
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNP NMR spectroscopy enabled direct characterization of polystyrene-supported catalyst species for synthesis of glycidyl esters by transesterification†
Shinji
Tanaka
 *ab,
Yumiko
Nakajima
a,
Atsuko
Ogawa
a,
Takashi
Kuragano
a,
Yoshihiro
Kon
a,
Masanori
Tamura
a,
Kazuhiko
Sato
a and
Christophe
Copéret
*b
*ab,
Yumiko
Nakajima
a,
Atsuko
Ogawa
a,
Takashi
Kuragano
a,
Yoshihiro
Kon
a,
Masanori
Tamura
a,
Kazuhiko
Sato
a and
Christophe
Copéret
*b
aInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology, Tsukuba, 305-8565, Japan. E-mail: shinji-tanaka@aist.go.jp
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1-5, Zürich, 8093, Switzerland. E-mail: ccoperet@ethz.ch
First published on 15th March 2022
Abstract
Polymer-supported catalysts have been of great interest in organic syntheses, but have suffered from the difficulty in obtaining direct structural information regarding the catalyst species embedded in the polymer due to the limitations of most analytical methods. Here, we show that dynamic nuclear polarization (DNP)-enhanced solid-state NMR is ideally positioned to characterize the ubiquitous cross-linked polystyrene (PS)-supported catalysts, thus enabling molecular-level understanding and rational development. Ammonium-based catalysts, which show excellent catalytic activity and reusability for the transesterification of methyl esters with glycidol, giving glycidyl esters in high yields, were successfully characterized by DNP 15N NMR spectroscopy at 15N natural abundance. DNP 15N NMR shows in particular that the decomposition of quaternary alkylammonium moieties to tertiary amines was completely suppressed during the catalytic reaction. Furthermore, the dilute ring-opened product derived from glycidol and NO3− was directly characterized by DNP 15N CPMAS and 1H–15N and 1H–13C HETCOR NMR using a 15N enriched (NO3) sample, supporting the view that the transesterification mechanism involves an alkoxide anion derived from an epoxide and NO3−. In addition, the detailed analysis of a used catalyst indicated that the adsorption of products on the cationic center is the major deactivation step in this catalysis.
Introduction
Immobilization of molecular catalysts on solid materials has been a strategy to intensify catalytic processes while keeping the advantages of homogeneous catalysts that are typically highly selective and more amiable to rational development.1–4 In this context, quaternary alkyl ammonium salts are one of the most well-investigated molecular organocatalysts for base-mediated organic transformations, where a counter anion together with its ammonium cation behaves as a key reactive centre (e.g. phase transfer catalysis,5 transesterification,6–8 carbonate synthesis from CO2 and epoxides,9–11etc.). Their immobilization has been investigated on cross-linked polystyrene (PS) because this support offers a high tolerance under basic conditions.12–17 However, the development of PS-supported alkylammonium salt catalysts has suffered from the lack of structural characterization of the catalytic sites and how they evolve under reaction conditions.12 For example, molecular quaternary alkylammonium salts are known to easily decompose into tertiary amines under basic conditions by Hofmann elimination and/or SN2 substitution,7,8 leading to significant suppression of the catalytic activity. Therefore, in order to develop highly efficient PS-supported quaternary alkylammonium salt catalysts, it is of great importance to directly trace subtle structural changes of both the cation and anion species under a PS-supported environment, although it is still a significant challenge owing to the difficulties in structural identification.In this context, solid-state NMR has emerged as a powerful tool to characterize the local structure of solid-supported catalysts in a non-destructive manner.18 However, its application range is often limited owing to the intrinsic low signal sensitivity of NMR. Recently, solid-state NMR enhanced by dynamic nuclear polarization (DNP) has attracted considerable attention because of its boosted signal sensitivity.19–23 In DNP NMR, the microwave-driven polarization transfer via a cross-effect with a biradical combined with further polarization transfer methods (e.g. cross polarization) enables the enhancement of the intensity of signals from less sensitive nuclei (e.g.13C, 15N, and 17O). With this benefit, DNP NMR is nowadays recognized as a powerful tool for the characterization of heterogeneous catalysts and has mostly been explored for catalysts dispersed on high surface area oxide supports.22,23 However, its application to organic polymer-supported materials is less explored, probably due to the lack of a versatile sample preparation method.24–30 In this context, we have recently reported a rational guideline for DNP sample preparation from cross-linked PS, a prototypical insoluble synthetic polymer.31 An appropriate choice of a DNP polarizing agent (PA) solution32,33 can be predicted by the swelling properties of each polymer, thus facilitating the uniform distribution of the PA over the polymer network to achieve natural abundance (0.37%) 15N NMR of quaternary alkylammonium salts in reasonable measurement time.
In this contribution, we have explored transesterification with glycidol (GD), giving glycidyl esters that are useful precursors of the epoxy resin, using solid organocatalysts based on PS-supported quaternary alkyl ammonium salts. Compared with the conventional epichlorohydrin process, this catalytic reaction is promising owing to the availability of biomass-derived feedstock (i.e. glycidol), thus contributing to the development of sustainable processes.35 Taking advantage of our recently developed DNP NMR protocol, we have performed detailed structural analysis of PS-supported quaternary alkyl ammonium salts at various stages of the catalytic process. In particular, the robustness of the quaternary alkyl ammonium fragment was clearly confirmed by DNP 15N NMR, consistent with the excellent activity and reusability for catalytic transesterification of methyl esters to glycidyl esters.36,37 Furthermore, we have also tracked the evolution of the active species embedded in the PS network, thanks to DNP-enhanced 1D and 2D NMR spectroscopy.
