
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2D-C3N4 encapsulated perovskite nanocrystals for efficient photo-assisted thermocatalytic CO2 reduction†
Hui
Bian
a,
Deng
Li
a,
Shengyao
Wang
 c,
Junqing
Yan
*a and
Shengzhong (Frank)
Liu
*ab
c,
Junqing
Yan
*a and
Shengzhong (Frank)
Liu
*ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Shaanxi Engineering Lab for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: junqingyan@snnu.edu.cn
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: szliu@dicp.ac.cn
cCollege of Science, Huazhong Agricultural University, Wuhan 430070, P. R. China
First published on 18th January 2022
Abstract
Very recently, halide perovskites, especially all-inorganic CsPbBr3, have received ever-increasing attention in photocatalysis owing to their superior optoelectronic properties and thermal stability. However, there is a lack of study on their application in thermocatalysis and photo-thermocatalysis. Herein, we rationally designed a core–shell heterojunction formed by encapsulating CsPbBr3 nanoparticles with the 2D C3N4 (m-CN) layer via a solid-state reaction (denoted as m-CN@CsPbBr3). A series of experiments suggest that abundant adsorption and active sites of CO2 molecules as well as polar surfaces were obtained by utilizing m-CN-coated CsPbBr3, resulting in significant improvement in CO2 capture and charge separation. It is found that the m-CN@CsPbBr3 effectively drives the thermocatalytic reduction of CO2 in H2O vapor. By coupling light into the system, the activity for CO2-to-CO reduction is further improved with a yield up to 42.8 μmol g−1 h−1 at 150 °C, which is 8.4 and 2.3 times those of pure photocatalysis (5.1 μmol g−1 h−1) and thermocatalysis (18.7 μmol g−1 h−1), respectively. This work expands the application of general halide perovskites and provides guidance for using perovskite-based catalysts for photo-assisted thermocatalytic CO2 reduction.
The reduction of CO2 into valuable hydrocarbon fuels via chemical catalytic processes to mitigate the greenhouse-effect has received wide attention.1,2 Basically, the CO2 molecule has a straight double-bonded non-polar structure, resulting in a huge activation energy barrier for the CO2 reduction reaction (CRR).3,4 Using a catalyst to adsorb and then activate the CO2 molecules with free electrons is thus imperative.5,6 Currently, there are three main types of catalytic CO2 reduction reactions, i.e. thermocatalysis,7 electrocatalysis8a,b and photocatalysis,4b,c,5c where the catalysts used are referred to as thermocatalysts, electrocatalysts and photocatalysts, respectively. Traditionally, thermocatalysis is regarded as the conventional CRR process where the CO2 molecules are well-activated and reduced by thermocatalysts using heat and the reductant agent H2. Hence, thermocatalysis usually shows a relatively higher CRR efficiency compared to the other two cases.7a,b,9a However, as it is very complex to safely transport and use H2 for this process, it is more advantageous to use safe and free H2O as the reductant if high thermocatalytic CRR efficiency can be achieved. Photocatalysis is regarded as an effective approach for CO2 reduction owing to the merits of utilizing solar energy directly and low energy consumption. However, the efficiency of the photocatalytic CRR is still limited by the sluggish kinetics of CO2 activation and H2O dissolving.4c,9b Very recently, Xu and co-workers reported a novel lead-free perovskite, Cs3Sb2I9, for CO2 reduction to CO and CH4via photothermal synergistic catalysis without using any sacrificial agents or cocatalysts.9c Unfortunately, the perovskite is not stable enough in the working solution. In addition, its activity is still limited. There is still some room to develop a simple method for synthesizing effective and stable photothermal catalysts.
All-inorganic perovskites, for example cesium lead tribromide (CsPbBr3), have emerged as a promising type of photocatalyst for the CRR, owing to their long photogenerated carrier diffusion length, tunable size, wide light-absorption range, etc.5c,10 CsPbBr3 perovskites have been widely explored in composites with other materials, such as g-C3N4,11 graphene oxide5a,12 and TiO2 (ref. 13) to construct heterojunctions for photocatalytic CO2 reduction. For example, Xu and his co-workers mixed CsPbBr3 with NHx-rich g-C3N4 nanosheets to construct a heterojunction with a fast carrier-transfer bridge for improved charge separation and hence enhanced photocatalytic activity for CO2 reduction.14a Similarly, Lu's group reported the coating of CsPbBr3 with graphdiyne as a physical protection layer to tackle the stability issue in photocatalytic CO2 reduction in H2O vapor, given that the perovskite shows a low tolerance towards water.14b It should be mentioned that most of the reported halide perovskite nanocrystals (NCs) were prepared by the solution method using organic solvents, which is troublesome for large-scale production.15a,b Besides, considering that thermocatalysis usually outperforms photocatalysis in CO2 reduction, it would be intriguing to explore the catalytic activity of CsPbBr3 under coupled thermal and irradiation effects. To the best of our knowledge, there are few reports of water-stable CsPbBr3-based thermal CRR with H2O as a reducing agent.
Herein, using a molten-salt method, we elaborately encapsulate CsPbBr3 with a 2D m-CN layer to construct a water-stable heterojunction for photo-assisted thermal catalytic CO2 reduction. We found that under pure thermocatalysis conditions, m-CN@CsPbBr3 showed the ability to drive the CRR reduction to CO using the CO2 and H2O as reactants. Moreover, with the introduction of simulated solar illumination, the corresponding reduction yield rises to 42.8 μmol g−1 h−1, which is 2.3 and 8.4 times those of pure thermocatalysts and photocatalysts, respectively. This is the first report on the fabrication of CsPbBr3 perovskite NCs by means of a solid-phase reaction. This work expands the application of CsPbBr3 perovskite and can help us better understand CO2 reduction by H2O.
