
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II)-catalyzed regioselective 1,6-conjugate addition of umpolung aldehydes as carbanion equivalents†
Hyotaik
Kang
 and
Chao-Jun
Li
*
and
Chao-Jun
Li
*
Department of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montréal, Québec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 29th November 2021
Abstract
One of the most efficient and reliable approaches to construct C–C bonds involves the conjugate addition of carbon nucleophiles to electron-deficient ketones. Yet, 1,6-conjugate additions of extended conjugated systems largely remain underexplored due to difficulties in controlling the regioselectivity. Herein, we report umpolung aldehydes as carbanion equivalents for highly regioselective 1,6-conjugate addition reactions to unsaturated ketones, with preliminary studies of the enantioselective variant. The synergy of ruthenium(II) catalyst and electron-rich, bidentate phosphine ligand is essential for the reactivity and selectivity under mild reaction conditions.
Introduction
Building molecular complexity via C–C bond formations is an invaluable tool in synthetic chemistry and often plays a pivotal role in reaction designs.1 In particular, the 1,4-addition of carbon nucleophiles to unsaturated carbonyl compounds is a common C–C bond formation strategy in the synthesis of fine chemicals and pharmaceuticals.2–5 However, controlling the regioselectivity of such nucleophiles can be challenging and has been a constant research interest.6 Additionally, this challenge is further elevated in the less common 1,6-conjugate addition, with comparably stagnant developments than the closely related 1,4-addition reactions. The complication is attributed to the presence of multiple electrophilic sites, resulting in different regioselectivities.7 Thus, strategies to adopt 1,6-addition includes: (1) modification of the electrophiles; and (2) adaptation of the carbon nucleophiles (Fig. 1a). The (1) modification of electrophiles involves using conjugated enynones8–11 and resorting to substitution at the β-position carbon to sterically suppress the 1,4-addition.12–19 Additionally, organocatalytic20,21 and Lewis-acid catalytic22,23 1,6-additions allow for different activation pathways of the electrophiles. The (2) adaptation of carbon nucleophiles commonly utilizes soft organometallic reagents via