
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect observation of protein structural transitions through entire amyloid aggregation processes in water using 2D-IR spectroscopy†
So Yeon
Chun‡
ab,
Myung Kook
Son‡
bcd,
Chae Ri
Park
bcd,
Chaiho
Lim
ab,
Hugh I.
Kim
 *bcd,
Kyungwon
Kwak
*ab and
Minhaeng
Cho
*ab
*bcd,
Kyungwon
Kwak
*ab and
Minhaeng
Cho
*ab
aCenter for Molecular Spectroscopy and Dynamics, Institute for Basic Science (IBS), Seoul 02841, Republic of Korea. E-mail: kkwak@korea.ac.kr; mcho@korea.ac.kr
bDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: hughkim@korea.ac.kr
cCenter for Proteogenome Research, Korea University, Seoul 02841, Republic of Korea
dSingle Cell Analysis Laboratory, Korea University, Seoul 02841, Republic of Korea
First published on 18th March 2022
Abstract
Amyloid proteins that undergo self-assembly to form insoluble fibrillar aggregates have attracted much attention due to their role in biological and pathological significance in amyloidosis. This study aims to understand the amyloid aggregation dynamics of insulin (INS) in H2O using two-dimensional infrared (2D-IR) spectroscopy. Conventional IR studies have been performed in D2O to avoid spectral congestion despite distinct H–D isotope effects. We observed a slowdown of the INS fibrillation process in D2O compared to that in H2O. The 2D-IR results reveal that different quaternary structures of INS at the onset of the nucleation phase caused the distinct fibrillation pathways of INS in H2O and D2O. A few different biophysical analysis, including solution-phase small-angle X-ray scattering combined with molecular dynamics simulations and other spectroscopic techniques, support our 2D-IR investigation results, providing insight into mechanistic details of distinct structural transition dynamics of INS in water. We found the delayed structural transition in D2O is due to the kinetic isotope effect at an early stage of fibrillation of INS in D2O, i.e., enhanced dimer formation of INS in D2O. Our 2D-IR and biophysical analysis provide insight into mechanistic details of structural transition dynamics of INS in water. This study demonstrates an innovative 2D-IR approach for studying protein dynamics in H2O, which will open the way for observing protein dynamics under biological conditions without IR spectroscopic interference by water vibrations.
Introduction
Amyloid proteins undergo self-assembly to form insoluble fibrillar aggregates with a common architecture of unbranched, highly ordered β-sheets. For many amyloid proteins, fibrils and intermediates such as oligomers are associated with various human diseases, known as amyloidosis.2,3 The complex fibrillation process of amyloid aggregation is affected by diverse biological factors, as it involves dynamic structural heterogeneity and diverging mechanisms, making it challenging to understand and control. Therefore, much effort has been undertaken to understand the structural details of the oligomeric species at an early stage of the fibrillation process at the molecular level.4–9 Among various amyloid proteins, insulin (INS), a critical hormone that regulates glucose homeostasis, is associated with injection localized amyloidosis in type II diabetes.10 INS is prone to form amyloid fibers under various pH, concentration, salt, buffering agent, and temperature conditions.11 INS is also known to form polymorphic amyloid fibrils depending on aggregation conditions.12,13 Therefore, understanding the mechanisms of INS fibrillation in various experimental conditions is necessary for developing therapeutic strategies for amyloidosis.To study amyloid-associated protein dynamics, tremendous efforts have been made to track the appropriate structural dynamics of the intermediates generated in the aggregation processes by using spectroscopic techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy,14–16 which monitor protein structural dynamics on timescales from nanoseconds to years. In particular, protein dynamics on subnanosecond timescales can be studied by infrared (IR) spectroscopy, utilizing nine characteristic IR absorption bands inherent in proteins, specifically amide A, B, and I–VII. Among these, the spectral lineshape and frequency of the amide I band, primarily the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch vibrations of the peptide linkages, are exceptionally sensitive to secondary structural elements of a protein. Therefore, time-resolved amide I IR spectroscopy with high temporal resolution (<ps) is one of the most incisive tools for studying protein dynamics in condensed phases. Despite these advantages, the spectral overlap between the H–O–H bending mode of H2O (δH–O–H, 1650 cm−1) and the amide I mode (νCO, 1600–1700 cm−1) prohibits the direct application of various IR spectroscopic techniques to the studies of proteins dissolved in ordinary water. Most IR studies have thus been performed in D2O to avoid spectral congestion, and they have been undoubtedly successful in determining and monitoring protein structure and dynamics.17,18 In addition, solution NMR, one of the most extensively used methods in protein dynamics studies, typically uses 10% D2O to stabilize the magnetic field strength and lock the field-dependent frequency. However, nuclear quantum effects19–21 arising from tunneling, nuclear delocalization, zero-point energy differences, and other H–D isotope effects22 could, in general, influence the properties of proteins.23,24 For example, it was reported that the stability of folded proteins in D2O differs from that in H2O, leading to different aggregation rates,22,23,25–27 especially in the fibrillation of amyloid proteins since water plays a key role in modulating the transition free energy of amyloid nucleation.28–30 Furthermore, the use of D2O could result in substantial changes in the couplings between heavy water and protein vibrations due to the frequency shifts of water bending and stretching modes upon H–D exchange,31,32 which would alter the energy transfer and vibrational relaxation processes. Therefore, the IR spectroscopy of a protein in heavy water could differ from that in ordinary water, implying that an extra layer of caution is necessary when generalizing the IR spectroscopic results on protein folding–unfolding or aggregation processes in D2O.
O stretch vibrations of the peptide linkages, are exceptionally sensitive to secondary structural elements of a protein. Therefore, time-resolved amide I IR spectroscopy with high temporal resolution (<ps) is one of the most incisive tools for studying protein dynamics in condensed phases. Despite these advantages, the spectral overlap between the H–O–H bending mode of H2O (δH–O–H, 1650 cm−1) and the amide I mode (νCO, 1600–1700 cm−1) prohibits the direct application of various IR spectroscopic techniques to the studies of proteins dissolved in ordinary water. Most IR studies have thus been performed in D2O to avoid spectral congestion, and they have been undoubtedly successful in determining and monitoring protein structure and dynamics.17,18 In addition, solution NMR, one of the most extensively used methods in protein dynamics studies, typically uses 10% D2O to stabilize the magnetic field strength and lock the field-dependent frequency. However, nuclear quantum effects19–21 arising from tunneling, nuclear delocalization, zero-point energy differences, and other H–D isotope effects22 could, in general, influence the properties of proteins.23,24 For example, it was reported that the stability of folded proteins in D2O differs from that in H2O, leading to different aggregation rates,22,23,25–27 especially in the fibrillation of amyloid proteins since water plays a key role in modulating the transition free energy of amyloid nucleation.28–30 Furthermore, the use of D2O could result in substantial changes in the couplings between heavy water and protein vibrations due to the frequency shifts of water bending and stretching modes upon H–D exchange,31,32 which would alter the energy transfer and vibrational relaxation processes. Therefore, the IR spectroscopy of a protein in heavy water could differ from that in ordinary water, implying that an extra layer of caution is necessary when generalizing the IR spectroscopic results on protein folding–unfolding or aggregation processes in D2O.