A molten-salt method, where ion salts act as a high-temperature liquid solvent to accelerate the dissolution of raw materials, the transport of reactants, and the directional assembly of basic units, was used to prepare perovskite catalysts with a specific morphology.15c It is worth noting that melem can be obtained by thermal condensation of urea below 450 °C, while g-C3N4 can be acquired in the temperature range between 480 °C and 550 °C (Fig. S1a†).16a Therefore, the composition of the coating layer may be assigned to an intermediate product (named m-CN) possessing many edge NHx groups between melem and g-C3N4, which is attributed to the higher local temperature (>450 °C) caused by the molten salt. To confirm this, a comparison study is performed at 420 °C and 470 °C for the synthesis of m-CN@CsPbBr3 catalysts. As revealed in Fig. S1b,† the best preparation temperature is 450 °C for m-CN@CsPbBr3 catalysts, while the phase transition of CsPbBr3 occurs at 470 °C. During the heat treatment, CsPbBr3 was changed into the solution state, i.e. molten salt (Fig. S2†). Meanwhile the polymerized monomers of carbon nitride adsorbed onto the surface of CsPbBr3, and then the micron-sized CsPbBr3 was “cut” into nanoparticles by encapsulation with m-CN coatings, as shown in Fig. 1a. To elucidate the formation process of m-CN@CsPbBr3, different coatings with CsPbBr3-to-urea mass ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:5 and 1:10 were synthesized and are denoted as m-CN@CsPbBr3-3, m-CN@CsPbBr3-5 and m-CN@CsPbBr3-10, respectively. The corresponding XRD and FTIR results also suggest the encapsulation of CsPbBr3 with m-CN in these samples, as shown in Fig. S3a and b.† It should be noted that the m-CN@CsPbBr3-3 sample gives the typical peaks of g-C3N4 at 2-theta of 13 and 27°, while the broad XRD peaks of m-CN@CsPbBr3-10 could be ascribed to melem, suggesting that a certain content of molten salt is required to improve the temperature profile for the thermal polymerization of the CN coating layers.16b Fourier transform infrared (FTIR) spectroscopy of m-CN@CsPbBr3-3 and m-CN@CsPbBr3-10 showed similar peaks, where the peaks in the wide range from 1200 to 1700 cm−1 are attributed to the skeleton signal of g-C3N4.16c The absorption bands from 3000 to 3500 cm−1 come from NHx and –OH groups.14a,16b In comparison to m-CN@CsPbBr3-10, the normalized intensity of the band from 3000 to 3500 cm−1 for m-CN@CsPbBr3-3 is evidently decreased, which can be ascribed to the stronger interactions of edge NHx groups and bromide anions of CsPbBr3.16 It is also observed that the m-CN@CsPbBr3-3 peaks shifted to lower values compared with m-CN@CsPbBr3-10, as shown in the magnified FTIR spectra of Fig. S3,† suggesting stronger interactions occurred with the introduction of more bromide anions.14a,16d When the relative mass ratio of urea to CsPbBr3 is changed, either the amount of loaded CsPbBr3 particles is lower and sporadic (Fig. S4a,† m-CN@CsPbBr3-10) or the CsPbBr3 particles are larger and exposed (Fig. S4b,† m-CN@CsPbBr3-3), whereas the m-CN@CsPbBr3-5 sample displays completely uniform and coated CsPbBr3 particles, suggesting that the appropriate ratio of urea to CsPbBr3 is critical for optimal m-CN@CsPbBr3 formation. Therefore, the m-CN@CsPbBr3-5 catalyst with an optimized coating ratio was chosen for further study because it may be endowed with a more heterogeneous interface and excellent stability. For the synthesis of pure CsPbBr3 for further composition, vacuum was employed to obtain less-defective CsPbBr3. We also attempted to treat the mixture of CsPbBr3 and urea in air. However, very little polymerization of urea to carbon nitride occurred, and thus, the N2 condition was chosen for m-CN@CsPbBr3 preparation. Therefore, we successfully encapsulated the perovskite CsPbBr3 with a 2D m-CN layer by utilizing CsPbBr3 as a molten salt under N2 conditions. It is greatly emphasized that this is the first report on the fabrication of CsPbBr3 perovskite NCs by means of a solid-phase reaction. Differing from the traditional synthesis approaches such as ball milling and solution processing strategies, this unique method can avoid some difficulties, including a complicated procedure and the formation of organic branches and undesired phases, showing the attractive application of this method for fabricating catalysts beyond perovskites.
:3, 1:5 and 1:10 were synthesized and are denoted as m-CN@CsPbBr3-3, m-CN@CsPbBr3-5 and m-CN@CsPbBr3-10, respectively. The corresponding XRD and FTIR results also suggest the encapsulation of CsPbBr3 with m-CN in these samples, as shown in Fig. S3a and b.† It should be noted that the m-CN@CsPbBr3-3 sample gives the typical peaks of g-C3N4 at 2-theta of 13 and 27°, while the broad XRD peaks of m-CN@CsPbBr3-10 could be ascribed to melem, suggesting that a certain content of molten salt is required to improve the temperature profile for the thermal polymerization of the CN coating layers.16b Fourier transform infrared (FTIR) spectroscopy of m-CN@CsPbBr3-3 and m-CN@CsPbBr3-10 showed similar peaks, where the peaks in the wide range from 1200 to 1700 cm−1 are attributed to the skeleton signal of g-C3N4.16c The absorption bands from 3000 to 3500 cm−1 come from NHx and –OH groups.14a,16b In comparison to m-CN@CsPbBr3-10, the normalized intensity of the band from 3000 to 3500 cm−1 for m-CN@CsPbBr3-3 is evidently decreased, which can be ascribed to the stronger interactions of edge NHx groups and bromide anions of CsPbBr3.16 It is also observed that the m-CN@CsPbBr3-3 peaks shifted to lower values compared with m-CN@CsPbBr3-10, as shown in the magnified FTIR spectra of Fig. S3,† suggesting stronger interactions occurred with the introduction of more bromide anions.14a,16d When the relative mass ratio of urea to CsPbBr3 is changed, either the amount of loaded CsPbBr3 particles is lower and sporadic (Fig. S4a,† m-CN@CsPbBr3-10) or the CsPbBr3 particles are larger and exposed (Fig. S4b,† m-CN@CsPbBr3-3), whereas the m-CN@CsPbBr3-5 sample displays completely uniform and coated CsPbBr3 particles, suggesting that the appropriate ratio of urea to CsPbBr3 is critical for optimal m-CN@CsPbBr3 formation. Therefore, the m-CN@CsPbBr3-5 catalyst with an optimized coating ratio was chosen for further study because it may be endowed with a more heterogeneous interface and excellent stability. For the synthesis of pure CsPbBr3 for further composition, vacuum was employed to obtain less-defective CsPbBr3. We also attempted to treat the mixture of CsPbBr3 and urea in air. However, very little polymerization of urea to carbon nitride occurred, and thus, the N2 condition was chosen for m-CN@CsPbBr3 preparation. Therefore, we successfully encapsulated the perovskite CsPbBr3 with a 2D m-CN layer by utilizing CsPbBr3 as a molten salt under N2 conditions. It is greatly emphasized that this is the first report on the fabrication of CsPbBr3 perovskite NCs by means of a solid-phase reaction. Differing from the traditional synthesis approaches such as ball milling and solution processing strategies, this unique method can avoid some difficulties, including a complicated procedure and the formation of organic branches and undesired phases, showing the attractive application of this method for fabricating catalysts beyond perovskites.