transition-metal catalysis.24–27 Thus, several copper-catalyzed works are exemplified by Feringa,28,29 Hoveyda,7,30 and others.31,32 Alternative transition-metal catalyzed 1,6-additions have been developed by Hayashi with rhodium,11 cobalt,33 and iridium catalysts.34,35 Expansion of metal-catalyzed 1,6-additions to boryl and silyl groups are represented by the works of Lam,36,37 Liao38 and Newhouse.39 While these advancements are very significant, most involve stoichiometric use of organometallic nucleophiles, which inevitably leads to sizable metallic waste and poor atom economy. With our group's continuous pursuit in the umpolung of hydrazones as “soft” alkyl carbanions (Fig. 1b), herein we report the first example of ruthenium-catalyzed 1,6-addition of hydrazones as a simple and effective nucleophile with excellent regioselectivity (Fig. 1c). The application of hydrazones as nucleophiles is beneficial as their precursors, aldehyde moieties, are ubiquitous, commercially available, and can be renewably sourced.40 Furthermore, the formation of carbanion species by carbonyl umpolung generates a “soft” nucleophile through polarity inversion of the carbonyl carbon.41 To capitalize on the soft property of the nucleophile, we speculated a favourable, soft–soft interaction following the HSAB theory.42 For the nucleophile, such an interaction can be realized with a late transition-metal, ruthenium(II)-catalyst. Likewise, the much softer δ-electrophilic position compared to the β-position favours the 1,6-addition over 1,4-addition.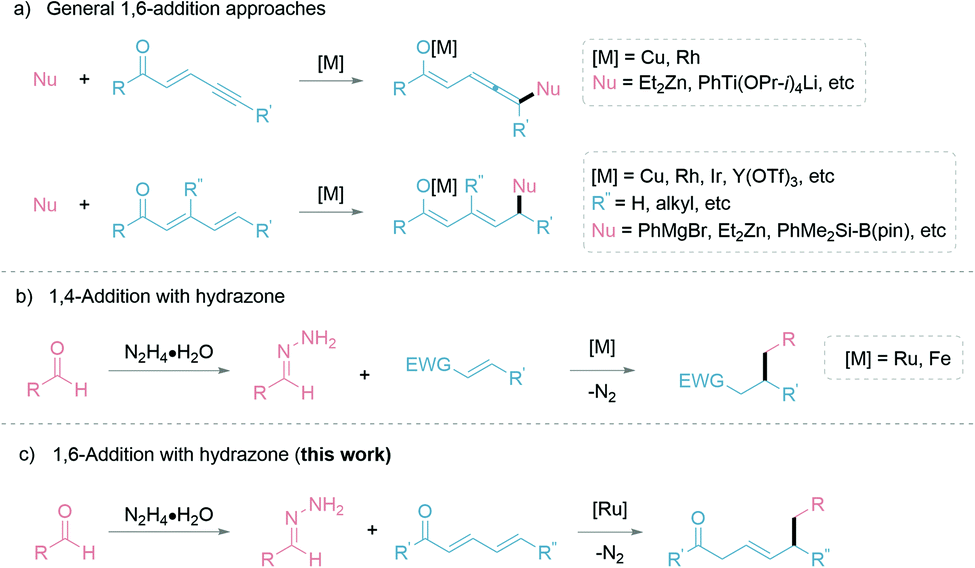 | ||
| Fig. 1 Strategies for various conjugate nucleophilic addition reactions. | ||
Our group previously developed ruthenium-catalyzed 1,2-43,44 and 1,4-addition45 of hydrazones and postulated a possible six-membered ring transition-state from the in situ generated ruthenium-coordinated hydrazone intermediate (Fig. 2, A and B).46 We speculated that the polarized double bonds in the extended conjugated system could adopt a bicyclic transition state (Fig. 2, C), delivering the nucleophile at the 1,6-position selectively.
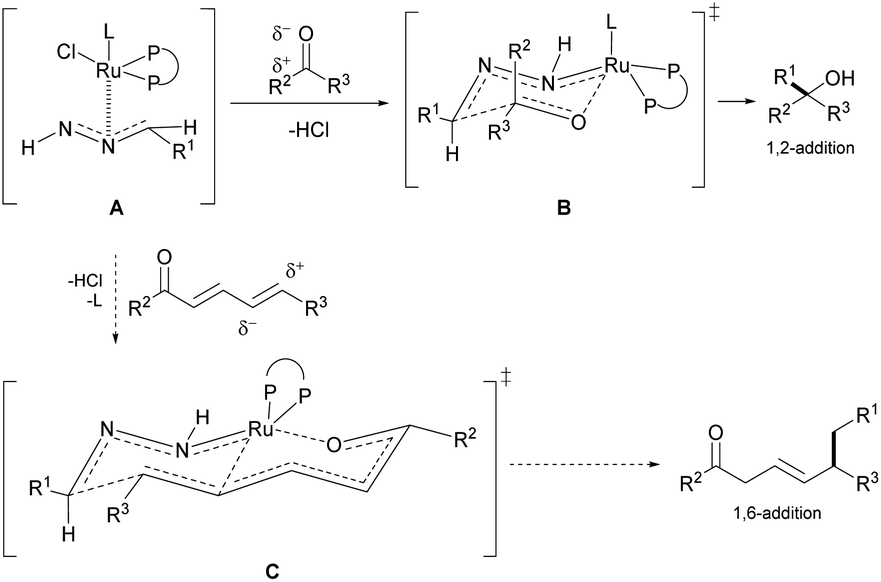 | ||
| Fig. 2 Proposed bicyclic TS (C) for the 1,6-conjugate addition and 6-membered ring chair-like TS (B) for 1,2-addition from the Ru-coordinated hydrazone intermediate (A). | ||
Results and discussion
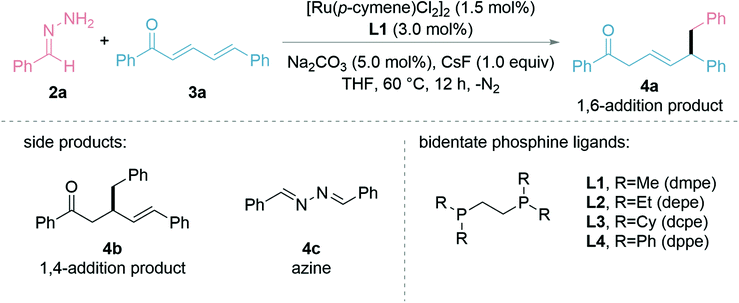