Over the past two decades, two-dimensional IR (2D-IR) spectroscopy, an IR analog of 2D-NMR, has become a powerful tool for studying protein dynamics.17,33 In a 2D-IR spectrum, off-diagonal peaks produced by vibrational couplings provide information about the spatial and chemical connectivity between two oscillators. Furthermore, the real-time monitoring of 2D-IR diagonal and off-diagonal peaks and their lineshapes enables the rates of conformational transitions, vibrational relaxation, and local solvent fluctuations. In addition, the nonlinear dependence of the 2D-IR signal on oscillator strengths makes the contrast between strongly and weakly IR-absorbing modes more prominent than those of linear IR spectroscopy, often resulting in an enhanced spectral resolution.18,34–36 Because of these advantages of 2D-IR technique with an improved spectral resolution, it was used to identify intermediates during amyloid protein aggregation processes and track the mechanism of amyloid fibril formation at the molecular level.37,38 More importantly, the improved temporal resolution enables the selective gating of slowly decaying signals in the time domain. For example, the vibrational lifetime of the protein amide I mode, which is in the range from 0.5 to 1 ps depending on secondary structures and the extent of solvent exposure,32,39–41 is substantially longer than that of the δH–O–H of H2O (∼170 fs). As a consequence, there is the Goldilocks time window during which the protein 2D-IR signal remains strong without interfering with water contributions, which allows one to measure the 2D-IR spectra of proteins selectively even in H2O.
In this work, we investigated the distinct amyloid fibrillation processes of human INS in H2O and D2O using 2D-IR spectroscopy. In addition to addressing fundamental questions regarding the isotope effects of D2O on protein dynamics, we aim to understand the causes of the different amyloid aggregation kinetics of INS in H2O and D2O. We observed a slowdown of the fibrillation process of INS in D2O compared to that in H2O. Here, we show that this difference in INS fibrillations originates from the distinctively different tertiary structures of INS in H2O and D2O. Various ancillary spectroscopic and biophysical techniques allowed us to gain further insights into the details of the structural transitions during the initial stage, nucleation and lag phase, and fibril elongation process of INS in water.
Results and discussion
Kinetics of insulin fibrillation
The fibrillation kinetics of INS (17 mg mL−1) in H2O and D2O were examined using the standard thioflavin T (ThT) fluorescence assay method (Fig. 1a). The stiff rise of ThT fluorescence intensity is an indication of the formation of fibrils. Interestingly, under our experimental conditions (INS-H2O at pH = 2.1, INS-D2O at pD = 2.1, 50 °C, and 25 rpm for incubation), the fibrillation (∼16 h) of INS in H2O occurs earlier than that (∼20 h) in D2O (ESI Note I). This observation was further confirmed by the measured circular dichroism (CD) spectra, which exhibited a slightly faster conversion of α-helix-rich INS into β-sheet fibrils in H2O than in D2O (Fig. 1b, c, S1, Tables S1 and S2†).42 However, no significant difference in fibril morphology was observed in TEM images of INS fibrils formed in H2O and D2O (Fig. 1d). We observed that the fibrillations of INS in H2O occur faster than those in D2O at different experimentally measured pH (pHM) values (HCl (DCl)–KCl buffer solution, pHM = 1.7, 2.1, and 2.5).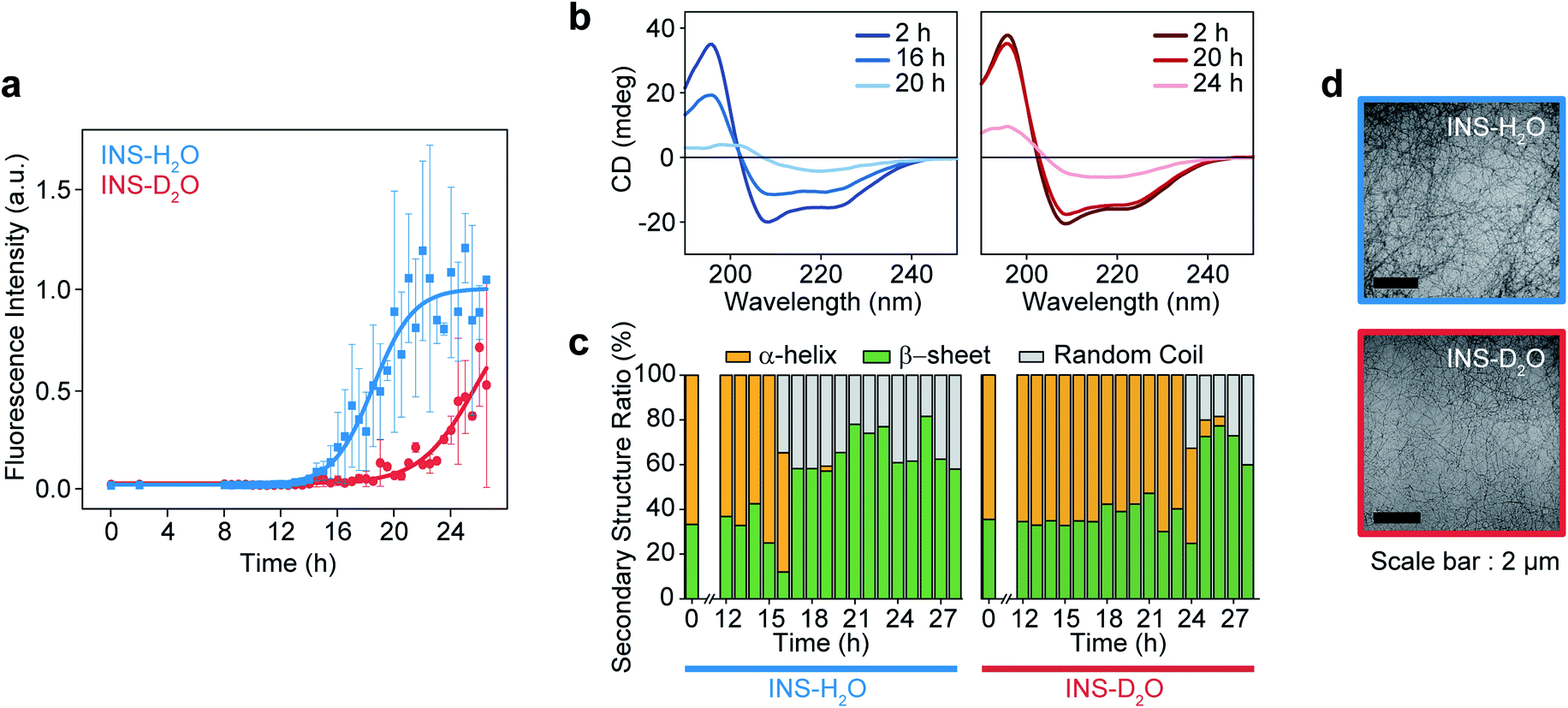 | ||