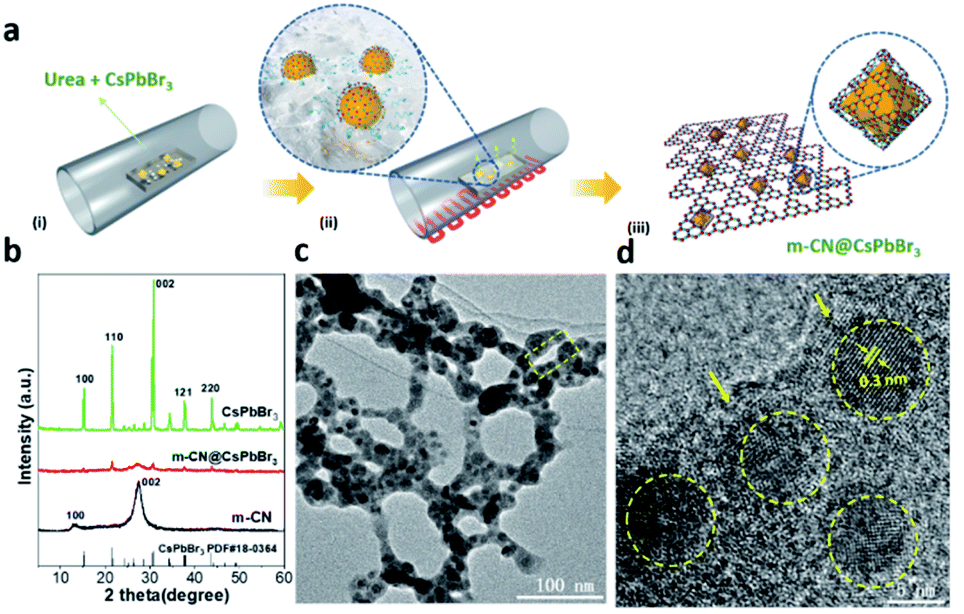 | ||
| Fig. 1 (a) Illustration of the synthesis of m-CN@CsPbBr3; (b) XRD patterns of m-CN@CsPbBr3, the reference m-CN and CsPbBr3; (c and d) TEM image and the corresponding HRTEM result of m-CN@CsPbBr3, where the yellow arrows mark the thin m-CN layers. | ||
As a representative material, m-CN@CsPbBr3-5 is selected to investigate the influence of encapsulating perovskite nanocrystals with 2D m-CN for boosting photo-assisted thermocatalytic CO2 reduction. The XRD patterns in Fig. 1b show that the intensity of the peaks ascribed to CsPbBr3 was obviously decreased after the m-CN encapsulation, suggesting that the crystallinity of CsPbBr3 is reduced. Magnification of the diffraction region from 21.2 to 22° suggests that the m-CN@CsPbBr3 sample exhibits a larger full-width at half maximum (FWHM) than bare CsPbBr3 (Fig. S5†), further indicating that the particle size was decreased by the calcination treatment. It should also be noted that the as-prepared CsPbBr3 formed in a vacuum also showed a crystal-lattice orientation, given that the texture coefficient of the [200] peak is much higher than the standard value of 1, while it is less than 1 for CsPbBr3 prepared in N2 and air (Fig. S6a and b†). It is noted that the texture coefficient of the [200] peak is in agreement with CsPbBr3 prepared in N2 after m-CN coating, which further proved the melt-crystallization process of CsPbBr3. The FTIR spectra of m-CN@CsPbBr3 and melem showed similar peaks (Fig. S7†), and the normalized intensity of the band from 3000 to 3500 cm−1 of m-CN@CsPbBr3 is obviously decreased compared to the reference m-CN. Furthermore, compared with the pure m-CN layer, the peaks for m-CN@CsPbBr3 located at 1250 cm−1, corresponding to the typical stretching mode of aromatic C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N heterocycles in m-CN, are systematically shifted to lower values in the magnified FTIR spectra in Fig. S7,† suggesting that the edge NHx groups interact with the bromide anion of CsPbBr3via ionic bonding.16,17aFig. 1c displays the transmission electron microscopy (TEM) image of the m-CN@CsPbBr3 sample. Clearly, it shows that the CsPbBr3 nanoparticles were uniformly encapsulated by the m-CN layer. The corresponding high-resolution transmission electron microscopy (HRTEM) images further show clear lattice fringes with a measured interplanar spacing of 0.3 nm, as shown in Fig. 1d, which can be assigned to the (002) plane of cubic CsPbBr3. Additionally, it shows that nano-sized (ca. 8 nm) CsPbBr3 particles were encapsulated by the m-CN layer in the m-CN@CsPbBr3 catalyst (Fig. S8†). In contrast, the reference CsPbBr3, which was prepared without urea, is micron-sized (Fig. S9†), further confirming that the particle size of CsPbBr3 was changed by the in situ encapsulation with the m-CN layer. Fig. S10† displays the scanning transmission electron microscopy (STEM) image with the corresponding elemental mapping results, which further confirm that the CsPbBr3 nanoparticles are wrapped by the m-CN layer. The TEM images and the corresponding elemental mapping of the reference synthesized melem are shown in Fig. S11 and S12,† respectively, and they resemble those of m-CN@CsPbBr3, further confirming that the coating layer can be attributed to m-CN.
