
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMigratory insertion of isocyanide into a ketenyl–tungsten bond as key step in cyclization reactions†
Christopher
Timmermann
a,
Paula
Thiem
a,
Dominik
Wanitschke
ab,
Mareike
Hüttenschmidt
a,
Johanna
Romischke
a,
Alexander
Villinger
 a and
Wolfram W.
Seidel
*ab
a and
Wolfram W.
Seidel
*ab
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Straße 3a, D-18059 Rostock, Germany. E-mail: wolfram.seidel@uni-rostock.de
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, D-18059 Rostock, Germany
First published on 6th December 2021
Abstract
Treatment of the side-on tungsten alkyne complex of ethinylethyl ether [Tp*W(CO)2(η2-C,C′-HCCOCH2CH3)]+ {Tp* = hydridotris(3,4,5-trimethylpyrazolyl)borate} (2a) with n-Bu4NI afforded the end-on ketenyl complex [Tp*W(CO)2(κ1-HCCO)] (4a). This formal 16 ve complex bearing the prototype of a ketenyl ligand is surprisingly stable and converts only under activation by UV light or heat to form a dinuclear complex [Tp*2W2(CO)4(μ-CCH2)] (6). The ketenyl ligand in complex 4a underwent a metal template controlled cyclization reaction upon addition of isocyanides. The oxametallacycles [Tp*W(CO)2{κ2-C,O-C(NHXy)C(H)C(Nu)O}] {Nu = OMe (7), OEt (8), N(i-Pr)2 (9), OH (10), O1/2 (11)} were formed by coordination of Xy-NC (Xy = 2,6-dimethylphenyl) at 4a and subsequent migratory insertion (MI) into the W-ketenyl bond. The resulting intermediate is susceptible to addition reactions with protic nucleophiles. Compounds 2a-PF6, 4a/b, and 7–11 were fully characterized including XRD analysis. The cyclization mechanism has been confirmed both experimentally and by DFT calculations. In cyclic voltammetry, complexes 7–9 are characterized by a reversible W(II)/W(III) redox process. The dinuclear complex 11 however shows two separated redox events. Based on cyclic voltammetry measurements with different conducting electrolytes and IR spectroelectrochemical (SEC) measurements the W(II)/W(III) mixed valent complex 11+ is assigned to class II in terms of the Robin-Day classification.
Introduction
Migratory insertion (MI) of carbon monoxide (CO) into a metal alkyl carbon bond is a fundamental reaction step in selective hydroformylation of aliphatic olefins to linear aldehydes, which is one of the large-scale industrial processes based on homogeneous catalysis.1–3 Isocyanides in turn are more versatile C1 building blocks, which are known for their remarkable potential as partners in multicomponent addition reactions, most notably through the work of Ugi.4 More recently, MI of isocyanides5,6 in Pd-catalyzed C/C coupling reactions has attracted much attention.7–9 Even double insertion into two C–H bonds of a η5-coordinated cyclopentadienyl group at Cu(I) leading to an 6-aminofulvene-2-aldimine complex has been reported.10 In contrast, MI of CO into metal acyl bonds has only rarely been reported and considered thermodynamically unfavorable, which is fundamental for the chemoselectivity of hydroformylation.11,12 The reactivity of related formyl complexes is governed by the high stability of either metal hydride (reverse MI) or the CO complex (hydride transfer).13–15 However, migration of an acyl group under CO pressure has been reported for an oxorhenium complex leading to cis-coordinated acetate and CO.16 Analogous experiments including metal bonded acyl groups and isocyanides have not been reported so far. By chance, we now found a MI step of isocyanide into a tungsten ketenyl bond.First ketenyl complexes were obtained by Lewis base induced C/C coupling of coordinated CO and a metal bound alkylidyne at the group VI metals Mo and W.17–21 Depending on the formal electron count at the metal either an end-on κ1 or a side-on η2 coordination mode is adopted (A or B, respectively, Chart 1).22–24 Later, a number of alternative access routes with W, Mn, Re, Fe and even La have been developed,25–29 which involve either reductive coupling of CO29 or use of Cn building blocks like carbon suboxide C3O2 or oxalyl chloride.30,31 The latter examples allowed the isolation of bridging μ-κ1-ketenyl (C) or ketenylidene32–34 ligands. The only known ketenyl complexes with the prototype –C(H)CO were successfully isolated within this dinuclear scaffold by addition of CO to a CH-alkylidene bridged diiron35 or a dimolybdenum36 complex. A related azaallenyl ligand –C(H)CNR at niobium could be obtained by reaction of a methylidyne complex with isocyanate leading to an Nb oxo species.37
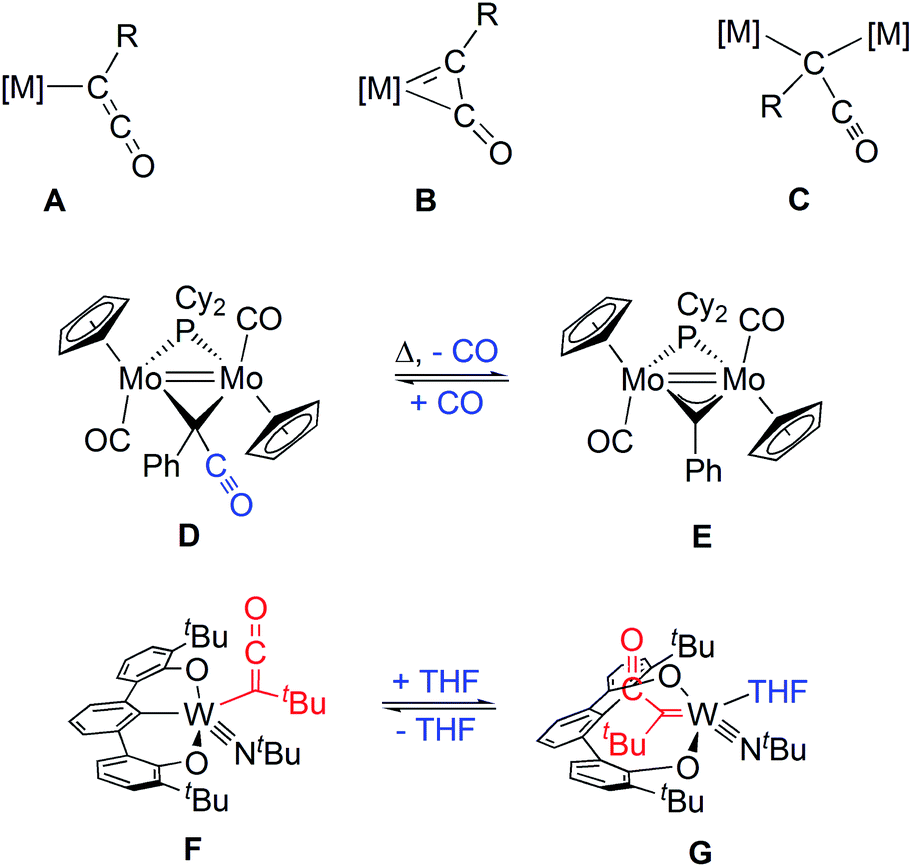 | ||
| Chart 1 Types of ketenyl complexes; reversible CO addition/release and reversible intramolecular nucleophilic addition. | ||
Investigations on the reactivity of ketenyl complexes comprise reactions with Lewis acids and alkylation at oxygen leading to the corresponding side-on alkyne complexes.38 However, reversible coupling reactions with ketenyl ligands are of particular mechanistic interest. Recently published dinuclear [(η5-C5H5)Mo] complexes, bridged likewise by an alkylidyne ligand, allowed a reversible addition of CO affording a μ-κ1-ketenyl ligand (D and E, Chart 1).36,39,40 Similarly, the coupling reaction of a ketenyl ligand with a phenyl group of a pincer ligand at tungsten is reversible depending on THF coordination (F and G, Chart 1).41 MI into a W-ketenyl bond has been reported for only a single case employing isocyanate, but the underlying mechanism remained open.42
Our research in turn is directed to supramolecular coordination chemistry based on donor-substituted alkynes. Side-on alkyne complexes with thiolate,43–45 amide46,47 or phosphine48,49 substitution in both α-positions represent chelates, which can serve as redox-active metallo-ligands.50 In the course of this studies aiming at alkyne complexes with direct oxygen-substitution we obtained a mononuclear W-complex with the prototypical κ1-ketenyl ligand C(H)CO. Reactivity studies uncovered a facile cyclisation reaction with isocyanides.