| Fig. 1 Fibrillation process kinetic analysis of INS in H2O and D2O at 17 mg mL−1 concentration. (a) The plot of ThT fluorescence spectra at the intensity of 490 nm. (b) CD spectra of INS in H2O and D2O during fibrillation. (c) The ratio of secondary structures (β-sheet, α-helix, and random coil) from analysis of CD spectrum using BeStSel server.1 (d) TEM image after the fibrillation process (∼28 h) of INS-H2O (top) and INS-D2O (bottom). | ||
To understand this result, we noted that D2O molecules could form a stronger deuterium bond (D-bond) between D2O molecules, which induces a greater number of D-bonds per molecule than H-bonds per H2O in water.22,23 Thus, the entropic cost of dissolving a solute molecule in D2O is, in general, higher than that in H2O. Accordingly, D2O effectively increases the hydrophobic interactions between amino acid residues in INS, thereby increasing the thermodynamic stability (ΔGu, free energy of unfolding) and enthalpy of unfolding (ΔHu) of INS.24,26 The helical propensity of INS in D2O lasts longer than that in H2O, which was observed in the time-resolved CD spectra and supports this supposition (Tables S1, S2 and Fig. S5†).25,26
Linear IR and 2D-IR spectroscopy of insulin fibrillation
To investigate the distinct structural dynamics of INS, which induce different fibrillation kinetics in H2O and D2O, we examined the amide I IR absorption spectra of INS after subtracting the solvent spectrum from the raw spectrum (ESI Note II†). For the INS-water solution, such solvent background correction should be performed carefully because the H2O bending signal substantially overlaps with the amide I band of INS and because the two vibrations could mix via H-bonding interactions. When INS is dissolved in D2O, the H–D exchange processes of the peptide N–H groups should be completed before the IR measurements (ESI Note III). The different fibrillation rates in H2O and D2O observed in the ThT fluorescence assay and CD spectroscopy could also be confirmed using linear IR spectroscopy. The time-resolved IR spectra of INS (Fig. 2a) suggest that the conformational transition from an α-helix-rich structure (∼1658 cm−1 in H2O and ∼1649 cm−1 in D2O) to a β-sheet-rich fibrillar structure (∼1629 cm−1 in H2O and ∼1620 cm−1 in D2O) occurs faster in H2O than in D2O.43–45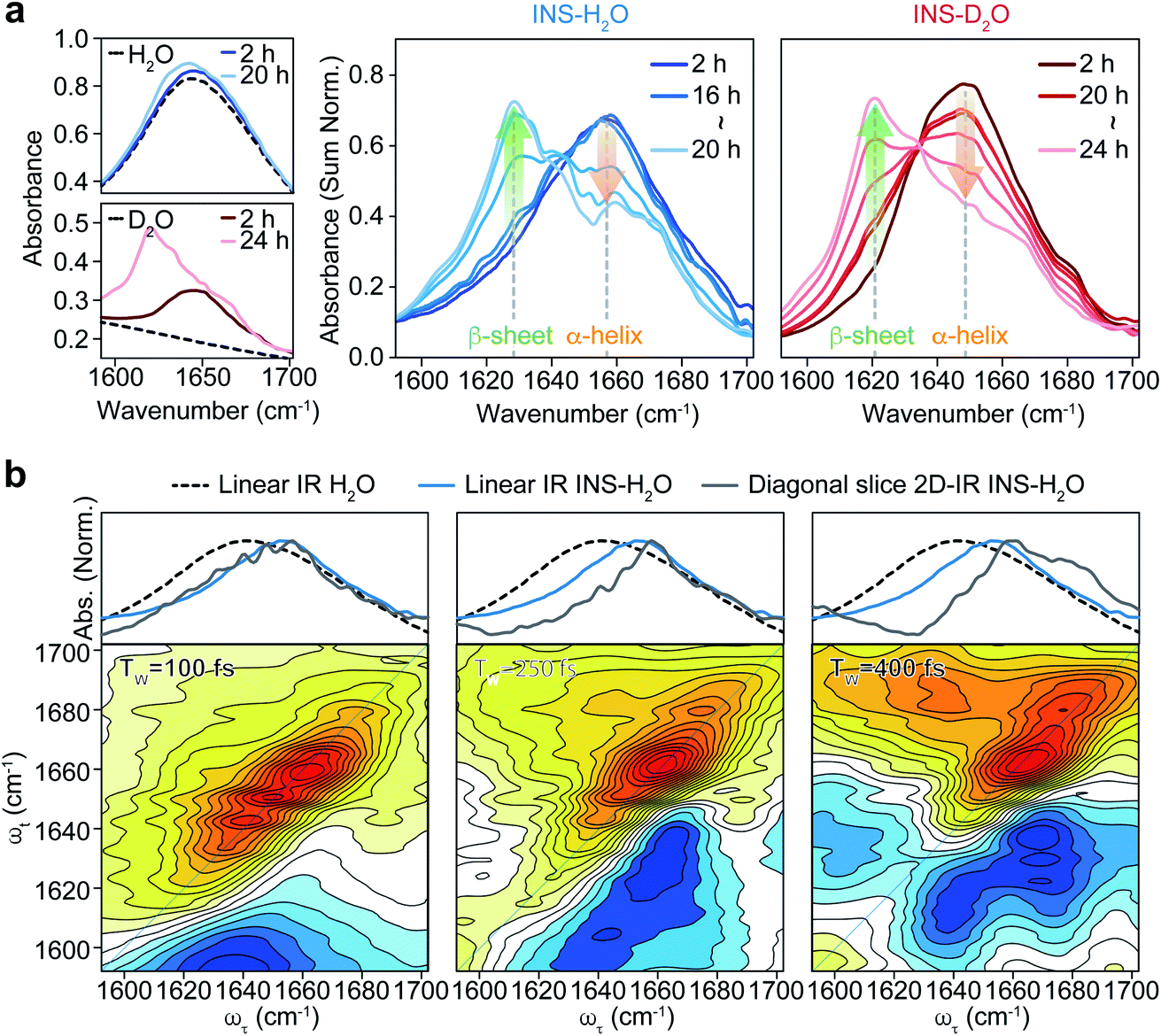 | ||
| Fig. 2 Linear IR of INS during fibrillation and waiting time (TW) dependent 2D-IR spectra of INS. (a) Area-normalized linear IR absorption spectra of INS in H2O and D2O obtained during incubation. The raw linear IR spectra of two solvents (H2O and D2O) and INS-solvent, showing the signal overlap of H–O–H bending mode of water and protein amide I band at 1650 cm−1, are shown in the left. (b) Spectra comparison of linear IR spectra of H2O, INS-H2O, and diagonal slice 2D-IR spectra of INS-H2O (top) and 2D-IR spectra of INS-H2O at a waiting time of 100, 250, and 400 fs (bottom). | ||