We started testing our hypothesis with the preformed hydrazone 2a from benzaldehyde and (E,E)-cinnamylideneacetophenone (3a) as the model substrates. The initial studies showed that the combination of bidentate phosphine ligand L1 (1.5 mol%) with [Ru(p-cymene)Cl2]2 (0.75 mol%), sodium carbonate (Na2CO3, 5.0 mol%), and caesium fluoride (CsF, 1.0 equiv.) in THF at 60 °C for 12 h gave a moderate yield of the desired 1,6-adduct 4a (Table 1, entry 1). Other inorganic bases such as KOtBu, K2CO3, and Cs2CO3 were not as efficient (entry 2) as Na2CO3. Ir- and Rh-catalysts proved less effective (entries 3 and 4) and (PPh3)4RuCl2 performed better (entry 5). Evaluation of different ligands (entries 6–8) showed that the more π-acidic, phenyl substituted ligand, 1,2-bis(diphenylphosphino)ethane (L4, dppe), which could lead to stronger coordination and more polarization at the 6-position, gave both greater 1,6-regioselectivity and product yield compared to the alkyl and cyclohexyl counterparts, which were known previously beneficial for 1,2- and 1,4-additions. The additive CsF at 1.0 equiv. was critical for the reaction to proceed (entries 9 and 10), which was consistent with our previous reports.43–45 Conducting the reaction at 40 °C, 80 °C or using 2-Me-THF as solvent diminished the product yield (entries 11–13). The yield of 4a could be increased to 96% by prolonging the reaction time to 16 h (entries 14). It is noteworthy that the reaction under the optimized conditions gave 4a exclusively, with no 1,2- or 1,4-addition (4b) being observed. The entries with a reduced formation of 4a were mainly attributed to the recovered starting material 3a and the formation of azine 4c.|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield% |
| a Conditions: 0.2 mmol scale. 2a (1.25 M THF, 1.5 equiv.), 3a (0.20 mmol), catalyst (1.5 mol%), ligand (3.0 mol%), base (5.0 mol%), CsF (1.0 equiv.), THF, 60 °C, 12 h under N2 atmosphere. Yields by 1H NMR with dibromomethane as the internal standard. b Isolated yield. | ||
| 1 | None | 62 |
| 2 | Other inorganic bases than Na2CO3 (KOtBu, K2CO3, and Cs2CO3) | 48–57 |
| 3 | [Cp*IrCl2]2 | 15 |
| 4 | [Cp*RhCl2]2 | 20 |
| 5 | (PPh3)4RuCl2 | 66 |
| 6 | (PPh3)4RuCl2, L2 | 51 |
| 7 | (PPh3)4RuCl2, L3 | 53 |
| 8 | (PPh3)4RuCl2, L4 | 86 |
| 9 | (PPh3)4RuCl2, L4, no CsF | — |
| 10 | (PPh3)4RuCl2, L4, 1.5 equiv. CsF | 78 |
