
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic synthesis of tetra-substituted furans promoted by carbon dioxide†
Ya-Ming
Tian
 ,
Huaiju
Wang
,
Ritu
and
Burkhard
König
*
,
Huaiju
Wang
,
Ritu
and
Burkhard
König
*
Institute of Organic Chemistry, Faculty of Chemistry and Pharmacy, University of Regensburg, 93040 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de
First published on 6th December 2021
Abstract
We report a simple protocol for the transition metal-free, visible-light-driven conversion of 1,3-diketones to tetra-substituted furan skeleton compounds in carbon dioxide (CO2) atmosphere under mild conditions. It was found that CO2 could be incorporated at the diketone enolic OH position, which was key to enabling the cleavage of a C–O bond during the rearrangement of a cyclopropane intermediate. This method allows for the same-pot construction of two isomers of the high-value tetra-substituted furan scaffold. The synthetic scope and preliminary mechanistic investigations are presented.
Introduction
Polysubstituted furans are of great importance in the flavor and fragrance industry, pharmaceutical industry and materials chemistry, and are also valuable building blocks in organic synthesis.1 However, the preparation of such polysubstituted furans is often complicated and circuitous. While direct functionalization of the furan C–H positions is possible (Scheme 1a), regioselectivity challenges and detrimental reactivity of the furan ring itself hampered such approaches. Classical strategies to construct fully functionalized furans include the Feist–Benary reaction2 and Paal–Knorr condensation3 (Scheme 1b). Annulation of unsaturated substrates with ketones or imines4 (Scheme 1c) as well as other cross-coupling approaches5 could also afford polysubstituted furans, but migratory cycloisomerization6 and rearrangements7 are the most straightforward and convergent routes (Scheme 1d).8 Furthermore, current methods to access such furans typically require the use of metal catalysts and multiple components. As such, an atom-efficient, transition metal-free, organocatalytic protocol to synthesize tetra-substituted furans from a single precursor is highly sought after.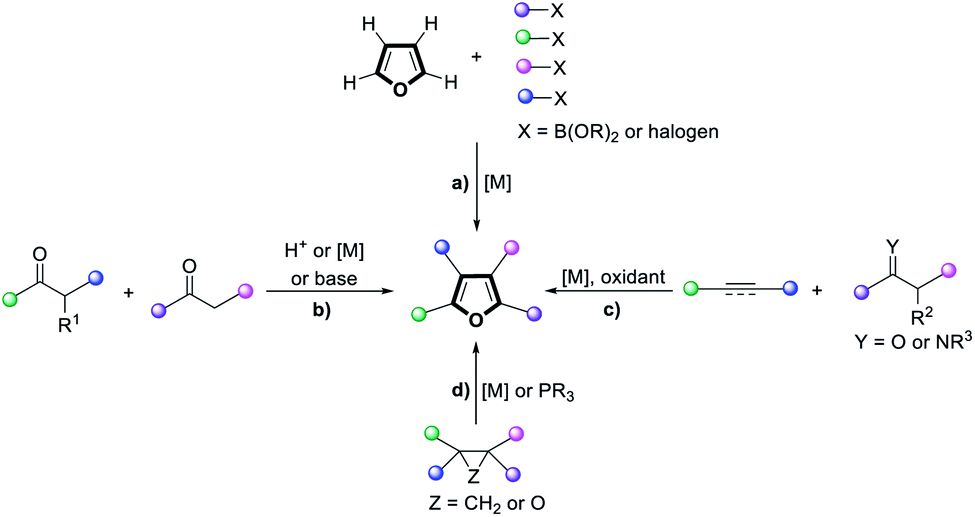 | ||
| Scheme 1 Approaches to access highly substituted furans via: (a) successive direct functionalization of furan C–H; (b) condensation; (c) annulation of unsaturated substrates with ketones or imines; (d) migratory cycloisomerization and rearrangements. | ||
Carbon dioxide as a natural, abundant, inexpensive, easy-to-separate, and recyclable C1 building block has become the focus of recent research,9–11 but its capability to catalytically promote reactions has not been widely explored. Shell Oil Company first patented the utilization of CO2 to facilitate the synthesis of propionaldehydes in 1968,12 but only in 2007 was a CO2-catalyzed rearrangement of propargyl alcohols to unsaturated ketones reported by Yamada (Scheme 2a).13 Proceeding via a carbonate intermediate generated between CO2 and the propargyl alcohol, the reaction regenerates the gas upon formation of the product. Similarly, Tunge showed the use of CO2 to engage allylic OH groups, producing better leaving groups for cross-coupling (Scheme 2b),11g and Das further reported the CO2-promoted oxidation of allylic alcohols to unsaturated aldehydes (Scheme 2c).10b More recently, it was also demonstrated that CO2 can act on amine substrates to activate α-C–H bonds for intermolecular hydrogen atom transfer.14,15 Taken together, while the study of CO2-catalysis is still in its infancy, it offers new opportunities to perform organocatalysis that can complement traditional metal-based chemistry.