Results and discussion
Synthesis and characterization of W-Ketenyl complexes
The synthesis of the κ1-ketenyl complex 4a (Scheme 1) succeeded by dealkylation of the corresponding alkyne complex [Tp*W(CO)2(η2-HC2OEt)]PF6, {Tp* = hydridotris(3,4,5-trimethylpyrazolyl)borate}, 2a-PF6. The latter was obtained by treatment of [Tp*W(CO)3] with FcPF6 and subsequently the ethinyl ether derivative HC2OEt. The identity of 2a-PF6 as side-on 4e donor alkyne complex was confirmed by 13C NMR resonances at 212.4 and 207.0 ppm for the W bound alkyne C atoms, respectively. The CO stretching frequencies are observed at 2074 and 1997 cm−1. | ||
| Scheme 1 Synthesis and resonance structures of ketenyl complex 4a. | ||
Treatment of cationic alkyne complexes of type 2a+ with halide ions usually leads to neutral halogen species by substitution of one CO ligand. This was indeed observed by conversion of 2a-PF6 to [Tp*W(CO)Cl(η2-HC2OEt)] 3, in dichloromethane irrespective if n-Bu4Cl or n-Bu4I was used. Such influence of the solvent in reactions with n-Bu4NI and carbonyl complexes has already been observed in similar reactions.51 In contrast, change of the conditions to n-Bu4I in THF afforded the ketenyl complex 4a after several hours. Compound 4a could be isolated by column chromatography in good yields (43% over two steps). The O-dealkylation by formation of ethyl iodide proceeds under mild conditions, while formation of the W-iodide derivative is obviously hampered. Given the redox potentials of corresponding W-alkyne complexes52 and the iodine/iodide couple, a radical mechanism with preceding electron transfer can be largely excluded. Interestingly, related metal-free ketene formation by rearrangement of silyloxyalkynes was reported by Ponomarev.53 The Tp′ congeners 2b-PF6 and 4b {Tp′ = hydridotris(3,5-dimethylpyrazolyl)borate} were synthesized accordingly for improvement of XRD data. The identity of 3, 4a and 4b was determined by single-crystal XRD analysis. The molecular structure of 4b is depicted in Fig. 1. Complex 4a exhibits a disorder of the ketenyl ligand in the crystal structure (see Fig. S1 and S2† for molecular structures of 3 and 4a).
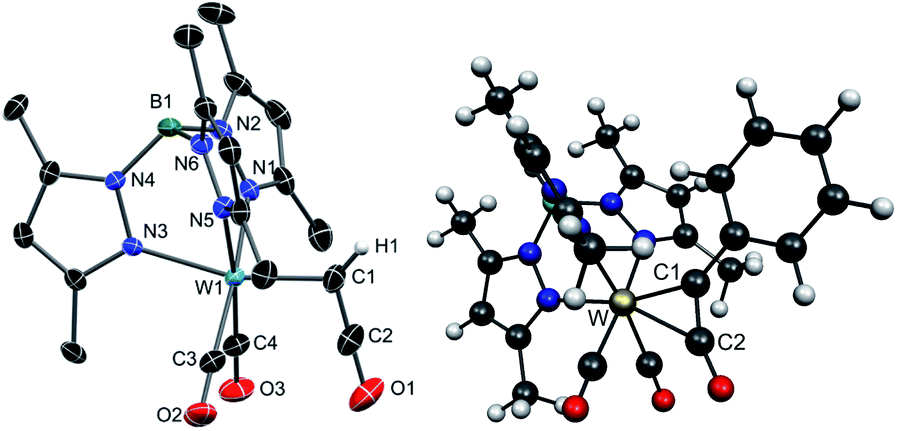 | ||
| Fig. 1 Left: molecular structure of 4b in the crystal with thermal ellipsoids set at 50% probability. Hydrogen atoms except for the ketenyl H have been omitted for clarity. Selected bond lengths [Å]: W1–C1 2.053(3), W1–C3 1.981(2), W1–C4 1.954(2), C1–C2 1.307(4), C2–O1 1.175(4), W1–N1 2.206(2), W1–N3 2.176(2), W1–N5 2.222(2). Right: geometry optimized structure of Ph-4b calculated by DFT; W–C1 2.032, W–C2 2.300. | ||
Compounds 4a/b are a novel type of mononuclear complexes with the rare prototype ketenyl ligand –C(H)CO, and represent formally W(II) 16 ve species. In the light of a potential formation of a 18 ve side-on ketenyl complex, which was indeed reported by Hill et al. for the related complex [TpW(CO)(PPh3){C(p-tolyl)CO}], F (Scheme 1),20 the connectivity of 4a/b is remarkable.
The W–C1 bond length of 2.053(3) Å is very short as compared with 2.27(2) Å in the related W(II) complex [Cp(CO)(PMe3)2W–C(R)CO],17 which is attributed to the strong Lewis acidity of the 16 ve tungsten center. Consistently, W–C1 rather matches the W ketenyl bond length in the W(VI) complex [L(O)W–C(t-Bu)CO] featuring the amidodioxolate ligand L3− = N{C6H3(Me)C(CF3)2O}2.54 Even the W carbene bonds in 7–11 (vide infra) with formal double bond show distinctively larger bond lengths. The resonance structure of 4a on the right in Scheme 1 takes the short metal carbon bond into account, representing now a 18 ve W(0) complex. However, the C1–C2 bond length of 1.307(4) Å is compatible with a double bond.
The characteristic ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) as(C
as(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) absorption band at 2030 cm−1 observed in the IR spectrum of 4a is higher in energy than the CO vibrations (1908, 1817 cm−1). Cyclic voltammetry with 4a showed a reversible W(II/I) redox process at a moderate potential of −1.56 V vs. Fc/Fc+, reflecting the 16 ve character (Fig. S7†). The 13C resonances of C1 at 173.0 ppm and of C2 at 172.1 ppm barely differ. Especially the value of C1 lies outside the range of W coordinated carbenes (δ > 200 ppm) and confirms the dominating single bond character of the ketenyl ligand to the metal. Compounds 4a/b are stable at room temperature under atmospheric conditions and the ketenyl ligand remains in its κ1-coordination mode in solution.
CO) absorption band at 2030 cm−1 observed in the IR spectrum of 4a is higher in energy than the CO vibrations (1908, 1817 cm−1). Cyclic voltammetry with 4a showed a reversible W(II/I) redox process at a moderate potential of −1.56 V vs. Fc/Fc+, reflecting the 16 ve character (Fig. S7†). The 13C resonances of C1 at 173.0 ppm and of C2 at 172.1 ppm barely differ. Especially the value of C1 lies outside the range of W coordinated carbenes (δ > 200 ppm) and confirms the dominating single bond character of the ketenyl ligand to the metal. Compounds 4a/b are stable at room temperature under atmospheric conditions and the ketenyl ligand remains in its κ1-coordination mode in solution.
The intrinsic reason for the structural difference between 4a/b and F was indicated by DFT calculations with 4a/b and its virtual Ph derivatives Ph-4a/b. Mere geometry optimization showed κ1 in 4a/b and η2 in Ph-4a/b being the more stable coordination mode (Fig. 1, S40 and S41†). Apparently, the higher polarity of the CH bond leaves a more electron rich metal bound carbon atom. In turn, a resulting stronger donation to tungsten prevents side-on coordination.
Alkylation of the ketenyl oxygen with Meerwein salt Me3OBF4 led to the side-on coordinated alkyne complex 5-BF4 (Scheme 2).38,55 In addition to spectroscopic characterization, the identity as alkyne complex was checked by single-crystal XRD analysis (Fig. S3†). The alkylation is reversible, because addition of n-Bu4NI to the methyl derivative 5-BF4 in THF restores compound 4a. In the course of weeks complex 4a is subjected to a slow conversion process by release of CO leading to compound 6, which turned out to be a formal dimerization product of the corresponding carbyne complex [Tp*(CO)2WCH]. The process could be accelerated by refluxing in toluene. Irradiation experiments resulted no reaction of 4a with visible light (450 nm), however, formation of the carbyne complex (CO = 1985, 1891 cm−1) and subsequent reaction to 6 was observed at ambient temperature under UV light (Hg lamp, Fig. S39†). Accordingly, direct release of CO is more likely than a preceding migration of CO to tungsten being supported by the 20 ve count of the presumed tricarbonyl carbyne complex. The structural identity of 6 as vinylidene bridged dimer was proven by single crystal XRD analysis, which however did not provide a satisfying data set (Fig. S4†). Furthermore, the identity of 6 is supported by the corresponding Tp' derivative [(Tp′)2W2(CO)4(μ-CCH2)], which was discovered on a different preparative route by Templeton.56 According to this work, the asymmetric bridging moiety represents a rare case of a side-on coordinated vinylidene. The IR spectroscopic characteristics of 6 (CO = 1972, 1913, 1876, 1827 cm−1) and the published Tp′ derivative (CO = 1977, 1916, 1876, 1825 cm−1) match very well. In addition, the 13C resonances of the bridging carbon atoms are detected at 304.4 and −2.9 ppm (Tp′ ref. 304.4, −3.3 ppm). The two methylene protons are equivalent (δH = 2.21 ppm) proving the C2 symmetry of the complex in solution.
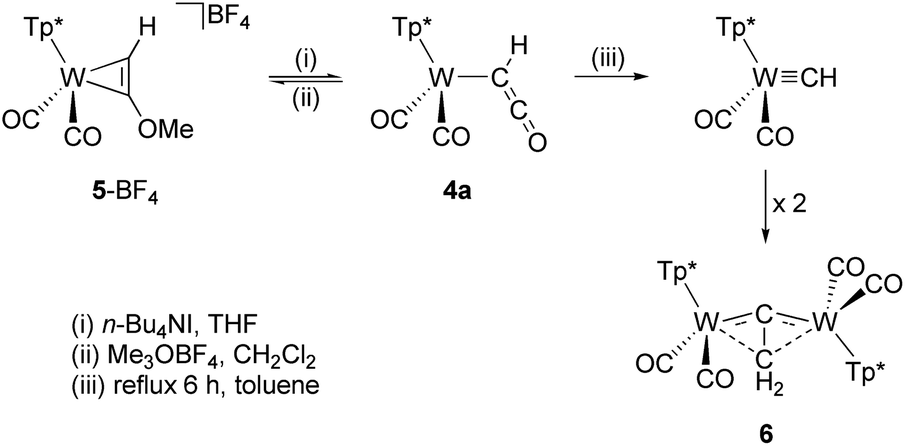 | ||
| Scheme 2 Reversible alkylation of 4a and CO release. | ||
Typical organic reactions of ketenes like 2 + 2 cycloaddition with imines (Staudinger synthesis)57 or reactions with protic nucleophiles such as alcohols and amines58 could not be observed with ketenyl complex 4a. However, the observed release of CO from 4a promoted a study aiming at substitution of CO by related σ-donor/π-acceptor ligands like isocyanides or phosphines. An intriguing reactivity was anticipated with isocyanides, because preliminary experiments uncovered a complex reaction behavior according to several consecutive color changes and indicative changes in IR spectra.
