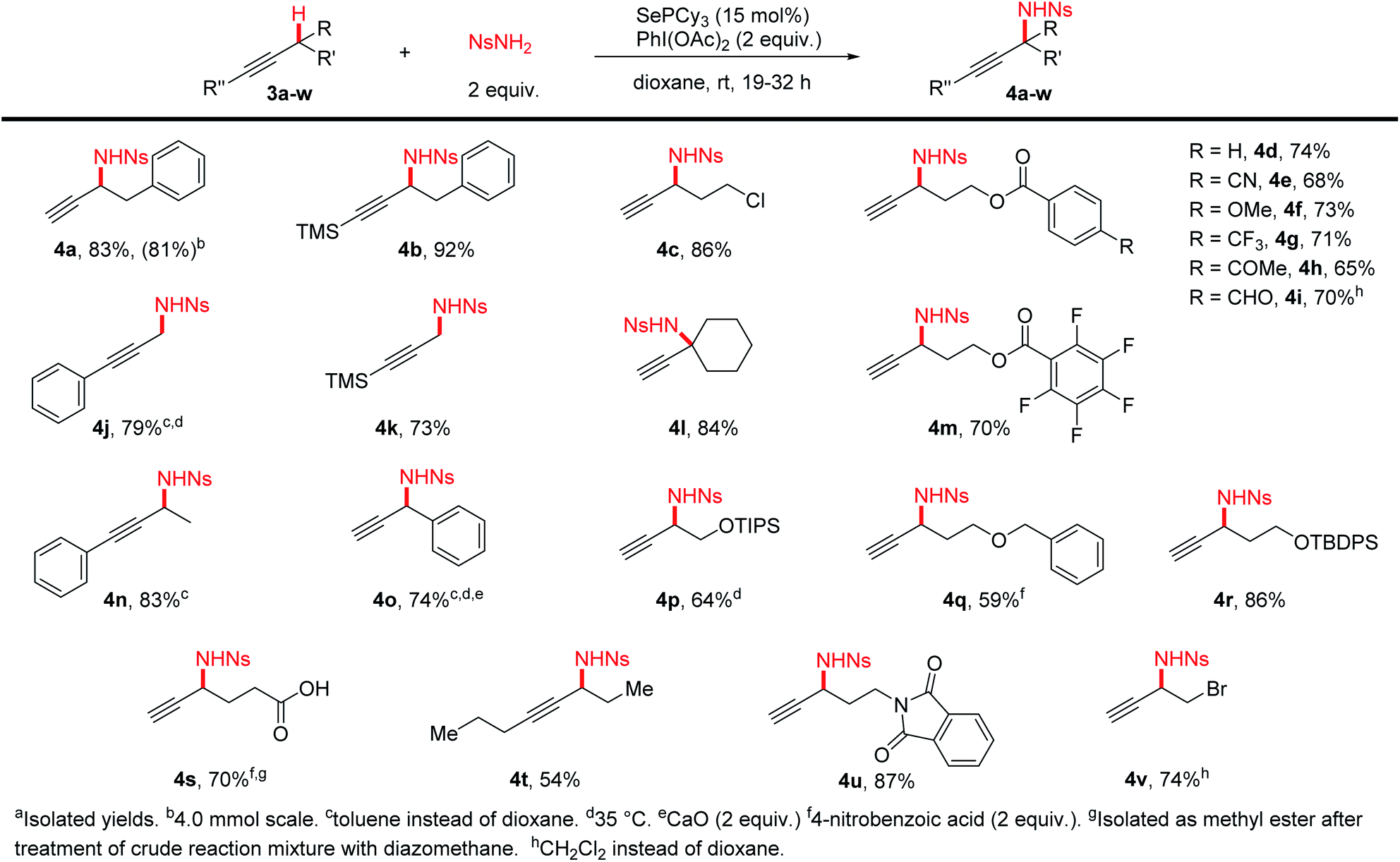 | ||
| Scheme 3 Terminal and silyl-substituted alkyne substrate scope. | ||
Having established the generality of this method for the introduction of propargylic nitrogen groups in a wide variety of alkyne substrates, we next examined the regioselectivity of amination for a series of internal alkynes (Scheme 4). Importantly, we found that good yields of monoaminated products could be obtained from a range of internal alkynes, though minor amounts of diamination were occasionally formed at high conversion. A comparison of the reactivity of primary, secondary and tertiary C–H bonds (5a,b) revealed that in each case the more highly substituted C–H bond was selectively activated (tertiary![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :secondary:primary ∼ 8:4:1). This is in contrast to the selectivity we observed in the allylic amination of alkenes, which followed the order secondary > tertiary ∼ primary.42 Reduced steric hindrance in the alkyne substrates relative to the alkenes may be revealing a more fundamental electronic preference in selenium-catalyzed C–H activation (see below for more discussion).
:secondary:primary ∼ 8:4:1). This is in contrast to the selectivity we observed in the allylic amination of alkenes, which followed the order secondary > tertiary ∼ primary.42 Reduced steric hindrance in the alkyne substrates relative to the alkenes may be revealing a more fundamental electronic preference in selenium-catalyzed C–H activation (see below for more discussion).
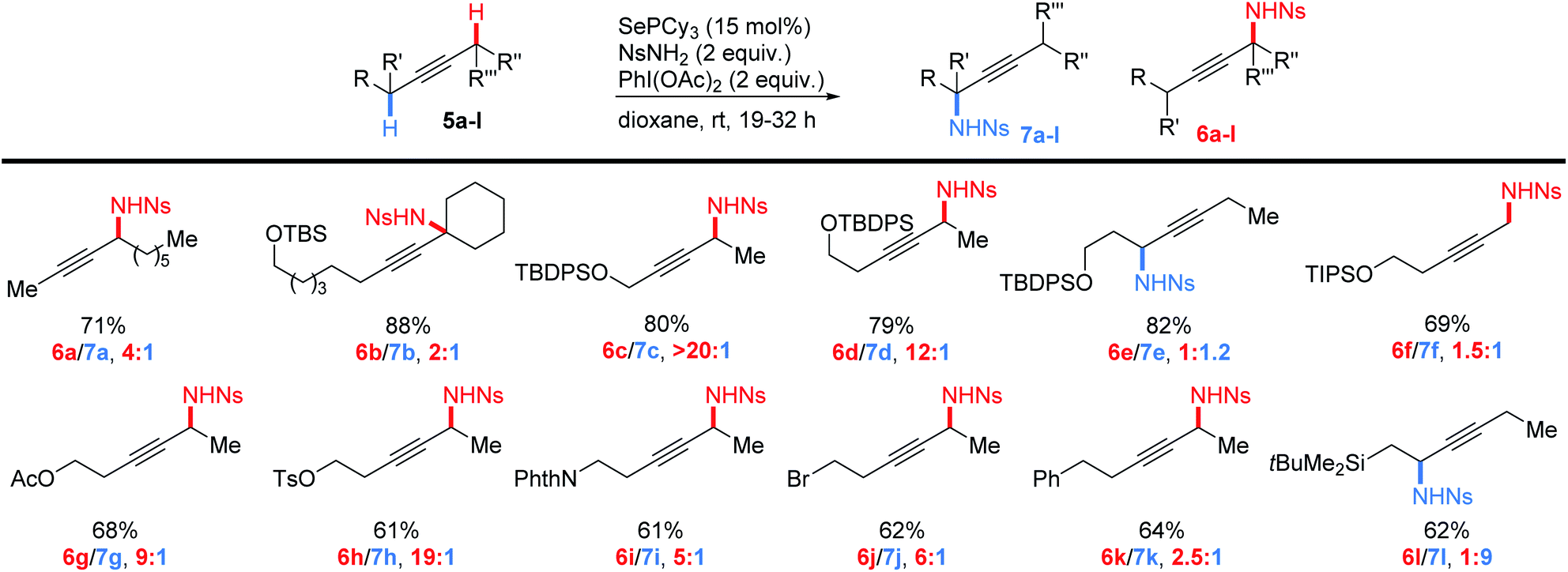 | ||
| Scheme 4 Regioselective amination of internal alkynes. | ||
We then examined the effects of electronegative substituents on the regioselectivity (5c–l). Placement of an OTBDPS group adjacent to the alkyne gave exclusively amination on the opposite side of the alkyne (5c). Gratifyingly, moving the siloxy group further away to the beta positions still resulted in high selectivity for C–H amination away from this group (5d). Interestingly, the gamma siloxy group appears to direct amination in the opposite direction, albeit with low selectivity (5e). Substrate 5f demonstrates that these electronic effects appear to be approximately multiplicative with substitution effects. Though this substrate would prefer to activate the secondary C–H bond (∼1:4), it prefers to activate away from the beta siloxy group more (∼12:1), thus leading to a moderate preference for product 6f (2:1). A range of other inductively electron-withdrawing groups (5g–k) also strongly directed amination to the distal side of the triple bond, including a simple phenyl group (2.5:1). Similar directing effects of inductively withdrawing groups have been previously observed in Ir-catalyzed allylic amination of alkenes,23 and in an enantioselective ene reaction of cis-alkenes with TsN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO,44 perhaps indicating that this may be a more general effect.
SO,44 perhaps indicating that this may be a more general effect.
Since inductively withdrawing groups direct C–H amination to the opposite side of the alkyne, we imagined that a more electropositive substituent may reverse that effect. To that end, we performed the amination on silyl alkyne 5l. We found that the β-silyl group indeed strongly directs amination towards the silyl substituent, consistent with the general trend in electronegativity.
To learn more about the relative rates of reactions of alkynes in the propargylic amination reaction, we performed a series of competition experiments using equimolar amounts of two competing alkyne substrates (Scheme 5). A simple terminal alkyne was found to be somewhat less reactive than the analogous terminal alkene (2.6:1). Competitions between alkynes bearing primary, secondary, and tertiary C–H bonds reveal that the general trend of reactivity is the same as that observed in the regioselectivity (i.e. tertiary > secondary > primary). Similarly, the effects of inductive withdrawing groups also follow the same trend, namely, that alkynes with closer electron-withdrawing groups react more slowly than those with more remote electron-withdrawing groups. These experiments reveal that regioselectivity and rate are fundamentally correlated in this reaction. The effects of substitution pattern on the regioselectivity and reactivity are both consistent with a model in which the alkyne is attacked by a highly electron-deficient selenium bis(imide) reagent in an initial ene reaction. This ene reaction would be expected to lead to increased positive charge at the carbon atom on the side of the C–H bond being activated, thus directing activation away from electron-withdrawing groups and towards electron-releasing groups.
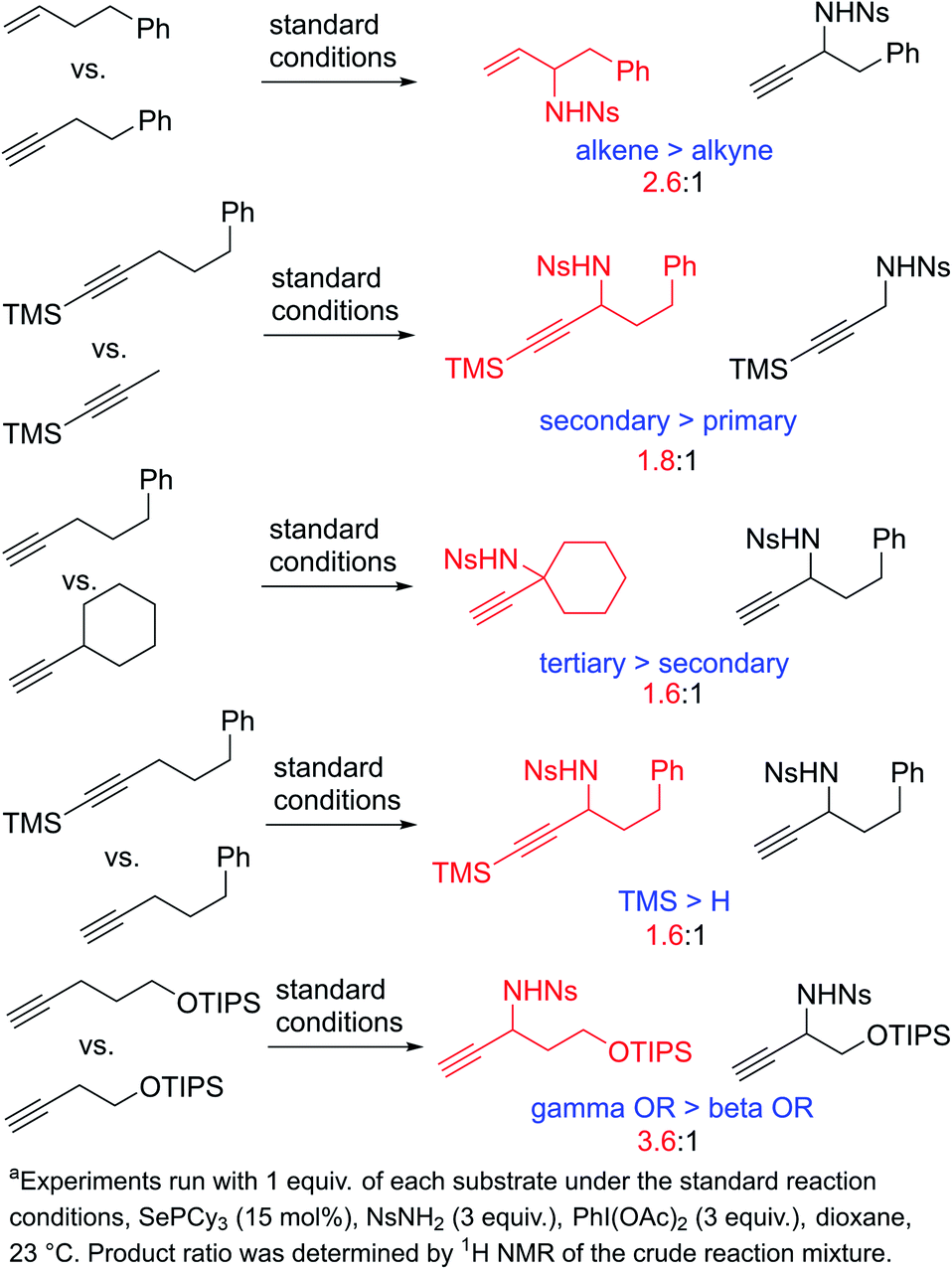 | ||
| Scheme 5 Relative rates of amination reactions. | ||
The proposed mechanism for propargylic C–H amination is presented in Scheme 6A. The phosphine selenide catalyst reacts with the in situ generated iminoiodinane to generate selenium bis(imide) A, possibly with a phosphine oxide coordinated to Se. Ene reaction with the alkyne generates allenylselenium species B, followed by [2,3]-sigmatropic rearrangement to form the new propargylic C–N bond. Oxidative turnover with in situ generated iminoiodinane regenerates the selenium bis(imide) and closes the catalytic cycle.
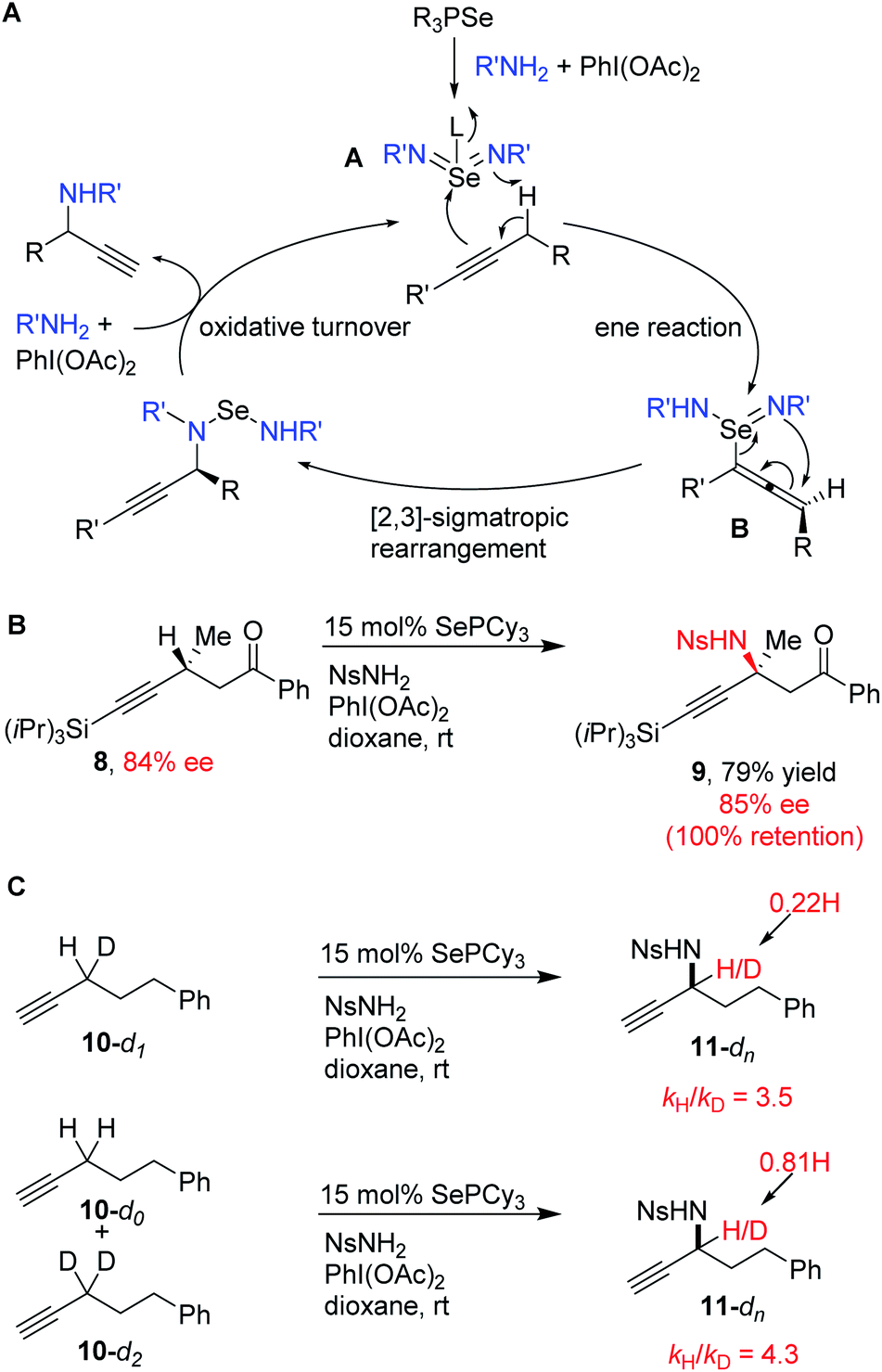 | ||
| Scheme 6 Reaction mechanism: (A) proposed mechanistic cycle, (B) stereoretention at alpha position, (C) kinetic isotope effects. | ||
Because of the suprafacial nature of both the ene reaction and [2,3]-sigmatropic rearrangements, we hypothesized that an alkyne bearing a chiral center at the alpha position should undergo amination with overall retention of stereochemistry via the intermediacy of chiral allene B. To test this, enantioenriched alkyne 8 was subjected to our standard conditions (Scheme 6B). The expected amination product 9 was obtained in good yield, despite the sterically bulky TIPS group on the alkyne. Complete retention of stereochemistry was observed.
To further investigate this mechanism, we measured the kinetic isotope effect for deuterium substitution in the alpha position via intramolecular competition (10-d1) and via intermolecular competition (10-d0vs.10-d2). Both gave primary KIEs (kH/kD = 3.5 and 4.3), consistent with C–H cleavage in the product determining step (Scheme 6C). These values are also roughly consistent with the magnitude of the KIE observed in the analogous reaction of alkenes with SeO2 (kH/kD = 2.6–3.7).45,46 The small difference in the KIE between the intra- and intermolecular competition experiments may reflect the contribution of a secondary isotope effect from the remaining H/D atom. The presence of a primary KIE in the intermolecular competition experiment also rules out the possibility of irreversible pre-coordination event occurring before C–H bond cleavage.
Finally, to more fully understand the origins of the high regioselectivity we observed, we performed DFT calculations on the initial ene reactions for several model alkyne substrates (Scheme 7).47 These calculations found plausible energies for the activation barriers (ΔG‡ = +11–17 kcal mol−1) and overall exergonicity of the ene reactions (ΔGrxn = −16 to 25 kcal mol−1). Such a highly exergonic reaction is unlikely to be reversible, which is consistent with the observed isotope effects and indicates that the ene reaction is the product determining step. The calculated activation barrier for the ene reaction of propyne is higher than that for propene (ΔG‡ = +16.5 vs. 11 kcal mol−1), consistent with the lower reactivity of the alkyne (see Scheme 5). The effects of changing substitution on the terminal alkyne are consistent with the observed rate differences, namely, increasing the substitution at the reacting C–H lowers the activation barrier, as does addition of a beta-SiMe3 group, whereas adding a beta-OAc group increases the barrier.
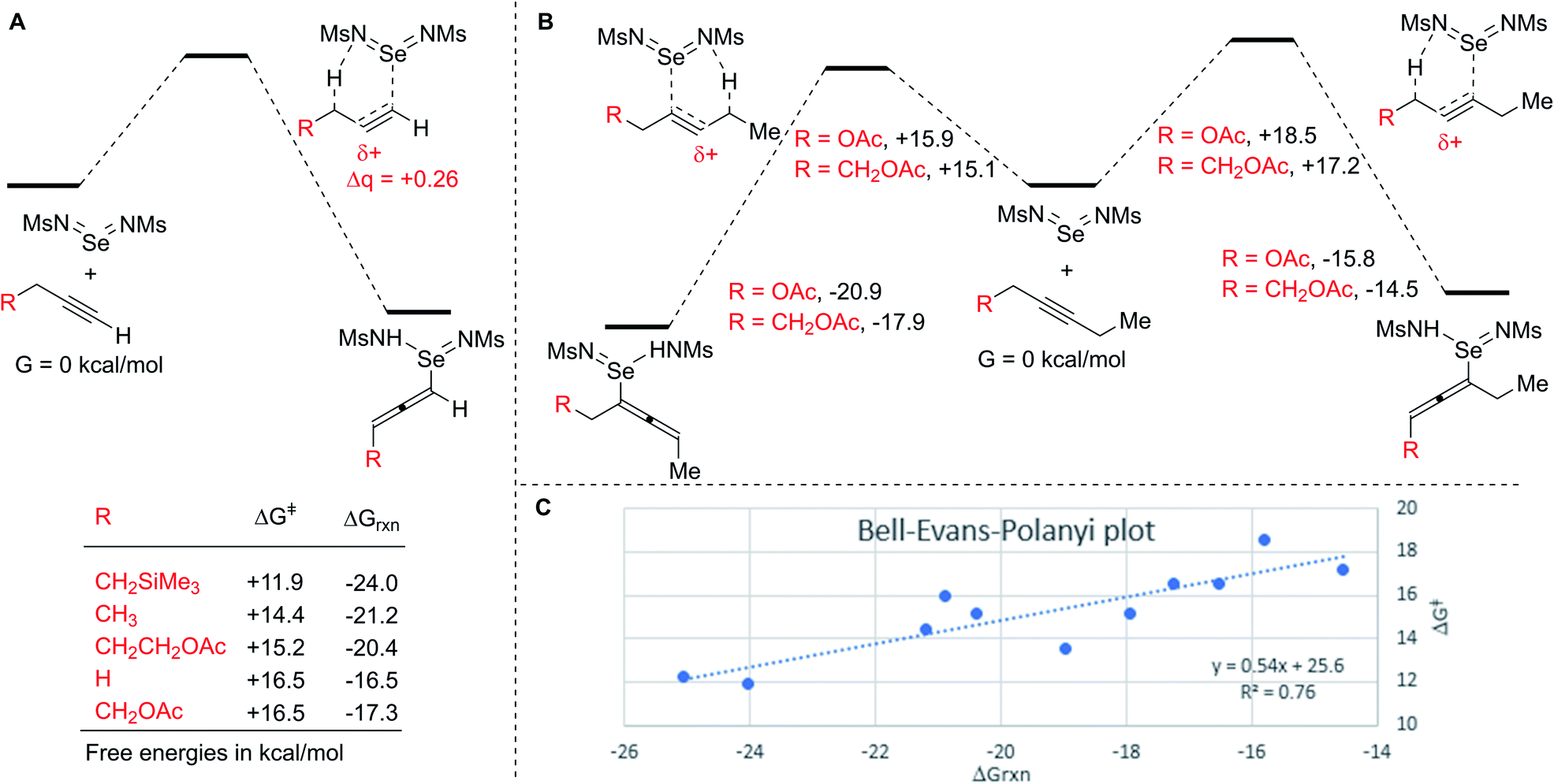 | ||
| Scheme 7 (A) Effect of substitution on terminal alkynes. (B) Effect of substitution on regioselectivity of internal alkynes. (C) Bell–Evans–Polanyi plot for calculated ene reactions. | ||
NBO charge analysis indicates that substantial positive charge develops on the central carbon in the ene transition state in all cases (Δq = +0.26). Inductive stabilization or destabilization of this developing positive charge is consistent with the observed regioselectivities of inductively withdrawing or donating substituents.
Both regioisomeric transition states for internal alkynes bearing alpha and beta acetoxy groups were found (Scheme 7B). The relative energies of these transition states are consistent with the observed regioselectivities, namely, ene reaction proximal to the acetoxy group has a higher activation barrier and is less exergonic, in a manner that decreases with distance. Notably, both distal transition states have similar energies to those without an OAc substituent, indicating that the observed regioselectivities are due to destabilization of the transition state leading to the disfavored isomer rather than stabilization of the transition state leading to the favored isomer.
Interestingly, for each of these regioisomeric pairs, the lower energy transition state also led to the more stable allene intermediate. Across all calculations, ene reactions with lower activation barriers generally tended to be more exergonic. To illustrate this correlation, a Bell–Evans–Polanyi (BEP) plot for all calculated ene reactions is given in Scheme 7C.48,49 The good fit of this line indicates that the energy of the transition states is strongly influenced by the relative stability of the ene reaction products. The slope of ∼0.5 is consistent with a transition state that is neither early nor late, but rather roughly halfway along the reaction coordinate. Consistent with this is the fact that the C–C bond distances in the transition states are nearly exactly halfway between those of the starting alkynes and the product allenes. Notably this slope is greater than observed in two other studies of C–H activation of weak C–H bonds (0.36 and 0.23).50,51 Our observed slope implies that any factors that stabilize the allenylselenium intermediate will substantially accelerate the reaction because the transition state develops substantial product-like character. Thus, the preference for activation of tertiary C–H bonds over secondary and primary can be explained by the fact that more substitution at the propargylic position results in formation of a more substituted, and thus more stable, allenylselenium intermediate.
Conclusions
In conclusion, we have developed a highly general intermolecular propargylic amination of alkynes. This reaction is operationally simple and requires no transition metal catalysts, relying instead on a phosphine selenide catalyst. A wide range of alkynes were aminated in high yields and good regioselectivity. Importantly, large directing effects from inductively withdrawing and donating functional groups were observed, where amination always took place on the more electron-rich side of the alkyne. Amination of propargylic chiral centers takes place with complete retention of stereochemistry. Mechanistic experiments and DFT calculations showed that an ene reaction between the alkyne and an in situ generated selenium bis(imide) is both the rate and product-determining step. The observed regioselectivities can be understood by the effects of substituents on the development of positive charge at the carbon adjacent to the C–H bond being activated, as well as the stabilizing effects of substitution on the allenylselenium intermediate.Data availability
Data for this paper, including characterization data for all new compounds and structures and energies for all DFT computations, are provided in the ESI.†Author contributions
T. P. M. and A. F. D. conducted all experiments. A. C. Z. performed all DFT calculations. T. P. M., A. F. D., and F. E. M. designed the experiments. F. E. M. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Washington and the National Science Foundation (CHE-1764450) for funding. Computations were facilitated through the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system at the University of Washington, funded by the Student Technology Fee.Notes and references
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492–5496 CrossRef CAS PubMed.
- Acetylene Chemistry, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2005 Search PubMed.
- K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Taylor & Francis Group, LLC, Boca Raton, 2007 Search PubMed.
- N. S. Dolan, R. J. Scamp, T. Yang, J. F. Berry and J. M. Schomaker, J. Am. Chem. Soc., 2016, 138, 14658–14667 CrossRef CAS PubMed.
- H. Lu, H. Jiang, Y. Hu, L. Wojtas and X. P. Zhang, Chem. Sci., 2011, 2, 2361–2366 RSC.
- K. Smith, C. D. Hupp, K. L. Allen and G. A. Slough, Organometallics, 2005, 24, 1747–1755 CrossRef CAS.
- R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS PubMed.
- S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS PubMed.
- G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS PubMed.
- H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS PubMed.
- T. Knecht, S. Mondal, J. H. Ye, M. Das and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 7117–7121 CrossRef CAS PubMed.
- J. S. Burman, R. J. Harris, C. M. B. Farr, J. Bacsa and S. Blakey, ACS Catal., 2019, 9, 5474–5479 CrossRef CAS.
- A. M. Kazerouni, T. A. F. Nelson, S. W. Chen, K. R. Sharp and S. B. Blakey, J. Org. Chem., 2019, 84, 13179–13185 CrossRef CAS PubMed.
- R. J. Harris, J. Park, T. A. F. Nelson, N. Iqbal, D. C. Salgueiro, J. Basca, C. E. MacBeth, M.-H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 5842–5851 CrossRef CAS PubMed.
- H. Lei and T. Rovis, Nat. Chem., 2020, 12, 725–731 CrossRef CAS PubMed.
- R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14, 280–283 CrossRef CAS PubMed.
- H. Liu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028–7032 CrossRef PubMed.
- M. Ju, E. E. Zerull, J. M. Roberts, M. Huang, I. A. Guzei and J. M. Schomaker, J. Am. Chem. Soc., 2020, 142, 12930–12936 CrossRef CAS PubMed.
- Q. Xing, C.-M. Chan, Y.-W. Yeung and W.-Y. Yu, J. Am. Chem. Soc., 2019, 141, 3849–3853 CrossRef CAS PubMed.
- Z. Zhou, S. Chen, Y. Hong, E. Winterling, Y. Tan, M. Hemming, K. Harms, K. N. Houk and E. Meggers, J. Am. Chem. Soc., 2019, 141, 19048–19057 CrossRef CAS PubMed.
- C. Li, K. Lang, H. Lu, Y. Hu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16837–16841 CrossRef CAS PubMed.
- H. Wang, Y. Park, Z. Bai, S. Chang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 7194–7201 CrossRef CAS PubMed.
- H. Lu, K. Lang, H. Jiang, L. Wojtas and X. P. Zhang, Chem. Sci., 2016, 7, 6934–6939 RSC.
- Y. Amaoka, S. Kamijo, T. Hoshikawa and M. Inoue, J. Org. Chem., 2012, 77, 9959–9969 CrossRef CAS PubMed.
- H. Wang, D. Zhang and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 5863–5866 CrossRef CAS PubMed.
- Z. Li, G. Vijaykumar, X. Li, C. Golz and M. Alcarazo, Org. Biomol. Chem., 2021, 19, 2941–2948 RSC.
- L.-M. Jin, P. Xu, J. Xie and X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20828–20836 CrossRef CAS PubMed.
- H. Lebel, C. Trudel and C. Spitz, Chem. Commun., 2012, 48, 7799–7801 RSC.
- While this manuscript was in review, an enzyme-catalyzed intermolecular propargylic C–H amination was reported: Z. Liu, Z.-Y. Qin, L. Zhu, S. V. Athavale, A. Sungupta, Z.-J. Jia, M. Garcia-Borrás, K. N. Houk and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 80–85 CrossRef CAS PubMed.
- J. Li, J. S. Cisar, C.-Y. Zhou, B. Vera, H. Williams, A. D. Rodriguez, B. F. Cravatt and D. Romo, Nat. Chem., 2013, 5, 510–517 CrossRef CAS PubMed.
- E. Keinan and E. Bosch, J. Org. Chem., 1986, 51, 4006–4016 CrossRef CAS.
- S. Ma and G. Wang, Angew. Chem., Int. Ed., 2003, 42, 4215–4217 CrossRef CAS PubMed.
- M. Baize, P. W. Blosser, V. Plantevin, D. G. Schimpff, J. C. Gallucci and A. Wojcicki, Organometallics, 1996, 15, 164–173 CrossRef CAS.
- W. P. Teh, D. C. Obenschain, B. M. Black and F. E. Michael, J. Am. Chem. Soc., 2020, 142, 16716–16722 CrossRef CAS PubMed.
- K. B. Sharpless, T. Hori, L. K. Truesdale and C. O. Dietrich, J. Am. Chem. Soc., 1976, 98, 269–271 CrossRef CAS.
- L. Bayeh, P. Q. Le and U. K. Tambar, Nature, 2017, 547, 196–201 CrossRef CAS PubMed.
- L. M. Stephenson and D. R. Speth, J. Org. Chem., 1979, 44, 4683–4689 CrossRef CAS.
- D. A. Singleton and C. Hang, J. Org. Chem., 2000, 65, 7554–7560 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. P. Bell, Proc. R. Soc. London, Ser. A, 1936, 154, 414–429 Search PubMed.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1938, 154, 11–24 RSC.
- M. Salamone, M. Galeotti, E. Romero-Montalvo, J. A. van Santen, B. D. Groff, J. M. Mayer, G. A. DiLabio and M. Bietti, J. Am. Chem. Soc., 2021, 143, 11759–11776 CrossRef CAS PubMed.
- F. Liu, Z. Yang, Y. Yu, Y. Mei and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 16650–16656 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07067c |
| This journal is © The Royal Society of Chemistry 2022 |