Results and discussion
Considering that the substituents of the ammonium fragment play an important role in transesterification with molecular catalysts,6,8 we first optimized the N-substituents of ammonium salts for transesterification of methyl esters with GD. Thus, 1-Cl, 2-Cl, 3-Cl, and 4-Cl were prepared by reactions of Merrifield resin with four types of alkyl amines, NMe3, NMe2nOct, NMenOct2, and NnOct3, respectively (Scheme 1). The amount of N-atoms in 1-Cl–4-Cl was quantified by combustion analysis (see the ESI†). Their FT-IR spectra exhibited strong peaks assignable to C–H groups at 2800–3000 cm−1, indicating the presence of alkyl groups (Fig. S1†). The conventional 13C CPMAS NMR spectra of the catalysts displayed signals at 15–35 ppm, which were assigned to n-octyl groups (Fig. S2†). The signals due to carbon atoms attached to a nitrogen atom were observed as broad signals at 50–70 ppm. Based on these observations, it was confirmed that nitrogen-based functional groups were successfully introduced into PS.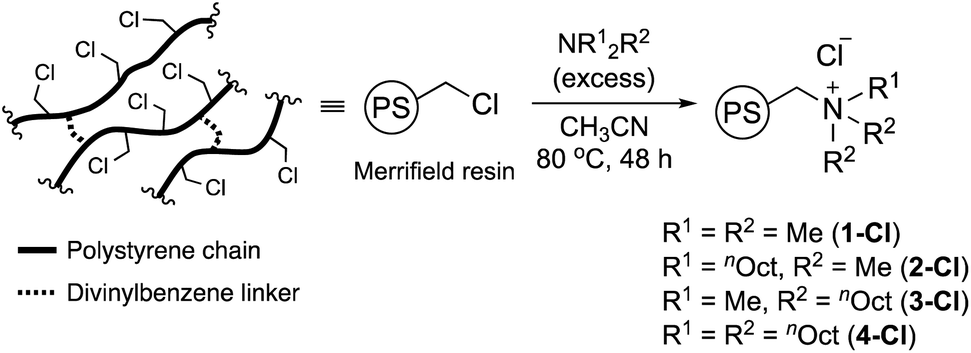 | ||
| Scheme 1 Preparation of PS-supported alkylammonium catalysts 1-Cl, 2-Cl, 3-Cl, and 4-Cl. | ||
Full identification of the structure around a nitrogen atom was achieved by using 15N DNP NMR signals using our NMR protocol developed for PS samples.31 Thus, the swelling test of PS-supported catalysts (1-Cl–4-Cl) with 1,1,2,2-tetrachloroethane (TCE) and dimethylsulfoxide (DMSO) was first conducted to predict the optimal PA solution (TEKPol/TCE or AMUPol/DMSO-d6) for efficient DNP signal enhancement of the immobilized catalysts. 2-Cl, 3-Cl, and 4-Cl were well swollen in both solvents, while DMSO was needed for 1-Cl; hence, AMUPol/DMSO-d6 was selected as the optimal PA solution (see the ESI, Table S1†). All measurements resulted in good sensitivity enhancement (εH = 35–56, Fig. S3(a)–(c)†); we could successfully identify 15N signals at natural abundance for each sample after a reasonable measurement time (3–6 hours) (see the ESI†). The DNP build-up curve indicated a homogeneous distribution of PAs giving TDNP values of 1.6 s (2-Cl), 1.4 s (3-Cl), and 1.6 s (3-NO3) (Fig. S4†). Fig. 1 shows the selected 15N CPMAS NMR spectra measured under DNP conditions at 102 K. 2-Cl exhibited a 15N signal assignable to a quaternary nitrogen atom at 57.5 ppm, while 3-Cl displayed a signal at 62.4 ppm along with a shoulder as the result of the presence of different conformers due to the sterically large alkyl substituents.31,34 These observations are consistent with our previous DNP 15N CPMAS NMR studies on 1-Cl (52.4 ppm) and 4-Cl (61.8 and 59.6 ppm).31 Overall, this clearly indicated that the quaternary alkylammonium moiety was introduced into the PS network.
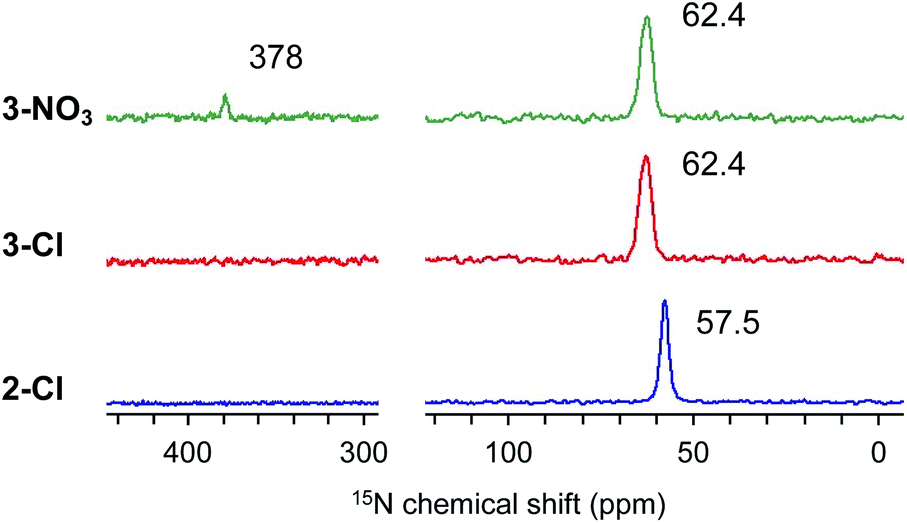 | ||
| Fig. 1 DNP 15N CPMAS NMR spectra of 2-Cl, 3-Cl, and 3-NO3. AMUPol/DMSO-d6 (16 mM) was used for all measurements. MAS frequency: 10 kHz (3.2 mm rotor), CP contact time: 3 ms, recycle delay: 2.2 s (2-Cl), 2.0 s (3-Cl) and 2.1 s (3-NO3), and number of scans (for 15N): 7900 (2-Cl), 3300 (3-Cl), and 4096 (3-NO3). | ||
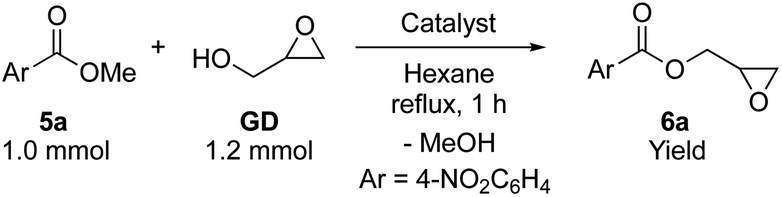
We then investigated the PS-supported ammonium salt catalysts (1-Cl–4-Cl) in transesterification of methyl 4-nitrobenzoate (5a) with a slight excess of GD under azeotropic reflux conditions in hexane (Table 1). Although the catalytic activity of 1-Cl was moderate (entry 2, 76% yield), 2-Cl, 3-Cl and 4-Cl showed higher catalytic activity affording glycidyl 4-nitrobenzoate (6a) in 92–96% yields (entries 3–5). Of all the immobilized catalysts, 3-Cl performed the best (entry 4, 96%). The higher activity of alkylammoniums with long-chain alkyl groups (2-Cl, 