Formation of oxametallacycles via MI
Treatment of complex 4a with 2,6-dimethylphenylisocyanide (Xy-NC) at low temperature afforded isolable products only, if protic compounds were subsequently added. Reaction of 4a with Xy-NC at −78 C, warming until a color change to orange is observed and subsequent addition of methanol, ethanol or di(isopropyl)amine afforded stable products. After purification by column chromatography and subsequent crystallization compounds 7–9 were isolated (Scheme 3, left).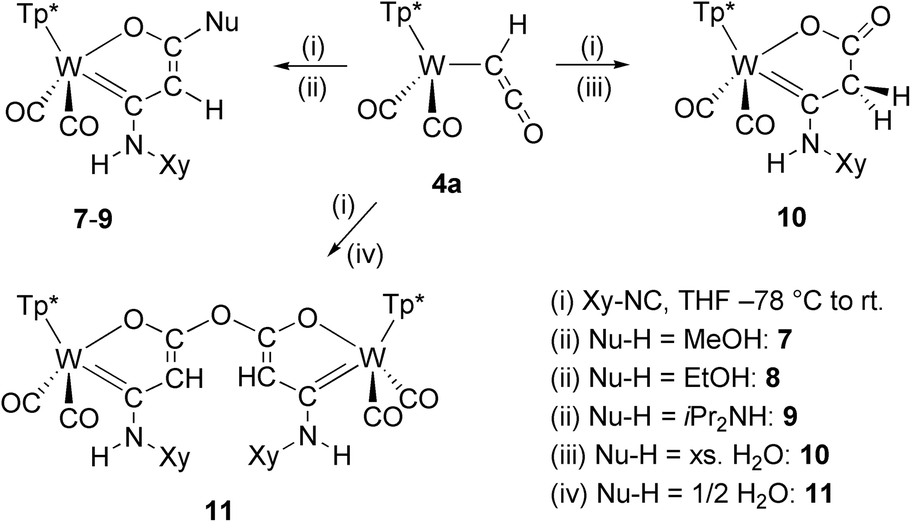 | ||
| Scheme 3 Cycloaddition reactions of ketenyl complex 4a. | ||
The connectivity was uncovered in all cases by single crystal XRD analysis. Compounds 7–9 represent cyclization products of the ketenyl moiety, the isocyanide and one equivalent of alcohol or amine at the metal template. The formed five-membered oxametallacycles are composed of three atoms of the ketenyl moiety (C2O), one carbon atom of the isocyanide and tungsten. Apparently, the nucleophile has attacked the carbonyl group, while the corresponding proton is bound to the nitrogen atom of the isocyanide. As a result, tungsten is coordinated by a Fischer type carbene enolate chelate ligand. If an excess of H2O was deliberately added as nucleophile, 10 was formed by keto–enol tautomerization (Scheme 3, right). The vibration of the resulting keto group can be clearly identified in the IR spectrum at 1659 cm−1.
The molecular structures of 8 and 10 are depicted in Fig. 2 (see Fig. S5 for 7 and Fig. S6† for 9) and selected bond lengths are compiled in Table 1. All structures of 7–10 have in common that the oxametallacycles are planar. The mean bond lengths of 7–9 within this ring amount to 1.40 Å for C1–C2, 1.38 Å for C2–C3 and 1.28 Å for C3–O1 noticeably indicating strong delocalization. As a result, the tungsten carbene bond W1–C1 of 2.18 Å is comparatively long within the normal range of 2.11–2.18 Å for Fischer type W(II) carbene complexes.59,60 In contrast, compound 10 clearly shows single bond character in C1–C2 of 1.50 Å and C2–C3 of 1.51 Å. In turn, the lack of delocalization causes a substantially shorter tungsten carbene bond W1–C1 of 2.11 Å. For comparison, an even shorter W–C bond of 1.94 Å was reported for the related cationic W(II) complex [Tp′(CO)2W = CPhMe]+, which clearly reflects the unsaturated nature in the latter 16 ve complex.61
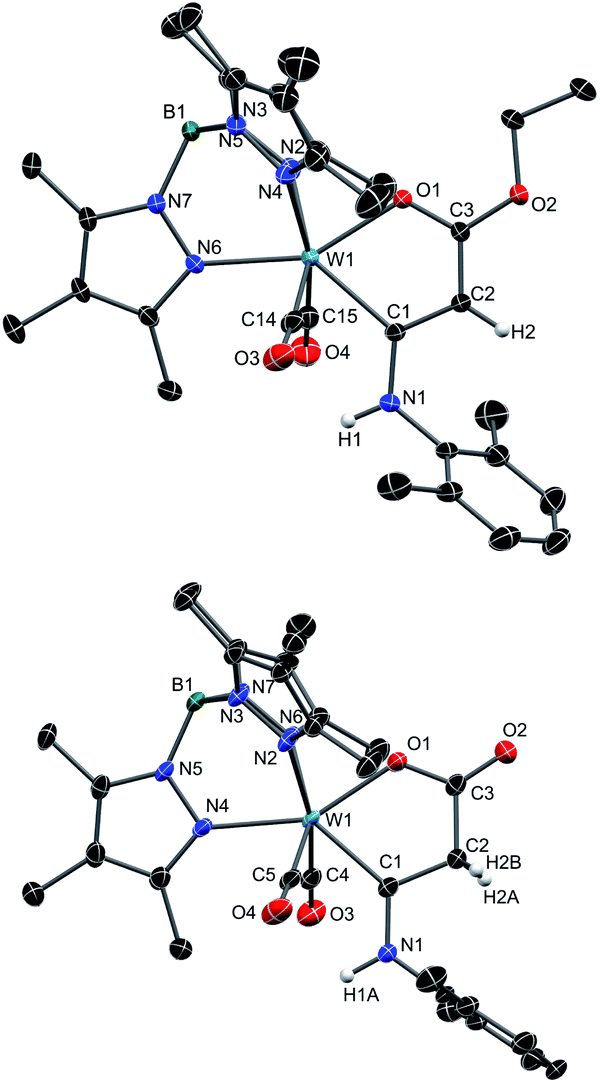 | ||
| Fig. 2 Molecular structures of 8 (top) and 10 (bottom) with thermal ellipsoids set at 50% probability. Co-crystallized CH2Cl2 molecules and hydrogen atoms except for the H-atoms on C2 have been omitted for clarity. Selected bond lengths are summarized in Table 1. | ||
| 7 | 8 | 9 | 10 | |
|---|---|---|---|---|
| W1–C1 | 2.183(4) | 2.189(3) | 2.1707(16) | 2.113(2) |
| W1–O1 | 2.115(3) | 2.110(2) | 2.0845(12) | 2.0929(19) |
| C1–C2 | 1.399(6) | 1.401(4) | 1.388(2) | 1.502(4) |
| C1–N1 | 1.357(5) | 1.366(4) | 1.369(2) | 1.340(3) |
| C2–C3 | 1.367(4) | 1.369(4) | 1.401(2) | 1.510(4) |
| C3–O1 | 1.272(5) | 1.281(3) | 1.290(2) | 1.292(3) |
| C3–O2 | 1.349(5) | 1.347(4) | — | 1.214(3) |
| C3–N2 | — | — | 1.359(2) | — |
The differences are also reflected in the 13C NMR data (Table 2). The carbene carbon atom (C1) of 10 at 245.2 ppm exhibits a downfield shift of about +25 ppm compared to the compounds 7 and 8. The carbon atom C2 of 10 detected at 49.3 ppm is strongly highfield shifted (−35 ppm) in accordance with sp3 hybridization. The chemical shifts of C3 come within the limits of 173.6 ppm for 9 and 180.3 ppm for the keto group in 10.
If the ketenyl complex 4a was treated with the isocyanide Xy-NC and just adventitious water under the condition described above, a double cyclisation process resulted in the dinuclear complex 11 (Scheme 3). Two formed oxametallacycles are now connected by a bis(enolate) anhydride bridging unit. The molecular structure of the dinuclear complex 11 is depicted in Fig. 3. An additional structural feature pertains to the torsion angle of the two five-membered chelate rings, which amounts to 67.5° leading to a intermetallic distance of 8.15 Å. The facile formation of the dinuclear complex 11 promoted us to use diprotic nucleophiles like ethanediol or hydroquinone in half-equivalent ratio to initiate formation of dimeric compounds based on more extended bridges between the monomeric W complex units. All these attempts failed due to the preferred formation of 11.