| 11 | (PPh3)4RuCl2, L4, 40 °C | 60 |
| 12 | (PPh3)4RuCl2, L4, 80 °C | 72 |
| 13 | (PPh3)4RuCl2, L4, 2-Me-THF | 70 |
| 14 | (PPh3)4RuCl2, L4, 16 h | 96b |
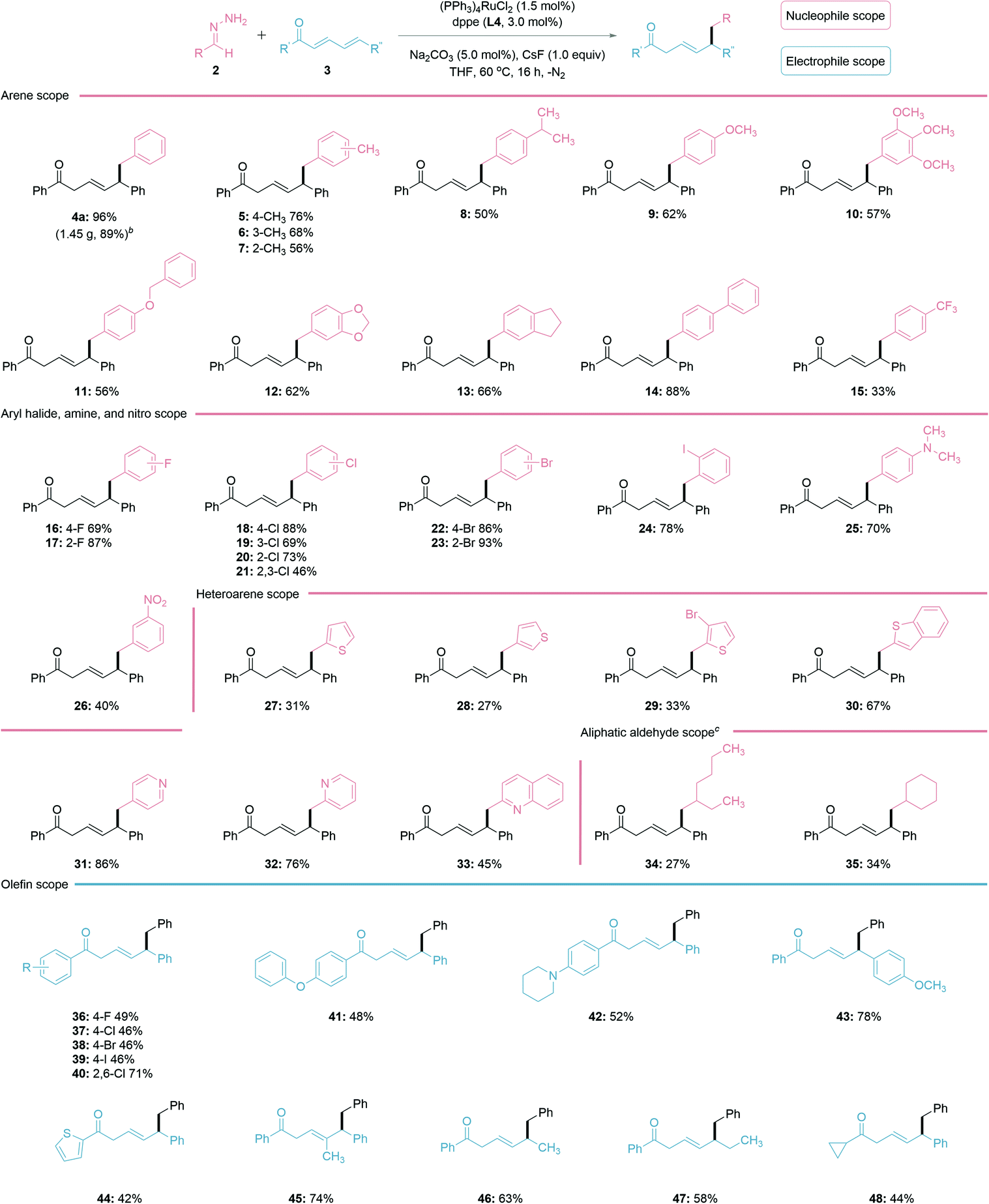
With the optimized reaction conditions in hand, the scope of the regioselective 1,6-conjugate addition was investigated (Table 2). In general, both electron-donating and withdrawing groups fared well under the reaction conditions. Alkyl, such as methyl and isopropyl substituted benzaldehyde hydrazones showed similar reactivities to the ether-substituted ones 5–12. Hydrazones generated from biphenyl-4-carboxaldehyde 14 and halobenzaldehydes 16–24 provided increased yields, possibly due to the stabilization of the in situ generated carbanion species by these substituents. The hydrazone bearing an aryl-amine functional group afforded a good yield of product 25, while the nitro-substituted one was less effective for generating 26. Delightfully, various heteroarene aldehydes, such as thiophene and pyridine-derived aldehydes (27–33) were all compatible with this reaction, albeit thiophene aldehydes and their derivatives (27–30) performed less efficiently. Surprisingly, the pyridine aldehyde and derivatives (31–33) gave good yields, despite being a possible chelating ligand in transition-metal catalysis.47 The utility of the reaction was further examined on linear and cyclic aliphatic aldehydes with increased base loading, generating the desired products 34–35 in lower yields. For the conjugated electrophile, both electron-withdrawing and donating groups on the aryl ketone provided the desired 1,6-addition products in moderate to good yields (36–43). The thiophene-derived heteroarene ketone also led to a moderate yield (44). It is noteworthy that ketones substituted at the γ-position, monoaryl substituted ketone, and a cyclopropyl ketone all generated the desired products in good yields (45–48). Importantly, a gram-scale synthesis of 4a (1.45 g, 89%) was performed to demonstrate the practicability of the reporting method.
| a General reaction conditions: 2 (1.25 M THF, 1.5 equiv.), 3 (0.20 mmol), (PPh3)4RuCl2 (1.5 mol%), dppe (L3, 3.0 mol%), Na2CO3 (5.0 mol%), CsF (1.0 equiv.), THF (100 μL) under N2 atmosphere at 60 °C for 16 h. The isolated yields are reported. b Gram-scale reaction for 4a: 3a (5 mmol), (PPh3)4RuCl2 (0.75 mol%) and dppe (1.5 mol%) in THF (100 μL); isolated yield (1.45 g, 89%). c Increased base loading to 1.2 equiv. for aliphatic aldehyde derivatives. |