We next carried out 2D-IR measurements of INS sampled intermittently from the incubation vessel (Fig. 2b, ESI Note IV†). It is known that the molar absorption coefficient of the amide I vibration is larger than that of H2O.46,47 Therefore, 2D-IR spectroscopy, whose signal is proportional to the fourth power of the vibrational transition dipole moment, is quite useful for distinguishing the strong amide I mode of INS from the H2O signal.48,49 However, the 2D-IR spectra at waiting times (Tw) shorter than 200 fs are still governed by the signal from H2O. Other undesired pump-induced heating signals contributed to the 2D-IR spectra after 300 fs.39 Because the lifetime of δH–O–H of H2O (∼170 fs) is much shorter than that of the amide I vibration (0.5–1 ps), the H2O signal rapidly decreases as the pump-probe delay time Tw increases. From these experimental observations, the ideal range of Tw for selective 2D-IR measurements of protein amide I modes is from 200 to 300 fs (Fig. 2b).50
The 2D-IR diagonal slice spectra at Tw = 250 fs reveal the distinctive difference in INS structures (Fig. 3). The dominant features in the 2D-IR spectra of INS in H2O and D2O in the early stage of incubation (∼2 h) are the broad peaks at 1660 cm−1 and 1651 cm−1, respectively, which correspond to the α-helix in native INS. The characteristic features of the β-sheets appear in the INS-H2O 2D-IR spectrum at ∼1627 cm−1 only after 16 h of incubation. However, D2O delays the conversion of INS from an α-helix to a β-sheet. More specifically, the 2D-IR peak associated with the β-sheets at ∼1616 cm−1 becomes noticeable after 20 h of incubation. Surprisingly, the 2D-IR diagonal slice spectra at an early stage of INS fibrillation in H2O and D2O differ from each other, suggesting distinctly different initial structures. In H2O, the broad featureless peak at 1660 cm−1 is due to the α-helix (Fig. 3a). The spectrum in D2O exhibits additional ν⊥ (∼1642 cm−1) and ν∥ (∼1680 cm−1) peaks that reflect the presence of β-strands in addition to the α-helix (Fig. 3b and S13†).51 These 2D-IR results strongly suggest that INS proteins tend to form dimers in D2O with antiparallel β-sheets.52–54
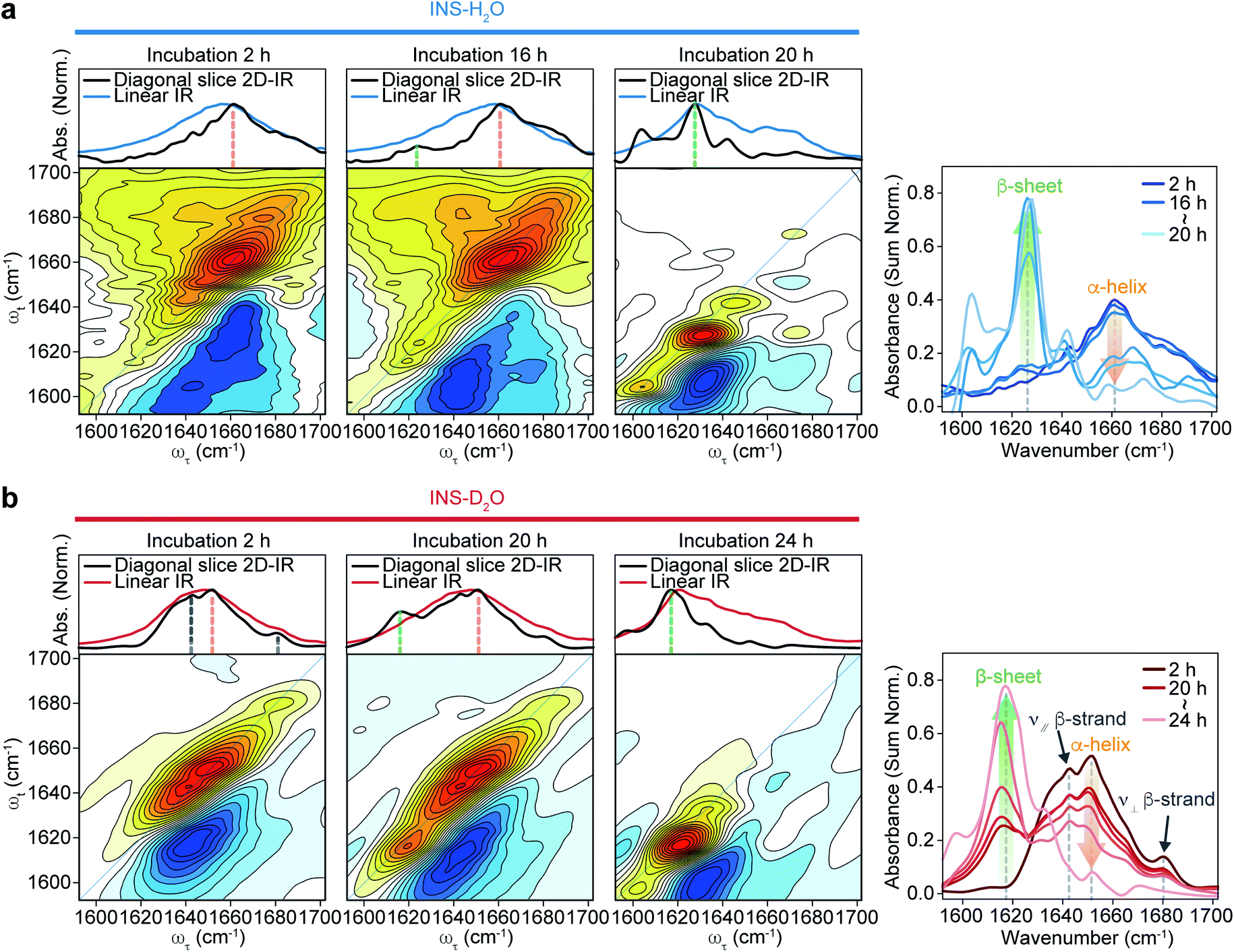 | ||
| Fig. 3 2D-IR spectra of INS during fibrillation. Spectra comparison of linear IR spectra and diagonal slice 2D-IR spectra (top) and 2D-IR spectra (bottom) of INS-H2O (a) and INS-D2O (b) obtained during incubation at a waiting time of 250 fs. The diagonal slice 2D-IR spectra of INS-H2O and INS-D2O obtained during incubation at a waiting time of 250 fs are shown in the right (INS-H2O: 2, 16–20 h, INS-D2O: 2, 20–24 h, 1 h interval). | ||
After the fibrillation process was completed, we confirmed the antiparallel β-sheet structures of INS fibrils in H2O and D2O using the 2D-IR technique; we used the main diagonal β-sheet peaks at 1627 and 1616 cm−1, respectively, weak high-frequency diagonal peaks at approximately 1673 and 1665 cm−1, respectively, and the cross-peaks (Fig. S14†).51,55,56 Thus, despite the different structures of INS in the early stages of fibrillation, we found no significant difference in the final fibril structures even with 2D-IR spectroscopy.