3-Cl and 4-Cl) compared to 1-Cl parallels what is observed in homogeneous catalysis.6 The catalytic activity of 4-Cl was slightly lower than that of 2-Cl and 3-Cl, owing to the absence of the smaller Me group(s).8 Yet, it should be noted that 1-Cl showed lower activity in spite of the presence of three Me groups bound to nitrogen. This is probably due to the higher hydrophilicity of the polymer network in 1-Cl, as indicated by the swelling test, which leads to the limited distribution of organic substrates.123-Cl showed high activity even with a reduced loading of the catalyst (0.5 mol%), affording the isolated product 6a in 87% yield (entry 6).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Yield (%)b |
| a Reaction conditions: a mixture of MNB (1.0 mmol), GD (1.2 mmol), and catalyst (5 mol%) in hexane (0.5 mL) was refluxed (heater temp: 80 °C) for 1 hour. b Yields were determined by 1H NMR analysis. Values are an average of 2 runs. c Yield of the isolated product. | |||
| 1 | None | — | 3 |
| 2 | 1-Cl | 5 | 76 |
| 3 | 2-Cl | 5 | 94 |
| 4 | 3-Cl | 5 | 96 |
| 5 | 4-Cl | 5 | 92 |
| 6 | 3-Cl | 0.5 | 90 (87)c |
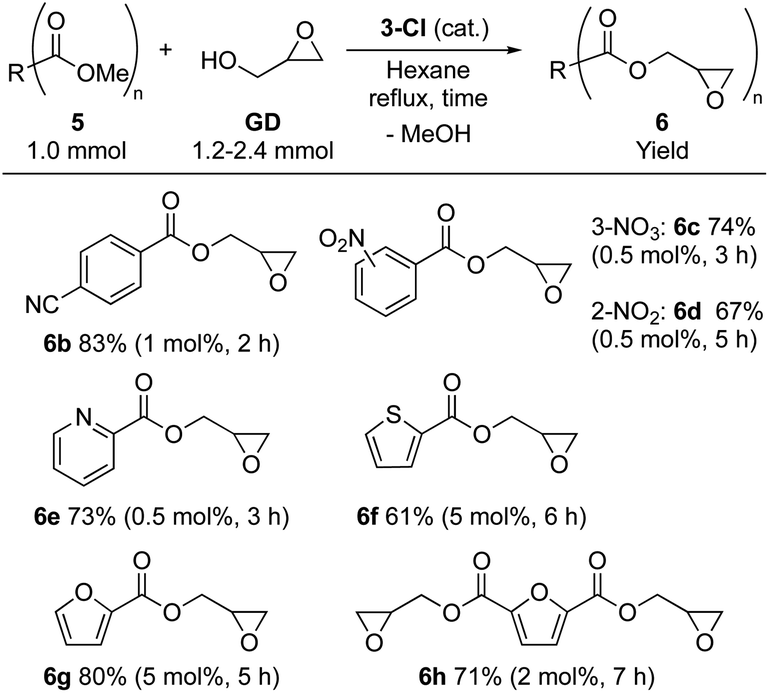
The substrate scope was investigated using 3-Cl (Table 2). Glycidyl 4-cyanobenzoate (6b) was isolated in high yield (83%). For benzoate derivatives with a substituent at other positions (2- and 3-), glycidyl 2-nitrobenzoate (6c) and glycidyl 3-nitrobenzoate (6d) were obtained in moderate yields (6c: 74%, 6d: 67%) with extended reaction time. The catalyst worked also for substrates having a heteroaromatic ring, affording glycidyl 2-pyridinecarboxylic acid (6e) and glycidyl 2-thiophenecarboxylic acid (6f) in moderate yields (6e: 73%, 6f: 61%). Moreover, methyl 2-furancarboxylate (5g) and dimethyl 2,5-furancarboxylate (5h), which are useful biomass-derived substrates,38–42 are also accessible through this approach: glycidyl esters 6g and 6h were isolated in good to moderate yields (6g: 80%, 6h: 71%). Overall, a wide range of value-added glycidyl esters is accessible using 3-Cl.
| a Reaction conditions were optimized for each substrate. Yields of the isolated product are shown. |
|---|
|
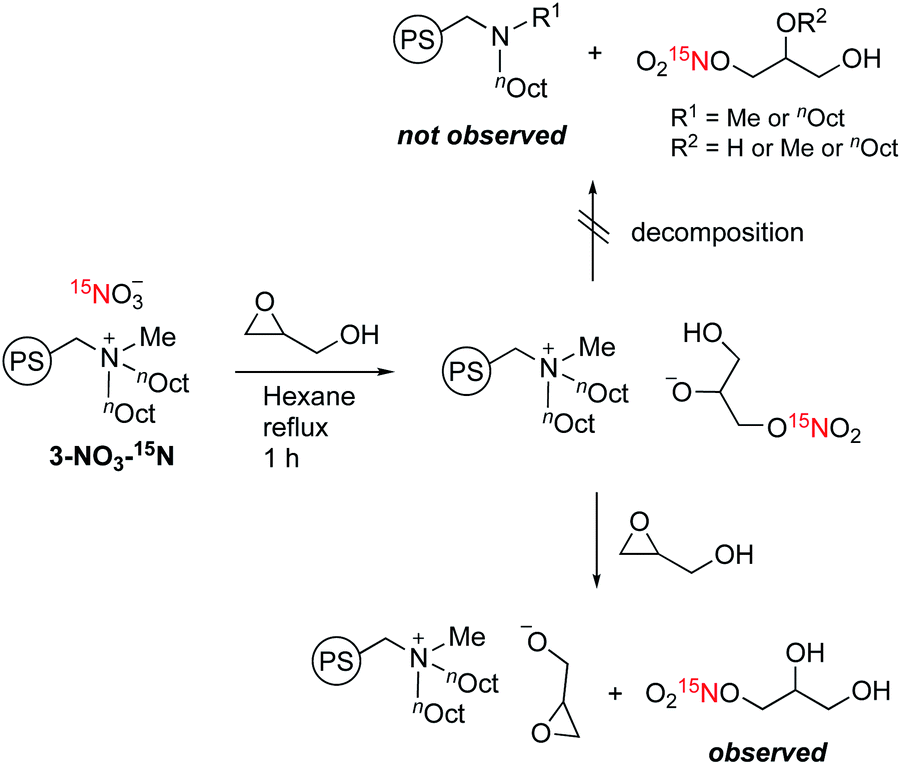
Next, we performed the anion exchange reaction of 3-Cl. Our previous work revealed that weakly basic anions facilitated transesterification while suppressing self-polymerization of GD, which might be a major side-reaction.43,44 Taking this result into account, we prepared 3-NO3 from 3-Cl by a sequential anion exchange via successive treatment with NaOH and HNO3 (Scheme 2(a)). Since the procedure was conducted by treatment with NaOH as a strong base, one should check whether the decomposition of alkyl ammoniums possibly proceeds during the anion exchange via SN2 substitution (paths A and B1) and/or Hofmann elimination (path B2) which results in the formation of tertiary amine moieties (Scheme 2(b)). To confirm this point, we performed DNP 15N NMR analysis to monitor 15N signals before and after the anion exchange reaction. As clearly shown from the DNP 15N NMR spectrum of 3-NO3, no additional signals except for NO3− at 378 ppm were observed after the anion exchange (Fig. 1). Furthermore, PS-supported tertiary amines 7 and 8 were alternatively synthesized by the reaction of Merrifield resin with secondary amines (see the ESI†). DNP 15N NMR spectra of 7 and 8 exhibited signals at 25–60 ppm, which appeared as broad signals presumably due to the existence of distinct conformers at cryogenic temperature due to the bulky tertiary amines45 (see the ESI, Fig. S5†). As a result, we confirmed that the quaternary ammonium fragment was maintained after the anion exchange procedure.