|---|
|
To explore the possible enantioselectivity of this transformation, we examined various chiral ligands (ESI† for details). To our satisfaction, 98% ee was obtained with ligand (S,S)-Ph-BPE under the modified reaction conditions (0 °C, 48 h), albeit with a lower yield of the desired 1,6-product and the recovery of 3a (Fig. 3a). We then turned our attention to chemoselectivity, being an important challenge faced in modern synthetic chemistry.48 To study the chemoselectivity, we designed a competition experiment with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of (E,E)-cinnamylideneacetophenone (3a), (E)-chalcone (3ab), and benzophenone (3ac) (Fig. 3b). The nucleophilic 1,6-addition product 4a was much more favoured over the 1,4-addition product 4ab under the standard conditions, whereas the 1,2-addition product 4ac was not detected. A deuterium-labelling experiment using deuterated hydrazone (2a–d, 90% D) was conducted with 3a under the standard reaction conditions (Fig. 3c). The observation of H/D scrambling exclusively at the benzylic and α-positions suggested that the hydrazone acts as both the alkyl nucleophile and hydrogen donor. Deuterium-labelled aryl-D5 hydrazone retained all the deuterium on the aryl ring during the reaction (Fig. 3d). Finally, a 13C-labelled hydrazone led to the synthesis of a 13C-labelled 1,6-conjugate addition product by this method (Fig. 3e).
:1:1 mixture of (E,E)-cinnamylideneacetophenone (3a), (E)-chalcone (3ab), and benzophenone (3ac) (Fig. 3b). The nucleophilic 1,6-addition product 4a was much more favoured over the 1,4-addition product 4ab under the standard conditions, whereas the 1,2-addition product 4ac was not detected. A deuterium-labelling experiment using deuterated hydrazone (2a–d, 90% D) was conducted with 3a under the standard reaction conditions (Fig. 3c). The observation of H/D scrambling exclusively at the benzylic and α-positions suggested that the hydrazone acts as both the alkyl nucleophile and hydrogen donor. Deuterium-labelled aryl-D5 hydrazone retained all the deuterium on the aryl ring during the reaction (Fig. 3d). Finally, a 13C-labelled hydrazone led to the synthesis of a 13C-labelled 1,6-conjugate addition product by this method (Fig. 3e).