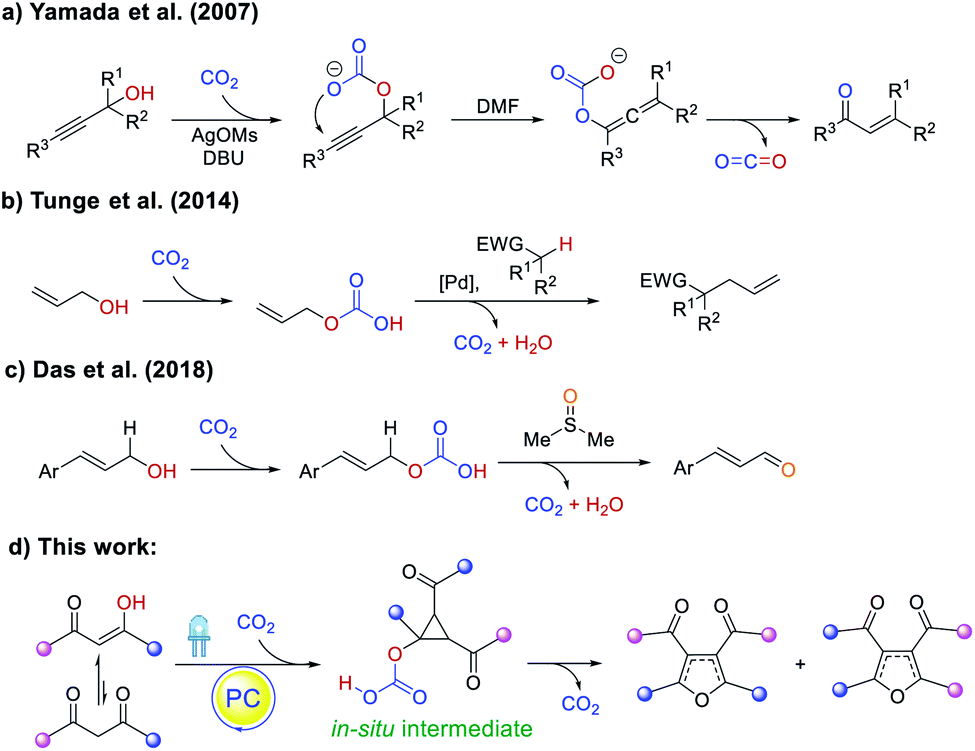 | ||
| Scheme 2 CO2-promoted organic transformations: (a) rearrangement of propargyl alcohols to unsaturated ketones; (b) cross-coupling using native allylic alcohol; (c) oxidation of allylic alcohols to unsaturated aldehydes. This work: (d) synthesis of tetra-substituted furans. | ||
Therefore, we envisioned utilizing CO2 together with organic dyes to catalyze the cyclizations of 1,3-diketones1b,16,17 and subsequent cyclopropane rearrangement events7en route to valuable tetra-substituted furan products, enabled by the reversible interaction of CO2 with the enol forms that can provide favorable carbonate leaving groups.10b,c,11g The optimization survey, substrate scope, and preliminary mechanistic studies of this transition metal-free, photocatalytic, and CO2-promoted furan synthesis strategy are presented herein.