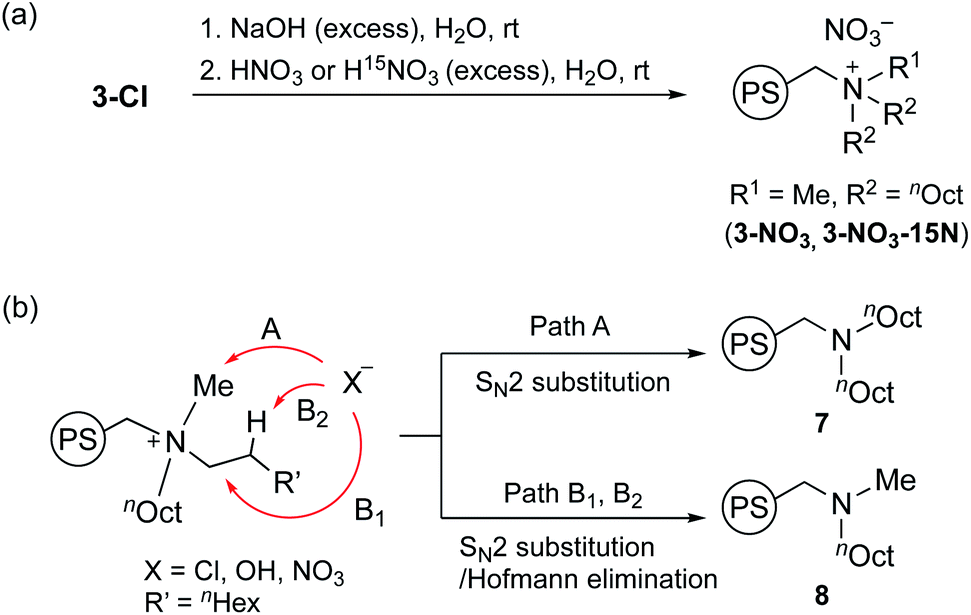 | ||
| Scheme 2 Anion exchange reaction of 3-Cl to 3-NO3 and 3-NO3-15N (a), and possible decomposition pathways of quaternary alkylammonium moieties to tertiary amines 7 and 8 (b). | ||
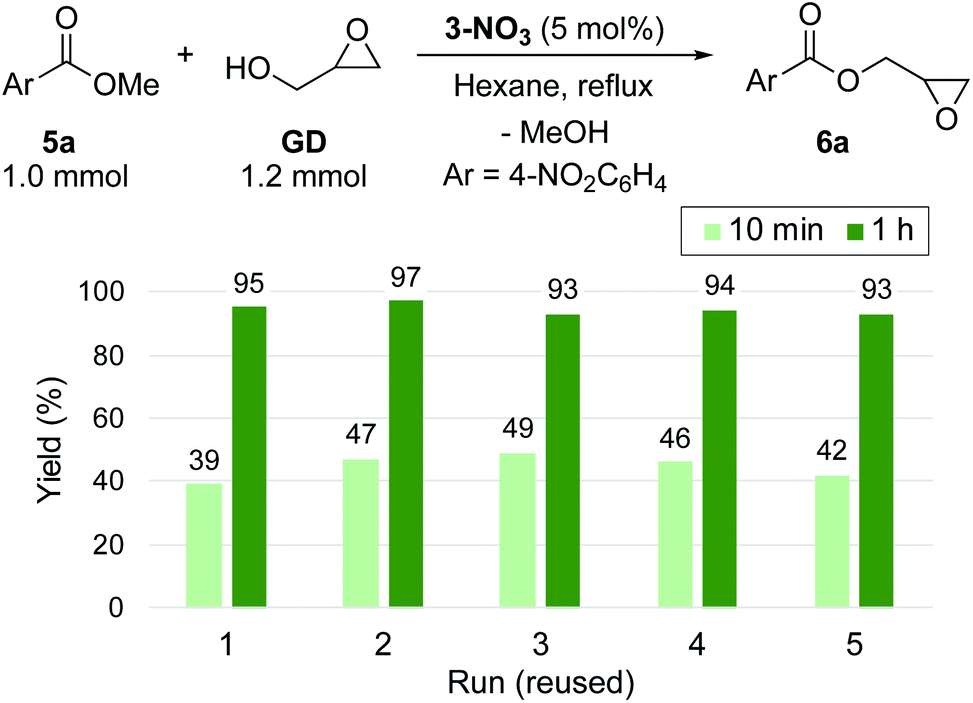
3-NO3 exhibited comparable high catalytic activity with 3-Cl towards transesterification (Scheme 3), enabling the efficient synthesis of glycidyl esters in a chlorine-free manner, which is crucial for the production of high-grade epoxy resins utilized for insulating materials.36 With this catalyst in hand, we further performed a reuse test for catalytic transesterification. Reusability is one of the key advantages of supported catalysts, generally relying on the stability of the catalyst under the reaction conditions. In this system, as described above, the decomposition of quaternary ammonium moieties into tertiary amines is a major pathway for catalyst deactivation (Scheme 2(b)). To reveal the catalytic behaviour of 3-NO3, we performed the reuse test with 5 mol% loading with a short reaction time (10 min). Under these conditions, 3-NO3 catalysed the transesterification of 5a with GD to give 6a in 39% yield in the first run. Interestingly, the activity was slightly improved in the second run (47%), and only slightly decreased after further reuse (5th: 42%) (Scheme 3). This result indicates that only a small structural transformation happened at the catalytically active site in PS. We note that the yield of 6a was excellent even in the fifth run under conditions with an extended reaction time (1 h), confirming the significant utility of this catalyst for the synthesis of glycidyl esters (Scheme 3).