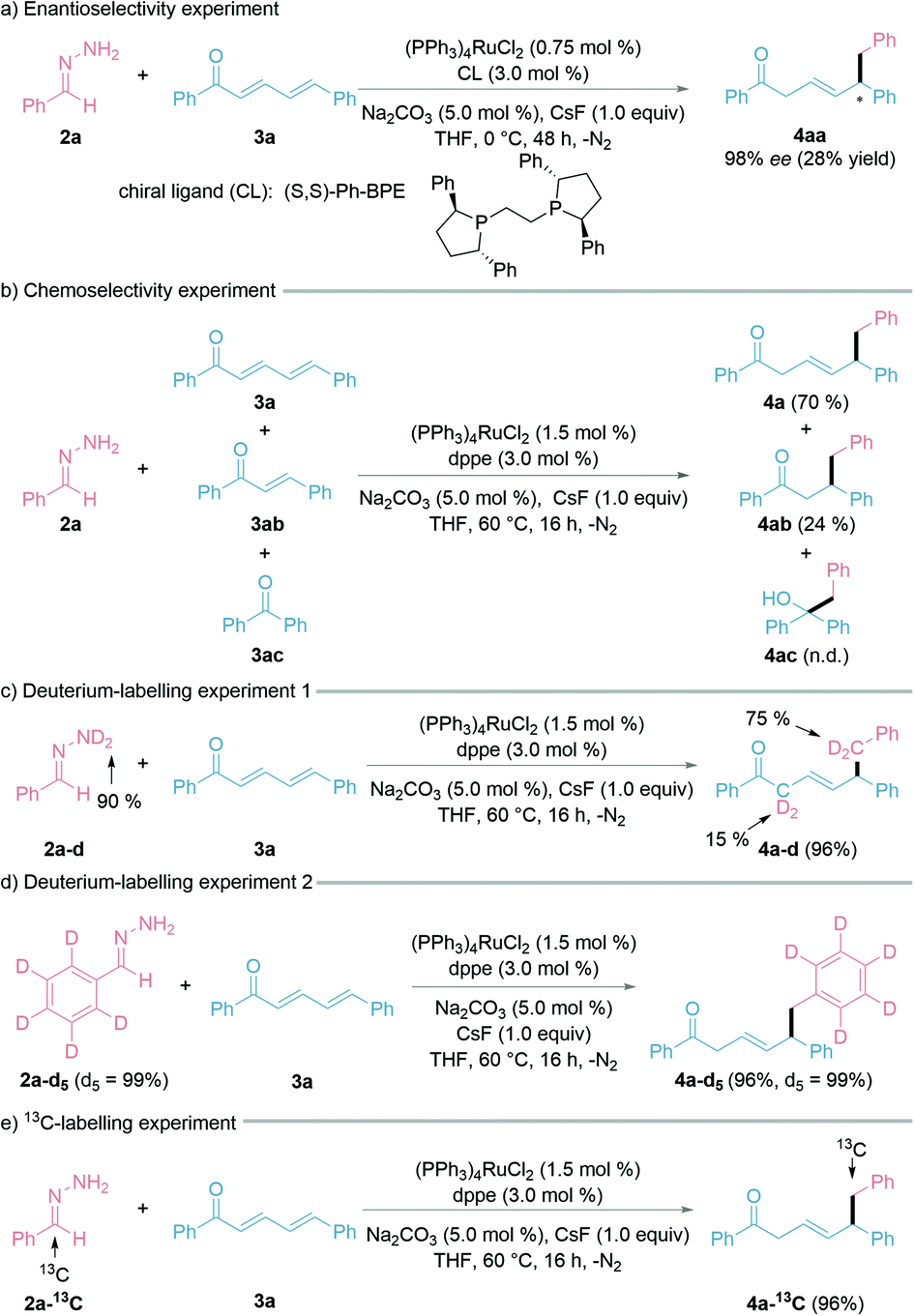 | ||
| Fig. 3 Enantioselectivity, chemoselectivity, and labelling experiments. See ESI† for more details. | ||
Conclusions
In conclusion, we have developed a highly regioselective 1,6-conjugate addition to extended conjugated ketones. The combination of umpolung aldehydes as carbanions and ruthenium-catalyst with bidentate phosphine ligand (dppe) is important to the regioselectivity as both exhibit the key “soft” property. The reaction proceeds under mild reaction conditions with various functional group tolerance. Our efforts in the expansion of enantioselectivity, coupling partners, and synthetic applications are currently ongoing.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes general information, general procedures, experimental details, characterizations, and copies of 1H and 13C NMR spectra.Author contributions
CJL was involved in the conceptualization and supervision of the project, with funding acquisition and writing – reviewing and editing. HK performed the experimental investigations, formal analysis of data, and writing original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Canada Research Chair Foundation (to C.-J. Li), the Canada Foundation for Innovation, the Fonds de Recherche du Québec – Nature et Technologies Centre in Green Chemistry and Catalysis, the Natural Sciences and Engineering Research Council of Canada, Killam Fellowship of the Canadian Council for the Arts, and McGill University for supporting our research. We would like to acknowledge the McGill Chemistry Characterization Facility for their contribution to the compound characterization. H. Kang is grateful for the discussion with group members and colleagues, especially C. Y. Huang (McGill University) for his guidance.Notes and references