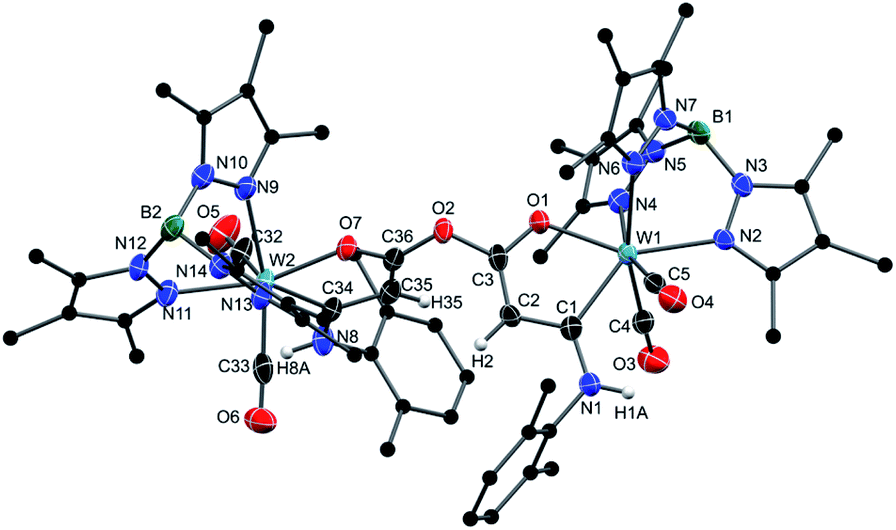 | ||
| Fig. 3 Molecular structure of 11 in the crystal of 11 3CH2Cl2 with thermal ellipsoids set at 50% probability. The carbon atoms of the Tp* and the Xy-group are displayed as ball and stick. Co-crystallized CH2Cl2 molecules and hydrogen atoms except for H1A, H2, H8A and H35 have been omitted for clarity. Selected bond lengths [Å] and torsion angle [°]: W1–C1 2.170(4), W1–C4 1.972(4), W1–C5 1.955(4), W1–O1 2.101(2), C1–C2 1.406(5), C1–N1 1.364(5), C2–C3 1.360(5), C3–O1 1.270(4), C3–O2 1.382(4), C2–C3–C36–C35 67.5(4). | ||
Mechanistic investigations
To elucidate the mechanism of the cyclization, we undertook IR monitoring of the formation of 8. Addition of the isocyanide and warming to ambient temperature resulted in a change of the ketenyl vibration in 4a (2031 cm−1) to a higher value (2055 cm−1). This intermediate IM2 showed an additional band for a CN double bond at 1611 cm−1. These bands decreased after addition of EtOH, while the product 8 with two CO-bands (1931, 1834 cm−1) and one NH-band (3412 cm−1) was finally formed. In a second experiment, we added CD3OD in order to trace the position of the acidic deuterium. The respective 1H NMR spectrum showed unequivocally full proton population at the ring CH position and all deuterium at nitrogen (Fig. S29†). Based on these observations, we suggest the reaction mechanism depicted in Scheme 4.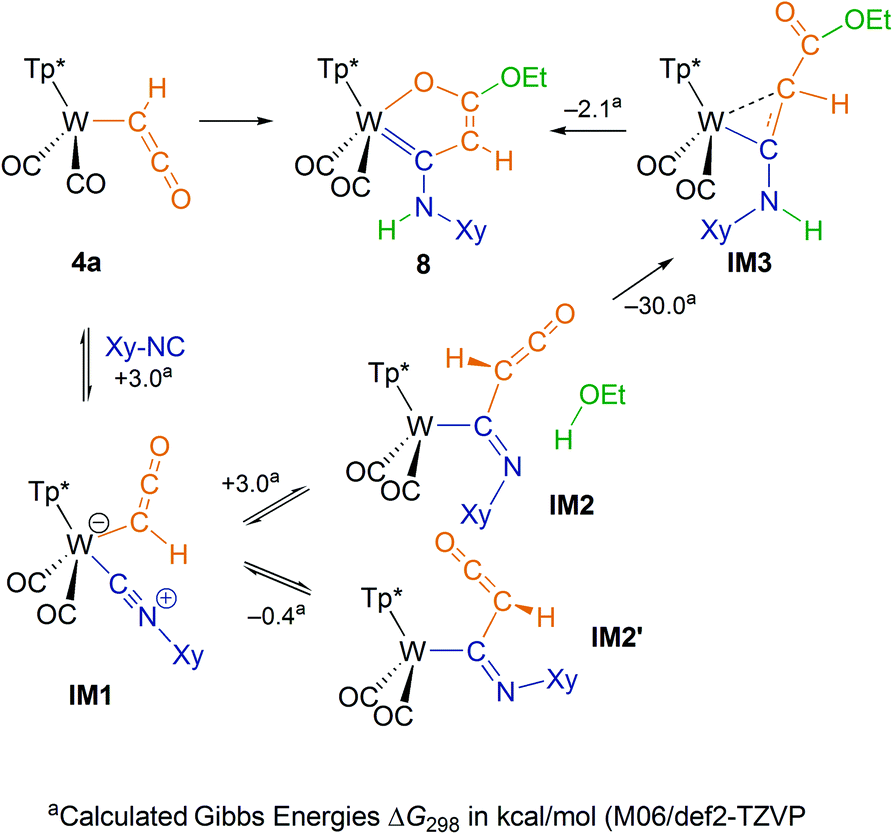 | ||
| Scheme 4 Mechanism for the formation of the oxametallacycle 8via MI. | ||
The reaction started by coordination of the isocyanide at the metal leading to a coordinatively saturated 18 ve complex IM1. Upon warming at about −20 °C a color change from green to orange was observed. MI of the isonitrile into the W-ketenyl bond resulted in intermediate IM2. In contrast to the metal-bound ketenyl ligand, the formed organic ketenyl group is now amenable to addition of protic agents. This step proceeds very likely as concerted addition by protonation at nitrogen and nucleophilic attack at the ketenyl carbonyl C-atom. An alternative addition over the ketenyl CC double bond and subsequent proton migration to nitrogen can be excluded by the labeling experiment with CD3OD. Finally, the coordinated acrylate derivative IM3 transforms into a chelate ring involving double bond isomerization.
The described mechanism could further be substantiated by DFT calculations. The Gibbs energy profile of potential intermediates (M06/def2-TZVP) depicted in Scheme 4 reveals small ΔG values for the isocyanide coordination as well as for migratory insertion supporting the equilibrium character of these steps, but proves the downhill character of the whole cyclization. Two rotational isomers IM2 and IM2′ were identified, while the planes of C(H)CO and WCN are oriented rather perpendicular to each other. Transition states were calculated for the migratory insertion and the ethanol addition step at BP86/def2-SVP level of theory (Fig. S43†). The activation barriers of 2.4 and 5.7 kcal mol−1, respectively, are small as well. The very low barrier for the MI step is reflected by the acute WCC′ angle of 71° between isocyanide and ketenyl ligand in the seven coordinate complex IM1. In the light of these low barriers, the observation of the MI intermediate by IR spectroscopy can be attributed to the higher stability of the kinetic dead end species IM2′ proving the concerted addition step between IM2 and ethanol, which is conceivable only in the higher energy rotamer. Attempts to isolate potential intermediates were not successful so far. Variation of the solvent from THF to acetonitrile did not prevent formation of 8, which shed light on the fact that acetonitrile does not effectively block the free coordination site at the metal.
Intermetallic electronic cooperativity in complex 11
All derivatives 7–11 were examined by cyclic voltammetry. Reversible W(II/III) redox events at E1/2 = −0.2 V were observed for 7–9, while 10 exhibits only an oxidative process at higher potential of +0.17 V (Fig. S8†). The dinuclear complex 11 exhibits two reversible clearly separated waves at E1/2 = +0.16 V and +0.40 V (Fig. S9†), which both can be assigned to W(II)/W(III). The potential difference proves the electronic cooperativity of both W centers, while the value of 0.24 V points to localized rather than delocalized states. Calculation of the comproportionation constant from the potential difference resulted Kkomp ∼ 104 being indicative of the stability of the mixed-valent W(II)/W(III) state 11+. Due to the medium potential difference the question of the cause of electronic cooperativity arises.62 A true delocalization of electronic states is rather excluded due to the torsion angle between the chelate planes in the molecular structure. In order to prove the contribution of sole electrostatic interaction, we performed cyclovoltammetric investigations with various conducting electrolytes based on n-Bu4N+ salts with anions of different size (Cl−, BF4− and PF6−).63 In doing so, a reduction of the potential difference was observed with decreasing anion size (Fig. S9†). Chloride as smallest anion tends to form a strong ion pair with 11+, which decreases the electrostatic interaction and hence the observed potential difference. Accordingly, electrostatic interaction is substantial.Finally, spectroelectrochemical (SEC) measurements were performed, at which the CO vibration bands served as a suitable IR probe (Fig. 4). During the first oxidation step the bands of the starting material 11 at 1936 und 1841 cm−1 decreased, while new bands of 11+ arose at 2026, 1944, 1851 cm−1. Apparently, within the IR time frame there are localized W(II) and W(III) centers, but the change of about 10 cm−1 for the CO ligands at the metal center being unaffected prove a mesomeric effect of the remote W(III). This state can also be generated by stoichiometric oxidation with [Fc]PF6, which exclusively resulted in 11+ (Fig. S11†).