From these experimental findings, we believe that the distinct initial structures of INS in H2O and D2O induce the difference in nucleation kinetics. INS proteins are known to exist in complex forms under physiological conditions. Therefore, the dissociation of dimers into monomers is required for nucleation and subsequent prefibril formation, followed by amyloid fibrillation under acidic conditions. Thus, the presence of INS dimers in D2O could be a factor delaying the initial nucleation step and the overall fibrillation process.
Structural dynamics of INS in the early stages of amyloid fibrillation
Although it is plausible that the enhanced hydrophobic interactions in D2O could stabilize the intermolecular β-sheet structure in the INS dimer,57,58 it is necessary to investigate the initial structures of INS at a low concentration of 2 mg mL−1 (25 °C) to estimate the average sizes using solution-phase small-angle X-ray scattering (SAXS). The 1D SAXS profiles of INS in H2O and D2O are shown in Fig. 4a. The radii of gyration (Rg) obtained from the Guinier analysis of I(q) are 12.41 ± 0.99 Å and 12.38 ± 0.92 Å for INS in H2O and D2O, respectively (Fig. 4b). Kratky analysis of the SAXS profiles (Fig. 4c), which provide information about the flexibility and structural disorder of a given protein, suggest that INS in H2O and D2O adopt similar globular conformations.59 The observations that the experimentally determined Rg values of INS are slightly larger than that of the INS monomer (Fig. S15,† ∼11.6 Å from the 3E7Y PDB structure)12 and that the Rg of INS in H2O is slightly larger than that in D2O indicate the presence of mixed forms of INS with complex structures.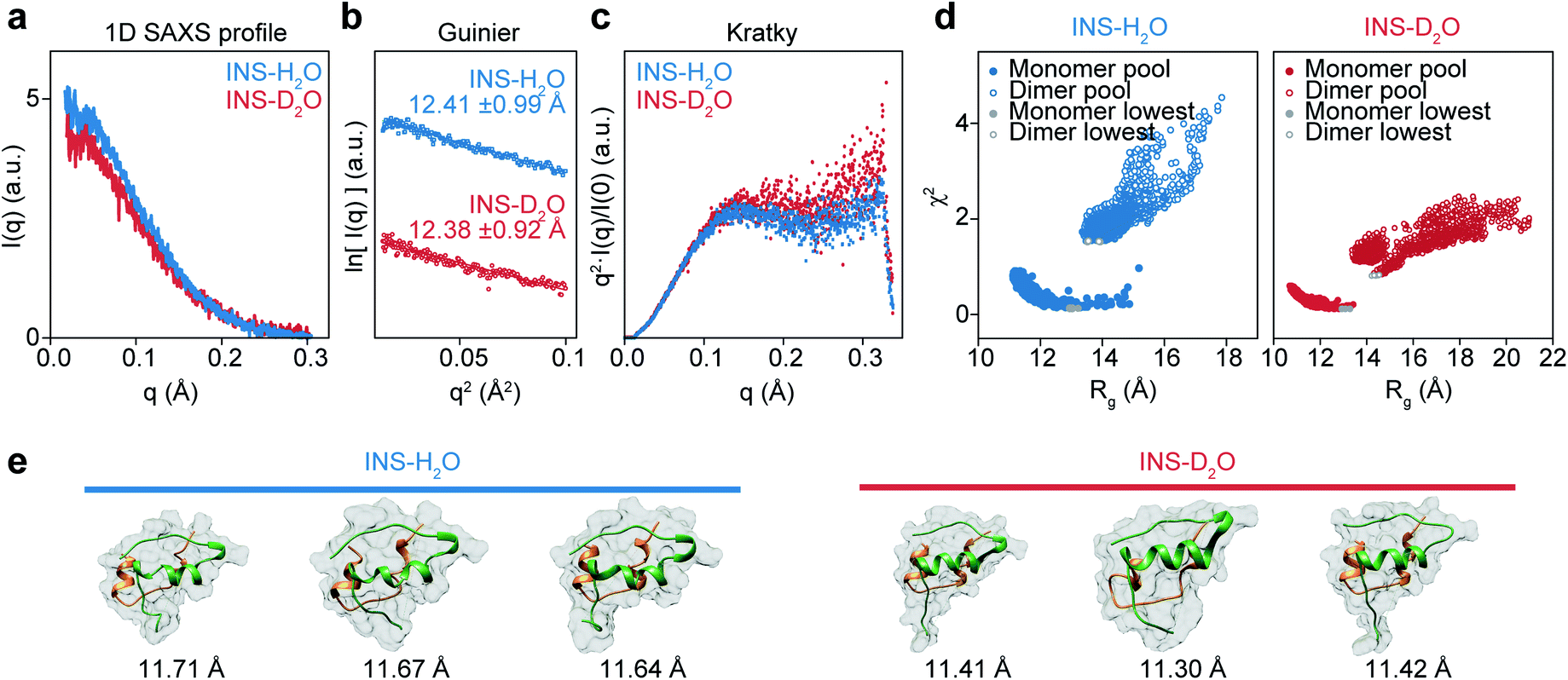 | ||
Fig. 4 Solution phase SAXS analysis of INS (INS-H2O (blue), INS-D2O (red)) and theoretical structures of INS obtained with MD-simulation. (a) Experimental SAXS scattering profiles of INS 2 mg mL−1 in pH (pD) = 2.1. (b) Guinier plot obtained from short q range of scattering profile. (c) Normalized Kratky analysis of INS. (d) χ2–Rg diagram of INS-H2O and INS-D2O from the MD-simulated 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 conformations, obtained with CRYSOL. Filled colored circles at the bottom left are the 14000 monomer conformations and filled red circles are the candidate structures of INS monomers with the smallest χ2 values. Empty colored circles at the top right are the 8000 dimer conformations and empty red circles are the INS dimers with the smallest χ2 values. (e) Candidate structures of INS-H2O and INS-D2O obtained with CRYSOL of MD-simulated monomer, dimer structure pools. 3 structures with the smallest χ2 structures were used as candidate structures. 000 conformations, obtained with CRYSOL. Filled colored circles at the bottom left are the 14000 monomer conformations and filled red circles are the candidate structures of INS monomers with the smallest χ2 values. Empty colored circles at the top right are the 8000 dimer conformations and empty red circles are the INS dimers with the smallest χ2 values. (e) Candidate structures of INS-H2O and INS-D2O obtained with CRYSOL of MD-simulated monomer, dimer structure pools. 3 structures with the smallest χ2 structures were used as candidate structures. | ||
Then, we studied the structures of INS by carrying out MD simulations. Noting that the SAXS results show the presence of a heterogeneous structural mixture of INS, we performed simulated annealing MD simulations to generate 14000 monomeric and 8000 dimeric structures. Then, the representative candidate structures were selected by comparing them with experimental scattering profiles (Fig. 4d).60 We found that the fraction of INS dimers in D2O was approximately 10%, whereas it was nearly zero in H2O (Table 1). Additionally, the simulated candidate structures of monomeric INS exhibited an enhanced helical propensity in D2O compared to those in H2O (Fig. 4e and Table 2). The dimers in D2O formed intermolecular β-sheet structures, which is consistent with our 2D-IR results. At a low INS concentration of 2 mg mL−1 in the SAXS measurements, a relatively small fraction of the proteins could form dimers, while increased dimer formation was expected at the high concentration of 17 mg mL−1 used in the 2D-IR experiments. As discussed earlier, the fibrillation of INS is still faster in H2O than D2O even at low concentrations (1 and 2 mg mL−1) of the protein (ESI Note I†), indicating that the difference in the fibrillation mechanisms of INS in two water solvents revealed by the 2D-IR spectroscopy remains valid even for comparatively low protein concentration solutions.