- B. H. Lipshutz, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 1, pp. 107–138 Search PubMed.
- A. G. Csákÿ, G. d. l. Herrán and M. C. Murcia, Chem. Soc. Rev., 2010, 39, 4080–4102 RSC.
- E. Vrancken, J.-M. Campagne and P. Mangeney, in Comprehensive Organic Synthesis, ed. P. Knochel and G. Molander, Elsevier, Oxford, 2014, vol. 1, pp. 74–123 Search PubMed.
- H. Nemoto, T. Kawamura and N. Miyoshi, J. Am. Chem. Soc., 2005, 127, 14546–14547 CrossRef CAS.
- Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS.
- B. M. Trost, Science, 1983, 219, 245–250 CrossRef CAS PubMed.
- F. Meng, X. Li, S. Torker, Y. Shi, X. Shen and A. H. Hoveyda, Nature, 2016, 537, 387–393 CrossRef CAS.
- T. Hayashi, N. Tokunaga and K. Inoue, Org. Lett., 2004, 6, 305–307 CrossRef CAS.
- M. Tissot, D. Poggiali, H. Hénon, D. Müller, L. Guénée, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2012, 18, 8731–8747 CrossRef CAS PubMed.
- N. Krause, Chem. Ber., 1990, 123, 2173–2180 CrossRef CAS.
- T. Nishimura, H. Makino, M. Nagaosa and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 12865–12867 CrossRef CAS PubMed.
- T. Hayashi, S. Yamamoto and N. Tokunaga, Angew. Chem., Int. Ed., 2005, 44, 4224–4227 CrossRef CAS.
- E. Fillion, A. Wilsily and E. T. Liao, Tetrahedron: Asymmetry, 2006, 17, 2957–2959 CrossRef CAS.
- H. Hénon, M. Mauduit and A. Alexakis, Angew. Chem., Int. Ed., 2008, 47, 9122–9124 CrossRef.
- E. J. Corey, C. U. Kim, R. H. K. Chen and M. Takeda, J. Am. Chem. Soc., 1972, 94, 4395–4396 CrossRef CAS.
- M. Magrez, J. Wencel-Delord, A. Alexakis, C. Crévisy and M. Mauduit, Org. Lett., 2012, 14, 3576–3579 CrossRef CAS PubMed.
- K.-S. Lee, H. Wu, F. Haeffner and A. H. Hoveyda, Organometallics, 2012, 31, 7823–7826 CrossRef CAS PubMed.
- K. Fukuhara and H. Urabe, Tetrahedron Lett., 2005, 46, 603–606 CrossRef CAS.
- J. Wencel-Delord, A. Alexakis, C. Crévisy and M. Mauduit, Org. Lett., 2010, 12, 4335–4337 CrossRef CAS PubMed.
- L. Bernardi, J. López-Cantarero, B. Niess and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 5772–5778 CrossRef CAS.
- L. Dell'Amico, Ł. Albrecht, T. Naicker, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2013, 135, 8063–8070 CrossRef.
- J. L. Brooks, P. A. Caruana and A. J. Frontier, J. Am. Chem. Soc., 2011, 133, 12454–12457 CrossRef CAS.
- J. L. Brooks and A. J. Frontier, J. Am. Chem. Soc., 2012, 134, 16551–16553 CrossRef CAS PubMed.
- F. Näf, P. Degen and G. Ohloff, Helv. Chim. Acta, 1972, 55, 82–85 CrossRef.
- Y. Yamamoto, S. Yamamoto, H. Yatagai, Y. Ishihara and K. Maruyama, J. Org. Chem., 1982, 47, 119–126 CrossRef CAS.
- F. Barbot, A. Kadib-Elban and P. Miginiac, J. Organomet. Chem., 1983, 255, 1–9 CrossRef CAS.
- M. Hulce, Tetrahedron Lett., 1988, 29, 5851–5854 CrossRef CAS.
- T. den Hartog, S. R. Harutyunyan, D. Font, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2008, 47, 398–401 CrossRef CAS PubMed.
- T. den Hartog, D. Jan van Dijken, A. J. Minnaard and B. L. Feringa, Tetrahedron: Asymmetry, 2010, 21, 1574–1584 CrossRef CAS.
- Y. Huang, S. Torker, X. Li, J. del Pozo and A. H. Hoveyda, Angew. Chem., Int. Ed., 2019, 58, 2685–2691 CrossRef CAS.
- M. Magrez-Chiquet, M. S. T. Morin, J. Wencel-Delord, S. DrissiAmraoui, O. Baslé, A. Alexakis, C. Crévisy and M. Mauduit, Chem.–Eur. J., 2013, 19, 13663–13667 CrossRef CAS PubMed.
- Y. Guo, J. Kootstra and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2018, 57, 13547–13550 CrossRef CAS PubMed.
- T. Sawano, A. Ashouri, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 18936–18939 CrossRef CAS PubMed.
- T. Nishimura, Y. Yasuhara and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 5164–5166 CrossRef CAS.
- T. Nishimura, Y. Yasuhara, T. Sawano and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 7872–7873 CrossRef CAS PubMed.
- Y. Luo, I. D. Roy, A. G. E. Madec and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 4186–4190 CrossRef CAS PubMed.
- Y. Luo, S. M. Wales, S. E. Korkis, I. D. Roy, W. Lewis and H. W. Lam, Chem.–Eur. J., 2018, 24, 8315–8319 CrossRef CAS.
- Y. Lou, P. Cao, T. Jia, Y. Zhang, M. Wang and J. Liao, Angew. Chem., Int. Ed., 2015, 54, 12134–12138 CrossRef CAS.
- C.-Y. Shi, J. Eun, T. R. Newhouse and L. Yin, Angew. Chem., Int. Ed., 2021, 60, 9493–9499 CrossRef CAS.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CAS.
- S. Wang and B. König, Angew. Chem., Int. Ed., 2021, 60, 21624–21634 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581–587 CrossRef CAS.
- N. Chen, X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6260–6263 CrossRef CAS.
- H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2017, 9, 374–378 CrossRef CAS.
- X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6302–6306 CrossRef CAS PubMed.
- X.-J. Dai and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5433–5440 CrossRef CAS.
- E. S. Raper, Coord. Chem. Rev., 1996, 153, 199–255 CrossRef CAS.
- N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262–310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03732c |
| This journal is © The Royal Society of Chemistry 2022 |