N heterocycles in m-CN, are systematically shifted to lower values in the magnified FTIR spectra in Fig. S7,† suggesting that the edge NHx groups interact with the bromide anion of CsPbBr3via ionic bonding.16,17aFig. 1c displays the transmission electron microscopy (TEM) image of the m-CN@CsPbBr3 sample. Clearly, it shows that the CsPbBr3 nanoparticles were uniformly encapsulated by the m-CN layer. The corresponding high-resolution transmission electron microscopy (HRTEM) images further show clear lattice fringes with a measured interplanar spacing of 0.3 nm, as shown in Fig. 1d, which can be assigned to the (002) plane of cubic CsPbBr3. Additionally, it shows that nano-sized (ca. 8 nm) CsPbBr3 particles were encapsulated by the m-CN layer in the m-CN@CsPbBr3 catalyst (Fig. S8†). In contrast, the reference CsPbBr3, which was prepared without urea, is micron-sized (Fig. S9†), further confirming that the particle size of CsPbBr3 was changed by the in situ encapsulation with the m-CN layer. Fig. S10† displays the scanning transmission electron microscopy (STEM) image with the corresponding elemental mapping results, which further confirm that the CsPbBr3 nanoparticles are wrapped by the m-CN layer. The TEM images and the corresponding elemental mapping of the reference synthesized melem are shown in Fig. S11 and S12,† respectively, and they resemble those of m-CN@CsPbBr3, further confirming that the coating layer can be attributed to m-CN.
X-ray photoelectron spectroscopy (XPS) was carried out to study the chemical environment of the elements in the m-CN@CsPbBr3 heterojunction. Fig. S13a† shows the C 1s XPS results of m-CN@CsPbBr3 and reference m-CN samples. Two main peaks located at ca. 284.8 and 288.2 eV can be detected, and the first is assigned to the C–C in graphitic carbon, while the second comes from the N–CN coordination of triazine rings.16,17b It should be noted that there is a slight shift of the N–CN signal of m-CN@CsPbBr3 to higher binding energy as compared with the bare m-CN, indicating there is an interaction between CsPbBr3 and m-CN. For the corresponding N 1s XPS curves, three main peaks centered at 398.7, 399.7 and 400.9 eV can be detected (Fig. S13b†), and they can be assigned to C–NC, N–(C)3 and C–N–H of the carbon nitride skeleton, respectively.17 However, the weak peak at ca. 404.4 eV is usually regarded as coming from positive charge localization in heterocycles.17c It almost disappears in m-CN@CsPbBr3 relative to bare m-CN, further suggesting that there is some chemical interaction between m-CN and CsPbBr3 for neutralizing the positive charge.14a Fig. S14a† gives the Cs 3d XPS results of m-CN@CsPbBr3 and bare CsPbBr3. Two main peaks at 724.5 and 738.4 eV can be detected and come from the Cs 3d5/2 and 3d3/2 signals.14,15 However, a slight shift to higher binding energy can be detected for m-CN@CsPbBr3, further confirming the above mutual interaction between m-CN and CsPbBr3. In the Pb 4f XPS result in Fig. S14b,† there are two peaks at ca. 138.4 and 143.3 eV. Similarly, there is a shift to higher binding energy for m-CN@CsPbBr3 compared with CsPbBr3. Fig. S14c† shows the Br 3d XPS results. Clearly, there are two fitted peaks located at ca. 68.3 and 69.3 eV. The sample of m-CN@CsPbBr3 exhibits a ca. 0.2 eV shift to higher binding energy compared with bare CsPbBr3. Both the shifts of Pb 4f and Br 3d in the XPS results suggest that chemical bonds of N–Br have been formed during the preparation process.11b,14a
The UV-vis absorption spectra of the pristine m-CN and micro-sized CsPbBr3 in Fig. 2a exhibit the typical absorption edges of CsPbBr3 and m-CN at ca. 553 and 430 nm, corresponding to bandgaps of 2.24 and 2.88 eV, respectively. For the sample of m-CN@CsPbBr3, two obvious edges were observed, which correspond to the light absorption edges of CsPbBr3 and m-CN, indicating the successful combination of the two materials. Fig. S15† displays the photoluminescence (PL) curves from 375 nm excitation. Strong emission at ca. 450 nm was marked for m-CN, and the peak at 524 nm is attributed to CsPbBr3. There are two peaks in the PL curve of m-CN@CsPbBr3, confirming the composition of the catalyst. Moreover, compared with the bare m-CN and CsPbBr3, the PL intensity of m-CN@CsPbBr3 is lower, which indicates suppressed carrier recombination in m-CN@CsPbBr3. The time-resolved PL spectrum was further obtained to explore the carrier dynamics of m-CN@CsPbBr3. Fig. S16a and b† exhibit the transient PL spectra at 440 and 523 nm, where the signals can be considered as coming from m-CN and CsPbBr3 (Fig. S15†), respectively, and the corresponding fitted results are shown in Tables S1 and S2.† Obviously, compared with pure m-CN at 440 nm (Fig. S16a†), m-CN@CsPbBr3 shows much faster decay after the introduction of CsPbBr3. Interestingly, with respect to pure CsPbBr3 at 523 nm (Fig. S16b†), the opposite trend could be observed. The results testify that the photogenerated electrons of m-CN and holes of CsPbBr3 will migrate towards each other under the effect of a built-in electric field, and the photogenerated electrons may be transported from CsPbBr3 to the m-CN layer. The steady-state photovoltages (SPV) of the different samples were further compared to evidence the promoted charge separation in the m-CN@CsPbBr3 heterojunction, as shown in Fig. 2b. Clearly, the samples of m-CN and CsPbBr3 exhibited negligible SPV signals. Meanwhile, the m-CN@CsPbBr3-5 sample demonstrated the highest positive photovoltage response compared with the other catalysts with coating layers (m-CN@CsPbBr3-10 and m-CN@CsPbBr3-3), suggesting that its surface is populated by a concentration of holes due to the high charge-separation efficiency (Fig. S17a†).18a Transient photovoltage (TPV) analysis was further carried out to study the migration dynamics of the photogenerated carriers.18 Two obvious positive response peaks (peaks 1 and 2) can be detected from the sample of m-CN@CsPbBr3 under 400 nm illumination (Fig. S17b†). Peak 1 can be assigned to a fast process related to the migration of photogenerated carriers in the built-in electric fields inside particles,18b and thus the stronger intensity of peak 1 of m-CN@CsPbBr3 suggests that more photogenerated carriers are transported to the interface between m-CN and CsPbBr3. Peak 2 is thought to arise from carrier transport between particles.18 A further step to study the carrier transfer dynamics was to perform picosecond transient absorption spectroscopy (TA). Both the samples of bare CsPbBr3 (Fig. S18a†) and m-CN@CsPbBr3 (Fig. S18b†) exhibit obvious negative ground-state bleaching. Fig. 2c shows the corresponding TA kinetic plots monitored at 520 nm with a distinct difference in the delays. As listed in Table S3,† the fitted average lifetime of m-CN@CsPbBr3 (197.7 ps) is much longer than that of bare CsPbBr3 (77.2 ps), consistent with the above transient PL and TPV results. It is concluded that the internal electric field between m-CN and CsPbBr3 would promote the carrier transfer and separation in the m-CN@CsPbBr3 heterojunction.19 According to the direction of carrier transfer and the strong built-in electric field, it can be inferred that an S-scheme heterojunction is formed between m-CN and CsPbBr3.