| χ 2 | Monomer fraction | Dimer fraction | |
|---|---|---|---|
| INS-H2O | 0.54 | 100.0% (±7.9%) | 0 |
| 0.56 | 100.0% (±0.4%) | 0 | |
| 0.68 | 86.6% (±1.5%) | 13.4% (±1.1%) | |
| 0.69 | 90.5% (±1.6%) | 9.5% (±1.1%) | |
| 0.71 | 89.1% (±1.6%) | 10.9% (±1.1%) | |
| INS-D2O | 0.49 | 93.4% (±1.9%) | 6.6% (±1.5%) |
| 0.5 | 88.9% (±2.3%) | 11.1% (±1.6%) | |
| 0.5 | 92.4% (±1.9%) | 7.6% (±1.5%) | |
| 0.51 | 90.6% (±2.1%) | 9.4% (±1.6%) | |
| 0.54 | 98.0% (±106.9%) | 2.0% (±2.4%) |
| INS monomer | H2O | D2O | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Exp. avg | MD-1 | MD-2 | MD-3 | Exp. avg | MD-1 | MD-2 | MD-3 | ||
| R g (Å) | 11.6 | 12.41 | 11.71 | 11.67 | 11.64 | 12.38 | 11.41 | 11.30 | 11.42 |
| Helicity (%) | 56 | — | 37.3 | 29.4 | 43.1 | — | 43.1 | 43.1 | 49.0 |
Kinetic control in INS fibril assembly by H2O and D2O
Many protein assemblies in vivo are diffusion-controlled reactions, where the transport rate of the protein (i.e., the diffusion coefficient of the protein) is critical for determining the reaction rate.61–63 As D2O is more viscous than H2O, the INS diffusion coefficient (DINS) is smaller in D2O than in H2O. Thus, we also considered the potential effects of the distinct viscosities of H2O and D2O on the fibrillation kinetics of INS (ESI Note V†). The fibrillation processes of INS (1 mg mL−1) in H2O and D2O were examined after the protein was fully denatured (i.e., monomeric highly disordered state, Fig. S17†) using sodium dodecyl sulfate (SDS 50 μM, 50 °C, 75 rpm) to confirm the effect of denaturation on protein fibrillation.Interestingly, we found that the fibrillation of INS was no longer significantly more accelerated in H2O than in D2O when the initial INS proteins were in a fully denatured state before incubation (Fig. S18†). This result indicates that the fibrillation kinetics of INS are dominantly affected by the unfolding transition of the INS monomer or dimer under our experimental conditions (H2O at pH = 2.1, D2O at pD = 2.1, 50 °C, 1–17 mg mL−1 INS) rather than by viscosity-dependent diffusion processes. In addition, time-resolved monitoring of INS fibrillation with FT-IR further supports our supposition that the initial denaturation process, which is independent of protein–protein collision, is the critical step during INS fibrillation (Fig. S19†).
Then, we measured the stability of INS before fibril formation using differential scanning calorimetry (DSC) (Fig. S20† and Table 3).64 The thermal transition temperature (T0), enthalpy (ΔHu), and entropy (ΔSu) upon INS unfolding in H2O were 59.55 °C, 42.53 kJ mol−1, and 128.3 J K−1, respectively. The T0 of INS in D2O (61.83 °C) was slightly higher than that of H2O, which implies that the protein was stabilized by enhanced hydrophobic interactions and dimer formation. The INS exhibited a slightly larger ΔHu (46.99 kJ mol−1) and ΔSu (140.8 J K−1) in D2O than in H2O, indicating the increased thermal stability and the propensity of INS dimer formation in heavy water. This result also confirms that the dissociation of the dimer to monomers followed by the formation of a partially unfolded INS intermediate in D2O requires a higher free energy cost than that in H2O. Based on the nucleation-growth mechanism, the formed nuclei interact with partially unfolded monomers to elongate and form prefibrils. This delayed formation of partially denatured monomeric intermediates further delays the elongation process of INS fibrillation in D2O, which is consistent with the 2D-IR observation that the slow fibrillation of INS in D2O results from the dissociation of INS dimers followed by the delayed denaturation of each INS monomer in D2O.