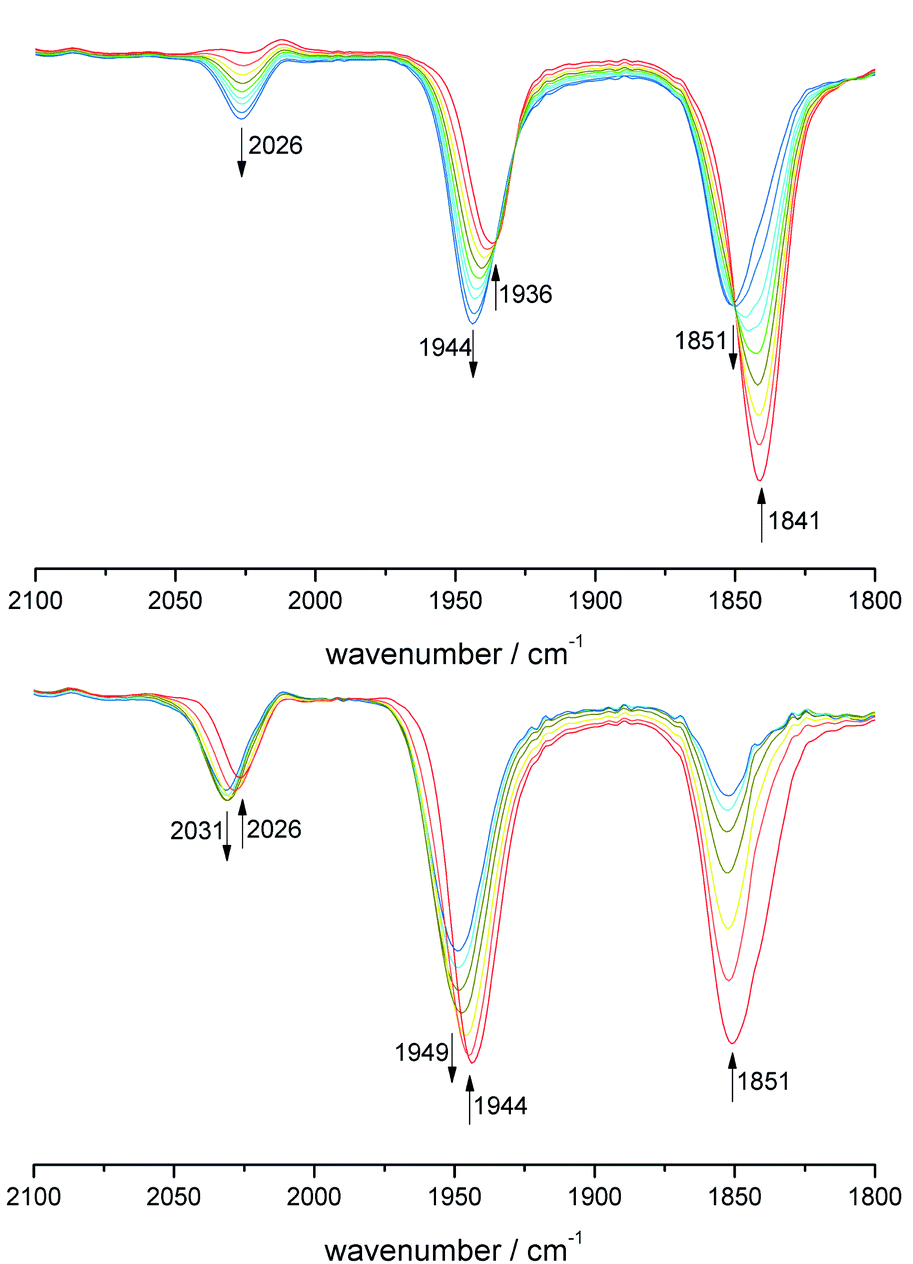 | ||
| Fig. 4 IR-SEC of 11: top: E = 0.21 V 11 (red) to 11+ (blue). Bottom: E = 0.45 V 11+ (red) to 112+ (blue). | ||
After passing through the second redox potential, SEC measurements show the appearance of only two CO bands at 2031 and 1949 cm−1 confirming the formation of 112+ with two W(III) centers. These wavenumbers show also a slight change of +5 cm−1 compared to the W(III) moiety in 11+. According to Geiger and Atwood, these changes can be used to calculate the partial delocalization of charge by setting these deviations into relation to the total change at the originally oxidized unit. In case of 11+ a value of 7% charge delocalization was obtained (for calculation see ESI†).64 According to all electrochemical and spectroscopic evidence, complex cation 11+ can be assigned to a Robin-Day class II type mixed-valence system.
Conclusions
We have reported the reversible formation of the end-on ketenyl tungsten complex [Tp*W(CO)2{κ1-C(H)CO}] from the corresponding side-on alkyne complex of ethinylethyl ether. The formal 16 ve complex, which is stable at room temperature, represents a first example of a prototype HCCO ligand devoid of bridging function. Reactivity studies uncovered loss of CO at elevated temperatures and subsequent dimerization with concomitant isomerization of the intermediate CH carbyne complex. In addition, a template controlled cyclization reaction of the ketenyl moiety with an isocyanide in the presence of protic nucleophiles was uncovered. According to IR monitoring a MI of the isocyanide into the tungsten ketenyl bond is involved. In contrast to the metal coordinated ketenyl ligand, the MI intermediate is susceptible to addition of alcohols, amines or water affording planar oxametallacycles. The mechanism could be derived from IR and NMR spectroscopic studies and supported by results of DFT calculations. The isolation of a mononuclear end-on complex with the lightest ketenyl ligand and its migration behavior is highlighted by the recent discovery that interstellar HCCO is a surprisingly abundant radical.65,66 The dual cyclometallated complex connected by a bis(enolate) anhydride moiety was investigated in detail by cyclic voltammetry and spectroelectrochemistry (SEC) in order to obtain information on the intermetallic cooperativity in the mixed valent compound. The separation of the W(II)/W(III) redox potentials of about 200 mV proves intermetallic cooperativity, which is mainly assigned to electrostatic forces. However, IR SEC measurements allowed an estimate of 7% charge delocalization. Accordingly, the singly-oxidized dinuclear W(II)/W(III) complex belongs to class II of the Robin and Day classification. Ongoing investigations are directed towards the use of cyclic carbenes for Wulff–Dötz type reactions67,68 in order to develop a synthetic pathway to complex organic derivatives. Future research must address the question, if the discovered MI reactivity reflects a general bias of the prototypical ketenyl group. Because the latter represents a fundamental organic building block, respective cyclization reactions in a catalytic routine might be of high interest in organic synthesis.Experimental section
Materials and methods
All operations were carried out in an atmosphere of dry argon using Schlenk and glovebox techniques. Solvents were dried and saturated with argon by standard methods and freshly distilled prior to use. One- and two-dimensional NMR spectra were recorded at 300 K with Bruker AVANCE 250, 300 or 500 MHz spectrometers, respectively. In 1H and 13C NMR, the chemical shifts were internally referenced to the solvent residual peak. IR spectroscopy was conducted on a Nicolet 380 FT-IR with a Smart Orbit ATR module. Elemental analyses were performed with a Thermo Finnigan Flash EA 1112 Series. Mass spectrometry by electrospray ionization (ESI) was obtained with an Agilent 6210 time-of-flight LC/MS. [Tp*W(CO)3] (1)48 was prepared according to literature methods. All other chemicals were obtained from commercial sources (at least of reagent grade quality) and used as received. Analytical thin layer chromatography was performed on silica gel (Silica 60 F254). Column chromatography was performed using silica gel 60 (pore size 0.063–0.2 mm) as stationary phase purchased from Merck.Cyclic voltammetry and spectroelectrochemistry
CV experiments were performed using a Princeton Applied Research VersaSTAT 3. A three-electrode arrangement with glassy carbon working electrode, platinum wire counter electrode and Ag/AgBF4/CH3CN reference electrode and 0.1 M n-Bu4NPF6, n-Bu4NBF4 or n-Bu4NCl in CH2Cl2 as supporting electrolyte were employed. The ferrocene/ferrocenium (Fc/Fc+) redox couple was used as internal standard. Spectroelectrochemistry experiments were performed using a Princeton Applied Research VersaSTAT 3 combined with a Bruker Alpha compact FT-IR spectrometer. The redox potential remained constant while IR spectra were recorded over the time until the conversion was complete. A three-electrode arrangement with platinum grid working electrode, platinum grid counter electrode and Ag/AgCl reference electrode and 0.25 M n-Bu4NPF6 in 1,2-dichloroethane (DCE) as supporting electrolyte were employed. The setup was placed in an IR cell with KBr windows.Synthesis of [Tp*,′W(CO)2(η2-HCCOEt)]PF6 (2a/b-PF6)
To a solution of 1a/b (a: 2.00 g, b: 1.86 g, 3.29 mmol) in 50 mL CH2Cl2 was added ethinylethyl ether (0.30 g, 4.28 mmol). After cooling to 0 °C [Fc]PF6 (1.09 g, 3.29 mmol) was added in portions over a period of 1 hour. Gas formation and color change from red to dark yellow was observed. After warming to r.t. and stirring for another hour complete formation of 2a/b-PF6 was confirmed by IR. All volatiles were removed in vacuo and the residue was dissolved in 10 mL CH2Cl2 and layered with 60 mL Et2O. Finally the yellow supernatant was decanted and 2a/b-PF6 was collected after drying in vacuo. Yield: 2a-PF6: 1.67 g (2.10 mmol, 64%), 2b-PF6: not determined. 2a-PF6: Anal. calcd for C24H34BF6N6O3PW 4CH2Cl2 (1133.9): C, 29.66; H, 3.73; N, 7.41. Found: C, 29.50; H, 3.23; N, 6.71. 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 13.08 (s, 1H, CCH), 4.79 (s, 2H, CH2O), 2.47 (s, 6H, CCH3), 2.43 (s, 3H, CCH3), 2.35 (s, 3H, CCH3), 1.96 (s, 6H, CCH3), 1.94 (s, 6H, CCH3), 1.90 (s, 3H, CCH3), 1.52 (s, 3H, CH2CH3). 13C{H} NMR (CDCl3, 62.9 MHz, 300 K): δ 225.5 (WCO), 212.4 (HCOCH2), 207.0 (WCH), 151.8, 151.1, 147.0, 144.3 (CCH3), 115.4, 114.7 (CCH3(CCH3)2), 81.9 (CH2), 15.1, 14.7, 11.6, 11.0 (CCH3), 8.3 (CH2CH3). 31P{H} NMR (CDCl3, 121.5 MHz, 298.2 K): δ −146.9 (sept, PF6). 19F{H} NMR (CDCl3, 282.4 MHz, 298.2 K): δ 75.51 (d, PF6). IR (CH2Cl2, cm−1): 2566 (w, BH), 2074 (s, CO), 1997 (s, CO). 2b-PF6: IR (CH2Cl2, cm−1): 2566 (w, BH), 2076 (s, CO), 2000 (s, CO).