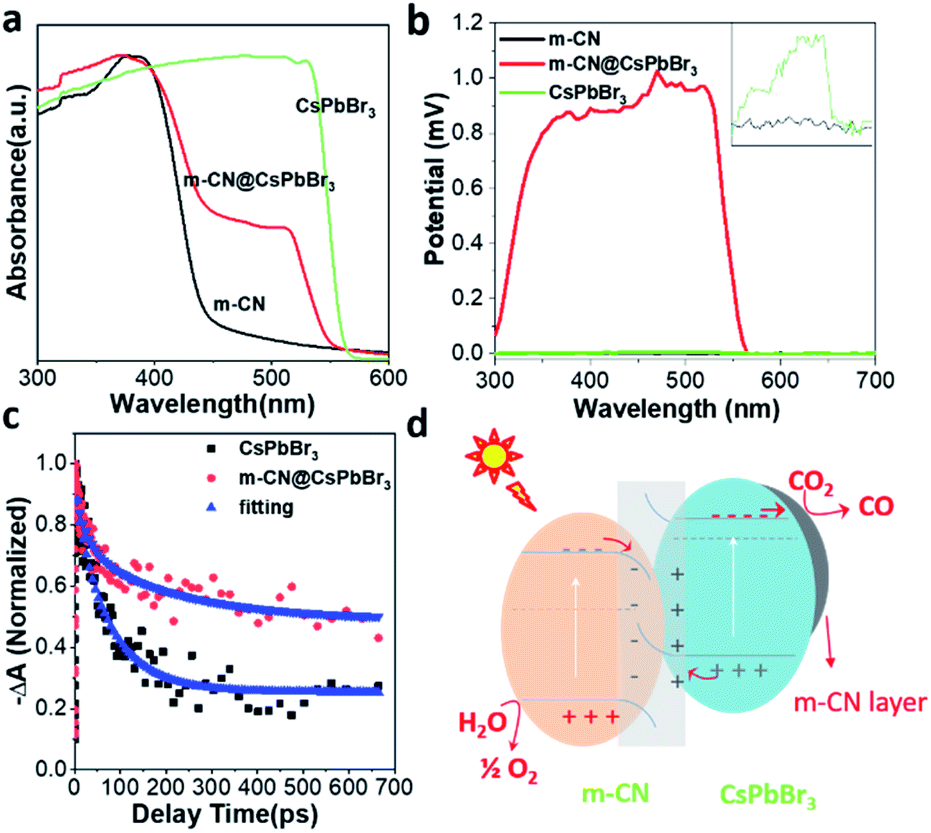 | ||
| Fig. 2 (a) UV-Vis absorption spectra of m-CN@CsPbBr3, the reference m-CN and CsPbBr3; (b) surface photovoltage plots of the m-CN, CsPbBr3 and m-CN@CsPbBr3 samples; (c) transient absorption spectroscopy curves and lifetime fittings at 520 nm; (d) band structure of the m-CN@CsPbBr3 heterojunction and the corresponding paths of CO2 photo-reduction. | ||
In order to acquire stronger confirmation of the direction of carrier transfer, ultraviolet photoelectron spectroscopy (UPS) was performed to check the valence band (VB) potential and conduction band (CB) potential of the m-CN and CsPbBr3 samples. The results and corresponding valence/conduction band potentials (vs. vacuum level) are given in Fig. S19† and Table S4.† The relative valence/conduction band positions of m-CN and CsPbBr3 are shown in Fig. 2d, and they can be attributed to an S-scheme heterojunction, agreeing well with the aforementioned analysis. There is an offset of the bands of the m-CN and CsPbBr3, and thus close contact between these two materials would cause band bending at the interface. Then, a built-in electric field will be formed making the m-CN and CsPbBr3 centers of positive and negative charges, respectively. Moreover, the unique conduction/valence band structure of this heterojunction results in polar surfaces of m-CN (holes) and CsPbBr3 (electrons). This built-in electric field would also facilitate the separation of photogenerated charges and thus suppress charge recombination.
Considering that the m-CN@CsPbBr3 catalyst possesses excellent photogenerated carrier separation ability, its photo-assisted thermocatalytic CO2 reduction activity was then investigated. We evaluated the catalytic CO2 reduction of the samples in a flow reactor with CO2 and H2O vapor as the reactants, such that the H2O would continuously pass through the catalyst bed. During the flow reaction process, light and heat were applied to the reactor. Thermogravimetric analysis (TGA) was performed to check the temperature tolerance of the m-CN@CsPbBr3 catalyst in nitrogen, as shown in Fig. S20.† It indicates that the thermocatalytic CRR can take place at less than 200 °C because of the absence of any decomposition below this temperature. The instability of CsPbBr3 to humidity has always been a huge obstacle for applications. The m-CN@CsPbBr3 catalyst with the protection of the m-CN layer was then treated in pure water to check its chemical stability. As shown in Fig. S21a and b,† after soaking in water for ∼0.5 and 17 h, there is almost no change in color; however, the bare CsPbBr3 turns white after being in water for less than 1 h. The corresponding XRD results show no change from before to after the water immersion for ∼0.5 and 17 h. Meanwhile, bare CsPbBr3 exhibited an obvious phase transition. Additionally, the water-treated samples were checked by TEM. As shown in Fig. S22,† there is almost no change in morphology after the treatment of ∼0.5 and 17 h, in accordance with the above XRD results. Fig. S23† shows the XPS results of the water-treated samples, and there are almost no changes for all the studied elements. These results suggest that m-CN@CsPbBr3 exhibited superior water stability due to the m-CN encapsulation. Fig. 3a compares the CO2 reduction activities of pristine m-CN, micro-sized CsPbBr3 and m-CN@CsPbBr3 under three different conditions, with our m-CN@CsPbBr3 catalyst benefiting from superior durability. Under the pure simulated sunlight condition (Fig. 3a), the sample of m-CN@CsPbBr3 displays the highest CO2-to-CO yield of 8.15 μmol g−1 h−1, followed by CsPbBr3 (4.5 μmol g−1 h−1) and m-CN (2.2 μmol g−1 h−1). For pure thermocatalysis at the temperature of 150 °C, the CO generation of m-CN@CsPbBr3 is 22.4 μmol g−1 h−1, and those of the bare CsPbBr3 and m-CN are 8.8 and 0 μmol g−1 h−1, respectively. More impressively, when the light and heat were coupled to drive photo-assisted thermocatalysis, the CO2-to-CO yield of m-CN@CsPbBr3 was increased to 42.8 μmol g−1 h−1, which is much higher than those of bare m-CN (5.1 μmol g−1 h−1) and CsPbBr3 (18.7 μmol g−1 h−1). It is suggested that