 | ||
| Scheme 3 Reuse test of 3-NO3 toward the synthesis of 2a. | ||
The peculiar catalytic behaviour in the recycling study prompted us to gain insights into the active species on PS in a direct manner using DNP NMR. Our previous experimental and computational studies have implied that the ring-opening of glycidol affords the active anion that initiates transesterification by abstracting a proton from the substrate alcohol.6 It is also reported that the ring-opened species from epoxides and weak anions behave as an intermediate for the catalytic cycle of cyclic carbonate and polycarbonate synthesis from epoxides and CO2.9–11 Because a high concentration of ring-opened species leads to the self-oligomerization of the epoxide, a dilute condition is necessary for achieving high yields of the product, yet such dilute species are not observable by conventional analytical methods. For an in-depth study of such dilute ring-opened species by DNP NMR spectroscopy,46 we prepared 3-NO3-15N using 15N-enriched HNO3. Moreover, the sample 3-NO3-15N-GD was synthesized by the reaction of 3-NO3-15N with 2 equiv. of GD in hexane (see the ESI†). The DNP 15N CPMAS NMR spectrum of 3-NO3-15N-GD with AMUPol/DMSO-d6 displayed an additional signal at 342 ppm along with the original signals of 3-NO3-15N (62.4 and 378 ppm) (Fig. 2). To identify the product that exhibits the signal at 342 ppm, we prepared 1-nitro-2,3-propanediol as a possible ring-opened compound derived from GD and NO3−.47 As expected, the DNP 15N CPMAS NMR spectrum of 1-nitro-2,3-propanediol under the same conditions (AMUPol/DMSO-d6) also showed a 15N signal at 342 ppm (Fig. 2). Thus, it was possible to prove that the ring-opened product 1-nitro-2,3-propanediol was formed during catalysis. For further characterization of ring-opened species, we performed DNP-enhanced 1H–15N and 1H–13C HETCOR NMR of 3-NO3-15N-GD. The 1H–15N HETCOR spectrum showed a correlation peak between the 15N signal at 342 ppm and the 1H signal at 4.1 ppm (Fig. 3(a)). In addition, the 13C signal at 73 ppm, which newly appeared after conversion of glycidol to 3-NO3-15N, was also correlated with the 1H signal at 4.1 ppm in the 1H–13C HETCOR spectrum (Fig. 3(b)). These observations strongly supported the presence of a ring-opened product bearing NO3 at the terminal position (δC ∼76 ppm) of GD rather than at an internal position (δC ∼86 ppm).47 This result is consistent with the general regio-selectivity of ring-opening of epoxides under basic conditions. Because the signal of the ring-opened product was not significantly correlated with either alkyl (1.2 ppm for 1H) or aromatic groups (7.0 ppm for 1H) under these conditions (CP contact time: 1 ms (15N), 0.2 ms (13C)), the ring-opened product likely exists in a protonated form,48 which is free from a cationic moiety in PS. Because decomposition of the quaternary ammonium fragment to a tertiary amine was not observed (Fig. 2), another GD molecule presumably worked as a proton source, generating a deprotonated GD (Scheme 4). As transesterification proceeds via an attack of the deprotonated GD on the carbonyl moiety of the ester, the presented results clearly support the mechanism that involves in situ generated active anion species. Overall, the observations by DNP NMR successfully evidenced the formation of ring-opened species derived from epoxides in an initiation step of the catalytic transesterification. The consumption of GD for the generation of active species likely explains the slightly lower product yield in the first run in the catalyst reuse test (Scheme 3).
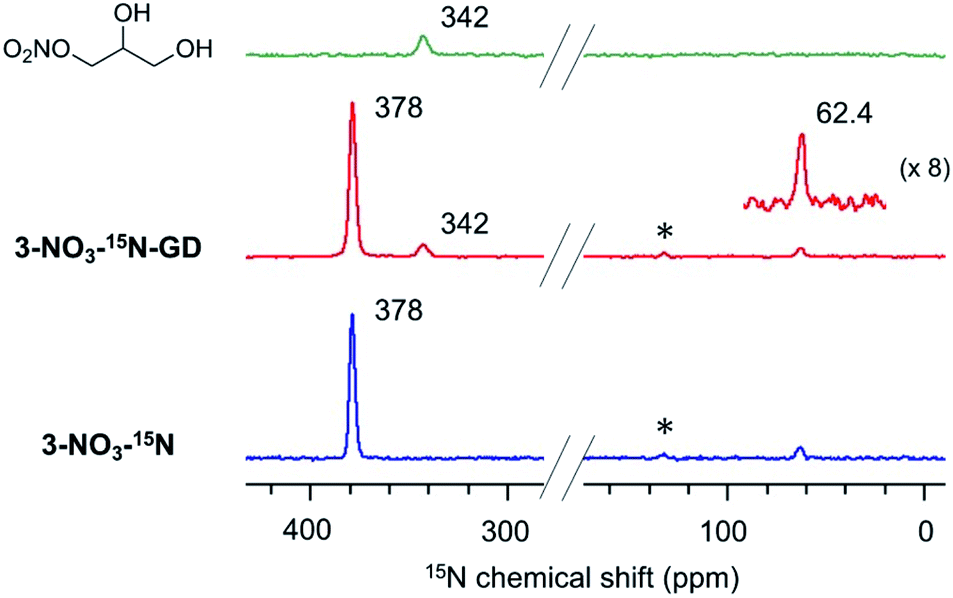 | ||
| Fig. 2 DNP 15N CPMAS NMR spectra of 1-nitro-2,3-propanediol (top), 3-NO3-15N-GD (middle) and 3-NO3-15N (bottom). AMUPol/DMSO-d6 (16 mM) was used as a PA. MAS frequency: 10 kHz (3.2 mm rotor), CP contact time: 3 ms (1-nitro-2,3-propanediol) and 8 ms (3-NO3-15N-GD, 3-NO3-15N), recycle delay: 3.3 s (1-nitro-2,3-propanediol), 2.1 s (3-NO3-15N-GD), and 2.5 s (3-NO3-15N), and number of scans: 128 (a), 512 (b), 2048 (3-NO3-15N-GD), and 1024 (3-NO3-15N). Peaks with an asterisk are spinning side-band signals. | ||
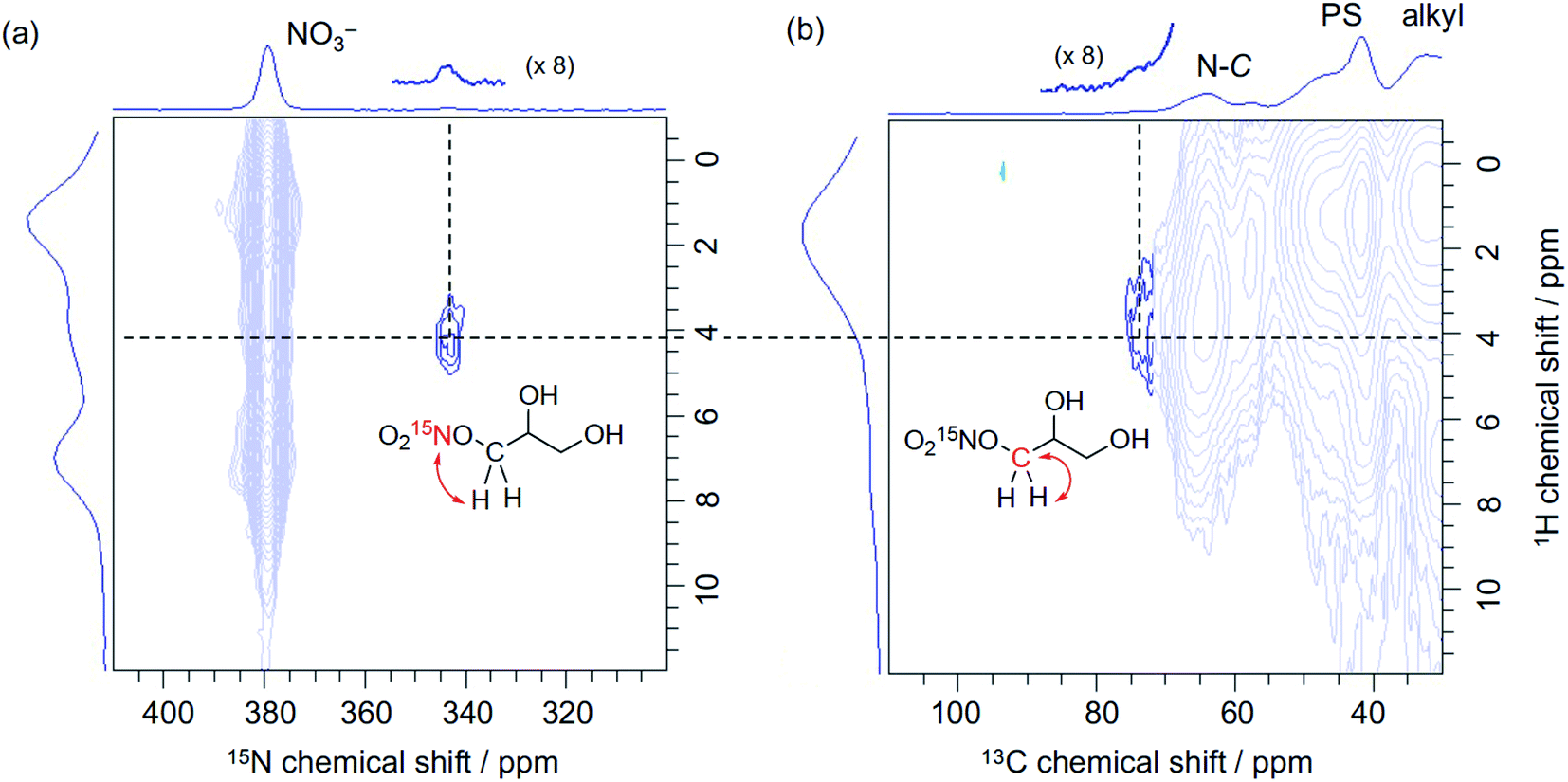 | ||
| Fig. 3 DNP 1H–15N HETCOR (a) and DNP 1H–13C HETCOR spectra (b) of 3-NO3-15N-GD. AMUPol/DMSO-d6 (16 mM) was used as a PA. FSLG (frequency-switched Lee-Goldburg) homonuclear decoupling was employed during the evolution of the 1H chemical shift. A decoupling field of 100 kHz was used. MAS frequency: 10 kHz (3.2 mm rotor for 15N and 1.9 mm rotor for 13C), CP contact time: 1 ms (for 15N) and 0.2 ms (for 13C), recycle delay: 2 s (for 15N) and 2.5 s (for 13C) and number of scans: 32 (for 15N) and 64 (for 13C). | ||
 | ||
| Scheme 4 Plausible mechanism of generation of ring-opened species from an epoxide and an NO3 anion and the reaction with proton sources. | ||
Next, our aim was to understand the deactivation step. To shed light on the deactivated species for this PS-immobilized material, we analysed the catalyst moieties embedded in PS after the catalytic reaction. The DNP 15N CPMAS NMR spectrum of 3-NO3 following five catalytic runs indicated that the signal of the quaternary ammonium fragment at 62.4 ppm remained unchanged, while no signal associated with tertiary amines was present. This confirms that the quaternary ammonium fragment remains intact even after five runs (Fig. 4(a)). Interestingly, a signal centered at 370 ppm was newly observed and was assigned to the nitro group (NO2). The DNP 13C CPMAS NMR spectrum showed signal(s) from the ester group at 158 ppm and broad shoulder signals due to a ring-opened epoxide moiety at 60–75 ppm (Fig. 4(b)). Consequently, it is likely that ring-opened species derived from 6a or its oligomerized forms are adsorbed during catalysis (Scheme 5). In other words, the major deactivation step of this catalysis is not the decomposition of the quaternary alkyl ammonium moiety itself, but is likely associated with the adsorption of products on a cationic moiety, inhibiting efficient catalyst turnover. This observation explains the slight loss of catalytic activity after several runs (Scheme 3). Understanding of the unusual deactivation mechanism of the alkylammonium salt catalyst, which was enabled with the help of DNP NMR spectroscopy, is certainly helpful toward the rational improvement of the catalyst.
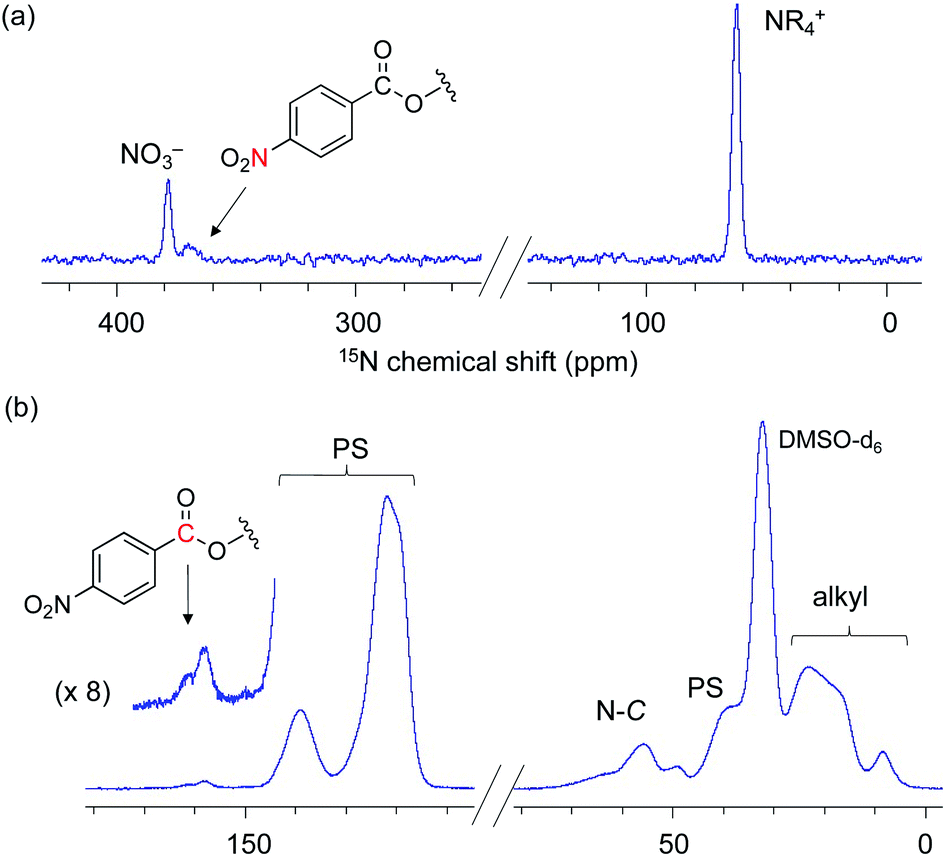 | ||
| Fig. 4 DNP 15N CPMAS NMR (a) and DNP 13C CPMAS NMR spectra (b) of 3-NO3 after five reuse cycles for the catalytic reaction. AMUPol/DMSO-d6 (16 mM) was used as a PA. MAS frequency: 10 kHz (3.2 mm rotor), CP contact time: 3 ms, recycle delay: 4.4 s, and number of scans: 6000 (a) and 512 (b). | ||
 | ||
| Scheme 5 Formation of ring-opened species derived from the product. | ||
Conclusions
In conclusion, we developed a well-defined PS-supported alkylammonium salt catalyst, which showed excellent catalytic activity and reusability for transesterification of methyl esters with GD, giving glycidyl esters in high yields. We also demonstrated that DNP NMR is a reliable analytical method to characterize and monitor the stability of PS-supported alkylammonium salt catalysts without the need for labelling strategies. The stability of the alkylammonium moiety, which is known to decompose into the inactive tertiary amine, was successfully monitored by DNP 15N NMR: the absence of formation of tertiary amines in these supported catalysts under the mild basic conditions used in this study is consistent with their very good stability and reusability. DNP NMR also helped us to obtain a mechanistic insight into the generation of reaction intermediates and active species. Furthermore, a ring-opened product derived from GD and NO3− was directly characterized by DNP 15N CPMAS and 1H–15N and 1H–13C HETCOR NMR using a 15N enriched (NO3) sample. The direct characterization of the used catalyst allowed us to gain a new insight into the deactivation mechanism, which is helpful for the rational improvement of catalysts. This study showcases how DNP NMR spectroscopy can provide unique molecular-level information regarding the active sites of polymer-supported catalysts. Further application of DNP NMR to various polymer-supported catalysts as well as high-performance polymer materials is ongoing in our laboratory.Data availability
All available data are included in ESI.†Author contributions
S. T.: conceptualization, investigation (NMR measurements), and writing – original draft. Y. N.: writing – review & editing. A. O. and T. K.: investigation (synthesis and catalysis). Y. K. and M. T.: supervision. K. S.: project administration and supervision. C. C.: conceptualization and writing – review & editing. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the New Energy and Industrial Technology Development Organization of Japan (NEDO) (Grant JPNP16010). Dr S. Tanaka acknowledges financial support by a Grant-in-Aid for Early-Career Scientists (JSPS KAKENHI Grant Number JP20K15329). Prof. C. Copéret is thankful for the JSPS program that allowed many discussions regarding the presented research. We acknowledge Mr Takuya Nakashima for preliminary studies on PS-supported catalysts.Notes and references
- M. R. Buchmeiser, Polymer-Supported Well-Defined Metathesis Catalysts, Chem. Rev., 2009, 109, 303–321 CrossRef CAS PubMed.
- M. P. Conley, C. Copéret and C. Thieuleux, Mesostructured Hybrid Organic–Silica Materials: Ideal Supports for Well-Defined Heterogeneous Organometallic Catalysts, ACS Catal., 2014, 4, 1458–1469 CrossRef CAS.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Immobilization of N-Heterocyclic Carbene Compounds: A Synthetic Perspective, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed.
- W. Wang, L. Cui, P. Sun, L. Shi, C. Yue and F. Li, Reusable N-Heterocyclic Carbene Complex Catalysts and Beyond: A Perspective on Recycling Strategies, Chem. Rev., 2018, 118, 9843–9929 CrossRef CAS PubMed.
- Asymmetric Phase Transfer Catalysis, ed. K. Maruoka, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Tanaka, T. Nakashima, T. Maeda, M. Ratanasak, J. Hasegawa, Y. Kon, M. Tamura and K. Sato, Quaternary Alkyl Ammonium Salt-Catalyzed Transformation of Glycidol to Glycidyl Esters by Transesterification of Methyl Esters, ACS Catal., 2018, 8, 1097–1103 CrossRef CAS.
- S. Tanaka, T. Nakashima, N. Satou, H. Oono, Y. Kon, M. Tamura and K. Sato, Epoxide as precatalyst for metal-free catalytic transesterification, Tetrahedron Lett., 2019, 60, 2009–2013 CrossRef CAS.
- M. Hatano, Y. Tabata, Y. Yoshida, K. Toh, K. Yamashita, Y. Ogura and K. Ishihara, Metal-free transesterification catalyzed by tetramethylammonium methyl carbonate, Green Chem., 2018, 20, 1193–1198 RSC.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Cyclic Carbonate Formation from Carbon Dioxide and Oxiranes in Tetrabutylammonium Halides as Solvents and Catalysts, Org. Lett., 2002, 4, 2561–2563 CrossRef PubMed.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, CO2 Cycloaddition Reactions Catalyzed by an Ionic Liquid Grafted onto a Highly Cross-Linked Polymer Matrix, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- F. Della Monica, A. Buonerba, A. Grassi, C. Capacchione and S. Milione, Glycidol: an Hydroxyl-Containing Epoxide Playing the Double Role of Substrate and Catalyst for CO2 Cycloaddition Reactions, ChemSusChem, 2016, 9, 3457–3464 CrossRef CAS PubMed.
- D. C. Sherrington, Preparation, structure and morphology of polymer supports, Chem. Commun., 1998, 2275–2286 RSC.
- A. R. Vaino and K. D. Janda, Solid-Phase Organic Synthesis: A Critical Understanding of the Resin, J. Comb. Chem., 2000, 2, 579–596 CrossRef CAS PubMed.
- J. Lu and P. H. Toy, Organic Polymer Supports for Synthesis and for Reagent and Catalyst Immobilization, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed.
- C. A. McNamara, M. J. Dixon and M. Bradley, Recoverable Catalysts and Reagents Using Recyclable Polystyrene-Based Supports, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS PubMed.
- S. Itsuno and M. M. Hassan, Polymer-immobilized chiral catalysts, RSC Adv., 2014, 4, 52023–52043 RSC.
- B. Altava, M. I. Burguete, E. García-Verdugo and S. V. Luis, Chiral catalysts immobilized on achiral polymers: effect of the polymer support on the performance of the catalyst, Chem. Soc. Rev., 2018, 47, 2722–2771 RSC.
- M. R. Hansen, R. Graf and H. W. Spiess, Interplay of Structure and Dynamics in Functional Macromolecular and Supramolecular Systems As Revealed by Magnetic Resonance Spectroscopy, Chem. Rev., 2015, 116, 1272–1308 CrossRef PubMed.
- Handbook of High-Field Dynamic Nuclear Polarization, ed. V. K. Michaelis, R. G. Griffin, B. Corzilius, and S. Vega, John Wiley & Sons, 2020 Search PubMed.
- Y. C. Su, L. Andreas and R. G. Griffin, Magic Angle Spinning NMR of Proteins: High-Frequency Dynamic Nuclear Polarization and H-1 Detection, Annu. Rev. Biochem., 2015, 84, 465–497 CrossRef CAS PubMed.
- A. J. Rossini, Materials Characterization by Dynamic Nuclear Polarization-Enhanced Solid-State NMR Spectroscopy, J. Phys. Chem. Lett., 2018, 9, 5150–5159 CrossRef CAS PubMed.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, Dynamic Nuclear Polarization Solid-State NMR in Heterogeneous Catalysis Research, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- W.-C. Liao, B. Ghaffari, C. P. Gordon, J. Xu and C. Copéret, Dynamic Nuclear Polarization Surface Enhanced NMR Spectroscopy (DNP SENS): Principles, Protocols, and Practice, Curr. Opin. Colloid Interface Sci., 2018, 33, 63–71 CrossRef CAS.