Synthesis of [Tp*WCl(CO)(η2-HCCOEt)] (3)
The crude product 2a-PF6 (prepared with 1.0 g, 1.65 mmol of 1 in 40 mL CH2Cl2) was used in a one-pot reaction. Addition of n-NBu4Cl (0.46 g, 1.65 mmol) caused gas evolution, while the solution turned green. After 1 h the completeness of the reaction was be checked by IR and the solvent was removed in vacuo. By chromatography a blue band was eluted with CH2Cl2. Crystallization by layering a CH2Cl2 solution with n-pentane gave 3 as dark blue crystals. Yield: 0.378 g (0.576 mmol, 35%). Anal. calcd for C23H34BClN6O2W·CH2Cl2 (741.6): C, 38.87; H, 4.89; N, 11.33. Found: C, 38.89; H, 4.75; N, 11.55. 1H NMR (CDCl3, 500 MHz, 298.2 K): δ 11.02 (s, 1H, HCC), 4.74 (dq, 1H, 2J = 10.1 Hz), 4.63 (dq, 1H, 2J = 10.1 Hz) (CH2CH3), 2.66, 2.52, 2.44, 2.38, 2.27, 1.98, 1.85, 1.83, 1.78 (s, 9 3H, CH3), 1.58 (t, 3H, CH2CH3). 13C{H} NMR (CDCl3, 125.8 MHz, 298.2 K): δ 230.4 (WCO), 216.5 (WCOCH2), 175.6 (WCH), 152.1, 151.9, 150.2, 143.3, 141.6, 140.8, 113.6, 113.1, 113.1 (CCH3), 77.3 (CH2CH3), 15.1 (CH2CH3), 14.5, 14.2, 13.8, 11.2, 11.1, 10.9, 8.4, 8.4, 8.2 (CCH3). IR (THF, cm−1): 2551 (w, BH), 1920 (s, CO).
Synthesis of [Tp*,′W(CO)2(κ1-HCCO)] (4a/b)
After synthesis of 2a/b-PF6 (with 2.00 g, 3.29 mmol of 1a, 1.86 g of 1b) the greyish green residue was dissolved in 40 mL THF. Rapid addition of n-NBu4I (1.21 g, 3.29 mmol) to the reaction solution was necessary, since 2a/b-PF6 catalyzes the polymerization of THF over time. After 2 h the solution was yellowish and complete conversion was confirmed by IR. All volatiles were removed in vacuo. By chromatography a green band was eluted with CH2Cl2. All volatiles were removed in vacuo and finally the green residue was washed twice with 20 mL n-pentane and dried in vacuo. Yield: 4a: 0.875 g (1.41 mmol, 43%), 4b: not determined. 4a: Anal. calcd for C22H29BN6O3W 0.5 CH2Cl2 (662.6): C, 40.78; H, 4.56; N, 12.68. Found: C, 40.51; H, 4.69; N, 12.47. 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 14.43 (s, 1H, HCCO), 2.51 (s, 3H, CCH3), 2.48 (s, 6H, CCH3), 2.30 (s, 3H, CCH3), 1.87 (s, 3H, CCH3), 1.86 (s, 6H, CCH3), 1.73 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 75.5 MHz, 298.2 K): δ 239.7 (WCO), 172.3 (HCCO), 171.8 (HCCO), 157.0, 149.7, 144.7, 142.1, 114.8, 112.7 (CCH3), 15.7, 14.9, 11.4, 10.9, 8.6, 8.3 (CCH3). IR (CH2Cl2, cm−1): 2547 (w, BH), 2031 (s, CCO), 1919 (s, CO), 1831 (s, CO); (ATR, cm−1): 2538 (w, BH), 2012 (s, CCO), 1908 (s, CO), 1817 (s, CO). MS (ESI-TOF) m/z calcd for C22H30BN6O3W+: 621.20 found: 621.20 [M+]. 4b: 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 14.54 (s, 1H, HCCO), 5.97 (s, 1H, CH), 5.87 (s, 2H, CH), 2.57 (s, 3H, CCH3), 2.55 (s, 6H, CCH3), 2.37 (s, 3H, CCH3), 1.79 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 62.9 MHz, 297.5 K): δ 239.8 (WCO), 171.9 (HCCO), 171.2 (HCCO), 159.5, 151.4, 147.0, 144.8 (CCH3), 109.1, 106.9 (CH), 17.4, 16.7, 13.0, 12.6 (CCH3). IR (THF, cm−1): 2552 (w, BH), 2020 (s, CCO), 1917 (s, CO), 1834 (s, CO).
Synthesis of [Tp*W(CO)2(η2-HCCOMe)]BF4 (5-BF4)
A mixture of 4a (0.2 g; 0.32 mmol) and Me3OBF4 (42 mg; 0.32 mmol) was dissolved in 20 mL CH2Cl2. The green solution was stirred overnight. The color changed from light green to dark green. After IR check of the completeness of the reaction the solvent was removed in vacuo. The crude product was recrystallized as dark blue crystals from CH2Cl2/n-pentane. Yield: 0.08 g (0.11 mmol, 34%). Anal. calcd for C23H32B2F4N6O3W (722.0): C, 38.26; H, 4.47; N, 11.64. Found: C, 38.06; H, 4.47; N, 11.24. 1H NMR (CDCl3, 500 MHz, 298.1 K): δ 13.38 (s, 1H, HCCO), 4.65 (s, 3H, OCH3), 2.42 (s, 6H, CCH3), 2.41 (s, 3H, CCH3), 2.33 (s, 3H, CCH3), 1.99 (s, 6H, CCH3), 1.89 (s, 6H, CCH3), 1.88 (s, 3H, CCH3). 13C{H} NMR (CDCl3, 125.8 MHz, 298.2 K): δ 228.0 (WCOCH3), 212.4 (WCO), 207.8 (WCH), 151.8, 151.1, 146.8, 144.1, 115.3, 114.6 (CCH3), 71.7 (OCH3), 14.7, 11.6, 11.0, 8.3, 8.2 (CCH3). 19F{H} NMR (CDCl3, 282.4 MHz, 298.7 K): δ −153.0 (s, 10BF4), −153.1 (s, 11BF4). IR (CH2Cl2, cm−1): 2569 (w, BH), 2075 (s, CO), 1997 (s, CO).
Synthesis of [Tp*2W2(CO)4(μ-CCH2)] (6)
A solution of 4a (0.2 g, 0.32 mmol) in 20 mL toluene was heated under reflux for 5 h. The color changed from green to brown. The solvent was removed at room temperature in vacuo. By chromatography using CH2Cl2 a yellow-green product was collected. The volatiles were evaporated in vacuo affording an olive-green solid, which can be crystallized by vapour diffusion of n-pentane into a toluene solution of 6. Yield: 0.12 g (0.10 mmol, 62%). Anal. calcd for C42H58B2N12O4W2 1.5 toluene (1322.5): C, 47.68; H, 5.34; N, 12.71. Found: C, 47.76; H, 5.23; N, 12.71. 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 2.44 (s, 6H, CCH3), 2.35 (s, 12H, CCH3), 2.21 (s, 2H, CH2), 2.20 (s, 6H, CCH3), 1.87 (br, 12H, CCH3), 1.81 (s, 6H, CCH3). 1H NMR (CDCl3, 500 MHz, 195.2 K): δ 4.63 (br, 2H, BH), 2.66 (s, 6H, CCH3), 2.44 (s, 6H, CCH3), 2.34 (s, 6H, CCH3), 2.32 (s, 6H, CCH3), 2.20 (s, 6H, CCH3), 2.15 (s, 2H, CH2), 1.92 (s, 6H, CCH3), 1.87 (s, 6H, CCH3), 1.81 (s, 6H, CCH3), 1.80 (s, 6H, CCH3). 13C NMR (CDCl3, 125.8 MHz, 298.2 K): δ 304.4 (W(μ-C)W), 152.0, 151.1, 141.9, 141.6, 112.5, 112.3 (CCH3), 14.2, 11.3, 11.1, 8.5 (CCH3),-2.9 (W(μ-CH2)W). IR (toluene, cm−1): 2547 (w, BH), 1972 (s, CO), 1913 (s, CO), 1876 (s, CO), 1827 (s, CO).
General procedure for the synthesis of [Tp*W(CO)2(η2-O,C-oxametallacycle)] (7–11)
One equivalent of Xy-NC was added to a solution of 4a in THF at −78 °C. Upon warming at approx. −20 °C a color change from green to orange was observed. At room temperature the protic nucleophile (7: MeOH, 1 equiv.; 8: EtOH, 1 equiv.; 9: i-Pr2NH, 1 equiv.; 10: H2O, 10 equiv.; 11: H2O, 0.5 equiv.) was added and the solution was stirred overnight. All volatiles were removed in vacuo. By chromatography a yellow to orange band was eluted. The volatiles were removed in vacuo and finally the residue was recrystallized from CH2Cl2/MeOH.
7: eluent: THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PET (1:3). Yield: 21%. Anal. calcd for C32H42BN7O4W (783.4): C, 49.06; H, 5.40; N, 12.52. Found: C, 48.61; H, 5.31; N, 11.51. 1H NMR (CDCl3, 500 MHz, 298.1 K): δ 7.15 (m, 3H, Xy-H), 7.02 (s, 1H, NH), 5.06 (s, 1H, CCH), 3.57 (s, 3H, OCH3), 2.45 (s, 6H, Xy-CH3), 2.40 (s, 3H, CCH3), 2.36 (s, 3H, CCH3), 2.33 (s, 6H, CCH3), 1.93 (s, 6H, CCH3), 1.90 (s, 3H, CCH3), 1.85 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 125.7 MHz, 298.2 K): δ 249.0 (WCO), 217.3 (WC), 175.0 (CHCOMe), 150.2, 149.8, 142.7, 140.8 (CCH3), 140.2 (Xy-Cipso), 137.1 (Xy-CCH3), 128.7, 127.1 (Xy-CH), 113.2, 112.4 (CCH3), 82.5 (CCHC), 54.4 (OCH3), 18.8 (Xy-CCH3), 14.8, 12.2, 11.6, 11.0, 8.7, 8.2 (CCH3). IR (THF, cm−1): 3403 (w, NH), 2556 (w, BH), 1933 (s, CO), 1836 (s, CO).
:PET (1:3). Yield: 21%. Anal. calcd for C32H42BN7O4W (783.4): C, 49.06; H, 5.40; N, 12.52. Found: C, 48.61; H, 5.31; N, 11.51. 1H NMR (CDCl3, 500 MHz, 298.1 K): δ 7.15 (m, 3H, Xy-H), 7.02 (s, 1H, NH), 5.06 (s, 1H, CCH), 3.57 (s, 3H, OCH3), 2.45 (s, 6H, Xy-CH3), 2.40 (s, 3H, CCH3), 2.36 (s, 3H, CCH3), 2.33 (s, 6H, CCH3), 1.93 (s, 6H, CCH3), 1.90 (s, 3H, CCH3), 1.85 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 125.7 MHz, 298.2 K): δ 249.0 (WCO), 217.3 (WC), 175.0 (CHCOMe), 150.2, 149.8, 142.7, 140.8 (CCH3), 140.2 (Xy-Cipso), 137.1 (Xy-CCH3), 128.7, 127.1 (Xy-CH), 113.2, 112.4 (CCH3), 82.5 (CCHC), 54.4 (OCH3), 18.8 (Xy-CCH3), 14.8, 12.2, 11.6, 11.0, 8.7, 8.2 (CCH3). IR (THF, cm−1): 3403 (w, NH), 2556 (w, BH), 1933 (s, CO), 1836 (s, CO).
8: eluent: THF:PET (1:3). Yield: 22%. Anal. calcd for C33H44BN7O4W (797.4): C, 49.71; H, 5.56; N, 12.30. Found: C, 49.42; H, 5.35; N, 11.78. 1H NMR (CDCl3, 300 MHz, 298.3 K): δ 7.13 (m, 3H, Xy-H), 7.00 (s, 1H, NH), 5.02 (s, 1H, CCH), 3.88 (q, 2H, CH2CH3), 2.43 (s, 6H, Xy-CH3), 2.38 (s, 3H, CCH3), 2.34 (s, 3H, CCH3), 2.30 (s, 6H, CCH3), 1.91 (s, 6H, CCH3), 1.88 (s, 3H, CCH3), 1.83 (s, 6H, CCH3), 1.13 (t, 3H, CH2CH3). 13C{H} NMR (CDCl3, 75.5 MHz, 298.2 K): δ 249.0 (WCO), 217.2 (WC), 174.8 (CHCOEt), 150.2, 149.7, 142.7, 140.7 (CCH3), 140.2 (Xy-Cipso), 137.2 (Xy-CCH3), 128.7, 127.0 (Xy-CH), 113.2, 112.4 (CCH3), 82.6 (CCHC), 63.1 (CH2CH3), 18.8 (Xy-CCH3), 14.8 (CH2CH3), 14.8, 12.3, 11.6, 11.0, 8.7, 8.2 (CCH3). IR (THF, cm−1): 3412 (w, NH), 2541 (w, BH), 1931 (s, CO), 1834 (s, CO).
9: eluent: THF. Yield: 28%. Anal. calcd for C37H53BN8O3W 0.5 CH2Cl2 (895.4): C, 50.32; H, 6.08; N, 12.52. Found: C, 50.69; H, 6.04; N 12.44. 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 7.13 (m, 3H, Xy-H), 6.71 (s, 1H, NH), 4.86 (s, 1H, CCH), 3.68 (br, 2H, CH(CH3)2), 2.46 (s, 6H, Xy-CH3), 2.37 (s, 3H, CCH3), 2.34 (s, 6H, CCH3), 2.33 (s, 3H, CCH3), 1.93 (s, 6H, CCH3), 1.86 (s, 3H, CCH3), 1.82 (s, 6H, CCH3), 0.94 (s, 12H, CH(CH3)2). 13C{H} NMR (CDCl3, 125.7 MHz, 298.1 K): δ 173.6 (CHCN(i-Pr)2), 149.9, 149.8, 142.4 (CCH3), 141.0 (Xy-Cipso),139.9 (CCH3), 137.0 (Xy-CCH3), 128.4, 126.2 (Xy-CH), 112.8, 111.8 (CCH3), 86.6 (CCHC), 45.5 (CH(CH3)2), 21.3 (CH(CH3)2), 18.9 (Xy-CCH3), 14.6, 12.6, 11.5, 11.0, 8.7, 8.2 (CCH3). IR (THF, cm−1): 3416 (w, NH), 2540 (w, BH), 1922 (s, CO), 1824 (s, CO).
10: eluent: THF:PET (1:1). Yield: 19%. Anal. calcd for C31H40BN7O4W (769.3): C, 48.40; H, 5.24; N, 12.74. Found: C, 48.87; H, 5.26; N, 11.93. 1H NMR (CDCl3, 300 MHz, 298.2 K): δ 7.91 (s, 1H, NH), 7.19 (m, 3H, Xy-H), 3.98 (d, 2H, CCH2), 2.39 (s, 6H, Xy-CH3), 2.38 (s, 3H, CCH3), 2.36 (s, 3H, CCH3), 2.27 (s, 6H, CCH3), 2.20 (s, 6H, CCH3), 1.90 (s, 3H, CCH3), 1.84 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 75.5 MHz, 298.2 K): δ 245.2 (WC), 240.2 (WCO), 180.3 (CH2CO2), 150.5, 149.0, 143.7, 141.2 (CCH3), 139.4 (Xy-Cipso),136.0 (Xy-CCH3), 129.5, 129.0 (Xy-CH), 113.8, 113.0 (CCH3), 49.3 (CCH2C), 18.8 (Xy-CCH3), 15.1, 13.2, 11.7, 10.9, 8.7, 8.2 (CCH3). IR (THF, cm−1): 3356 (w, NH), 2557 (w, BH), 1956 (s, CO), 1858 (s, CO), 1659 (m, CO).
11: eluent: CH2Cl2. Yield: 21%. Anal. calcd for C62H78B2N14O7W2 1.5 CH2Cl2 (1648.0): C, 46.28; H, 4.95; N, 11.90. Found: C, 46.44; H, 5.03; N, 11.78. 1H NMR (CDCl3, 300 MHz, 300 K): δ 7.15 (m, 3H, Xy-H), 6.99 (s, 1H, NH), 5.13 (s, 1H, CCH), 2.35 (s, 3H, CCH3), 2.32 (s, 3H, CCH3), 2.30 (s, 6H, CCH3), 2.25 (s, 6H, Xy-CH3), 1.87 (s, 3H, CCH3), 1.77 (s, 6H, CCH3), 1.62 (s, 6H, CCH3). 13C{H} NMR (CDCl3, 62.9 MHz, 298.0 K): δ 246.0 (WCO), 219.8 (WC), 170.6 (CHCO1.5), 150.3, 150.3, 142.9, 141.0 (CCH3), 140.0 (Xy-Cipso), 137.1 (Xy-CCH3), 128.7, 127.4 (Xy-CH), 113.3, 112.6 (CCH3), 87.5 (CCHC), 18.7 (Xy-CCH3), 14.8, 12.5, 11.6, 10.9, 8.7, 8.1 (CCH3). IR (THF, cm−1): 3408 (w, NH), 2542 (w, BH), 1936 (s, CO), 1841 (s, CO).
Data availability
Crystallographic data for 3, 4b, 5-BF4, 7, 8, 9, 10 and 11 has been deposited at the CCDC under accession numbers 2061110–2061117 respectively, and can be obtained from http://www.ccdc.cam.ac.uk. Cyclic voltammetry and spectroelectrochemistry data, NMR spectra, DFT calculation details, molecular structure representations of 4a and 6 and Cartesian coordinates of calculated complexes and intermediates supporting this article have been uploaded as part of the ESI.†Author contributions
The manuscript was written through contributions of C. T. and W. W. S. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (SE 890/7-1) is gratefully acknowledged.Notes and references
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed.
- C. S. Yeung and V. M. Dong, Angew. Chem., Int. Ed., 2011, 50, 809–812 CrossRef CAS.
- I. Ugi, B. Werner and A. Dömling, Molecules, 2003, 8, 53–66 CrossRef.
- M. Bach, T. Beweries, V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2007, 26, 4592–4597 CrossRef CAS.
- G. Poszmik, P. J. Carroll and B. B. Wayland, Organometallics, 1993, 12, 3410–3417 CrossRef CAS.
- T. Vlaar, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 CrossRef CAS PubMed.
- Q. Yang, C. Li, M.-X. Cheng and S.-D. Yang, ACS Catal., 2016, 6, 4715–4719 CrossRef CAS.
- A. H. Vahabi, A. Alizadeh, H. R. Khavasi and A. Bazgir, Eur. J. Org. Chem., 2017, 2017, 5347–5356 CrossRef CAS.
- A. L. Johnson, A. M. Willcocks, P. R. Raithby, M. R. Warren, A. J. Kingsley and R. Odedra, Dalton Trans., 2009, 922–924 RSC.
- J. B. Sheridan, S.-H. Han and G. L. Geoffroy, J. Am. Chem. Soc., 1987, 109, 8097–8098 CrossRef CAS.
- J. B. Sheridan and G. L. Geoffroy, J. Am. Chem. Soc., 1987, 109, 1584–1586 CrossRef CAS.
- J. A. Gladysz, Adv. Organomet. Chem., 1982, 20, 1–38 CrossRef CAS.
- W. W. Ellis, A. Miedaner, C. J. Curtis, D. H. Gibson and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1926–1932 CrossRef CAS PubMed.
- G. H. Imler, M. J. Zdilla and B. B. Wayland, J. Am. Chem. Soc., 2014, 136, 5856–5859 CrossRef CAS.
- J. L. Smeltz, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2011, 133, 13288–13291 CrossRef CAS PubMed.
- F. R. Kreißl, A. Frank, U. Schubert, T. L. Lindner and G. Huttner, Angew. Chem., Int. Ed. Engl., 1976, 15, 632–633 CrossRef.
- K. C. Stone, G. M. Jamison, P. S. White and J. L. Templeton, Inorg. Chim. Acta, 2002, 330, 161–172 CrossRef CAS.
- F. R. Kreißl, W. J. Sieber and H. G. Alt, Chem. Ber., 1984, 117, 2527–2530 CrossRef.
- A. F. Hill, J. M. Malget, A. J. P. White and D. J. Williams, Chem. Commun., 1996, 721–722 RSC.
- A. F. Hill, J. M. Malget, A. J. P. White and D. J. Williams, Eur. J. Inorg. Chem., 2004, 818–828 CrossRef CAS.
- F. R. Kreißl, P. Friedrich and G. Huttner, Angew. Chem., Int. Ed. Engl., 1977, 16, 102–103 CrossRef.
- H. Wadepohl, U. Arnold, H. Pritzkow, M. J. Calhorda and L. F. Veiros, J. Organomet. Chem., 1999, 587, 233–243 CrossRef CAS.
- W. Uedelhoven, K. Eberl and F. R. Kreißl, Chem. Ber., 1979, 112, 3376–3389 CrossRef CAS.
- J. Martin-Gil, J. A. K. Howard, R. Navarro and F. G. A. Stone, J. Chem. Soc., Chem. Commun., 1979, 1168–1169 RSC.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2006, 25, 1317–1320 CrossRef CAS.
- M. Akita, M.-C. Chung, M. Terada, M. Miyauti, M. Tanaka and Y. Moro-oka, J. Organomet. Chem., 1998, 565, 49–62 CrossRef CAS.
- O. Orama, U. Schubert, F. R. Kreißl, E. O. Fischer and Z. Naturforsch, Z. Naturforsch., B: J. Chem. Sci., 1980, 35, 82–85 CrossRef.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed.
- G. L. Hillhouse, J. Am. Chem. Soc., 1985, 107, 7772–7773 CrossRef CAS.
- C. E. Sumner Jr, J. A. Collier and R. Pettit, Organometallics, 1982, 1, 1350–1360 CrossRef.
- E. T. Blues, D. Bryce-Smith, I. W. Lawston and G. D. Wall, J. Chem. Soc., Chem. Commun., 1974, 513–514 RSC.
- K. D. Wells, R. McDonald, M. J. Ferguson and M. Cowie, Inorg. Chem., 2011, 50, 3523–3538 CrossRef CAS.
- G. L. Geoffroy and S. L. Bassner, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Elsevier, 1988, pp. 1–83 Search PubMed.
- C. P. Casey and P. J. Fagan, J. Am. Chem. Soc., 1982, 104, 7360–7361 CrossRef CAS.
- M. A. Alvarez, M. E. Garcia, D. Garcia-Vivo, M. E. Martinez and M. A. Ruiz, Organometallics, 2011, 30, 2189–2199 CrossRef CAS.
- T. Kurogi, B. Pinter and D. J. Mindiola, Organometallics, 2018, 37, 3385–3388 CrossRef CAS.
- F. R. Kreißl, W. J. Sieber, M. Wolfgruber and Z. Naturforsch, Z. Naturforsch., B: J. Chem. Sci., 1983, 38, 1419–1423 CrossRef.
- M. A. Alvarez, M. E. Garcia, M. E. Martinez, S. Menendez and M. A. Ruiz, Organometallics, 2010, 29, 710–713 CrossRef CAS.
- M. E. Garcia, D. Garcia-Vivo, S. Menendez and M. A. Ruiz, Organometallics, 2016, 35, 3498–3506 CrossRef CAS.
- V. Jakhar, D. Pal, I. Ghiviriga, K. A. Abboud, D. W. Lester, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2021, 143, 1235–1246 CrossRef CAS PubMed.
- K. Weiss, U. Schubert and R. R. Schrock, Organometallics, 1986, 5, 397–398 CrossRef CAS.
- W. W. Seidel, M. Schaffrath and T. Pape, Angew. Chem., Int. Ed., 2005, 44, 7798–7800 CrossRef CAS PubMed.
- W. W. Seidel, M. J. Meel, S. R. Hughes, F. Hupka and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 12617–12620 CrossRef CAS.
- W. W. Seidel, M. Schaffrath and T. Pape, Chem. Commun., 2006, 3999–4000 RSC.
- J. Rüger, C. Timmermann, A. Villinger, A. Hinz, D. Hollmann and W. W. Seidel, Chem.–Eur. J., 2016, 22, 11191–11195 CrossRef.
- J. Rüger, C. Timmermann, A. Villinger and W. W. Seidel, Inorg. Chem., 2019, 58, 9270–9279 CrossRef PubMed.
- K. Helmdach, S. Ludwig, A. Villinger, D. Hollmann, J. Kösters and W. W. Seidel, Chem. Commun., 2017, 53, 5894–5897 RSC.
- K. Helmdach, S. Dörk, A. Villinger and W. W. Seidel, Dalton Trans., 2017, 46, 11140–11144 RSC.
- S. Ludwig, K. Helmdach, M. Hüttenschmidt, E. Oberem, J. Rabeah, A. Villinger, R. Ludwig and W. W. Seidel, Chem.–Eur. J., 2020, 26, 16811–16817 CrossRef CAS PubMed.
- W. W. Seidel, M. Schaffrath, M. J. Meel, T. Hamilton, S. C. Ariza, T. Pape and Z. Naturforsch, Z. Naturforsch., B: J. Chem. Sci., 2007, 62, 791–798 CrossRef CAS.
- W. W. Seidel, M. D. Ibarra Arias, M. Schaffrath, M. C. Jahnke, A. Hepp and T. Pape, Inorg. Chem., 2006, 45, 4791–4800 CrossRef CAS PubMed.
- S. V. Ponomarev, A. S. Zolotareva, R. N. Ezhov, Y. V. Kuznetsov and V. S. Petrosyan, Russ. Chem. Bull., 2001, 50, 1093–1096 CrossRef CAS.
- S. A. Gonsales, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2016, 45, 15783–15785 RSC.
- K. R. Birdwhistell, T. L. Tonker, J. L. Templeton and W. R. Kenan, J. Am. Chem. Soc., 1985, 107, 4474–4483 CrossRef CAS.
- G. M. Jamison, A. E. Bruce, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1991, 113, 5057–5059 CrossRef CAS.
- H. Staudinger, Justus Liebigs Ann. Chem., 1907, 356, 51–123 CrossRef CAS.
- H. R. Seikaly and T. T. Tidwell, Tetrahedron, 1986, 42, 2587–2613 CrossRef CAS.
- A. C. Filippou, C. Völkl and P. Kiprof, J. Organomet. Chem., 1991, 415, 375–394 CrossRef CAS.
- R. Stumpf, N. Burzlaff, B. Weibert and H. Fischer, J. Organomet. Chem., 2002, 651, 66–71 CrossRef CAS.
- S. G. Feng, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1992, 114, 2951–2960 CrossRef CAS.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef.
- C. G. Atwood and W. E. Geiger, J. Am. Chem. Soc., 2000, 122, 5477–5485 CrossRef CAS.
- M. Agundez, J. Cernicharo and M. Guelin, Astron. Astrophys., 2015, 577, L5/1–6 CrossRef PubMed.
- C. H. Hu, H. F. Schaefer III, Z. Hou and K. D. Bayes, J. Am. Chem. Soc., 1993, 115, 6904–6907 CrossRef CAS.
- K. H. Dötz, Angew. Chem., Int. Ed. Engl., 1975, 14, 644–645 CrossRef.
- W. D. Wulff, P. C. Tang and J. S. McCallum, J. Am. Chem. Soc., 1981, 103, 7677–7678 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray diffraction data, molecular structures of 3, 4a, 5-BF4, 6, 7 and 9, cyclic voltammetry and spectroelectrochemistry data, NMR spectra, DFT calculation details, molecular structure representations and Cartesian coordinates of calculated complexes and intermediates. CCDC 2061110–2061117. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06149f |
| This journal is © The Royal Society of Chemistry 2022 |