the light excitation would generate electrons and holes and these electrons and holes would migrate to the surfaces of CsPbBr3 and m-CN, facilitating the thermocatalytic CRR process. Under all three conditions, i.e., photocatalysis, thermocatalysis and photo-thermocatalysis, the sample of bare CsPbBr3 gives the lowest activity, further suggesting the significance of the heterojunction for promoting the CRR performance. The photo-thermocatalytic performance of the sample of m-CN@CsPbBr3 is also comparable to those of the other systems, but exceeds those of most reported CsPbBr3-based photocatalysts (Table S5†). Moreover, we checked the impact of coating content on the catalytic activity of m-CN@CsPbBr3 at 150 °C and 3 suns. As depicted in Fig. S24,† in comparison with m-CN@CsPbBr3-3 and m-CN@CsPbBr3-10, m-CN@CsPbBr3-5 delivers the highest CO2-to-CO yield, possibly due to the existence of the enriched heterogeneous interface and strong built-in electric field. Meanwhile, the catalytic stability of m-CN@CsPbBr3-5 was also tested during the 6 h period of operation (Fig. S25†), and the result reveals that its reduction ability at the end is comparable to its original performance. Further, the XPS spectra before and after testing m-CN@CsPbBr3-5 manifest that all elements are almost unchanged (Fig. S26†), once again verifying its excellent catalytic stability, which is promising for practical application.
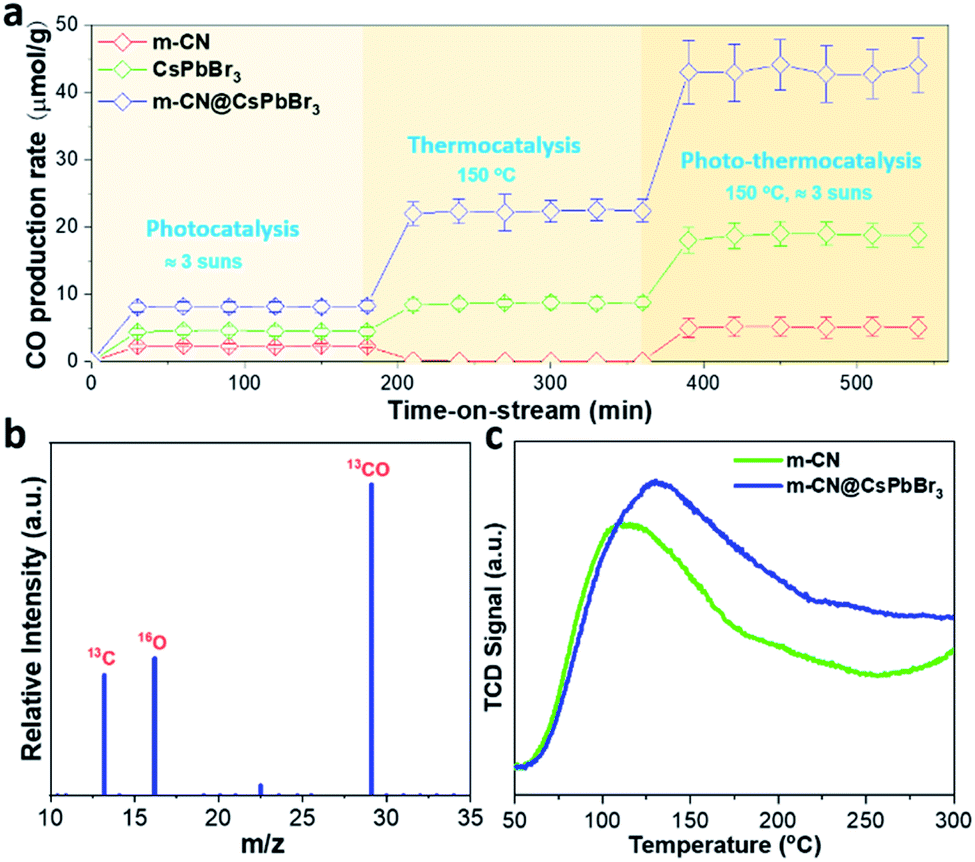 | ||
| Fig. 3 (a) The CO2 reduction to CO performance under the three conditions of photocatalysis, thermocatalysis and photo-thermocatalysis. For the latter two, the temperature is set to 150 °C; (b) mass spectrometry (MS) result of 13CO produced over m-CN@CsPbBr3 from the 13CO2 isotope experiment under thermocatalysis (m/z, mass/charge ratio); (c) CO2-TPD results of the reference m-CN and the m-CN@CsPbBr3 catalyst. | ||
In addition, a small amount of H2 was also measured as the reductive by-product from m-CN@CsPbBr3 samples at different temperatures (Fig. S27†). These results indicate that the coupling of photo and thermal effects profoundly promotes CO2 reduction. Fig. 3b shows the result of an isotopic 13CO2 labeling experiment under the thermocatalytic conditions. A clear peak at m/z = 29 (13CO) is observed, confirming that the generated CO originates from the thermocatalytic reduction of 13CO2 rather than from contaminants. The corresponding screen shots of the raw mass spectra data are displayed in Fig. S28.†
To understand the promoted performance of m-CN@CsPbBr3, the adsorption abilities of CO2 and CO on m-CN and m-CN@CsPbBr3 were studied. Fig. 3c exhibits the CO2 temperature-programmed desorption (TPD) curves of m-CN and m-CN@CsPbBr3, which yield the adsorbed amounts of CO2 according to the integrated areas of the curves. It is found that m-CN@CsPbBr3 has a higher adsorbed amount and thus more sites for CO2 adsorption and activation compared to m-CM, which could be attributed to the polar surfaces of m-CN and CsPbBr3. Moreover, the desorption peak of the m-CN@CsPbBr3 catalyst is situated at higher temperature, suggesting that the corresponding adsorption sites can facilitate the CO2 reduction due to a stronger interaction with the CO2 molecules. In the CO2-TPD curve of CsPbBr3 in Fig. S29,† a higher desorption temperature of ∼320 °C is observed compared with that of m-CN, which can be responsible for the enhancement of desorption for the m-CN@CsPbBr3 catalyst. CO-TPD was also performed, and the results are shown in Fig. S30.† Clearly, both the m-CN and m-CN@CsPbBr3 samples showed weak CO adsorption, which is thought to benefit CO evolution. Measurements were further performed at two other temperatures (100 and 200 °C) to determine the effect of heating on the CRR efficiency. As shown in Fig. S31† (100 °C) and Fig. S32† (200 °C), the yields of CO2-to-CO were lower than at 150 °C, which may be ascribed to the difference of CO2 and CO adsorption at different temperatures. The result suggests that there is an optimal reaction temperature for suitable balance of adsorption/desorption of CO2 and CO to obtain the highest performance of CO2 reduction.