| T 0 (°C) | ΔHu (kJ mol−1) | ΔSu (J mol−1 K−1) | |
|---|---|---|---|
| INS-H2O | 59.55 | 42.53 | 128.3 |
| INS-D2O | 61.38 | 46.99 | 140.8 |
Conclusions
We investigated the effects of deuterated aqueous environments on the protein dynamics associated with amyloid fibrillation of INS. The fibrillation process occurs slowly in D2O compared with that in H2O. Using the advantage of 2D-IR, the amide I band of INS, which is a diagnostic signature of protein secondary structure, can be measured in H2O without spectral congestion and interference by water bending vibrations. The ability to suppress the water background signal using time-selective 2D-IR spectroscopy enabled investigations into different structures of INS that were hard to resolve by other spectroscopic measurements (e.g., ThT fluorescence, CD, and IR spectroscopy). Dimer formation of INS in D2O induces a delayed structural transition required for the subsequent protein nucleation process at an early stage of fibrillation (Fig. 5). The SAXS, MD, and DSC results provide additional insight into mechanistic details of the protein structural transitions involved in each fibrillation step. Unexpectedly, we found substantial kinetic isotope effects on the amyloid fibrillation of INS. This result highlights the importance of using H2O instead of D2O in various time-resolved IR spectroscopic studies of protein dynamics. We anticipate that the present work will shed light on the mechanism of amyloidosis under physiological conditions through the use of nonlinear IR microspectroscopy with improved spatial and temporal resolution.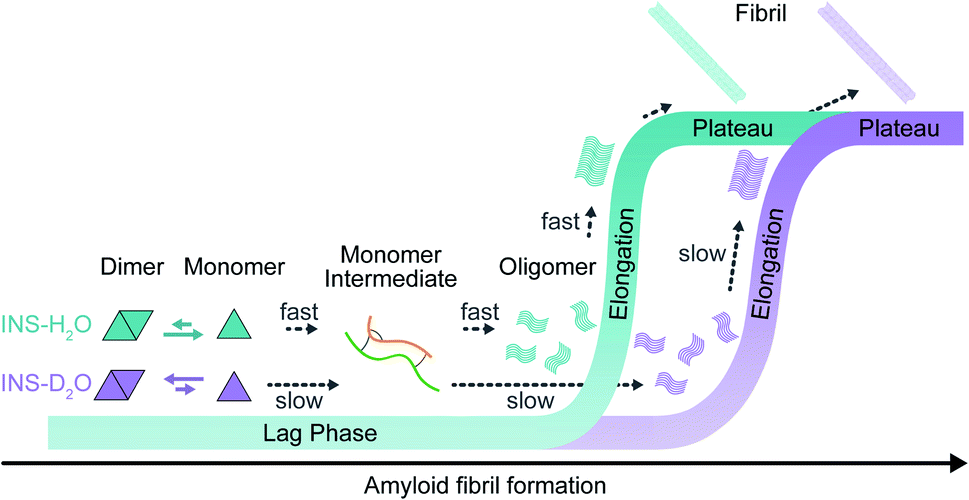 | ||
| Fig. 5 Fibrillation kinetics diagram of INS, based on kinetic and structural analysis. INS-H2O (blue) and INS-D2O (purple). | ||
Author contributions
MC, KK, and HIK designed the experiments; SYC, MKS, CRP, and CL carried out various experiments; SYC, MKS, HIK, and KK analyzed experimental data; SYC, MKS, HIK, KK, and MC wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a grant from the Institute for Basic Science (IBS-R023-D1) and by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2020R1A5A1019141). This research was also supported by the Korea Basic Science Institute (KBSI) National Research Facilities & Equipment Center (NFEC), funded by the Korean government (Ministry of Education) (2019R1A6C1010028). This work was also supported by Basic Research Program No. 2019R1H1A2079867 and No. 2019R1A2C2086193 through the National Research Foundation (NRF) of Korea, funded by the Ministry of Science, ICT, and Future Planning (MSIP). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2021R1A4A1032114). The synchrotron X-ray scattering measurements at the 4C SAXS II beamline of the Pohang Accelerator Laboratory were supported by the Ministry of Education and Science Technology. The authors thank the National Center for Seoul National University Research Facilities for their assistance with the TEM measurements.Notes and references
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- P. J. McLean, H. Kawamata and B. T. Hyman, Neuroscience, 2001, 104, 901–912 CrossRef CAS PubMed.
- E. E. Cawood, T. K. Karamanos, A. J. Wilson and S. E. Radford, Biophys. Chem., 2021, 268, 106505 CrossRef CAS PubMed.
- B. R. Sahoo, S. J. Cox and A. Ramamoorthy, Chem. Commun., 2020, 56, 4627–4639 RSC.
- M. I. Ivanova, Y. Lin, Y.-H. Lee, J. Zheng and A. Ramamoorthy, Biophys. Chem., 2021, 269, 106507 CrossRef CAS PubMed.
- S. A. Kotler, J. R. Brender, S. Vivekanandan, Y. Suzuki, K. Yamamoto, M. Monette, J. Krishnamoorthy, P. Walsh, M. Cauble, M. M. B. Holl, E. N. G. Marsh and A. Ramamoorthy, Sci. Rep., 2015, 5, 11811 CrossRef PubMed.
- G. J. Morgan, Biophys. Chem., 2022, 281, 106711 CrossRef CAS PubMed.
- D. Milardi, E. Gazit, S. E. Radford, Y. Xu, R. U. Gallardo, A. Caflisch, G. T. Westermark, P. Westermark, C. L. Rosa and A. Ramamoorthy, Chem. Rev., 2021, 121, 1845–1893 CrossRef CAS PubMed.
- A. Ahmad, V. N. Uversky, D. Hong and A. L. Fink, J. Biol. Chem., 2005, 280, 42669–42675 CrossRef CAS PubMed.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS PubMed.
- T. S. Choi, J. W. Lee, K. S. Jin and H. I. Kim, Biophys. J., 2014, 107, 1939–1949 CrossRef CAS PubMed.
- D. Bernson, A. Mecinovic, M. T. Abed, F. Lime, P. Jageland, M. Palmlof and E. K. Esbjorner, Eur. Biophys. J., 2020, 49, 145–153 CrossRef CAS PubMed.
- J. Chugh, S. Sharma and R. V. Hosur, Protein Sci., 2008, 17, 1319–1325 CrossRef CAS PubMed.
- I. R. Kleckner and M. P. Foster, Biochim. Biophys. Acta, 2011, 1814, 942–968 CrossRef CAS PubMed.
- R. B. Fenwick, H. van den Bedem, J. S. Fraser and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E445–E454 CrossRef CAS PubMed.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, 2011 Search PubMed.
- Z. Ganim, H. S. Chung, A. W. Smith, L. P. DeFlores, K. C. Jones and A. Tokmakoff, Acc. Chem. Res., 2008, 41, 432–441 CrossRef CAS PubMed.
- A. Berger, G. Ciardi, D. Sidler, P. Hamm and A. Shalit, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2458–2463 CrossRef CAS PubMed.
- B. R. Shrestha, S. Pillai, A. Santana, S. H. Donaldson Jr, T. A. Pascal and H. Mishra, J. Phys. Chem. Lett., 2019, 10, 5530–5535 CrossRef CAS PubMed.
- J. Guo, X.-Z. Li, J. Peng, E.-G. Wang and Y. Jiang, Prog. Surf. Sci., 2017, 92, 203–239 CrossRef CAS.
- Y. Marcus and A. Ben-Naim, J. Chem. Phys., 1985, 83, 4744–4759 CrossRef CAS.
- G. C. Kresheck, H. Schneider and H. A. Scheraga, J. Phys. Chem., 1965, 69, 3132–3144 CrossRef CAS PubMed.
- S. S. Stadmiller and G. J. Pielak, Protein Sci., 2018, 27, 1710–1716 CrossRef CAS PubMed.
- S.-Y. Sheu, E. W. Schlag, H. Selzle and D.-Y. Yang, J. Phys. Chem. A, 2008, 112, 797–802 CrossRef CAS PubMed.
