
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric hydrometallation of cyclobutenes with salicylaldehydes†
F. Wieland
Goetzke
 a,
Mireia
Sidera
b and
Stephen P.
Fletcher
*a
a,
Mireia
Sidera
b and
Stephen P.
Fletcher
*a
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stephen.fletcher@chem.ox.ac.uk
bVertex Pharmaceuticals (Europe) Ltd, 86–88 Jubilee Avenue, Milton Park, Abingdon, OX14 4RW, UK
First published on 10th December 2021
Abstract
Chiral, substituted cyclobutanes are common motifs in bioactive compounds and intermediates in organic synthesis but few asymmetric routes for their synthesis are known. Herein we report the Rh-catalyzed asymmetric hydrometallation of a range of meso-cyclobutenes with salicylaldehydes. The ortho-phenolic group promotes hydroacylation and can be used as a handle for subsequent transformations. The reaction proceeds via asymmetric hydrometallation of the weakly activated cyclobutene, followed by a C–C bond forming reductive elimination. A prochiral, spirocyclic cyclobutene undergoes a highly regioselective hydroacylation. This report will likely inspire the development of other asymmetric addition reactions to cyclobutenes via hydrometallation pathways.
Introduction
Substituted cyclobutanes can be found in many natural products,1 and are useful building blocks in synthetic chemistry but their asymmetric synthesis is often challenging.2 The cyclobutane motif has also received increased attention as a rigid scaffold and as an isostere in medicinal chemistry.3 So far 8 compounds that bear a cyclobutane ring have been approved by the FDA but their cyclobutyl moieties are generally simply substituted like in Boceprevir or Apalutamide (Fig. 1a) and none of them bear any stereogenic centres – which may be due to a scarcity of generally useful methods for the synthesis of complex, chiral cyclobutanes.4,5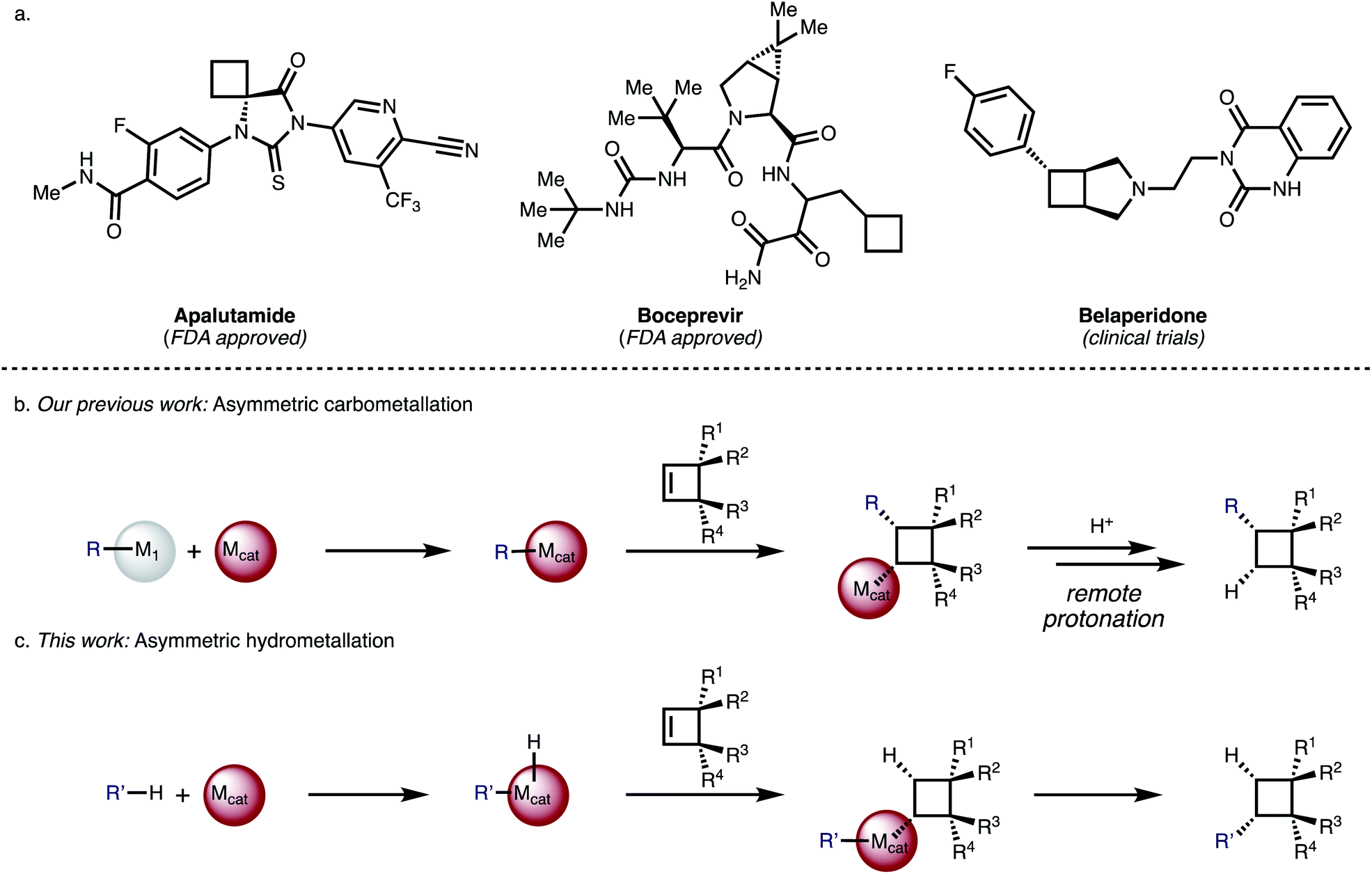 | ||
| Fig. 1 (a) Examples of bioactive cyclobutanes. (b) Asymmetric carbometallation of cyclobutenes. (c) Asymmetric hydrometallation of cyclobutenes. | ||
Most existing catalytic asymmetric approaches for cyclobutane synthesis rely either on ring-closure,6–13 or the functionalization of pre-formed four-membered rings using activating or directing groups.14–16 Direct addition reactions to unactivated cyclobutenes are rare,17–19 but attractive as they offer a modular entry to functionalized cyclobutanes and are generally not limited to specific substitution patterns.
We have reported Rh-catalysed asymmetric addition reactions of arylboronic acids to various cyclobutenes (Fig. 1b).20 These reactions proceed via an asymmetric carbometallation step, followed by remote protonation or elimination to give a diverse range of arylated cyclobutanes. For cyclobutenes, the carbometallation step is associated with a very small release of olefinic strain (1.9 kcal mol−1) compared to the olefinic strain of other small, cyclic molecules like cyclopropene (27.7 kcal mol−1).21 We became interested in the question if related but mechanistically distinct cyclobutene functionalization reactions with carbon-nucleophiles would be possible and identified an asymmetric hydrometallation-reductive elimination sequence as a viable strategy (Fig. 1c). A key advantage of this strategy is its high-atom economy and the avoidance of sensitive organometallic coupling partners.
Metal-catalysed hydroacylation,22 and especially Rh-catalysed hydroacylation reactions between alkenes and aldehydes are powerful tools for the synthesis of ketones and operate via a hydrometallation mechanism.23 While intramolecular hydroacylation reactions are well established, the intermolecular Rh-catalysed hydroacylation is often associated with an undesired reductive decarbonylation, and many specific solutions for this problem involving chelating groups have been developed.23 The use of ortho-hydroxybenzaldehydes (salicylaldehydes) represents one of the strategies and several useful asymmetric hydroacylations with terminal alkenes have been reported.24–26
Internal alkenes represent significantly more challenging substrates in Rh-catalysed intermolecular hydroacylations, but a few reactions between norbornadienes or cyclopropenes and salicylaldehydes have been reported by the groups of Bolm and Dong, which are likely driven by the release of olefinic-strain.27,28 Catalytic asymmetric carbofunctionalization reactions of strained alkenes that proceed via hydrometallation have also been reported with Nickel.29 We wondered if a related process would be possible with cyclobutenes – despite the very small release of ring strain in the hydrometallation step.
Results and discussion
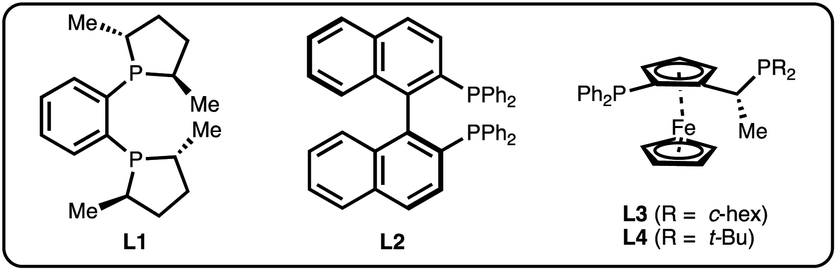
We chose salicylaldehyde 1a and cyclobutene 2 as our model substrates as they provide an entry to the bicyclic core of Belaperidone (see Fig. 1a). Our optimisation studies revealed that a catalytic system generated from [Rh(cod)OH]2 and MeDuphos (L1) gives 3a in excellent yield, enantio- and diastereoselectivity (Table 1, entry 1). Prolonged reaction times led to a slight decrease in diastereomeric ratio. Under these conditions, salicylaldehyde 1a was fully consumed, and decarbonylation of the aldehydes served as the major side-reaction (indicated by the disappearance of CHO signals in the 1H NMR spectra).30 The reaction does not proceed in absence of either Rh or phosphine ligand (Table 1, entries 2 and 3), while other ligand scaffolds provided only poor levels of enantioinduction in this reaction despite inducing high diastereomeric ratios (Table 1, entries 4–6). Slightly lower diastereoselectivity and yield was obtained with THF instead of toluene (Table 1, entry 7). Using benzaldehyde instead of salicylaldehyde did not provide the desired hydroacylation product (Table 1, entry 8) – highlighting the importance of a chelating group.|
|
|||||
|---|---|---|---|---|---|
| Entry | Variation from standard conditions | Time (h) | Yieldb (%) | eec (%) | drd |
| a [Rh(cod)OH]2 (2.5 mol%), ligand (6 mol%), cyclobutene 2 (0.3 mmol), salicylaldehyde 1a (0.2 mmol), PhMe (0.2 M), 1–20 h. b Isolated yield of the major diastereoisomer. c The ee values were determined by SFC analysis on a chiral non-racemic stationary phase. d The dr values were estimated by non-calibrated SFC analysis of the unpurified reaction mixture. e Performed on 0.4 mmol scale. | |||||
| 1 | Nonee | 1 | 81 | 98 | 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | No Rh | 20 | 0 | — | — |
| 3 | No ligand | 20 | ≤1 | — | — |
| 4 | L2 instead of L1 | 20 | 86 | 32 | >20:1 |
| 5 | L3 instead of L1 | 2 | 86 | −74 | >20:1 |
| 6 | L4 instead of L1 | 20 | 31 | −12 | >20:1 |
| 7 | THF instead of PhMe | 1 | 72 | 98 | 7:1 |
| 8 | PhCHO instead of 1a | 20 | 0% | — | — |
|
|||||
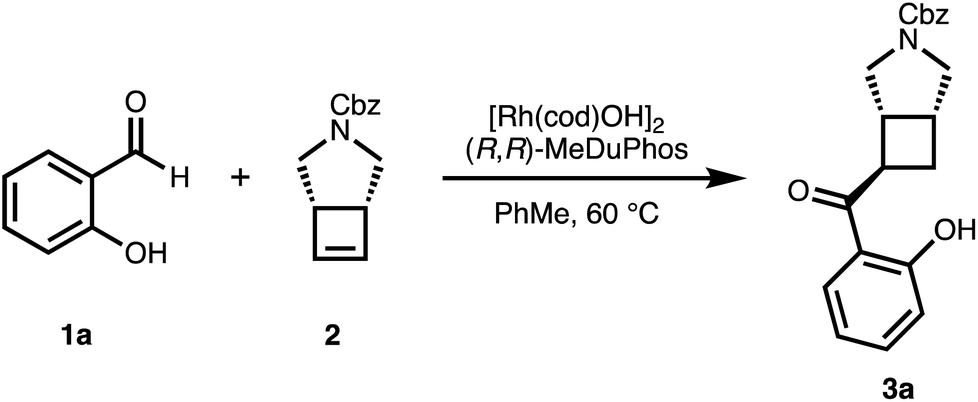
Having optimized conditions for 1a and 2 in hand, we subsequently explored the scope of the transformation on both the salicylaldehyde and the cyclobutene component. Several substitution patterns, electron-withdrawing and electron-donating functional groups, and halides are well tolerated with consistently excellent levels of enantioinduction (Scheme 1, 3a–3i). In all cases, we isolated the pure cis–trans isomer.‡ Remarkably, our catalytic system shows high chemoselectivity for the hydroacylation of the cyclobutene over a terminal alkene (3b).24 For more electron-deficient salicylaldehydes 1c, 1d, 1g and 1h, we observed unreacted aldehyde accompanied with decarbonylation under our standard reaction conditions. In these cases, better results and full conversion of the aldehyde was achieved with an increased catalyst loading of 5% of dimeric [Rh(cod)OH]2.
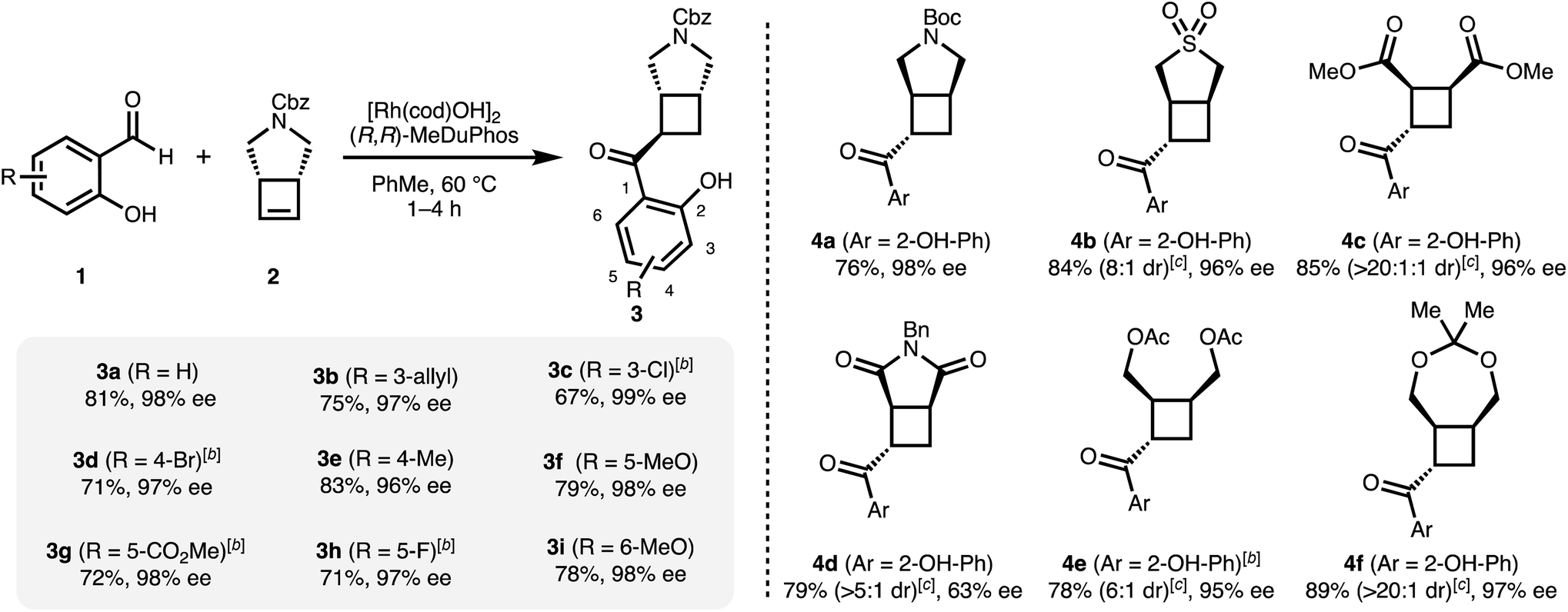 | ||
| Scheme 1 Asymmetric hydroacylation of cyclobutenes with different salicylaldehydes.a a[Rh(cod)OH]2 (2.5 mol%), MeDuphos (6 mol%), cyclobutene 2 (0.6 mmol), salicylaldehyde 1 (0.4 mmol), PhMe (0.2 M), 1–4 h. bIncreased catalyst loading of [Rh(cod)OH]2 (5 mol%) and MeDuphos (12 mol%). cDiastereomeric ratios of the unpurified reaction mixtures determined by 1H NMR spectroscopy. All yields refer to isolated yields of the major trans–cis diastereomer. Enantiomeric excesses determined by SFC analysis on a chiral non-racemic stationary phase. | ||
A range of different mono- and bicyclic meso-cyclobutenes are suitable substrates and give acylated cyclobutanes in good yields and in most cases with high enantiomeric excesses (Scheme 1, 4a–4f). For all compounds, the major trans–cis diastereomer was isolated in pure form. The absolute configuration of 4b was determined via X-ray crystallographic analysis. In our previous carbometallation study, (cis-cyclobut-3-ene-1,2-diyl)bis (methylene) diacetate (2e) underwent homo-allylic substitution reactions instead of hydroarylations with arylboronic acids.20 Using the same substrate under our hydroacylation conditions we obtain the hydroacylation product 4e – highlighting the difference between the carbometallation and hydrometallation pathways (cf.Fig. 1b and c).
Under related conditions using achiral ligand 1,1′-bis(diphenylphosphino)ferrocene (dppf), the achiral, spirocyclic cyclobutane 6 is obtained from 5 as a single regioisomer (Scheme 2).31 Good yields were obtained with a small set of functionalized salicylaldehydes.
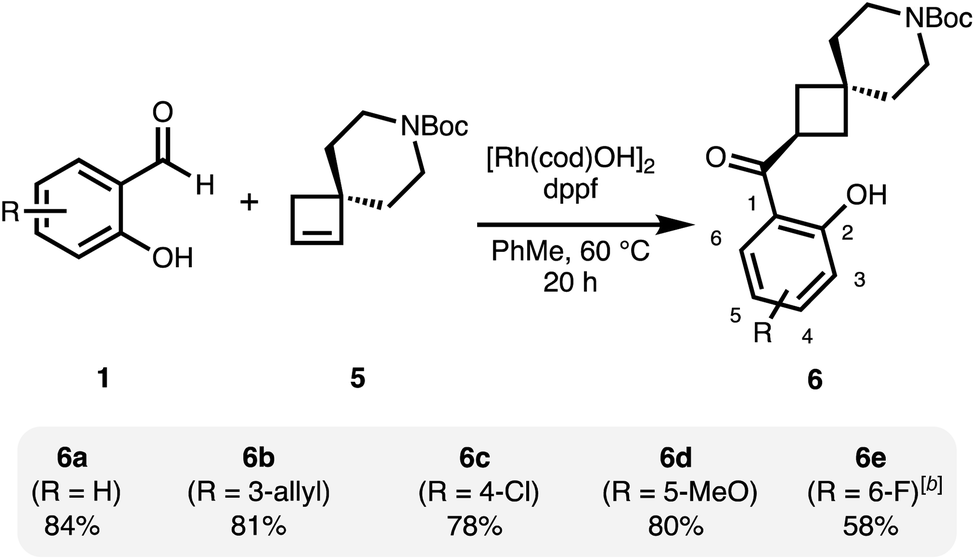 | ||
| Scheme 2 Regioselective hydroacylation of a spirocyclic, prochiral cyclobutene.a a[Rh(cod)OH]2 (2.5 mol%), dppf (6 mol%), cyclobutene 5 (0.6 mmol), salicylaldehyde 1 (0.4 mmol), PhMe (0.2 M), 20 h. bIncreased catalyst loading of [Rh(cod)OH]2 (5 mol%) and dppf (12 mol%). | ||
Also here, more electron-deficient salicylaldehydes were prone to an undesired decarbonylation pathway and therefore gave diminished yields (6e). The regioselectivity in this reaction is likely under steric control and is set in the initial hydrometallation step.
The reaction of 1 with 2a proceeds nicely at a 4 mmol scale gram-scale providing 1.2 g (84%, 98% ee) of 3a (Scheme 3a) while lowering the excess of cyclobutene from 1.5 to 1.2 equivalents. The 2-hydroxybenzoyl moiety could serve as a handle for subsequent functionalization reactions (7a, 7b) and the phenolic OH group can be removed in a two-step protocol (7c) (Scheme 3b). Furthermore, reduction of the benzoyl group provides an entry to benzylated cyclobutanes (7d) (Scheme 3b).
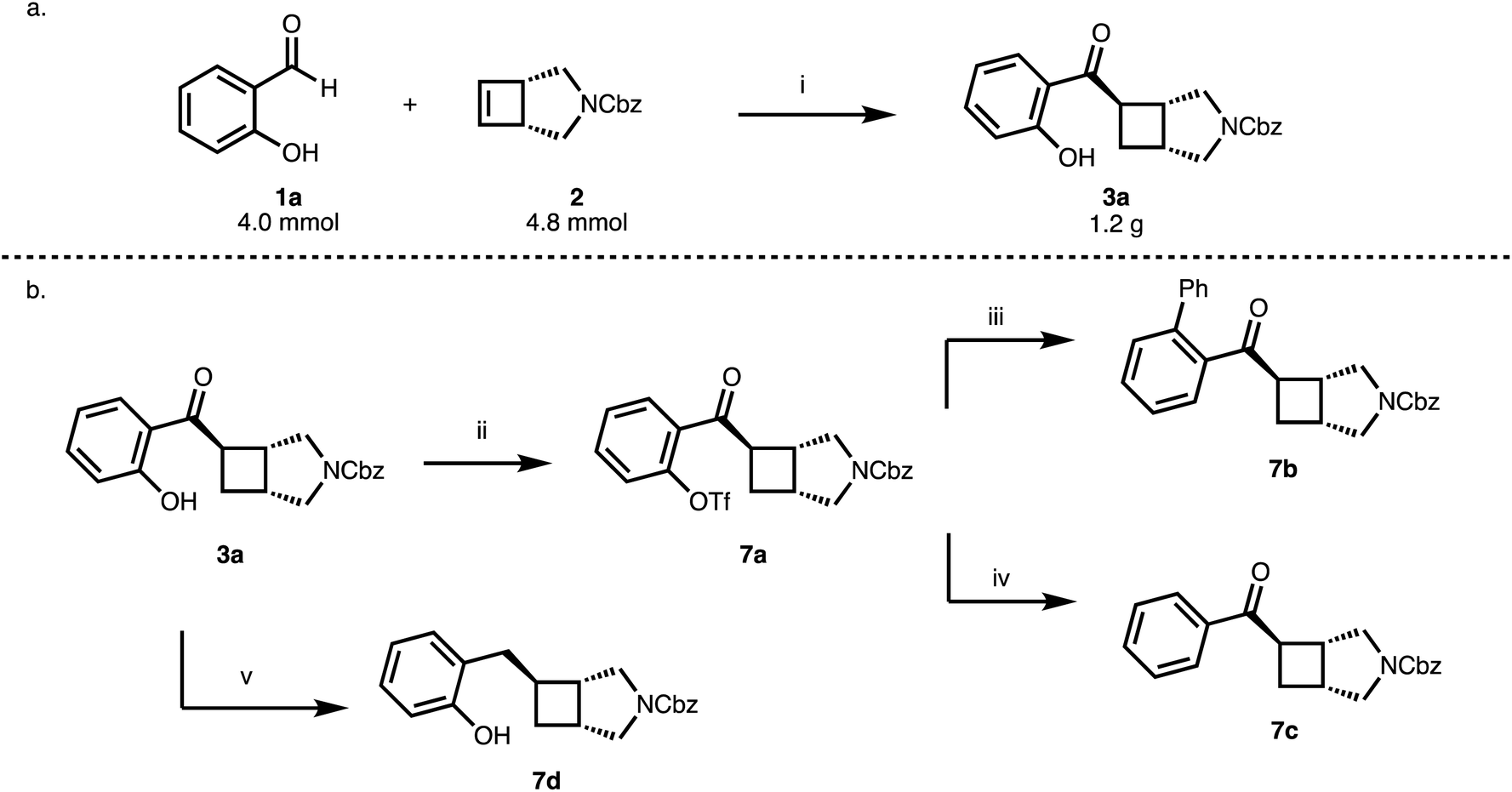 | ||
| Scheme 3 (a) Up-scale to gram-scale. (b) Functionalization of the phenol and benzoyl moiety.a a(i) [Rh(cod)OH]2 (2.5 mol%), MeDuphos (6 mol%), 2 (4.8 mmol), 1a (4.0 mmol), PhMe, 1 h, 60 °C, 84%, 98% ee; (ii) 2-PyrNTf2, DMAP (10 mol%), NEt3, CH2Cl2, 23 h, 23 °C, 92%; (iii) PhB(OH)2, K2CO3, [Pd(PPh3)4] (5 mol%), PhMe, 16 h, 110 °C, 67%; (iv) (CH3)2NH·BH3, K2CO3, [Pd(PPh3)4] (5 mol%), CH3CN, 6 h, 40 °C, 67%; (v) Et3SiH, TFA, 5 h, 0–23 °C, 65%. | ||
Conclusions
In summary, we have shown that the addition of Rh–H to weakly activated cyclobutenes is possible – exemplified with an asymmetric hydroacylation reaction of meso-cyclobutenes with salicylaldehydes. Furthermore, the hydroacylation of a spirocyclic cyclobutene proceeds with excellent regioselectivity. These reactions provide a modular entry to stereochemically complex, acylated cyclobutanes. Likely, other asymmetric Rh-catalysed addition reactions, that proceed via hydrometallation pathways, are feasible with cyclobutenes and those will be investigated in the future.Data availability
Crystallographic data for 4b has been deposited at the CCDC under 2116804 and can be obtained from http://www.ccdc.cam.ac.uk. All other data supporting this article have been uploaded as part of the supplementary material.Author contributions
F. W. G. performed all experiments and conceived the study. S. P. F. and M. S. guided the research. F. W. G. and S. P. F. wrote the manuscript with contributions from M. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Curtis Moore (Ohio State University) for X-ray crystallographic analysis of compound 4b. Financial support from the UK Engineering and Physical Sciences Research Council (EP/N022246/1) is gratefully acknowledged. F. W. G. is grateful to the National Research Fund, Luxembourg, for an AFR PhD Grant (11588566); the EPSRC Doctoral Training Partnership (DTP) for a studentship (EP/N509711/1); and Vertex Pharmaceuticals for financial support.Notes and references
- V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CrossRef CAS PubMed.
- E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS.
- M. R. Bauer, P. di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC.
- https://go.drugbank.com/, accessed Aug 23, 2021.
- S. O. Scholz, J. B. Kidd, L. Capaldo, N. E. Flikweert, R. M. Littlefield and T. P. Yoon, Org. Lett., 2021, 23, 3496–3501 CrossRef CAS PubMed.
- Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS.
- R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS.
- Y. M. Wang, N. C. Bruno, Á. L. Placeres, S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 10524–10527 CrossRef CAS PubMed.
- D. K. Kim, J. Riedel, R. S. Kim and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 10208–10211 CrossRef CAS.
- V. V. Pagar and T. v. RajanBabu, Science, 2018, 361, 68–72 CrossRef CAS.
- S. C. Patel, M. W. Smith, J. A. M. Mercer, K. Suzuki and N. Z. Burns, Org. Lett., 2021, 23, 6530–6535 CrossRef CAS PubMed.
- M. M. Parsutkar, V. V. Pagar and T. V. RajanBabu, J. Am. Chem. Soc., 2019, 141, 15367–15377 CrossRef CAS.
- M. Luparia, M. T. Oliveira, D. Audisio, F. Frebault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631–12635 CrossRef CAS PubMed.
- K. J. Xiao, D. W. Lin, M. Miura, R. Y. Zhu, W. Gong, M. Wasa and J. Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS.
- C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Angew. Chem., Int. Ed., 2020, 59, 2750–2754 CrossRef CAS.
- M. Guisán-Ceinos, A. Parra, V. Martín-Heras and M. Tortosa, Angew. Chem., Int. Ed., 2016, 55, 6969–6972 CrossRef.
- L. Nóvoa, L. Trulli, A. Parra and M. Tortosa, Angew. Chem., Int. Ed., 2021, 60, 11763–11768 CrossRef.
- S. Feng, H. Hao, P. Liu and S. L. Buchwald, ACS Catal., 2020, 10, 282–291 CrossRef CAS PubMed.
- F. W. Goetzke, A. M. L. Hell, L. van Dijk and S. P. Fletcher, Nat. Chem., 2021, 13, 880–886 CrossRef CAS PubMed.
- K. B. Wiberg, Angew. Chem., Int. Ed., 1986, 25, 312–322 CrossRef.
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS.
- R. T. Davison, E. L. Kuker and V. M. Dong, Acc. Chem. Res., 2021, 54, 1236–1250 CrossRef CAS.
- M. von Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef CAS PubMed.
- M. M. Coulter, K. G. M. Kou, B. Galligan and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16330–16333 CrossRef CAS PubMed.
- Y. Inui, M. Tanaka, M. Imai, K. Tanaka and H. Suemune, Chem. Pharm. Bull., 2009, 57, 1158–1160 CrossRef CAS.
- R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185–1198 CrossRef CAS.
- D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354–16355 CrossRef CAS.
- R. Kumareswaran, M. Nandi and T. V. RajanBabu, Org. Lett., 2003, 23, 4345–4348 CrossRef.
- C. M. Beck, S. E. Rathmill, Y. L. Park, J. Chen, R. H. Crabtree, L. M. Liable-Sands and A. L. Rheingold, Organometallics, 1999, 18, 5311–5317 CrossRef CAS.
- L. Nóvoa, L. Trulli, I. Fernández, A. Parra and M. Tortosa, Org. Lett., 2021, 23, 7434–7438 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2116804. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06035j |
| ‡ A precise determination of the diastereomeric ratios of the unpurified reaction mixture by 1H NMR was not possible due to the broad (rotameric) peak shapes. However, these crude NMR spectra suggest similar diastereomeric ratios for compounds 3a–3i and 4a (approximately 7:1 to 10:1). |
| This journal is © The Royal Society of Chemistry 2022 |