In situ FTIR spectroscopy was further carried out to investigate the possible reaction pathways of the CRR under photo-assisted thermocatalysis conditions. It is evident that increased IR peaks emerge with increasing irradiation time from 0 to 30 minutes (Fig. 4a and b), in which the peaks at 1457 cm−1 and 1646 cm−1 could be assigned to b-HCO3−, while the peaks at 1248 and 1695 cm−1 came from the vibration of the carboxylate (CO2−), and the peak at 1337 cm−1 can be attributed to the bidentate carbonate (b-CO32−). Also, the peaks located at 1379 and 1507 cm−1 matched well with monodentate carbonate groups (m-CO32−).20 Meanwhile, for the photo-assisted thermocatalysis in Fig. 4b, almost the same peaks were observed except with relatively stronger peak intensities, corresponding to the improved CRR efficiency under the coupled photo-thermal effect. Note that the peak at 1695 increased in intensity and the peaks at 1457 cm−1 and 1646 cm−1 disappeared compared with the spectrum under bare thermocatalytic conditions, indicating that b-HCO3− ions on the surface are transformed to surface CO2− species instead with the assistance of light.
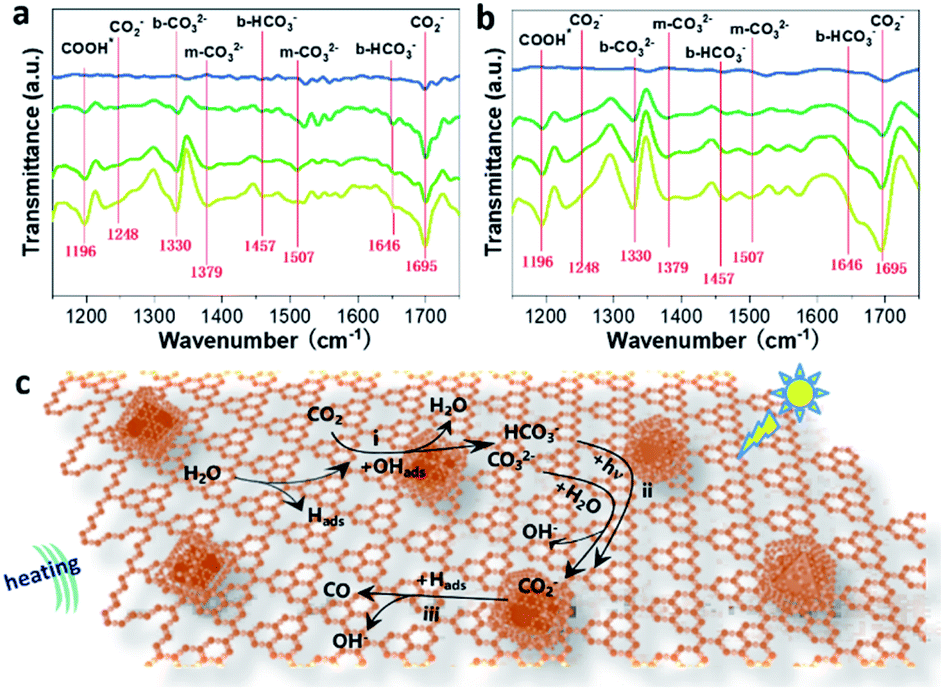 | ||
| Fig. 4 In situ FTIR spectra of m-CN@CsPbBr3 under (a) bare thermocatalytic and (b) photo-thermal conditions at ten-minute increments from 0 to 30 min (from top to bottom). (c) Schematic diagram of the possible reaction pathways of the CRR for the sample of m-CN@CsPbBr3 under photo-thermal conditions. | ||
In addition, the m-CO32− signals at 1507 cm−1 were more obvious compared to those under bare thermocatalytic conditions, suggesting that the adsorption of the reaction intermediates was adjusted by light irradiation. This can be attributed to the strong light absorption of CsPbBr3 according to the FTIR curves of the bare CsPbBr3 sample under the above two conditions (Fig. S33a and b†). This finding indicates the importance of encapsulating CsPbBr3 for altering the formation of intermediates and improving CRR activity under photo-assisted thermocatalysis conditions. Moreover, the FTIR curves of the reference m-CN sample are similar to those of m-CN@CsPbBr3 under bare thermocatalytic conditions (Fig. S34†). Thus, the possible CRR pathways can be summarized as follows (Fig. 4c): (a) the H2O molecules are first dissociated into Hads and OHads groups when meeting hot carriers, i.e., holes generated from thermal or photo excitation; (b) the adsorbed CO2 encounters OHads to generate HCO3− and m-CO32− (step i); (c) then the conversion from surface m-CO32− (step i) to CO2− species in the presence of H2O is proposed, and the surface HCO3− (step i) species can be converted conveniently into CO2− under photo-assisted thermocatalysis conditions (step ii); for (c), the CO2− finally releases CO gas when it encounters Hads and free electrons (step iii).
Herein, a water-stable m-CN@CsPbBr3 heterojunction was synthesized via a solid-state reaction, where bulk CsPbBr3 micro-sized particles were melted and converted to nanoparticles with encapsulation by an m-CN coating during the calcination. The intimate contact between m-CN and CsPbBr3 would induce band bending at the interface and form a built-in electric field, which would separate holes and electrons to m-CN and CsPbBr3, respectively. As such a heterojunction with two charge poles, m-CN@CsPbBr3 exhibited an excellent thermocatalytic CO2-to-CO yield of 42.8 μmol g−1 h−1 under the assistance of irradiation, higher than that of pure photocatalysis (5.1 μmol g−1 h−1) or thermocatalysis (18.7 μmol g−1 h−1). This is the first report of photo-assisted thermocatalysis using CsPbBr3-based materials. Our work thus expands the application of halide perovskites in CO2 reduction.