- D. Le, G. Casano, T. N. T. Phan, F. Ziarelli, O. Ouari, F. Aussenac, P. Thureau, G. Mollica, D. Gigmes, P. Tordo and S. Viel, Optimizing Sample Preparation Methods for Dynamic Nuclear Polarization Solid-state NMR of Synthetic Polymers, Macromolecules, 2014, 47, 3909–3916 CrossRef CAS.
- O. Ouari, T. Phan, F. Ziarelli, G. Casano, F. Aussenac, P. Thureau, D. Gigmes, P. Tordo and S. Viel, Improved Structural Elucidation of Synthetic Polymers by Dynamic Nuclear Polarization Solid-State NMR Spectroscopy, ACS Macro Lett., 2013, 2, 715–719 CrossRef CAS.
- G. N. Manjunatha Reddy, J. A. Gerbec, F. Shimizu and B. F. Chmelka, Nanoscale Surface Compositions and Structures Influence Boron Adsorption Properties of Anion Exchange Resins, Langmuir, 2019, 35, 15661–15673 CrossRef CAS PubMed.
- F. Blanc, S. Y. Chong, T. O. McDonald, D. J. Adams, S. Pawsey, M. A. Caporini and A. I. Cooper, Dynamic Nuclear Polarization NMR Spectroscopy Allows High-Throughput Characterization of Microporous Organic Polymers, J. Am. Chem. Soc., 2013, 135, 15290–15293 CrossRef CAS PubMed.
- N. J. Brownbill, R. S. Sprick, B. Bonillo, S. Pawsey, F. Aussenac, A. J. Fielding, A. I. Cooper and F. Blanc, Structural Elucidation of Amorphous Photocatalytic Polymers from Dynamic Nuclear Polarization Enhanced Solid State NMR, Macromolecules, 2018, 51, 3088–3096 CrossRef CAS.
- S. Grätz, M. de Olivera Junior, T. Gutmann and L. Borchardt, A comprehensive approach for the characterization of porous polymers using 13C and 15N dynamic nuclear polarization NMR spectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 23307–23314 RSC.
- T. Kanda, M. Kitawaki, T. Arata, Y. Matsuki and T. Fujiwara, Structural analysis of cross-linked poly(vinyl alcohol) using high-field DNP-NMR, RSC Adv., 2020, 10, 8039–8043 RSC.
- S. Tanaka, W.-C. Liao, A. Ogawa, K. Sato and C. Copéret, DNP NMR spectroscopy of cross-linked organic polymers: rational guidelines towards optimal sample preparation, Phys. Chem. Chem. Phys., 2020, 22, 3184–3190 RSC.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwalder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Coperet, A. Lesage, P. Tordo and L. Emsley, Large Molecular Weight Nitroxide Biradicals Providing Efficient Dynamic Nuclear Polarization at Temperatures up to 200 K, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- V. B. Luzhkov, F. Österberg, P. Acharya, J. Chattopadhyaya and J. Åqvist, Computational and NMR study of quaternary ammonium ion conformations in solution, Phys. Chem. Chem. Phys., 2002, 4, 4640–4647 RSC.
- D. Cespi, R. Cucciniello, M. Ricciardi, C. Capacchione, I. Vassura, F. Passarini and A. Proto, A simplified early stage assessment of process intensification: glycidol as a value-added product from epichlorohydrin industry wastes, Green Chem., 2016, 18, 4559–4570 RSC.
- J. P. Pascault, and R. J. Williams, In Epoxy Polymers: New Materials and Innovations, ed.Pascault, J. P. and Williams, R. J. J., Wiley-VCH, Weinheim, Germany, 2010, pp. 1–12 Search PubMed.
- S. Zhang, T. Liu, C. Hao, A. Mikkelsen, B. Zhao and J. Zhang, Hempseed Oil-Based Covalent Adaptable Epoxy-Amine Network and Its Potential Use for Room-Temperature Curable Coatings, ACS Sustainable Chem. Eng., 2020, 8, 14964–14974 CrossRef CAS.
- L. Zhang, X. Luo, Y. Qin and Y. Li, A novel 2,5-furandicarboxylic acid-based bis(cyclic carbonate) for the synthesis of biobased non-isocyanate polyurethanes, RSC Adv., 2017, 7, 37–46 RSC.
- A. Marotta, V. Ambrogi, P. Cerruti and A. Mija, Green approaches in the synthesis of furan-based diepoxy monomers, RSC Adv., 2018, 8, 16330–16335 RSC.
- S. Nameer, D. B. Larsen, J. Ø. Duus, A. E. Daugaard and M. Johansson, Biobased Cationically Polymerizable Epoxy Thermosets from Furan and Fatty Acid Derivatives, ACS Sustainable Chem. Eng., 2018, 6, 9442–9450 CrossRef CAS.
- J. Meng, Y. Zeng, P. Chen, J. Zhang, C. Yao, Z. Fang and K. Guo, New ultrastiff bio-furan epoxy networks with high Tg: Facile synthesis to excellent properties, Eur. Polym. J., 2019, 121, 109292 CrossRef.
- G. Wang, L. Lopez, M. Coile, Y. Chen, J. M. Torkelson and L. J. Broadbelt, Identification of Known and Novel Monomers for Poly(hydroxyurethanes) from Biobased Materials, Ind. Eng. Chem. Res., 2021, 60, 6814–6825 CrossRef CAS.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed.
- R. Haag, A. Sunder and J.-F. Stumbe, J. Am. Chem. Soc., 2000, 122, 2954–2955 CrossRef CAS.
- C. H. Bushweller, S. H. Fleischman, G. L. Grady, P. McGoff, C. D. Rithner, M. R. Whalon, J. G. Brennan, R. P. Marcantonio and R. P. Dominigue, Stereodynamics of diethylmethylamine and triethylamine, J. Am. Chem. Soc., 1982, 104, 6224–6236 CrossRef CAS.
- F. Ziarelli, M. Casciola, M. Pica, A. Donnadio, F. Aussenac, C. Sauvee, D. Capitani and S. Viel, Dynamic nuclear polarisation NMR of nanosized zirconium phosphate polymer fillers, Chem. Commun., 2014, 50, 10137–10139 RSC.
- K. Lange, A. Koenig, C. Roegler, A. Seeling and J. Lehmann, NO donors. Part 18: Bioactive metabolites of GTN and PETN—Synthesis and vasorelaxant properties, Bioorg. Med. Chem. Lett., 2009, 19, 3141–3144 CrossRef CAS PubMed.
- A. Leal-Duaso, M. Caballero, A. Urriolabeitia, J. A. Mayoral, J. I. García and E. Pires, Synthesis of 3-alkoxypropan-1,2-diols from glycidol: experimental and theoretical studies for the optimization of the synthesis of glycerol derived solvents, Green Chem., 2017, 19, 4176–4185 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00274d |
| This journal is © The Royal Society of Chemistry 2022 |