Results and discussion
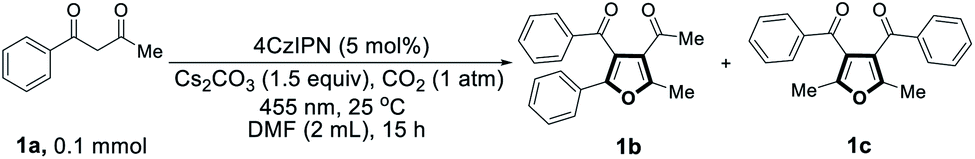
We started our investigation by employing 1-phenyl-1,3-butanedione (1a) as the model substrate in the presence of CO2. At first, a range of commercially available organic photocatalysts and bases were screened (Table S1†). Gratifyingly, the desired tetra-substituted furans were produced by using 4CzIPN as the photocatalyst, N,N-dimethylformamide (DMF) as solvent, Cs2CO3 as a base, at 25 °C under 455 nm light irradiation, and in CO2 atmosphere. Surprisingly, two different tetra-substituted isomers can be formed in the same reaction system, giving 48% and 50% yield of products 1b and 1c, respectively (Table 1, entry 1). Temperature of the photocatalytic reaction seemingly has an effect on the regioselectivity between 1b and 1c (Table 1, entry 2). The yields of the products are influenced by the amount of base, with 1.5 equivalents of Cs2CO3 providing the best yields (Table S2†). We then screened a range of solvents, with DMF proving to be optimal (Table S3†). Light sources at different wavelengths were found to be similarly effective (Table S4†). However, when the CO2 atmosphere was replaced with N2, no desired products were formed (Table 1, entry 3). Only trace amounts of products 1b and 1c were detected when the reaction was carried out under air (Table 1, entry 4), indicating that CO2 is an indispensable component in this reaction. In the dark, no reaction occurred at either 25 °C or 60 °C (Table 1, entries 5 and 6), ruling out a base-catalyzed thermal pathway. Similarly, no detectable products were formed when the reaction was carried out in the absence of photocatalyst or base (Table 1, entries 7 and 8).|
|
|||
|---|---|---|---|
| Entry | Deviations from standard conditions | Yield of 1ba | Yield of 1ca |
| a Yields were determined by GC-MS analysis against an internal standard and are the average of two runs; n.d., product was not detected. | |||
| 1 | None | 48% | 50% |
| 2 | 60 °C | 24% | 75% |
| 3 | N2 (1 atm) instead of CO2 | n.d. | n.d. |
| 4 | Air (1 atm) instead of CO2 | <1% | <1% |
| 5 | No light, 25 °C | n.d. | n.d. |
| 6 | No light, 60 °C | n.d. | n.d. |
| 7 | No 4CzIPN | n.d. | n.d. |
| 8 | No Cs2CO3 | n.d. | n.d. |
With the optimized reaction conditions in hand, we evaluated the scope and limitations of this method (Scheme 3). Both electron-rich and electron-deficient phenyl rings were well tolerated (2–4, 6), but a mesityl group provided deleterious steric hindrance (5). Diaryl-1,3-diketones reacted smoothly to afford the desired products (7–9). Notably, the reaction of 9a could produce three different products when the two phenyl groups contain different substituents. Other benzene systems such as biphenyl (10), phenyl ether (11), naphthalene (12) and benzodioxane (13) were also compatible with our reaction conditions. The preference for the symmetric regioisomers in 2, 6, and 10 cannot be explained at the current stage. Furthermore, this method was successfully extended to heterocyclic substituents, such as furan and thiophene (14, 15). Surprisingly, when the 1,3-diketone motif was replaced with 3-oxo-ester, the reaction yielded 2,5-dihydrofurans (16b, 17) and unsymmetric 2,3-dihydrofurans (16c, dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) that resisted regioisomerization and further oxidation to furans even after extensive exposure to air (vide infra). We speculate that the less hydridic C2 hydrogens and the overall less electron-rich conjugation systems made 16 and 17 products stable to oxidation, while the electronic effects of the benzene ring could affect the equilibrium between different dihydrofuran isomers (16cvs.17c). Unfortunately, no reaction occurred with acetylacetone 18a (vide infra). The structure of product 1b was unambiguously confirmed via single crystal X-ray analysis. A gram-scale reaction of 1a was also conducted under standard conditions, giving 40% and 47% yield of products 1b and 1c, respectively.
:1) that resisted regioisomerization and further oxidation to furans even after extensive exposure to air (vide infra). We speculate that the less hydridic C2 hydrogens and the overall less electron-rich conjugation systems made 16 and 17 products stable to oxidation, while the electronic effects of the benzene ring could affect the equilibrium between different dihydrofuran isomers (16cvs.17c). Unfortunately, no reaction occurred with acetylacetone 18a (vide infra). The structure of product 1b was unambiguously confirmed via single crystal X-ray analysis. A gram-scale reaction of 1a was also conducted under standard conditions, giving 40% and 47% yield of products 1b and 1c, respectively.
 | ||
| Scheme 3 Substrate scope of tetra-substituted furan from 1,3-diketone. aReaction conditions: 1,3-diketone (0.5 mmol), 4CzIPN (0.025 mmol), Cs2CO3 (0.75 mmol), CO2 (5 atm) in DMF (5 mL) at 25 °C under irradiation with a 455 nm LED for 15 h. Reported yields are isolated yields unless stated otherwise. bIsolated yields of gram-scale reaction. cYields were determined by GC-MS analysis against an internal standard and are the average of two runs. Products were not isolated due to their low yields. dCrystal structure of 1b as determined by X-ray crystallography at 123 K (see ESI†). | ||
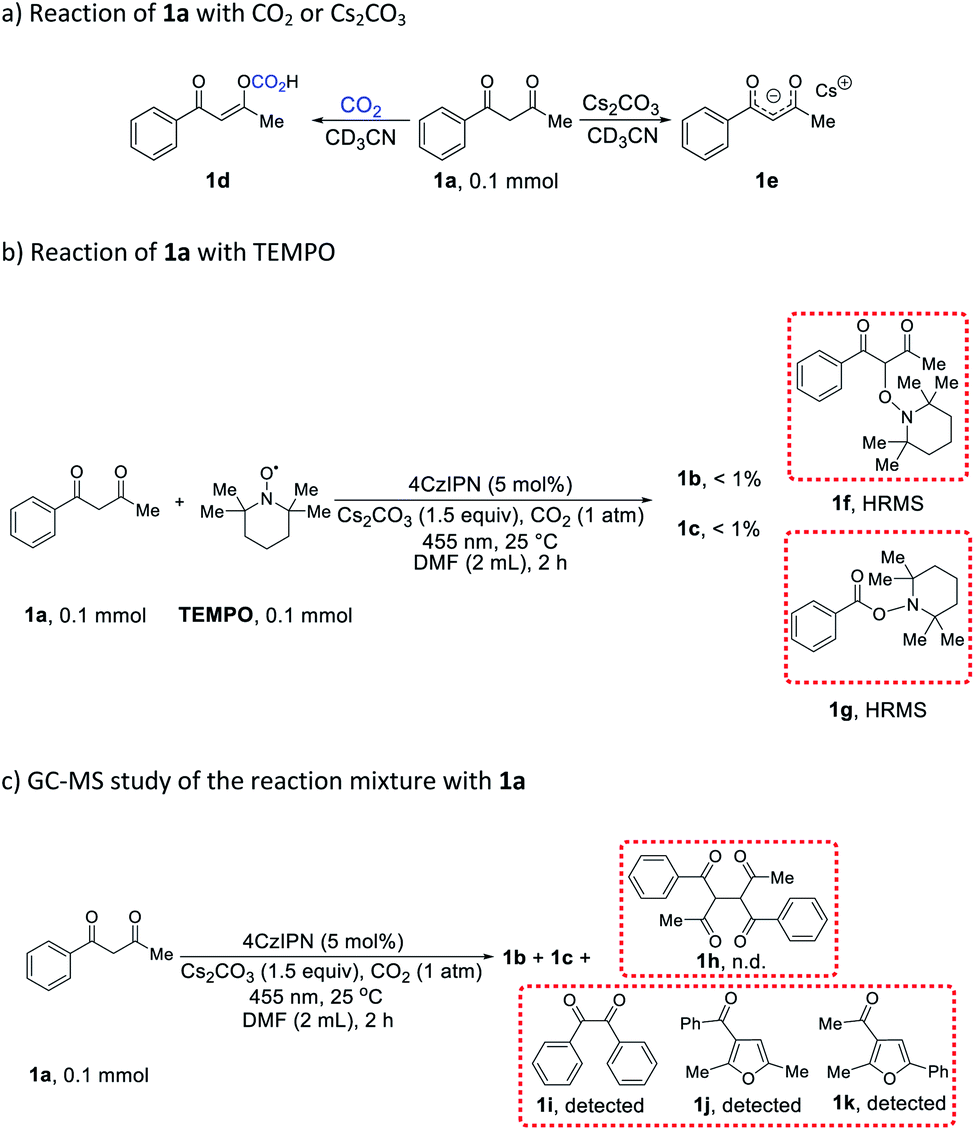
In order to gain insight into the aforementioned transformation, a series of mechanistic studies was conducted. To confirm whether the reaction proceeds via a radical process, in situ electron paramagnetic resonance (EPR) spectra of different reactions were recorded (Table S6† and Fig. S1–S14†). No signal was observed for the mixture of 1a, 4CzIPN, and CO2, either with or without blue light irradiation (Table S6,† entries 1 and 2). Similarly, the mixture of 1a, Cs2CO3, and CO2 did not show any signal with or without blue light irradiation (Table S6,† entries 3 and 4). In addition, there was no EPR signal when mixing 4CzIPN, Cs2CO3, and CO2 (Table S6,† entries 5 and 6), or 1a with CO2 alone (Table S6,† entries 7 and 8). However, a significant EPR signal was observed when a composition of 1a, 4CzIPN, Cs2CO3, and CO2 was irradiated (Table S6,† entry 10). When the CO2 atmosphere was replaced with N2, the same signal was still present (Table S6,† entry 12), indicating that CO2 did not participate in the generation of this radical species. To identify the origin of the radical resonance signal, we employed 7a instead of 1a in the EPR measurement, obtaining an identical signal (Fig. S7–S9†). Speculating that the signal arose from a 4CzIPN radical anion, we recorded the EPR spectrum of this species by adding NEt3 as an electron donor to a solution of 4CzIPN (Fig. S14†), which gave the same resonance signal as did the catalysis system.18 Accordingly, we concluded that 1a, 4CzIPN, Cs2CO3 and light irradiation together resulted in the formation of 4CzIPN radical anion. Then, the redox behavior of 1a and its anion 1a− were investigated. For 1a, no oxidation peak was observed within the electrochemical window of DMF as solvent (Fig. S15†). The anion of 1a, synthesized independently, showed an irreversible one-electron oxidation (Eox = −0.09 V vs. SCE), which is able to reduce the photoexcited state of 4CzIPN (E1/2[PC*/PC−] = +1.35 V vs. SCE)19 to provide the EPR-observed radical anion and a transient alkyl radical of 1a. Stern–Volmer luminescence quenching experiments (Fig. S17 and S18†) revealed efficient quenching of photoexcited 4CzIPN* upon addition of 1a and Cs2CO3 (KSV = 113 M−1). In situ NMR studies demonstrated that 1a react with CO2 to produce compound 1d (Scheme 4a and Fig. S20–S31,† observing the carbonic acid carbon peak), and naturally, Cs2CO3 can deprotonate 1a to give salt 1e (Scheme 4a and Fig. S32†). While no change was observed when 1a or 1d was subjected to irradiation together with 4CzIPN (Fig. S33–S40†), the 1H spectrum of 1e was altered under these conditions (Fig. S41–S44†).
 | ||
| Scheme 4 Mechanistic studies. | ||
When the reaction of 1a was carried out in the presence of 1 equiv. 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) as a radical trap, the desired reactivity was almost completely shut down, while adducts 1f and 1g were formed, hinting at the presence of C(sp3)-centered alkyl radicals generated from 1a, as well as benzoyl radicals (Scheme 4b). The detection of benzil 1i lends further support to the formation of benzoyl radicals in the course of the reaction (Scheme 4c). Correspondingly, tri-substituted furans 1j and 1k were also present in the reaction mixture (Scheme 4c). It is worth noting that benzoyl radical PhC(O)˙ (Ered = −1.13 V vs. SCE)20 is capable of oxidizing 4CzIPN radical anion (E1/2[PC/PC−] = −1.21 V vs. SCE),19 whereas CH3C(O)˙ (Ered = −1.75 V vs. SCE)20 is not, giving a possible explanation to the lack of reactivity with acetylacetone 18a. In addition, the dimer (1h) of starting material 1a was not formed in any appreciable amount (Scheme 4c), suggesting that alkyl radicals originating from 1a did not undergo a simple dimerization. Light “on–off” experiments indicate that the reaction needs continuous light irradiation to proceed (Fig. S19†), ruling out a radical-chain mechanism.
Based on the above observations and previous studies,7 a mechanism for the CO2-promoted photocatalytic activation of 1,3-diketones to afford tetra-substituted furans is proposed (Scheme 5). The starting material 1a is deprotonated by Cs2CO3 to generate enolate 1e, which is oxidized by photoexcited 4CzIPN* through a single electron transfer process, forming 4CzIPN˙− and radical Int2. In addition, 1a also equilibrates with CO2 to form adduct 1d, which reacts with radical Int2, giving the dimeric Int4. Subsequently, Int4 ejects benzoyl radical PhC(O)˙ (Int5) to form acylcyclopropane Int6via an intramolecular cyclization process (see ESI† section XII for discussion). Density functional theory (DFT) calculations reveal that the formation of Int5 and Int6 from Int4 (ΔG = +10 kcal mol−1, see ESI† section XIII) is most likely the rate-determining step. Thereafter, benzoyl radical Int5 oxidizes 4CzIPN˙− to close the catalytic cycle and produce benzoyl anion Int7. The ring-opening rearrangement of Int6 results in furanoid structures, Int8 and Int9, via two different pathways.7c,d Computations suggest that the ring-opening processes are exergonic and favorable. The protonation and deprotonation of Int8 and Int9 lead to the formation of conjugated Int10 and Int11. Cleavage of the C–O bonds and the accompanying nucleophilic attack by Int7 generate tetra-substituted 2,3-dihydrofurans Int12 and Int13 (isolated as product 16c). We postulated that 1j and 1k detected by GC-MS came from the direct C–O cleavage of Int10 and Int11 with further aromatization in air. Intermediates Int12 and Int13 undergo additional protonation and deprotonation to afford the more conjugated Int14 and Int15 (isolated as products 16b, 17b, 17c). At last, aromatization-driven oxidation processes yield the desired tetra-substituted furans.
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusion
In conclusion, we have developed an unusual, transition metal-free, CO2-promoted, visible-light-induced photocatalytic synthesis of highly substituted furan derivatives using 1,3-diketones as the only starting material. Mechanistic investigations indicated that CO2 was catalytically incorporated in order to create a better leaving group from the enolic OH group. The reaction proceeds under mild conditions via diketone radical additions and acylcyclopropane rearrangements, leading to the formation of two isomeric but differently substituted furan products. From 3-oxo-ester starting materials, partially hydrogenated furan scaffolds could also be obtained. This protocol expands the scope of the photocatalytic de novo synthesis of heterocyclic compounds as well as the catalytic use of CO2 as a reaction promoter.Data availability
All experimental, computational, and crystallographic data are available in the ESI.†Author contributions
Y.-M. T. and B. K. conceived the project. Y.-M. T. performed and analyzed the experiments. H. W. performed the DFT and CBS calculations. R. synthesized some materials. Y.-M. T., H. W., and B. K. prepared the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Science Foundation (DFG) (KO 1537/18-1). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement 741623). We thank Dr Rudolf Vasold (University of Regensburg) for his assistance in GC-MS measurements, Regina Hoheisel (University of Regensburg) for her assistance in cyclic voltammetry measurements and Birgit Hischa (University of Regensburg) for her assistance in single crystal X-ray measurements.Notes and references
- (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed; (b) A. Blanc, V. Bénéteau, J.-M. Weibel and P. Pale, Org. Biomol. Chem., 2016, 14, 9184–9205 RSC; (c) W. Zhang, W. Xu, F. Zhang and Y. Li, Chin. J. Org. Chem., 2019, 39, 1277–1283 CrossRef CAS.
- F. Feist, Chem. Ber., 1902, 35, 1537–1545 CrossRef CAS.
- V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301–307 CrossRef CAS.
- (a) Y. Yang, J. Yao and Y. Zhang, Org. Lett., 2013, 15, 3206–3209 CrossRef CAS PubMed; (b) B. Lu, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598–11601 CrossRef CAS PubMed; (c) S. Manna and A. P. Antonchick, Org. Lett., 2015, 17, 4300–4303 CrossRef CAS PubMed; (d) T. Naveen, A. Deb and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 1111–1115 CrossRef CAS PubMed; (e) S. Agasti, T. Pal, T. K. Achar, S. Maiti, D. Pal, S. Mandal, K. Daud, G. K. Lahiri and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 11039–11043 CrossRef CAS PubMed.
- (a) K. Miki, F. Nishino, K. Ohe and S. Uemura, J. Am. Chem. Soc., 2002, 124, 5260–5261 CrossRef CAS PubMed; (b) K. Miki, Y. Washitake, K. Ohe and S. Uemura, Angew. Chem., Int. Ed., 2004, 43, 1857–1860 CrossRef CAS PubMed; (c) J. Barluenga, L. Riesgo, R. Vicente, L. A. López and M. Tomás, J. Am. Chem. Soc., 2008, 130, 13528–13529 CrossRef CAS PubMed; (d) G. Zhang, X. Huang, G. Li and L. Zhang, J. Am. Chem. Soc., 2008, 130, 1814–1815 CrossRef CAS PubMed; (e) J. González, J. González, C. Pérez-Calleja, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2013, 52, 5853–5857 CrossRef PubMed; (f) L. Zhou, M. Zhang, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5853–5857 Search PubMed; (g) Z.-M. Zhang, P. Chen, W. Li, Y. Niu, X.-L. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350–4354 CrossRef CAS PubMed; (h) Y. Wang, P. Zhang, D. Qian and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14849–14852 CrossRef CAS PubMed; (i) J. S. Clark, F. Romiti, K. F. Hogg, M. H. S. A. Hamid, S. C. Richter, A. Boyer, J. C. Redman and L. J. Farrugia, Angew. Chem., Int. Ed., 2015, 54, 5744–5747 CrossRef CAS PubMed; (j) M.-Y. Huang, J.-M. Yang, Y.-T. Zhao and S.-F. Zhu, ACS Catal., 2019, 9, 5353–5357 CrossRef CAS.
- R. K. Shiroodi, O. Koleda and V. Gevorgyan, J. Am. Chem. Soc., 2014, 136, 13146–13149 CrossRef CAS PubMed.
- (a) S. Pratapan, K. Ashok, K. R. Gopidas, N. P. Rath, P. K. Das and M. V. George, J. Org. Chem., 1990, 55, 1304–1308 CrossRef CAS; (b) M. C. Sajimon, D. Ramaiah, M. Muneer, E. S. Ajithkumar, N. P. Rath and M. V. George, J. Org. Chem., 1999, 64, 6347–6352 CrossRef CAS; (c) J. Barluenga, H. Fanlo, S. López and J. Flórez, Angew. Chem., Int. Ed., 2007, 46, 4136–4140 CrossRef CAS PubMed; (d) J. Shao, Q. Luo, H. Bi and S. R. Wang, Org. Lett., 2021, 23, 459–463 CrossRef CAS PubMed; (e) X. He, Y. Tang, Y. Wang, J.-B. Chen, S. Xu, J. Dou and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 10698–10702 CrossRef CAS PubMed.
- H. Jin and A. Fürstner, Angew. Chem., Int. Ed., 2020, 59, 13618–13622 CrossRef CAS PubMed.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933–5948 CrossRef PubMed; (c) X. He, L.-Q. Qiu, W.-J. Wang, K.-H. Chen and L.-N. He, Green Chem., 2020, 22, 7301–7320 RSC; (d) Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS; (e) Z. Fan, Z. Zhang and C. Xi, ChemSusChem, 2020, 13, 6201–6218 CAS; (f) J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed; (g) B. Cai, H. W. Cheo, T. Liu and J. Wu, Angew. Chem., Int. Ed., 2021, 60, 18950–18980 CrossRef CAS PubMed.
- (a) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed; (b) D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030–3034 CrossRef CAS; (c) D. Riemer, W. Schilling, A. Goetz, Y. Zhang, S. Gehrke, I. Tkach, O. Hollóczki and S. Das, ACS Catal., 2018, 8, 11679–11687 CrossRef CAS.
- (a) P. Braunstein, D. Matt and D. Nobel, Chem. Rev., 1988, 88, 747–764 CrossRef CAS; (b) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC; (c) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (d) R. Martín and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef PubMed; (e) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC; (f) P. G. Jessop, S. M. Mercer and D. J. Heldebrant, Energy Environ. Sci., 2012, 5, 7240–7253 RSC; (g) S. B. Lang, T. M. Locascio and J. A. Tunge, Org. Lett., 2014, 16, 4308–4311 CrossRef CAS PubMed; (h) F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52–61 CrossRef CAS; (i) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739–6749 CrossRef PubMed; (j) S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666–3676 RSC; (k) P. Hirapara, D. Riemer, N. Hazra, J. Gajera, M. Finger and S. Das, Green Chem., 2017, 19, 5356–5360 RSC; (l) W. Schilling and S. Das, Tetrahedron Lett., 2018, 59, 3821–3828 CrossRef CAS; (m) Z. Zhang, J.-H. Ye, D.-S. Wu, Y.-Q. Zhou and D.-G. Yu, Chem.–Asian J., 2018, 13, 2292–2307 CrossRef CAS PubMed; (n) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed; (o) L. Wang, W. Sun and C. Liu, Chin. J. Chem., 2018, 36, 353–362 CrossRef CAS; (p) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644–659 CrossRef CAS; (q) C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5492–5502 CrossRef CAS PubMed; (r) Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed; (s) M. Schmalzbauer, T. D. Svejstrup, F. Fricke, P. Brandt, M. J. Johansson, G. Bergonzini and B. König, Chem, 2020, 6, 2658–2672 CrossRef CAS; (t) S. Wang, B.-Y. Cheng, M. Sršen and B. König, J. Am. Chem. Soc., 2020, 142, 7524–7531 CrossRef CAS PubMed; (u) Y. Zhang, T. Zhang and S. Das, Green Chem., 2020, 22, 1800–1820 RSC; (v) K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242–252 CrossRef CAS PubMed; (w) R. Cauwenbergh and S. Das, Green Chem., 2021, 23, 2553–2574 RSC; (x) P. K. Sahoo, Y. Zhang and S. Das, ACS Catal., 2021, 11, 3414–3442 CrossRef CAS.
- E. F. Lutz (Shell Oil Co.), US Pat. 3518310A, 1970.
- Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- M. Kapoor, P. Chand-Thakuri and M. C. Young, J. Am. Chem. Soc., 2019, 141, 7980–7989 CrossRef CAS PubMed.
- (a) J. Song, H. Zhang, X. Chen, X. Li and D. Xu, Synth. Commun., 2010, 40, 1847–1855 CrossRef CAS; (b) Y. Yue, Y. Zhang, W. Song, X. Zhang, J. Liu and K. Zhuo, Adv. Synth. Catal., 2014, 356, 2459–2464 CrossRef CAS; (c) P.-J. Zhou, C.-K. Li, S.-F. Zhou, A. Shoberu and J. P. Zou, Org. Biomol. Chem., 2017, 15, 2629–2637 RSC.
- (a) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766–5769 CrossRef CAS PubMed; (b) Y. Ma, S. Zhang, S. Yang, F. Song and J. You, Angew. Chem., Int. Ed., 2014, 53, 7870–7874 CrossRef CAS PubMed.
- For the EPR signals of similar isophthalonitrile spieces, see: (a) E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend and N. C. Greenham, J. Phys. Chem. Lett., 2018, 9, 4053–4058 CrossRef CAS PubMed; (b) J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef PubMed.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2069 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2113371. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06403g |
| This journal is © The Royal Society of Chemistry 2022 |