Data availability
Experimental data have been made available as ESI.†Author contributions
J. Y. designed this study. J. Y. and S. L. supervised and directed the experiments and reviewed and revised the manuscript. H. B. performed the experiments, analyzed the data and wrote the manuscript. D. L. and S. W. helped perform the analysis with constructive discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Key Research Program of China (2017YFA0204800), the National Natural Science Foundation of China (22072081 and 22002084), and the Fundamental Research Funds for the Central Universities (2019TS005, GK202003042, and GK202103111).References
- (a) B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC; (b) G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L.-Z. Wu, C.-H. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed; (c) H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- (a) A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed; (b) S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS PubMed.
- (a) S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS; (b) A. Akhundi, A. Habibi-Yangjeh, M. Abitorabi and S. Rahim Pouran, Catal. Rev., 2019, 61, 595–628 CrossRef CAS.
- (a) J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS; (b) V. K. Ravi, S. Saikia, S. Yadav, V. V. Nawale and A. Nag, ACS Energy Lett., 2020, 5, 1794–1796 CrossRef CAS; (c) Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- (a) Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed; (b) S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S.-m. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed; (c) Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- (a) J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed; (b) Y. Jiang, H.-Y. Chen, J.-Y. Li, J.-F. Liao, H.-H. Zhang, X.-D. Wang and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- (a) B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS; (b) Y. Bai, J. Zhao, S. Feng, X. Liang and C. Wang, Chem. Commun., 2019, 55, 4651–4654 RSC; (c) X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed.
- (a) J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC; (b) J. Liu, L. Zhang, D. Zang and H. Wu, Adv. Funct. Mater., 2021, 31, 2105018 CrossRef CAS.
- (a) L. Zhang, G. Kong, Y. Meng, J. Tian, L. Zhang, S. Wan, J. Lin and Y. Wang, ChemSusChem, 2017, 10, 4709–4714 CrossRef CAS PubMed; (b) L. Lin, K. Wang, K. Yang, X. Chen, X. Fu and W. Dai, Appl. Catal., B, 2017, 204, 440–455 CrossRef CAS; (c) Y. Wang, Q. Zhou, Y. Zhu and D. Xu, Appl. Catal., B, 2021, 294, 120236 CrossRef CAS.
- (a) Z. Zhang, M. Shu, Y. Jiang and J. Xu, Chem. Eng. J., 2021, 414, 128889 CrossRef CAS; (b) J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 2100154 CrossRef CAS; (c) J.-F. Liao, Y.-T. Cai, J.-Y. Li, Y. Jiang, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Energy Chem., 2021, 53, 309–315 CrossRef.
- (a) Q. Chen, X. Lan, Y. Ma, P. Lu, Z. Yuan and J. Shi, Sol. RRL, 2021, 5, 2100186 CrossRef CAS; (b) Y. Wang, Z. Liu, X. Tang, P. Huo, Z. Zhu, B. Yang and Z. Liu, New J. Chem., 2021, 45, 1082–1091 RSC.
- Y.-F. Mu, W. Zhang, G.-X. Dong, K. Su, M. Zhang and T.-B. Lu, Small, 2020, 16, 2002140 CrossRef CAS PubMed.
- (a) F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed; (b) Y.-F. Xu, X.-D. Wang, J.-F. Liao, B.-X. Chen, H.-Y. Chen and D.-B. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- (a) M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 ( Angew. Chem. , 2018 , 130 , 13758–13762 ) CrossRef CAS PubMed; (b) K. Su, G.-X. Dong, W. Zhang, Z.-L. Liu, M. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 50464–50471 CrossRef CAS PubMed.
- (a) Q. Chen, Y. Ma, L. Wang, X. Lan and J. Shi, Sol. RRL, 2021, 5, 2000755 CrossRef CAS; (b) Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CrossRef CAS; (c) J. Fu, Y. Hou, X. Liu, M. Zheng and M. Zhu, J. Mater. Chem. C, 2020, 8, 8704–8731 RSC.
- (a) P. Kumar, E. Vahidzadeh, U. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mahdi, K. Cui, G. M. Bernard, V. K. Michaelis and K. Shankar, J. Am. Chem. Soc., 2019, 141, 5415–5436 CrossRef CAS PubMed; (b) Y. Wang, Y. Zhang, S. Zhao, Z. Huang, W. Chen, Y. Zhou, X. Lv and S. Yuan, Appl. Catal., B, 2019, 248, 44–53 CrossRef CAS; (c) S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed; (d) J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- (a) X. Li, Y. Wang, H. Sun and H. Zeng, Adv. Mater., 2017, 29, 1701185 CrossRef PubMed; (b) J. Zhang, M. Zhang, L. Lin and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 6297–6301 ( Angew.Chem. , 2015 , 127 , 6395–6399 ) CrossRef CAS PubMed; (c) D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 ( Angew. Chem. , 2014 , 126 , 9394–9399 ) CrossRef CAS PubMed.
- (a) X. Zhang, K. Hu, X. Zhang, W. Ali, Z. Li, Y. Qu, H. Wang, Q. Zhang and L. Jing, Appl. Surf. Sci., 2019, 492, 125–134 CrossRef CAS; (b) C. Han, R. Zhang, Y. Ye, L. Wang, Z. Ma, F. Su, H. Xie, Y. Zhou, P. K. Wong and L. Ye, J. Mater. Chem. A, 2019, 7, 9726–9735 RSC.
- (a) X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed; (b) J. Liu, M. Liu, X. Yang, H. Chen, S. F. Liu and J. Yan, ACS Sustainable Chem. Eng., 2020, 8, 6055–6064 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, S. Jin, Y. Xiang, P. Wang, B. Jiang, F. Ichihara, M. Oshikiri, X. Meng, Y. Li, W. Matsuda, J. Ma, S. Seki, X. Wang, H. Huang, Y. Wada, H. Chen and J. Ye, Nat. Commun., 2020, 11, 1149 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06131c |
| This journal is © The Royal Society of Chemistry 2022 |