- B. W. Chellgren and T. P. Creamer, J. Am. Chem. Soc., 2004, 126, 14734–14735 CrossRef CAS PubMed.
- M. Reslan and V. Kayser, Pharm. Dev. Technol., 2018, 23, 1030–1036 CrossRef CAS PubMed.
- L. Jean, C. F. Lee, P. Hodder, N. Hawkins and D. J. Vaux, Sci. Rep., 2016, 6, 32124 CrossRef CAS PubMed.
- J. Roche, Y. Shen, J. H. Lee, J. Ying and A. Bax, Biochemistry, 2016, 55, 762–775 CrossRef CAS PubMed.
- A. D. Stephens, J. Kölbel, R. Moons, M. T. Ruggerio, N. Mahmoudi, T. A. Shmool, T. M. McCoy, D. Nietlispach, A. F. Routh, F. Sobott, J. A. Zeitler and G. S. K. Schierle, bioRxiv, 2021 Search PubMed.
- D. Czurlok, J. Gleim, J. Lindner and P. Vöhringer, J. Phys. Chem. Lett., 2014, 5, 3373–3379 CrossRef CAS PubMed.
- J. Tan, J. Zhang, C. Li, Y. Luo and S. Ye, Nat. Commun., 2019, 10, 1–6 CrossRef PubMed.
- M. Cho, Two-Dimensional Optical Spectroscopy, CRC press, 2009 Search PubMed.
- N. Demirdöven, C. M. Cheatum, H. S. Chung, M. Khalil, J. Knoester and A. Tokmakoff, J. Am. Chem. Soc., 2004, 126, 7981–7990 CrossRef PubMed.
- L. P. DeFlores, Z. Ganim, R. A. Nicodemus and A. Tokmakoff, J. Am. Chem. Soc., 2009, 131, 3385–3391 CrossRef CAS PubMed.
- A. M. Woys, A. M. Almeida, L. Wang, C.-C. Chiu, M. McGovern, J. J. De Pablo, J. L. Skinner, S. H. Gellman and M. T. Zanni, J. Am. Chem. Soc., 2012, 134, 19118–19128 CrossRef CAS PubMed.
- Y. L. Ling, D. B. Strasfeld, S.-H. Shim, D. P. Raleigh and M. T. Zanni, J. Phys. Chem. B, 2009, 113, 2498–2505 CrossRef CAS PubMed.
- L. E. Buchanan, E. B. Dunkelberger, H. Q. Tran, P. N. Cheng, C. C. Chiu, P. Cao, D. P. Raleigh, J. J. De Pablo, J. S. Nowick and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19285–19290 CrossRef CAS PubMed.
- S. Hume, G. M. Greetham, P. M. Donaldson, M. Towrie, A. W. Parker, M. J. Baker and N. T. Hunt, Anal. Chem., 2020, 92, 3463–3469 CrossRef CAS PubMed.
- S. Hume, G. Hithell, G. M. Greetham, P. M. Donaldson, M. Towrie, A. W. Parker, M. J. Baker and N. T. Hunt, Chem. Sci., 2019, 10, 6448–6456 RSC.
- C. T. Middleton, L. E. Buchanan, E. B. Dunkelberger and M. T. Zanni, J. Phys. Chem. Lett., 2011, 2, 2357–2361 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- L. N. Garriques, S. Frokjaer, J. F. Carpenter and J. Brange, J. Pharm. Sci., 2002, 91, 2473–2480 CrossRef CAS PubMed.
- M. Bouchard, J. Zurdo, E. J. Nettleton, C. M. Dobson and C. V. Robinson, Protein Sci., 2000, 9, 1960–1967 CrossRef CAS PubMed.
- F. Piccirilli, S. Mangialardo, P. Postorino, L. Baldassarre, S. Lupi and A. Perucchi, Soft Matter, 2012, 8, 11863–11870 RSC.
- A. A. Laogun, R. J. Sheppard and E. H. Grant, Phys. Med. Biol., 1984, 29, 519–524 CrossRef CAS PubMed.
- S. Y. Venyaminov and F. G. Prendergast, Anal. Biochem., 1997, 248, 234–245 CrossRef CAS PubMed.
- N. T. Hunt, Chem. Soc. Rev., 2009, 38, 1837–1848 RSC.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- N. Huse, S. Ashihara, E. T. J. Nibbering and T. Elsaesser, Chem. Phys. Lett., 2005, 404, 389–393 CrossRef CAS.
- A. W. Smith and A. Tokmakoff, J. Chem. Phys., 2007, 126, 045109 CrossRef PubMed.
- Z. Ganim, K. C. Jones and A. Tokmakoff, Phys. Chem. Chem. Phys., 2010, 12, 3579–3588 RSC.
- W. G. Turnell and J. T. Finch, J. Mol. Biol., 1992, 227, 1205–1223 CrossRef CAS PubMed.
- C. F. Hjorth, M. Norrman, P.-O. Wahlund, A. J. Benie, B. O. Petersen, C. M. Jessen, T. Å. Pedersen, K. Vestergaard, D. B. Steensgaard, J. S. Pedersen, H. Naver, F. Hubálek, C. Poulsen and D. Otzen, J. Pharm. Sci., 2016, 105, 1376–1386 CrossRef CAS PubMed.
- S. Hahn, S.-S. Kim, C. Lee and M. Cho, J. Chem. Phys., 2005, 123, 084905 CrossRef PubMed.
- S. J. Roeters, A. Iyer, G. Pletikapić, V. Kogan, V. Subramaniam and S. Woutersen, Sci. Rep., 2017, 7, 41051 CrossRef CAS PubMed.
- V. N. Uversky, L. N. Garriques, I. S. Millett, S. Frokjaer, J. Brange, S. Doniach and A. L. Fink, J. Pharm. Sci., 2003, 92, 847–858 CrossRef CAS PubMed.
- Y. Pocker and S. B. Biswas, Biochemistry, 1981, 20, 4354–4361 CrossRef CAS PubMed.
- C. D. Putnam, M. Hammel, G. L. Hura and J. A. Tainer, Q. Rev. Biophys., 2007, 40, 191–285 CrossRef CAS PubMed.
- D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS.
- M. Oliveberg and P. G. Wolynes, Q. Rev. Biophys., 2005, 38, 245–288 CrossRef CAS PubMed.
- M. Schlosshauer and D. Baker, Protein Sci., 2004, 13, 1660–1669 CrossRef CAS PubMed.
- P. E. Schavemaker, A. J. Boersma and B. Poolman, Front. Mol. Biosci., 2018, 5, 93 CrossRef CAS PubMed.
- L. Liu, C. Yang and Q.-X. Guo, Biophys. Chem., 2000, 84, 239–251 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental methods and supplementary results. See DOI: 10.1039/d1sc06047c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |